

Abstract
The glycoprotein CD44 is barely detected in normal mouse and human glomeruli, but is increased in glomerular parietal epithelial cells following podocyte injury in focal segmental glomerulosclerosis (FSGS). To determine the biological role and regulation of CD44 in these cells, we employed an in vivo and in vitro approach. Experimental FSGS was induced in CD44 knockout and wildtype mice with a cytotoxic podocyte antibody. Albuminuria, focal and global glomerulosclerosis (periodic acid-Schiff stain) and collagen IV staining were lower in CD44 knockout compared with wild type mice with FSGS. Parietal epithelial cells had lower migration from Bowman’s capsule to the glomerular tuft in CD44 knockout mice with disease compared with wild type mice. In cultured murine parietal epithelial cells, overexpressing CD44 with a retroviral vector encoding CD44 was accompanied by significantly increased collagen IV expression and parietal epithelial cells migration. Because our results showed de novo co-staining for activated ERK1/2 (pERK) in parietal epithelial cells in experimental FSGS, and also in biopsies from patients with FSGS, two in vitro strategies were employed to prove that pERK regulated CD44 levels. First, mouse parietal epithelial cells were infected with a retroviral vector for the upstream kinase MEK-DD to increase pERK, which was accompanied by increased CD44 levels. Second, in CD44 overexpressing parietal epithelial cells, decreasing pERK with U0126 was accompanied by reduced CD44. Finally, parietal epithelial cell migration was higher in cells with increased and reduced in cells with decreased pERK. Thus, pERK is a regulator of CD44 expression and increased CD44 expression leads to a pro-sclerotic and migratory parietal epithelial cells phenotype.
Keywords: podocyte, glomerulosclerosis, Mitogen-activated protein kinases, collagen, glomerulus, FSGS
INTRODUCTION
Glomerular parietal epithelial cells (PECs) are increasingly recognized for their pathogenic role in fibrotic glomerular diseases,1–3 and in the aged kidney.4 PECs are pivotal in forming the pathognomonic bridge (synechial attachment) between Bowman’s capsule and the glomerular tuft in FSGS, considered an initial podocyte disorder.5 Studies show that PEC derived extracellular matrix increase in FSGS,1, 6 diabetes7 and the aging kidney,4 contributing to glomerular scarring. Several extracellular matrix proteins also co-localize to PECs in aged kidneys.4
Smeets and Moeller were the first to show de novo expression of CD44 in PECs in glomerular diseases.8 CD44 is a family of trans-membrane glycoproteins consisting of different variants (CD44v) due to alternative splicing.9 CD44 is the main receptor for hyaluronic acid10 but binds other molecules, mostly components of extracellular matrix.11 Different biological functions have been described and include proliferation,12 inflammation,13 tumor progression/ metastasis,14, 15 embryogenesis16 and cell migration.17, 18 CD44 is barely expressed in normal mouse and human glomeruli, being detected in only 0.8% of glomeruli in normal human biopsies.19 In contrast, CD44 is markedly increased in PECs in patients with FSGS, which might distinguish this podocyte disease from minimal change disease.20 CD44 is increased in PECs in mice with FSGS,1, 19–22 advanced age,4 and human IgA nephropathy.23 The de novo expression of CD44 in PECs coincides with an increase in extracellular matrix accumulation.1, 6 and in a subset of PECs that have migrated on to the glomerular tufts in FSGS.21
The mechanism(s) underlying increased CD44 in PECs in diseases where podocytes are injured is unclear, and the precise biological role remains to be proven. The purpose of the current studies was two-fold: first, to determine if the de novo increase in CD44 was responsible in part for increased PEC matrix production and/or migration in FSGS, considered primarily a podocyte disease. Second, because we recently reported that a subset of PECs co-expressed CD44 and active ERK1/2,21 we also determined if de novo CD44 was mediated by activated ERK1/2.
RESULTS
The increase in CD44 in PECs is not detected in CD44−/− mice with experimental FSGS
We began identifying if specific variants of CD44 was increased in experimental FSGS by performing staining with antibodies directed to the following: the standard variant CD44s (lacking exon sequences v1–10), CD44 variants v3, v7 and v10. Staining for CD44s was readily detected in CD44+/+ mice following the onset of FSGS, but was absent in CD44−/− mice, as expected (Supplementary Figure 1). Staining for CD44 variants v3, v7 and v10 was neither detected in the glomerulus of diseased CD44+/+ nor diseased CD44−/− mice (Supplementary Figure 1). This was not a false negative, because staining was detected in CD44+/+ mice outside the glomerulus. These results show the CD44s variant was increased in PECs in this FSGS model.
There were no histological differences on light microscopy in kidneys between CD44+/+ or CD44−/− mice at baseline prior to disease induction (Figure 1A, C). CD44 staining (all variants) was detected in 1–2% of all glomeruli at baseline in CD44+/+ mice (Figure 1A). Following induction of FSGS in CD44+/+ mice, there was de novo staining for CD44 in PECs and on the glomerular tuft at d14 (Figure 1B). These data were similar to our recent report.21 As expected, CD44 staining was absent in CD44−/− mice at baseline, and after experimental FSGS induction (Figure 1C, D).
Figure 1. FSGS in CD44+/+ and CD44−/− mice.

(A–D) CD44 and PAS co-staining. (A) CD44 staining is not detected in parietal epithelial cells (PECs) in CD44+/+ mice at baseline. (B) In CD44+/+ mice at day (d) 14 of FSGS, CD44 staining (brown) is detected in cells lining Bowman’s capsule (arrow heads), and also along the outer aspect of the glomerular tuft (thin arrows). (C) CD44 staining was not detected in glomeruli of CD44−/− mice at baseline, nor at d14 of FSGS (D). These results show that CD44 was de novo expressed in PECs and podocytes only in CD44+/+ mice at d14 of FSGS.
(E–H) β-Galactosidase (βGal) and DAPI staining. βGal staining (red color) was used to detect LacZ that was knocked into exon 1 and intron 1 of CD44, and DAPI stained nuclei (blue color). (E) and (F) shows that βGal staining was not detected in CD44+/+ mice at baseline nor d14 FSGS respectively. (G) βGal staining was not detected in CD44−/− mice at baseline. (H) βGal staining was present in cells along Bowman’s capsule in CD44−/− mice at FSGS d14. These results confirmed that the CD44 promoter was activated in PECs in CD44 null/knockin mice with FSGS.
(I–L) Sheep IgG and DAPI staining. Because a sheep anti-podocyte antibody was used to induce FSGS, sheep IgG staining (green color) was performed to assess antibody deposition at d14 of FSGS. (I) Low power view showing sheep IgG staining in cortical glomeruli in CD44+/+ mice. (J) High power view of typical sheep IgG pattern of staining along the glomerular basement membrane in CD44+/+ mice. (K) and (L) shows that in CD44−/− mice, sheep IgG staining at low and high magnification respectively was indistinguishable from CD44+/+ mice. (M) Urinary albumin to creatinine ratio (ACR). ACR were very low, but similar in CD44+/+ (circles) and CD44−/− (squares) mice at baseline. ACR was statistically higher in CD44+/+ mice with FSGS at d7 and d14 compared to CD44−/− mice.
In CD44−/− mice, a neomycin resistance/LacZ cassette was used to disrupt exon 1 and intron 1, which are shared between all CD44 variants As a result, there is no expression of CD44 mRNA or protein, but rather, the expression of LacZ under the control of endogenous regulatory regions of CD44. βGal staining (red color), used to detect LacZ, was absent in glomeruli of CD44+/+ mice at baseline (Figure 1E), and in FSGS (Figure 1F). βGal staining was not detected in CD44−/− mice at baseline (Figure 1G), but was in glomeruli at d14 of FSGS mimicking the pattern seen for CD44 staining in CD44+/+ mice (Figure 1H). These results show that CD44 increases in cells along Bowman’s capsule, but not in diseased CD44−/− mice. Rather, LacZ is de novo expressed in diseased CD44−/− mice in PECs where CD44 is normally expressed.
No differences in antibody deposition, glomerular infiltrating cells, glomerular hyaluronic acid or endothelial injury between diseased CD44+/+ and CD44−/− mice
Abrupt podocyte depletion underlying this model of FSGS was induced by a cytopathic anti-podocyte antibody, which binds to podocytes, and not to PECs as we have reported.24 Sheep IgG staining used to detect the disease-inducing antibody was indistinguishable between diseased CD44+/+ and CD44−/− mice and limited to podocytes (Figure 1I–L).
Experimental and clinical FSGS is typically considered a non-inflammatory glomerular disease. However, because of the potential effect of CD44 on circulating inflammatory cells, staining was performed with antibodies specific for CD3 (activated T cells), B220 (B cells), F4/80 (macrophages) and LG-6G (neutrophils). Supplementary Figure 2 shows no statistical differences in the small number of infiltrating cells in glomeruli between diseased CD44+/+ and CD44−/− mice.
CD44 is a cell surface receptor for hyaluronic acid (HA), a component of extracellular matrix often found in wound repair.25,10 To make certain differences between CD44+/+and CD44−/− mice were not due to differences in HA, staining with hyaluronic acid binding protein was performed. The percentage of glomeruli with staining for HA increased in FSGS, but there were no differences in glomerular staining between CD44+/+ and CD44−/− mice at baseline or in FSGS (Supplementary Figure 3).
Finally, in order to determine if differences in endothelial/vascular injury may account for differences between CD44+/+ and CD44−/− mice, staining for the glomerular endothelial cell marker CD31 was performed. There were no striking differences in glomerular CD31 staining between CD44+/+ and CD44−/− mice at the time points studies (not shown).
Taken together, any differences in CD44+/+ and CD44−/− mice with FSGS were unlikely to be mediated by differences in cytopathic antibody binding, infiltrating cells , HA or endothelial damage.
Albuminuria was lower in CD44−/− mice with experimental FSGS compared to CD44+/+ mice
Urinary albumin-to-creatinine ratios (ACR) were measured on all mice, and the results are shown in Figure 1M. No differences were detected at baseline between CD44+/+ and CD44−/− mice, as we have previously shown.21 ACR increased in CD44+/+ mice at d7 (23.03 ± 1.7, n=8 vs, 0.074 ± 0.011 n=5 at baseline, p< 0.0001), but was significantly lower in CD44−/− mice at d7 (7.7 ± 1.6 CD44−/− vs. 23.03 ± 1.7, n=8, CD44−/− vs. CD44+/+ p< 0.0001) and d14 (1.4 ± 0.15 CD44−/− vs. 3.5 ± 0.69, n=8, CD44−/− vs. CD44+/+ p=0.013).
Glomerulosclerosis and collagen IV staining are lower in CD44−/− mice with experimental FSGS despite similar reductions in podocyte density
Because of the well-established impact of podocyte depletion on the onset and magnitude of glomerular scarring, podocyte density, identified by p57 staining, was measured in both mouse strains.26 Podocyte density was similar in CD44+/+ and CD44−/− mice at baseline (365±23 CD44+/+ vs. 327±25 CD44−/− pods/x106 um3, CD44+/+ vs. CD44−/− p=0.35) and the extent of depletion was similar at D14 (308±20 CD44+/+ vs. 280±32 CD44−/− pods/x106 um3, CD44+/+ vs. CD44−/− p=0.69) and (451±30 CD44+/+ vs. 341±53 CD44−/− pods/x106 um3, CD44+/+ vs. CD44−/− p=0.15) D28 of FSGS (Supplementary Table 3).
The impact of CD44 on glomerular sclerosis and extracellular matrix accumulation was measured and quantitated by periodic-acid Schiff (PAS) staining (Figure 2A–F) and collagen IV staining (Figure 2 G–M). Figure 2 panels A–F show histological changes by PAS staining and quantification. We evaluated glomerular changes based on three categories: (i) Pseudocrescents (Figure 2A, D): The percentage of glomeruli with pseudocrescents was higher in CD44+/+ mice at D14 FSGS (25±2.1% vs. 14±0.9%, P<0.001 CD44+/+ vs. CD44−/−); (ii) Focal glomerulosclerosis (Figure 2B, E): The percentage of glomeruli with focal glomerulosclerosis was higher in CD44+/+ mice at both D14 (7±1.2% vs. 3±0.9%, P<0.05 CD44+/+ vs. CD44−/−) and D28 (18±3.0% vs. 7±1.3%, P<0.05 CD44+/+ vs. CD44−/−); (iii) Global glomerulosclerosis (Figure 2C, F): The percentage of glomeruli with global glomerulosclerosis was higher in CD44+/+ mice at D28 (5±0.8% vs. 0.3±0.25%, P<0.01 vs. CD44−/−).
Figure 2. Focal and global glomerulosclerosis and Collagen IV staining is lower in CD44−/− mice with FSGS.

(A–F) Quantification and representative images of p57 stained, PAS counterstained glomeruli. Podocytes were detected by p57 staining (brown, nuclear), and PAS staining was used to better identify glomerular structure, cellularity and sclerosis. Quantitation and corresponding representative images are shown for pseudocrescents, focal sclerosis and global sclerosis.
(A, D) Pseudocrescents. The percentage of glomeruli with pseudocrescents was higher in CD44+/+ mice at D14 compared to CD44−/− mice.
(B, E) Focal sclerosis. Focal sclerosis was higher at D14 and D28 of FSGS in CD44+/+ mice compared to CD44−/− mice..
(C, F) Global sclerosis. Global sclerosis was only detected in CD44+/+ mice at D28 of FSGS.
(G–M) Quantification and representative images of collagen IV staining. (G) Collagen IV staining was similar in CD44+/+ (circles) and CD44−/− (squares) mice at baseline. The percentage of glomeruli with increased collagen IV staining was higher in CD44+/+ mice at both d14 and d28 FSGS compared to CD44+/+ mice with FSGS.
(H–M) Representative images of collagen IV staining (red). (H–J) CD44+/+ mice: Compared to baseline CD44+/+ mice (H), collagen IV staining was higher at d14 (I) and d28 (J) CD44+/+ mice with FSGS, in the glomerular tuft, along Bowman’s capsule (arrow heads represent examples) and synechial attachments were present. (K–M) CD44−/− mice: (K) Collagen IV staining in glomeruli of CD44−/− mice was in lower abundance. Mild collagen staining was detected in CD44−/− mice with FSGS at d14 (L) and d28 (M).
We also measured the extracellular matrix protein collagen IV, which is constitutively expressed in Bowman’s capsule, and increases in PECs exposed to high glucose, AGEs (advanced glycation end-products) and TGFβ.7 Figure 2G shows collagen IV quantification, and Figures 2H–M show representative images. Collagen IV staining was similar at baseline in CD44+/+ and CD44−/− mice along Bowman’s capsule, within the mesangium and tubular basement membranes (Figure 2H & K). The percentage of glomeruli with increased Collagen IV staining along Bowman’s capsule and within synechial attachments significantly increased in CD44+/+ mice at d14 (27±1.2%, P<0.0001 vs. baseline), and d28 (20±1.4%, P<0.0001 vs. baseline; P=0.004 vs. d14). Collagen IV staining was significantly lower in CD44−/− mice at d14 (17±0.4% vs. 27±1.2%; P<0.0001 vs. CD44+/+) and d28 (11±1.4% vs. 20±1.4%; P=0.0015 vs. CD44+/+)(Figure 2G).
Taken together, despite similar decreases in podocyte density, pseudocrescents, focal glomerulosclerosis, global glomerulosclerosis and collagen IV staining were lower in CD44−/− compared to CD44+/+ FSGS mice, consistent with a role for CD44 in glomerular scarring.
PEC migration to the glomerular tuft is lower in CD44−/− mice with FSGS
We21, 27 and others3, 5, 28–31 have reported that a subset of CD44 stained PECs, migrate from Bowman’s capsule to the glomerular tuft following podocyte injury. In the current study, PECs were identified by staining for PAX832 and SSeCKS.27 In baseline CD44+/+ and CD44−/− mice, PAX8 staining localized to cells lining Bowman’s capsule, consistent with PEC distribution (Figure 3B &E). In CD44+/+ mice, the percentage of glomeruli with PAX8 stained cells on the glomerular tuft increased significantly at d14 (20±1.3% vs. 7.8±1.3%; P<0.0001 vs. baseline)(Figure 3A), but was similar to baseline at d28 (9.6±0.7% vs. 7.8±1.3%; P=0.25 vs. baseline). By contrast, the percentage was significantly lower in CD44−/− mice at d14 (10±0.75% vs. 20±1.3%; P<0.0001 vs. CD44+/+), but not at d28 (Figure 3A).
Figure 3. Migration of parietal epithelial cells (PECs) from Bowman’s capsule to the glomerular tuft is lower in CD44−/− mice with FSGS.

(A) Quantification of PAX8 staining. PAX8 staining was used to identify PECs that had migrated to the glomerular tuft. PEC migration increased in CD44+/+ mice with FSGS at d14, but not in CD44−/− mice with disease at this time point.
(B–G) Representative images of PAX8 staining in CD44+/+and CD44−/− mice. (B) PAX8 staining (brown, nuclear) is restricted to Bowman’s capsule at baseline, but can be detected in the glomerular tuft at FSGS d14 (C) and d28 (D) in CD44+/+ mice (arrow heads indicate examples). (E) PAX8 is detected along Bowman’s capsule in baseline in CD44−/− mice. An occasional PAX8 stained cell is detected in the glomerular tuft in CD44−/− mice with FSGS at d14 (F) and d28 (G).
(H) Quantification of SSeCKS staining. SSeCKS staining, a marker of PECs, was increased on the glomerular tuft in CD44+/+ at d14 of FSGS compared to CD44−/− mice at the same time point.
(I–N) Representative images of SSeCKS staining in CD44+/+ mice and CD44−/− mice. (I) SSeCKS staining (green) in CD44+/+ mice at baseline was limited to cells lining Bowman’s capsule, consistent with PECs. (J) In CD44+/+ mice at d14 FSGS, PECs had migrated to the glomerular tuft (arrow heads represent examples). (K) SSeCKS staining at d28 in CD44+/+ mice showing less PEC migration. (L) SSeCKS staining was typically limited to cells lining Bowman’s capsule in CD44−/− mice at baseline (I), d14 (M) and d28 (N).
SSeCKS staining was typically restricted to PECs along Bowman’s capsule in baseline CD44+/+ (Figure 3I) and CD44−/− (Figure 3L) mice. The percentage of glomeruli with cells staining for SSeCKS on the glomerular tuft was higher in CD44+/+ mice at d14 (19±1.1% vs. 5.4±0.6%; P<0.0001 vs. baseline) and d28 (9.2±0.9% vs. 5.4±0.6%; P=0.007 vs. baseline). By contrast, the percentage of glomeruli with SSeCKS stained cells on the glomerular tuft was lower in CD44−/− mice at d14 (Figure3A, 9.6±0.6% vs. 19±1.1%; P<0.0001 vs. CD44+/+), but not at d28 (Figure 3A).
Results in CD44+/+ mice are consistent with our previous reports in this model.21, 27 However, in the absence of CD44, PEC migration from Bowman’s capsule to the glomerular tuft was reduced, suggesting a role for CD44 in migration.
In order to determine if CD44 positive cells located on the glomerular tuft expressed PEC or a podocyte markers, double staining was performed for CD44 with either PAX8 (PEC marker) or synaptopodin (podocyte marker). In CD44+/+ mice with FSGS, the majority of CD44 staining cells on the tuft co-stained for PAX8 (Figure 4A). Likewise, in human FSGS kidney, CD44 staining cells also co-stained for PAX8 (Figure 4B). In contrast, the majority of CD44 positive cells on the tuft were negative for synaptopodin, and in most cases CD44 and synaptopodin staining was mutually exclusive (Figure 4 C–H).
Figure 4. CD44 positive cells on the glomerular tuft express PAX8 but not synaptopodin in mice and humans with FSGS.

(A & B) Representative images of CD44-PAX8 co-staining in CD44+/+mice and Humans. (A) CD44 (all variants) staining (red) and PAX8 staining (green, nuclear) co-localized in most of cells on the glomerular tuft in CD44+/+ mice with FSGS (white arrow heads indicate examples). (B) CD44 staining (red) and PAX8 staining (brown, nuclear) also co-localized in cells on the glomerular tuft in humans with FSGS (black arrow heads indicate examples).
(C–H) Representative images of CD44-Synaptopodin co-staining in CD44+/+mice. (C) CD44 (all variants) staining (red), (D) synaptopodin staining (green) and (E) merged images show that CD44 and synaptopodin did not co-localize in most of cells on the glomerular tuft in CD44+/+ mice with FSGS. (F–H) higher magnification images of the insets indicated by the white dashed squares in images C–E. CD44 and synaptopodin did not co-localize (white arrow heads indicate examples).
Overexpressing CD44 in cultured mouse PECs increases collagen IV levels
To validate these findings in vitro, we utilized immortalized mouse PECs previously characterized.33 Protein levels for CD44 were barely detected by western blot analysis (Figure 5A). To mimic the in vivo increase in CD44, PECs were infected with a retroviral pBABE-puro vector encoding for the standard isoform of CD44 (the isoform that increases in vivo), or with an empty vector as control. Densitometry on western blot analysis (Fig 5A’) using β-actin as a loading control, shows successful overexpression of CD44 (P=0.013 vs. empty vector infected), which was not detected in control-infected PECs (P=0.810 vs. non infected)(Figure 5A). The lower levels of collagen IV detected by western blot analysis in non-infected and empty vector infected PECs was increased in CD44 infected cells (Figure 5B). Using β-actin as a loading control, densitometry on western blot analysis showed higher levels of Collagen IV in PECs infected with CD44 compared with control infected PECs (P=0.044 vs. empty vector infected)(Figure 5B’). These results show expressing CD44 in cultured PECs increases collagen IV levels, consistent with the increase in collagen IV in FSGS.
Figure 5. CD44 Infected PECs in culture have higher Collagen IV expression and increased migration.

(A) Western blot analysis for CD44. CD44 protein is not detected in non-infected immortalized mouse PECs (western blot analysis lane 1), nor in empty vector infected cells (lane 2). CD44 protein is present in cultured PECs infected with CD44 vector (third lane). The lower western blot analysis shows the protein loading control β-Actin. (A’) Densitometry confirms the significant increase in CD44 in CD44 vector infected PECs.
(B) Collagen IV western blot analysis. Low levels of collagen IV protein are detected in non-infected (lane 1) and empty vector infected (lane 2) immortalized mouse PECs. Following CD44 infection, collagen IV protein levels are increased (lane 3). The lower panel shows β-Actin used as a protein loading control. (B’) Densitometry shows that collagen IV is higher in the presence of CD44.
(C) Quantification of PEC migration. The number of cells that migrated in to the wound was quantitated in PECs infected with CD44 vector (solid line) or empty vector (dashed line). Following a scratch, PEC migration was higher in CD44 infected cells at 24h and 48h compared with empty vector infected cells. These results show that overexpressing CD44 increased the migration of cultured PECs.
(D–I) Migration assay following scratch test. Column 1 shows a representative phase contrast images of cultured mouse PECs infected with CD44 vector (upper panels) or empty vector (lower panels) following the initiation of a scratch test at time zero (D–E, column 1), 24 hours (F–G, column 2) and 48 hours (H–I, column 3). The black line is drawn to demarcate the boundary of each side of the scratch. Examples of migrating cells are indicated by a red colored X.
The rate of migration is increased in cultured PECs by CD44
An in vitro scratch migration assay was performed to measure migration in cultured PECs.34 Following scratch-induced denuding of PECs infected with CD44 and control vector, migration of PECs into denuded areas were followed over time (Figure 5C, D–I). PEC migration was significantly higher in CD44 infected PECs, at 24h (24.4±1.4 vs. 9.6±0.51 PECs/2mm scratch, P<0.0001 vs. empty vector), and 48h (27.9±0.98 vs. 19.7±0.75 PECs/2mm scratch, P<0.0001 vs. empty vector). These results show that overexpressing CD44 in cultured PECs significantly increases their migration rate, consistent with the in vivo findings in FSGS.
De Novo CD44 staining in PECs co-localizes with active ERK1/2 in experimental FSGS
We next sought to understand the mechanism for increased CD44. Our recent report21 showed staining for phosphorylated-ERK1/2 (pERK1/2), the activated form of ERK1/2, increased in PECs in FSGS, and often co-stained with CD44.
Accordingly, we validated the co-staining of CD44 and pERK1/2 in PECs in CD44+/+ mice. CD44 and pERK1/2 staining were not present at baseline (Figure 6A–C). CD44 and pERK1/2 increased and co-localized in PECs at day 14 (Figure 6D–F), similar to our previous report.21
Figure 6. pERK co-localizes with CD44 in mouse and man with FSGS.

(A–C) Co-staining for CD44, pERK, and DAPI in CD44+/+ mice at baseline. (A) An image of staining for CD44 (red) and DAPI (blue) in CD44+/+ mice at baseline shows DAPI staining only. (B) An image of staining for pERK (green) and DAPI (blue) in CD44+/+ mice at baseline shows DAPI staining only. (C) A merged image of staining for CD44 (red), pERK (green) and DAPI (blue) in CD44+/+ mice at baseline shows DAPI staining only.
(D–F) Co-staining for CD44, pERK, and DAPI in CD44+/+ mice at d14 FSGS. (D) An image of staining for CD44 (red) and DAPI (blue) in CD44+/+ mice at d14 of FSGS shows CD44 staining along Bowman’s capsule and within the glomerular tuft. (E) An image of staining for pERK (green) and DAPI (blue) in CD44+/+ mice at d14 of FSGS also shows pERK staining along Bowman’s capsule and within the glomerular tuft. (F) A merged image of staining for CD44 (red), pERK (green) and DAPI (blue) in CD44+/+ mice at d14 of FSGS shows co-staining of CD44 and pERK in many cells (yellow/orange, arrowheads indicate examples).
(G–I) Co-staining for βGal, pERK, and DAPI in CD44−/− mice at baseline. (G) An image of staining for, βGal (red) and DAPI (blue) in CD44−/− mice at baseline shows DAPI staining only. (H) An image of staining for pERK (green) and DAPI (blue) in CD44−/− mice at baseline shows DAPI staining only in glomeruli. (I) A merged image of staining for βGal (red), pERK (green) and DAPI (blue) in CD44−/− mice at baseline shows DAPI staining only in glomeruli.
(J–L) Co-staining for βGal, pERK, and DAPI in CD44−/− mice at d14 FSGS. (J) An image of staining for βGal (red) and DAPI (blue) in CD44−/− mice at d14 of FSGS shows CD44 staining along Bowman’s capsule and within the glomerular tuft. (K) An image of staining for pERK (green) and DAPI (blue) in CD44−/− mice at d14 of FSGS also shows pERK staining along Bowman’s capsule and within the glomerular tuft. (L) A merged image of staining for βGal (red), pERK (green) and DAPI (blue) in CD44−/− mice at d14 of FSGS shows co-staining of βGal and pERK in cells (yellow/orange, arrowheads indicate examples). Because pERK increases in PECs in FSGS mice with and without CD44 deletion, these results are consistent with pERK being upstream of CD44.
(M–P) CD44 and pERK staining in serial sections of a human biopsy from a patient with FSGS. (M) CD44 staining (magenta) is detected in a segment of a glomerulus (top left quadrant, dashed box) along Bowman’s capsule and within the glomerular tuft in an area with synechial attachment. (O) The inset in M is shown at higher power magnification (examples of staining indicated by arrow heads) (N) pERK staining (brown) is detected in the same quadrant of the same glomerulus in a serial section, where staining co-localizes to the same cells staining for CD44. (P) The inset in N is shown at higher power magnification (examples of staining indicated by arrow heads) in the same cells that stain for CD44.
To prove CD44 was not upstream of ERK1/2 as has been described in another cell line,35 pERK1/2 staining was performed in CD44−/− mice (Figure 6). Glomerular pERK1/2 staining was not detected in CD44−/− mice at baseline (Figure 6H), but increased in CD44−/− mice at day 14 (Figure 6K). Moreover, pERK1/2 staining typically co-localized with βGal staining (which replaced CD44) (Figure 6L). As expected, CD44 staining was absent, and did not overlap with pERK1/2 (not shown). Staining for pERK1/2 in PECs was comparable in CD44+/+ and CD44−/− mice (P>0.05) and consistent with three findings: de novo pERK1/2 is independent of CD44 expression, CD44 is downstream of pERK1/2, and differences between CD44+/+ and CD44−/− mice are likely attributed to CD44 levels in PECs, not pERK1/2.
De Novo CD44 staining co-localizes with pERK1/2 in patient biopsies with FSGS
To prove CD44 was up-regulated in human FSGS and mediated by pERK1/2, we investigated serial sections from human biopsies diagnosed for secondary and primary FSGS. CD44 was detected in glomeruli along Bowman’s capsule and within FSGS lesions of the glomerular tuft (Figure 6M, O). In addition, CD44 co-localized with pERK1/2 staining in glomeruli and cells lining Bowman’s capsule (Figure 6N, P; arrowheads). Therefore, in analogy with the mouse model of FSGS, up-regulation of CD44 in PECs in human FSGS is also associated with ERK1/2 activation.
Activated ERK1/2 governs CD44 levels in cultured PECs
The in vivo results showed temporal association and co-expression of activated phospho-ERK1/2 (pERK) and CD44 in PECs in experimental and clinical FSGS. Accordingly, we next used two strategies to test p-ERK as a regulator for CD44 in cultured PECs: (i) pERK inhibitor UO12635; DMSO served as vehicle control (Figures 7A, B’); (ii) infecting PECs with MEK-DD, the upstream activator of pERK, or control empty vector (Figures 7 C–D’). As reported above, CD44 protein was not detected by western blot analysis in empty vector infected PECs, and exposing cells to UO126 had no effect (Figure 7A, Lanes 1 & 2). Infecting PECs with CD44 markedly increased CD44, which was not affected by DMSO (P=0.013 vs. empty vector)(Fig 7A, lane 3; Fig 7A’). Densitometry results are shown in Fig 7A’. However, in CD44 infected PECs, CD44 levels were decreased substantially by pERK inhibition with U012635 (P=0.023 vs. DMSO)(Figure 7, lane 4). The inhibition of ERK1/2 phosphorylation by U0126 was confirmed by densitometry using total ERK levels as the loading control (Figure 7B, B’).
Figure 7. pERK is a regulator of CD44 in cultured mouse parietal epithelial cells (PECs).

(A) Inhibiting ERK activity lowers CD44 protein levels. Western blot analysis of PEC lysates shows that CD44 protein was not detected in PECs transfected with sham empty vector in the presence of either DMSO vehicle control (lane 1) or the pERK inhibitor U0126 (lane 2). CD44 protein in PECs infected with CD44 vector was not altered by exposing cells to DMSO (lane 3), but was significantly reduced when pERK was inhibited by UO126. The lower panel shows protein loading with β-Actin. (A’) Densitometry confirms that the significant increase in CD44 in PECs overexpressing CD44 is markedly reduced when ERK activity is inhibited. These results show that pERK is upstream of CD44.
(B) CD44 infection has no effect on ERK. The upper panel shows a western blot analysis for the phosphorylated active form of ERK (pERK), and the lower panel shows protein loading using total ERK protein. Active pERK (lane 1) is reduced by UO126 (lane 2) in sham empty vector infected PECs, and also in CD44 infected PECs (lane 4). (B’) Densitometry shows that U0126 significantly reduces pERK levels compared to DMSO..
(C) MEK-DD increases pERK. Western blot analyses for phosphorylated ERK (pERK) and total ERK are shown in the upper and lower western blots respectively. (C’) Densitometry shows that pERK1/2 protein levels increased in PECs infected with MEK-DD, an upstream kinase of ERK1/2 (lane 2).
(D) Forced pERK activity increases CD44 levels. The upper western blot analysis shows that when pERK is increased by infecting PECs with MEK-DD, CD44 protein is detected (lane 2), whereas CD44 is not detected in empty vector infected PECs (Lane 1). The lower panel shows protein loading using β-Actin, the denominator for the densitometry results (D’) Densitometry shows that that CD44 was increased relative to β-Actin. Taken together, these results show that inhibiting pERK activity reduces CD44 protein levels, and that enhancing pERK increases CD44 levels..
We next overexpressed MEK-DD in PECs, an upstream kinase of ERK1/2.36, 37 As expected, MEK-DD overexpression enhanced pERK1/2, but did not change total ERK levels (Figure 7C, C’). CD44 protein levels were increased by MEK-DD (Figure 7D); densitometry confirms the increase compared to control empty vector (P=0.041)(Fig 7D’).
Taken together, when pERK1/2 is reduced in PECs, CD44 levels are reduced; conversely when pERK1/2 is increased, CD44 levels are increased. These results are consistent with active pERK1/2 as a regulator of CD44 in PECs.
pERK activity regulates PEC migration
We next asked if interfering with ERK1/2 activation affected PEC migration by performing an in vitro scratch assay.34 First, ERK1/2 activity was reduced by U0126,38 and second, ERK1/2 was activated by overexpressing MEK-DD. In control infected PECs, inhibiting ERK1/2 activation with U0126 reduced migration at both 24 hours (2.3±0.3 vs. 6.5±0.5, P<0.0001 vs. DMSO used as vehicle control), and 48 hours (4.1±0.4 vs. 15.7±1, P<0.0001 vs. DMSO)(Figure 8A–M). In CD44 infected PECs, inhibiting ERK1/2 activity also reduced PEC migration at 24 hours (2.1±0.3 vs. 9.6±0.5, P<0.0001, vs. DMSO), and 48 hours (4.4±0.5 vs. 25.7±1.3, P<0.0001 vs. DMSO)(Figure A8-M).
Figure 8. pERK is a regulator of PEC Migration.

(A) Quantification of PEC migration following ERK inhibition in CD44 infected PECs. Following a scratch induced wound, PEC migration is higher in CD44 infected PECs exposed to DMSO (vehicle for UO126) compared to empty vector infected PECs exposed to DMSO at 24h and 48h. However, in the presence of the ERK inhibitor U0126, scratch induced PEC migration was substantially inhibited in CD44 and empty vector infected PECs at 24h and 48h. These results show that active pERK is required for both CD44 dependent and independent PEC migration.
(B–M) Representative images of scratch assay. Cultured PECs. immediately following the scratch induced wound at time zero (B–E, first column), at 24h (F–I, second column) and 48h (J–M, third column) in CD44 infected and empty vector infected PECs in the presence of either the vehicle DMSO or the ERK inhibitor U0126. The black lines are drawn to demarcate the boundary of each side of the scratch. PECs that have migrated in to the wound are indicated by red circles.
(N) Quantification of PEC migration following ERK activation in MEK-DD infected PECs. Following scratch induced wound, migration is higher at 24h and 48h in PECs infected with MEK-DD (solid line), which activates pERK, compared to empty transfected cells (dashed line).
(O–T) Representative images of scratch assay. Cultured PECs immediately following the scratch induced wound at time zero (O–P, first column), and at 24h (Q–R, second column) and 48h (S–T, third column) in MEK-DD infected and empty vector infected PECs. The black lines show the edge of the wound, and examples of PECs that have migrated are indicated by red circles. These results show that pERK augments PEC migration.
Increasing ERK1/2 activation by overexpressing MEK-DD increased PEC migration at 24 hours (27.4±1.7 vs. 9.8±1.1, P<0.0001 vs. PECs infected empty vector), and 48 hours (47±2.3 vs. 17.5±1.1, P<0.0001 vs. PECs infected empty vector)(Figure 8N–T).
Taken together, when ERK1/2 activation was inhibited, PEC migration was reduced and when ERK1/2 was activated, PEC migration increased.
DISCUSSION
Increasing literature shows that following injury to podocytes in glomerular diseases, neighboring glomerular parietal epithelial cells (PECs) are not innocent bystanders.39, 40 Rather, a subset begin to de novo express CD44,1,4,19–22,23 a biological state called “activated” PECs.3, 41 Activated PECs have been reported to distinguish FSGS from minimal change disease on human kidney biopsies.19, 42 We have extended these discoveries in the current study, by showing activated ERK1/2 increases CD44 and two biological roles include increased PEC extracellular matrix production and migration.
The first finding was that CD44 is a regulator of extracellular matrix accumulation by PECs, in vivo and in vitro. Studies by our group and others in mouse and man,3, 21 show CD44 is not expressed in PECs under non-stressed conditions. CD44 increased markedly in PECs in FSGS in CD44+/+ mice, coinciding with increased collagen IV staining, consistent with previous reports.1,19–22,7 These associations suggested, but did not prove, CD44 is linked with increased matrix accumulation. Thus, to prove glomerulosclerosis is lower in mice lacking CD44, we utilized CD44−/− mice. The results showed kidney histology and albumin-to-creatinine measures from CD44−/− mice were indistinguishable from CD44+/+ mice at baseline. Following FSGS, there were no differences in disease inducing antibody deposition and podocyte number was not different at the time points studied. However, CD44−/− mice had reduced focal and global glomerulosclerosis, as well as fewer pseudocrescents. Similarly, collagen IV was reduced in CD44−/− mice. These results support that de novo expression of CD44 in PECs is associated with increased matrix proteins and glomerulosclerosis. The study as designed could not differentiate if glomeruli with proliferative PECs might recover.
PEC culture was used to test the hypothesis that CD44 governs matrix production. PECs express very low levels of CD44 protein, so CD44 was overexpressed. Compared to control, CD44 overexpressing PECs displayed higher levels of collagen IV. Smeets and Moeller have shown this association by double-staining for CD44 and LKIV69, an antibody raised against extra-cellular matrix produced by PECs.42, 43 Holderied et al. showed cultured PECs increase several extracellular matrix proteins when exposed to high glucose, AGEs and TGFs.7 Yet, the current study is the first direct proof that CD44 regulates matrix production by PECs both in vivo and in vitro.
The second finding was that migration increased in PECs expressing CD44. Glomerular staining for the PEC markers PAX8 and SSeCKS was limited to cells lining Bowman’s capsule in both CD44+/+ and CD44−/− mice at baseline. In CD44+/+ mice with FSGS, 20% of glomeruli had evidence of PECs migration to the glomerular tuft, consistent with previous studies.1, 21, 22, 42 In CD44−/− mice with FSGS, only 9% of glomeruli had evidence of PEC migration. PEC migration was also measured by an in vitro scratch assay34 following CD44 overexpression. Compared to control, cells overexpressing CD44 had increased migration. These in vivo-in vitro results are consistent with CD44 being a regulator of PEC migration. This raised the question if PEC activation is a beneficial or detrimental. The current studies were not designed to directly test this question. We speculate that the biological consequences of PEC activation may be context dependent, perhaps being reparative initially, but harmful over time.
The third finding was that pERK1/2, the activated form, is a regulator of CD44 levels in PECs. The rationale derives from our recent mouse studies in FSGS21 and aged kidneys,4 showing pERK1/2 and CD44 often co-localize in PECs. Co-localization of pERK1/2 and CD44 was validated in the current study.21. Active pERK1/2 is also increased in PECs in human biopsies from patients with FSGS, supporting the mouse data. To prove pERK1/2 is a regulator of CD44, CD44 was overexpressed in PECs. Inhibiting ERK1/2 activation in CD44 overexpressing cells using the inhibitor U012638 reduced CD44 protein levels. By contrast, increasing pERK1/2 by overexpressing MEK-DD,36 increased CD44 protein levels. These results differ from a subset of cancer cells, where CD44 is upstream of pERK1/2.35, 44, 45 However, in endothelial cells, knocking down CD44 had no effect on ERK1/2.46 Similarly, in the current studies, CD44 had no effect on ERK1/2 activation. Our results showed a spectrum of staining for both CD44/β-gal and for pERK. While some cells express both, some have only pERK or CD44/β gal. These results are likely because activation and migration are not synchronous processes.. Our results suggest pERK decreases once PEC migration has happened. Some PECs also express CD44/β-gal, but do not migrate.
The fourth finding was that ERK1/2 phosphorylation regulates PEC migration. When pERK1/2 was inhibited PEC, migration was reduced. Moreover, inhibiting pERK1/2 in CD44 overexpressing PECs further reduced migration. Conversely, overexpressing pERK1/2, substantially increased PEC migration. Several lines of evidence suggest pERK1/2 has additional roles in PECs. We have shown pERK1/2 is increased in PECs exposed to high albumin concentrations.37 Inhibiting ERK1/2 phosphorylation enhanced apoptosis, and increased pERK1/2 enhanced PEC survival.37 When mice with FSGS are treated with enalapril, the number of PECs expressing pERK1/2 increased.47 This correlated with an overall increase in PEC number, and a subpopulation that co-expressed WT-1.47,ERK1/2 activation increases under various conditions, and its biological roles are likely context dependent, similar to other cell types.48
We acknowledge several limitations in the current study. We used a global CD44−/− mouse, rather than PEC specific deletion, because to date there is no unique PEC gene identified.. CD44 has been shown in other systems to have biological effects on inflammation. Although a bone marrow transplant study might tease out such effects, we showed no differences in the number of activated T cells, B cells, macrophages and neutrophils in glomeruli of CD44+/+ and CD44−/−. This is not surprising, as FSGS has low abundance of such inflammatory cells in glomeruli. We cannot rule out the effects of CD44 on other glomerular structures such as endothelial cells and glomerular basement membrane at different times points that might explain differences in proteinuria. We recognize that we have not explained how ERK1/2 is activated in PECs following podocyte injury. Several scenarios are under active investigation, including the release of growth factors from PECs and/or injured podocytes. Contact between denuded GBM and PECs at sites of synechial attachment and proteinuria needs to be considered too. Noteworthy was that pERK1/2 was increased similarly in CD44+/+ and CD44−/− mice, suggesting that whatever cause(s) for ERK1/2 activation, was not different in the two strains. Finally, we recognize that timed urine collections might be more sensitive than spot urines to measure ACR.
In summary, CD44 is not just a biomarker of PEC activation, but has important biological roles. De novo expression of CD44 in PECs following podocyte injury is mediated in part by ERK1/2 activation, which increases PEC matrix production and migration (Figure 9). Future studies should consider how to limit CD44 levels and/or ERK1/2 activation specifically in PECs.
Figure 9. Schema of study findings.
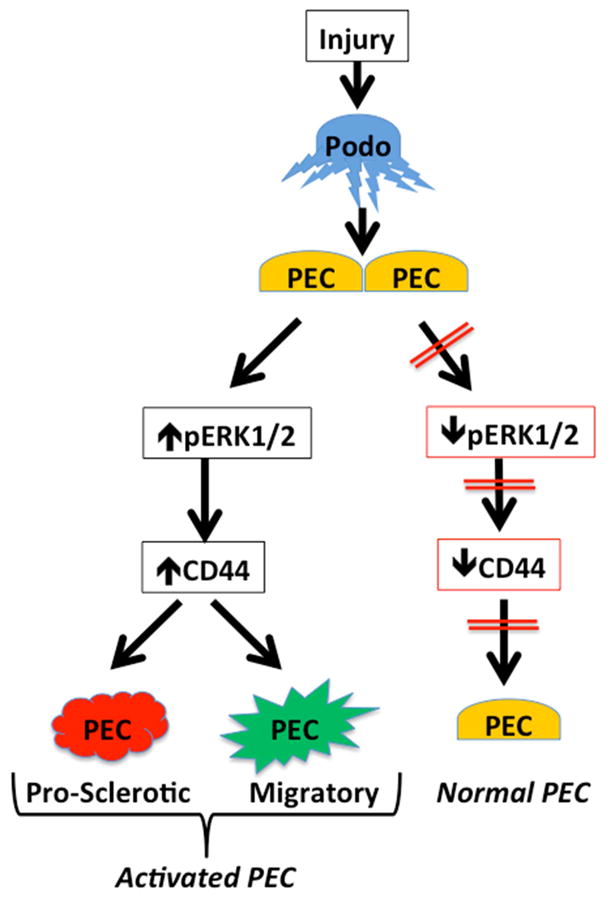
Podocyte injury leads to increased ERK1/2 activation in PECs, which increases CD44 expression and causes matrix production and PEC migration to the glomerular tuft resulting in increased glomerular sclerosis.
METHODS
CD44 Null Mice
CD44 knockout/ LacZ knockin (CD44−/−) and wildtype control (CD44+/+) mice were obtained from The Jackson Laboratory (Bar Harbor, ME).49 In brief, a neomycin/LacZ cassette was used to abolish CD44 function by disrupting exon 1 and intron 1 which are common to all CD44 isoforms, resulting in the expression of the LacZ gene where CD44 would otherwise be expressed. Mice were maintained in the animal care facility at the University of Washington under specific pathogen-free conditions with ad libitum food and water. Animal protocols were approved by the University of Washington Institutional Animal Care and Use Committee (2968-04).
Experimental FSGS
An age and sex matched cohort of CD44+/+ (n=5) and CD44−/− mice (n=5) that did not get disease served as baseline. Sheep anti-podocyte antibody was administered to adult male CD44+/+ (n=14) and CD44−/− (n=16) mice to induce FSGS as previously described.47, 50, 51 Two doses of sheep IgG, at 11mg/20g bodyweight, were administered via IP injection 24 hours apart. Mice were then randomly sacrificed at d14 (n=17), and d28 (n=13). At sacrifice animals were processed as previously described.21
Assessment of Albuminuria
Spot urines were collected from all animals at baseline, 7, 14 and 28 days of FSGS. Urinary mouse albumin concentration was measured by radial immunodiffusion assay (RID) as previously described.52 Creatinine was measured in the urine via a colorimetric assay (Cayman Chemical, Ann Arbor, MI) and an albumin to creatinine ratio was calculated.
Immunostaining and Quantification
Indirect immunoperoxidase and immunofluorescence staining were performed on 4μm tissue sections from frozen (Sheep IgG, CD44 variant 3, 7 & 10, hyaluronic acid binding protein and leukocytes staining) or formalin fixed paraffin embedded (all other) renal biopsies as described previously21, 24, 53–55 In brief, optimum cutting temperature compound was removed from frozen sections by washing in PBS followed by post fixation in −20°C methanol and a subsequent wash in PBS. Paraffin was removed from formalin-fixed sections using Histoclear (National Diagnostics, Atlanta, GA, USA) and rehydrated in a graded series of ethanol. Antigen retrieval was accomplished by boiling sections in 10mM citric acid buffer pH 6.0. Nonspecific protein binding was blocked using Background Buster (Accurate Chemical & Scientific, Westbury, NY, USA). Endogenous biotin activity was suppressed with an Avidin/Biotin Blocking Kit (Vector Laboratories, Burlingame, CA, USA). Antibodies were diluted in 1% IgG free BSA in PBS. Visualization of immunoperoxidase staining was by precipitation of diaminobenzidine (DAB; Sigma-Aldrich , St. Louis, Missouri, USA). Slides were dehydrated in ethanol and mounted with Histomount. Immunofluorescence samples were mounted in Vectashield with DAPI (Vector Laboratories). For a list of primary and secondary antibodies used please see Supplementary Tables 1 and 2.
To assess glomerular injury, immunostaining was performed for p57 with Periodic Acid Schiff (PAS) counterstaining, as previously reported.50, 54–56
For quantification, 50 glomeruli were examined on a Leica DMRB microscope or an EVOS FL Cell Imaging System. Images were collected using confocal microscopy on a Leica DMI400B. Scale bars were applied to each image by calibrating set scale at each magnification to a slide micrometer and applying with the scale bar tool in Image J 1.44o (NIH).
Mouse Parietal Epithelial Cells in Culture
A previously generated and characterized33 conditionally immortalized mouse glomerular parietal epithelial cell line was used for in vitro experiments. Cells were plated on Primaria dishes (Corning Inc, Corning, NY) coated with collagen-I (BD Bioscience, Bedford, MA) and cultured in RMPI-1640 medium (Thermo Fisher Scientific, Waltham, MA) supplemented with 2% fetal bovine serum (Gemini Bioproducts, West Sacramento, CA), 1% penicillin/streptomycin (Sigma Aldrich) and 0.1mM sodium pyruvate (Thermo Fisher Scientific). For propagation, PECs were cultured under growth permissive conditions (33°C and 5% CO2) and supplemented with 50U/ml INF-γ (Roche Diagnostics, Indianapolis, IN). After 14 days in growth restrictive conditions (37°C and 5% CO2) in the absence of INF-γ PECs were considered differentiated.
Infecting cultured PECs
Retroviral pBABE-puro vectors encoding for the standard isoform of CD44 (Addgene #19127,57 for MEK-DD (Addgene #15268,58) and an empty pBABE-puro vector (Addgene #1764,59) were transfected into Phoenix Eco packaging cells (Gary Nolan, The Baxter Laboratory of Genetic Pharmacology, Stanford, CA) using the calcium phosphate precipitation method. Retrovirus containing supernatant was harvested, filtered and applied to growth permissive, proliferating PECs for infection. Infected cells were selected by passaging in medium containing 2,5μg/ml puromycin. Transfected PECs were maintained under growth restrictive condition for 14 days for all experiments.
Inhibition of ERK phosphorylation
Phosphorylation of Thr202- and Tyr204-residues of MAPK/ERK1/2 was inhibited as previously described.37 In brief, PECs were incubated in standard medium (see above) supplemented with either 10μM U0126 (Cell Signaling, Danvers, MA) or with DMSO (vehicle control). Due to the short half-life of U0126, the reagent was applied every 12h.
In vitro PEC migration assay
To assess PEC migration, an in vitro scratch assay was performed following a modified standard protocol.34 Briefly, denuded areas of consistent width were created in a confluent monolayer of growth restricted PECs by scraping the culture dish with a sterile p200 pipet tip. Pictures of denuded areas were acquired immediately, 24 and 48 hours later. A minimum of 30 images of denuded areas 2mm in length were assessed.
Western Blot Analysis
PECs lysates were collected in RIPA lysis buffer (50mM Tris-HCl, pH 8.0, 5mM EDTA, 150mM NaCl, 1% IP-40, 1% Trion X-100, 50mM NaF, 1mM Na-orthovanadate (Sigma-Aldrich) and protease inhibitors (Roche). Protein concentrations were determined using the BCA protein assay (Pierce). SDS-PAGE gels (8–12%) were electrophoresed and electroblotted onto PVDF membranes (Sigma-Aldrich). Membranes were blocked in 5% non-fat milk in TBST (10mM Tris-HCl, pH 8.0, 150mM NaCl, 0.05% Tween 20) and incubated with the appropriate primary antibodies and horseradish peroxidase conjugated secondary antibodies (supplemental tables 1 & 2). Immunostaining was visualized using enhanced chemiluminescent substrate (Thermo Scientific). In some cases, membranes were incubated in stripping buffer (100mM glycine, 1% SDS, pH 2.5) and re-stained. Images of stained membranes were obtained on a Chemidoc XRS+ System (Bio-Rad Laboratories).
Human kidney biopsies
FFPE material60 from the archive of the Department of Nephropathology, FAU Erlangen-Nürnberg of primary (n=20) and secondary FSGS (n=20) were used. The use of kidney biopsies was approved by the Ethics Committee of the Friedrich-Alexander-University of Erlangen-Nürnberg, waiving the need for retrospective consent for the use of archived rest material (Re.-No.4415). Staining was performed on 2μm serial tissue sections. Following paraffin removal and rehydration sections were boiled in Target Retrieval Solution (Dako, Hamburg, Germany) and blocked with normal goat serum (Jackson ImmunoResearch Europe Ltd., Suffolk, UK) diluted in 5% nonfat dry milk (Biorad, Hercules, CA). Slides were incubated with primary antibodies (Supplementary Table 1) diluted in 1% IgG free BSA (Sigma-Aldrich). pERK staining was detected using a secondary biotinylated anti-rabbit antibody (Supplementary Table 2), Vectastain-ABC-Kit (Vector) and diaminobenzidine (Sigma-Aldrich) as a substrate. CD44 staining was detected using the POLAP-100 Polymer Kit (Zytomed Systems, Berlin, Germany) with FastRed (Zytomed Systems). Slides were counterstained with hematoxylin (Merck Millipore, Darmstadt, Germany) and mounted with Aquatex (Merck Millipore). Images of were obtained by scanning and digitalizing the whole sample with a Pannoramic MIDI slide scanner (3D Histech Ltd., Budapest, Hungary) followed by alignment with Pannoramic Viewer software (3D Histech Ltd.) and merging with Photoshop software (Adobe, San Jose, CA).
Statistical analysis
Results are presented as mean ± SEM. Statistical analysis was performed using the GraphPad Prism 6.0 software (La Jolla, CA). Normal distribution was tested using Kolmogorov-Smirnov Test. A two-tailed unpaired Student’s t-test was applied to compare means of groups, and P<0.05 was considered statistically significant.
Supplementary Material
(A, B) CD44 variant 3 is not detected in glomeruli of CD44+/+ (A) or CD44−/− (B) mice with FSGS (white arrow heads indicate glomeruli). Variant 3 (red) is detected in cells in the tubular interstitial space in CD44+/+ mice.
(C, D) CD44 isoform 10 is not detected in glomeruli of CD44+/+ (C) or CD44−/− (D) mice with FSGS (white arrow heads indicate glomeruli). Variant 10 (red) is detected in subset of cells in the tubular interstitial space (red) in CD44+/+ mice.
(E–H) CD44 variant 7 is not detected in glomeruli of CD44+/+ (E) or CD44−/− (F) mice with FSGS (dashed white squares indicate glomeruli). (G & H) higher magnification images of the glomeruli in images E & F. Variant 7 (red) is detected in a small subset of cells in the tubular interstitial space (red) in CD44+/+ mice.
(I, J) CD44 standard variant (brown), which excludes variants 1–10, is detected within tubular and interstitial cells as well as the glomerulus (indicated by black dashed square) in CD44+/+ mice. (J) higher magnification image of glomerulus in image I showing CD44 standard variant staining in PECs (black arrow heads).
Representative images of staining for (A) CD3 (activated T cell marker), (B) B220 (B cell marker), (C) F4/80 (macrophage marker) and (D) LY-6G (neutrophil marker) in diseased mice. White arrow heads indicate examples of positive staining in cells. The accompanying table shows quantification for the number of positive cells per glomerulus for each cell type . The number were extremely small, did not change much with disease (no significant glomerular influx) and showed no statistical differences between CD44+/+ and CD44−/− mice with this experimental model of FSGS.
Representative images of staining for hyaluronic acid binding protein in (A) CD44+/+ mice and (B) CD44−/− mice. The accompanying table shows quantification for the percentage of glomeruli with positive HABP staining. There was no difference between CD44+/+ and CD44−/− mice at baseline. The percentage of glomeruli with positive HABP staining increased with disease, but there was no statistical difference between CD44+/+ and CD44−/− mice.
Acknowledgments
Grant Support: 5 R01 DK 056799-10, 5 R01 DK 056799-12, 1 R01 DK097598-01A1 This work was also supported by an Emerging Fields Initiative (EFI) for Cell Cycle in Disease and Regeneration (CYDER) from the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) (Germany).
The excellent technical assistance of Stefan Söllner, Miriam Reutelshöfer and Bairbre McNicholas is acknowledged. The present work was performed in (partial) fulfillment of the requirements for obtaining the degree ‘Dr. med.’ from the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU). S. Roeder was supported by a scholarship of the Interdisciplinary Center for Clinical Research (IZKF) of the FAU Erlangen-Nürnberg.
Abbreviations
- PECs
parietal epithelial cells
- pERK1/2
phospho-extracellular signal-related kinase 1/2
- β-Gal
Beta galactosidase
- FSGS
focal segmental glomerulosclerosis
Footnotes
COI: None of the authors have any financial or other conflicts of interest. The results presented in this paper have not been published previously, in whole or part.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kuppe C, Grone HJ, Ostendorf T, et al. Common histological patterns in glomerular epithelial cells in secondary focal segmental glomerulosclerosis. Kidney Int. 2015;88:990–998. doi: 10.1038/ki.2015.116. [DOI] [PubMed] [Google Scholar]
- 2.Shankland SJ, Smeets B, Pippin JW, et al. The emergence of the glomerular parietal epithelial cell. Nat Rev Nephrol. 2014;10:158–173. doi: 10.1038/nrneph.2014.1. [DOI] [PubMed] [Google Scholar]
- 3.Smeets B, Moeller MJ. Parietal epithelial cells and podocytes in glomerular diseases. Semin Nephrol. 2012;32:357–367. doi: 10.1016/j.semnephrol.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 4.Roeder SS, Stefanska A, Eng DG, et al. Changes in glomerular parietal epithelial cells in mouse kidneys with advanced age. Am J Physiol Renal Physiol. 2015;309:F164–178. doi: 10.1152/ajprenal.00144.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smeets B, Uhlig S, Fuss A, et al. Tracing the Origin of Glomerular Extracapillary Lesions from Parietal Epithelial Cells. J Am Soc Nephrol. 2009;12:2604–2615. doi: 10.1681/ASN.2009010122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smeets B, Kuppe C, Sicking E-M, et al. Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22:1262–1274. doi: 10.1681/ASN.2010090970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holderied A, Romoli S, Eberhard J, et al. Glomerular parietal epithelial cell activation induces collagen secretion and thickening of Bowman's capsule in diabetes. Lab Invest. 2015;95:273–282. doi: 10.1038/labinvest.2014.160. [DOI] [PubMed] [Google Scholar]
- 8.Fatima H, Moeller MJ, Smeets B, et al. Parietal Epithelial Cell Activation Marker in Early Recurrence of FSGS in the Transplant. Clin J Am Soc Nephrol. 2012;7:1852–1858. doi: 10.2215/CJN.10571011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ponta H, Wainwright D, Herrlich P. The CD44 protein family. Int J Biochem Cell Biol. 1998;30:299–305. doi: 10.1016/s1357-2725(97)00152-0. [DOI] [PubMed] [Google Scholar]
- 10.Aruffo A, Stamenkovic I, Melnick M, et al. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990;61:1303–1313. doi: 10.1016/0092-8674(90)90694-a. [DOI] [PubMed] [Google Scholar]
- 11.Goodison S, Urquidi V, Tarin D. CD44 cell adhesion molecules. Mol Pathol. 1999;52:189–196. doi: 10.1136/mp.52.4.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikolic-Paterson DJ, Jun Z, Tesch GH, et al. De novo CD44 expression by proliferating mesangial cells in rat anti-Thy-1 nephritis. J Am Soc Nephrol. 1996;7:1006–1014. doi: 10.1681/ASN.V771006. [DOI] [PubMed] [Google Scholar]
- 13.Qiaoling Z, Xiaoyun J, Wei W, et al. Altered P-selectin and CD44 expression in the renal tissues and peripheral blood of children with IgA nephropathy. Int Urol Nephrol. 2009;41:703–711. doi: 10.1007/s11255-008-9512-y. [DOI] [PubMed] [Google Scholar]
- 14.Gvozdenovic A, Arlt MJ, Campanile C, et al. CD44 enhances tumor formation and lung metastasis in experimental osteosarcoma and is an additional predictor for poor patient outcome. J Bone Miner Res. 2013;28:838–847. doi: 10.1002/jbmr.1817. [DOI] [PubMed] [Google Scholar]
- 15.Cho SH, Park YS, Kim HJ, et al. CD44 enhances the epithelial-mesenchymal transition in association with colon cancer invasion. Int J Oncol. 2012;41:211–218. doi: 10.3892/ijo.2012.1453. [DOI] [PubMed] [Google Scholar]
- 16.Fanni D, Fanos V, Gerosa C, et al. CD44 immunoreactivity in the developing human kidney: a marker of renal progenitor stem cells? Ren Fail. 2013;35:967–970. doi: 10.3109/0886022X.2013.808955. [DOI] [PubMed] [Google Scholar]
- 17.Deboux C, Ladraa S, Cazaubon S, et al. Overexpression of CD44 in neural precursor cells improves trans-endothelial migration and facilitates their invasion of perivascular tissues in vivo. PLoS One. 2013;8:e57430. doi: 10.1371/journal.pone.0057430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiscox S, Baruha B, Smith C, et al. Overexpression of CD44 accompanies acquired tamoxifen resistance in MCF7 cells and augments their sensitivity to the stromal factors, heregulin and hyaluronan. BMC Cancer. 2012;12:458. doi: 10.1186/1471-2407-12-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fatima H, Moeller MJ, Smeets B, et al. Parietal epithelial cell activation marker in early recurrence of FSGS in the transplant. Clin J Am Soc Nephrol. 2012;7:1852–1858. doi: 10.2215/CJN.10571011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sicking EM, Fuss A, Uhlig S, et al. Subtotal Ablation of Parietal Epithelial Cells Induces Crescent Formation. Journal of the American Society of Nephrology : JASN. 2012;23:629–640. doi: 10.1681/ASN.2011050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eng DG, Sunseri MW, Kaverina NV, et al. Glomerular parietal epithelial cells contribute to adult podocyte regeneration in experimental focal segmental glomerulosclerosis. Kidney Int. 2015;88:999–1012. doi: 10.1038/ki.2015.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okamoto T, Sasaki S, Yamazaki T, et al. Prevalence of CD44-positive glomerular parietal epithelial cells reflects podocyte injury in adriamycin nephropathy. Nephron Exp Nephrol. 2013;124:11–18. doi: 10.1159/000357356. [DOI] [PubMed] [Google Scholar]
- 23.Kim S, Kim YH, Choi KH, et al. Glomerular epithelial CD44 expression and segmental sclerosis in IgA nephropathy. Clin Exp Nephrol. 2015 doi: 10.1007/s10157-015-1222-z. [DOI] [PubMed] [Google Scholar]
- 24.Pippin JW, Sparks MA, Glenn ST, et al. Cells of Renin Lineage Are Progenitors of Podocytes and Parietal Epithelial Cells in Experimental Glomerular Disease. American Journal Of Pathology. 2013;183:542–557. doi: 10.1016/j.ajpath.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lesley J, Hyman R, Kincade PW. CD44 and its interaction with extracellular matrix. Adv Immunol. 1993;54:271–335. doi: 10.1016/s0065-2776(08)60537-4. [DOI] [PubMed] [Google Scholar]
- 26.Venkatareddy M, Wang S, Yang Y, et al. Estimating podocyte number and density using a single histologic section. J Am Soc Nephrol. 2014;25:1118–1129. doi: 10.1681/ASN.2013080859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burnworth B, Pippin J, Karna P, et al. SSeCKS sequesters cyclin D1 in glomerular parietal epithelial cells and influences proliferative injury in the glomerulus. Lab Invest. 2012;92:499–510. doi: 10.1038/labinvest.2011.199. [DOI] [PubMed] [Google Scholar]
- 28.Ueno T, Kobayashi N, Nakayama M, et al. Aberrant Notch1-dependent effects on glomerular parietal epithelial cells promotes collapsing focal segmental glomerulosclerosis with progressive podocyte loss. Kidney Int. 2013;83:1065–1075. doi: 10.1038/ki.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hackl MJ, Burford JL, Villanueva K, et al. Tracking the fate of glomerular epithelial cells in vivo using serial multiphoton imaging in new mouse models with fluorescent lineage tags. Nat Med. 2013;19:1661–1666. doi: 10.1038/nm.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagata M, Hattori M, Hamano Y, et al. Origin and phenotypic features of hyperplastic epithelial cells in collapsing glomerulopathy. Am J Kidney Dis. 1998;32:962–969. doi: 10.1016/s0272-6386(98)70070-8. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki T, Matsusaka T, Nakayama M, et al. Genetic podocyte lineage reveals progressive podocytopenia with parietal cell hyperplasia in a murine model of cellular/collapsing focal segmental glomerulosclerosis. Am J Pathol. 2009;174:1675–1682. doi: 10.2353/ajpath.2009.080789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohse T, Chang AM, Pippin JW, et al. A new function for parietal epithelial cells: a second glomerular barrier. American Journal of Physiology-Renal Physiology. 2009;297:F1566–1574. doi: 10.1152/ajprenal.00214.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohse T, Pippin JW, Vaughan MR, et al. Establishment of Conditionally Immortalized Mouse Glomerular Parietal Epithelial Cells in Culture. Journal of the American Society of Nephrology. 2008;19:1879–1890. doi: 10.1681/ASN.2007101087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nature protocols. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 35.Yu S, Cai X, Wu C, et al. Adhesion glycoprotein CD44 functions as an upstream regulator of a network connecting ERK, AKT and Hippo-YAP pathways in cancer progression. Oncotarget. 2015;6:2951–2965. doi: 10.18632/oncotarget.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–20416. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang AM, Ohse T, Krofft RD, et al. Albumin-induced apoptosis of glomerular parietal epithelial cells is modulated by extracellular signal-regulated kinase 1/2. Nephrol Dial Transplant. 2012;27:1330–1343. doi: 10.1093/ndt/gfr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Favata MF, Horiuchi KY, Manos EJ, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 39.Shankland SJ, Smeets B, Pippin JW, et al. The emergence of the glomerular parietal epithelial cell. Nat Rev Nephrol. 2014 doi: 10.1038/nrneph.2014.1. [DOI] [PubMed] [Google Scholar]
- 40.Swetha G, Chandra V, Phadnis S, et al. Glomerular parietal epithelial cells of adult murine kidney undergo EMT to generate cells with traits of renal progenitors. Journal of cellular and molecular medicine. 2009 doi: 10.1111/j.1582-4934.2009.00937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smeets B, Angelotti ML, Rizzo P, et al. Renal Progenitor Cells Contribute to Hyperplastic Lesions of Podocytopathies and Crescentic Glomerulonephritis. Journal of the American Society of Nephrology. 2009;20:2593–2603. doi: 10.1681/ASN.2009020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smeets B, Stucker F, Wetzels J, et al. Detection of activated parietal epithelial cells on the glomerular tuft distinguishes early focal segmental glomerulosclerosis from minimal change disease. Am J Pathol. 2014;184:3239–3248. doi: 10.1016/j.ajpath.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wijnhoven TJ, Lensen JF, Rops AL, et al. Aberrant heparan sulfate profile in the human diabetic kidney offers new clues for therapeutic glycomimetics. Am J Kidney Dis. 2006;48:250–261. doi: 10.1053/j.ajkd.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 44.Herishanu Y, Gibellini F, Njuguna N, et al. Activation of CD44, a receptor for extracellular matrix components, protects chronic lymphocytic leukemia cells from spontaneous and drug induced apoptosis through MCL-1. Leuk Lymphoma. 2011;52:1758–1769. doi: 10.3109/10428194.2011.569962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bourguignon LY, Gilad E, Rothman K, et al. Hyaluronan-CD44 interaction with IQGAP1 promotes Cdc42 and ERK signaling, leading to actin binding, Elk-1/estrogen receptor transcriptional activation, and ovarian cancer progression. J Biol Chem. 2005;280:11961–11972. doi: 10.1074/jbc.M411985200. [DOI] [PubMed] [Google Scholar]
- 46.Olofsson B, Porsch H, Heldin P. Knock-down of CD44 regulates endothelial cell differentiation via NFkappaB-mediated chemokine production. PLoS One. 2014;9:e90921. doi: 10.1371/journal.pone.0090921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Pippin JW, Vaughan MR, et al. Retinoids Augment the Expression of Podocyte Proteins by Glomerular Parietal Epithelial Cells in Experimental Glomerular Disease. Nephron Exp Nephrol. 2012;121:e23–e37. doi: 10.1159/000342808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacological research : the official journal of the Italian Pharmacological Society. 2012;66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 49.Protin U, Schweighoffer T, Jochum W, et al. CD44-deficient mice develop normally with changes in subpopulations and recirculation of lymphocyte subsets. J Immunol. 1999;163:4917–4923. [PubMed] [Google Scholar]
- 50.Zhang J, Pippin JW, Krofft RD, et al. Podocyte Repopulation by Renal Progenitor Cells Following Glucocorticoids Treatment in Experimental FSGS. Am J Physiol Renal Physiol. 2013;304:F1375–1389. doi: 10.1152/ajprenal.00020.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohse T, Vaughan MR, Kopp JB, et al. De novo expression of podocyte proteins in parietal epithelial cells during experimental glomerular disease. American Journal of Physiology-Renal Physiology. 2010;298:F702–711. doi: 10.1152/ajprenal.00428.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marshall CB, Krofft RD, Blonski MJ, et al. Role of smooth muscle protein SM22alpha in glomerular epithelial cell injury. Am J Physiol Renal Physiol. 2011;300:F1026–1042. doi: 10.1152/ajprenal.00187.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pippin JW, Kaverina NV, Eng DG, et al. Cells of renin lineage are adult pluripotent progenitors in experimental glomerular disease. Am J Physiol Renal Physiol. 2015;309:F341–358. doi: 10.1152/ajprenal.00438.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J, Yanez D, Floege A, et al. ACE-inhibition increases podocyte number in experimental glomerular disease independent of proliferation. Journal of the reninangiotensin-aldosterone system : JRAAS. 2015;16:234–248. doi: 10.1177/1470320314543910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pippin JW, Glenn ST, Krofft RD, et al. Cells of renin lineage take on a podocyte phenotype in aging nephropathy. Am J Physiol Renal Physiol. 2014;306:F1198–1209. doi: 10.1152/ajprenal.00699.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pichaiwong W, Hudkins KL, Wietecha T, et al. Reversibility of structural and functional damage in a model of advanced diabetic nephropathy. J Am Soc Nephrol. 2013;24:1088–1102. doi: 10.1681/ASN.2012050445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Godar S, Ince TA, Bell GW, et al. Growth-inhibitory and tumor-suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boehm JS, Zhao JJ, Yao J, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 59.Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schlote J, Schroder A, Dahlmann A, et al. Cardiovascular and renal effects of high salt diet in GDNF+/− mice with low nephron number. Kidney Blood Press Res. 2013;37:379–391. doi: 10.1159/000355716. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A, B) CD44 variant 3 is not detected in glomeruli of CD44+/+ (A) or CD44−/− (B) mice with FSGS (white arrow heads indicate glomeruli). Variant 3 (red) is detected in cells in the tubular interstitial space in CD44+/+ mice.
(C, D) CD44 isoform 10 is not detected in glomeruli of CD44+/+ (C) or CD44−/− (D) mice with FSGS (white arrow heads indicate glomeruli). Variant 10 (red) is detected in subset of cells in the tubular interstitial space (red) in CD44+/+ mice.
(E–H) CD44 variant 7 is not detected in glomeruli of CD44+/+ (E) or CD44−/− (F) mice with FSGS (dashed white squares indicate glomeruli). (G & H) higher magnification images of the glomeruli in images E & F. Variant 7 (red) is detected in a small subset of cells in the tubular interstitial space (red) in CD44+/+ mice.
(I, J) CD44 standard variant (brown), which excludes variants 1–10, is detected within tubular and interstitial cells as well as the glomerulus (indicated by black dashed square) in CD44+/+ mice. (J) higher magnification image of glomerulus in image I showing CD44 standard variant staining in PECs (black arrow heads).
Representative images of staining for (A) CD3 (activated T cell marker), (B) B220 (B cell marker), (C) F4/80 (macrophage marker) and (D) LY-6G (neutrophil marker) in diseased mice. White arrow heads indicate examples of positive staining in cells. The accompanying table shows quantification for the number of positive cells per glomerulus for each cell type . The number were extremely small, did not change much with disease (no significant glomerular influx) and showed no statistical differences between CD44+/+ and CD44−/− mice with this experimental model of FSGS.
Representative images of staining for hyaluronic acid binding protein in (A) CD44+/+ mice and (B) CD44−/− mice. The accompanying table shows quantification for the percentage of glomeruli with positive HABP staining. There was no difference between CD44+/+ and CD44−/− mice at baseline. The percentage of glomeruli with positive HABP staining increased with disease, but there was no statistical difference between CD44+/+ and CD44−/− mice.