

Abstract
Background
Early studies established that certain lipids were lower in acute myeloid leukemia (AML) cells than normal leukocytes. Because lipids are now known to play an important role in cell signaling and regulation of homeostasis, and are often perturbed in malignancies, we undertook a comprehensive lipidomic survey of plasma from AML patients at time of diagnosis and also healthy blood donors.
Methods
Plasma lipid profiles were measured using three mass spectrometry platforms in 20 AML patients and 20 healthy blood donors. Data were collected on total cholesterol and fatty acids, fatty acid amides, glycerolipids, phospholipids, sphingolipids, cholesterol esters, coenzyme Q10 and eicosanoids.
Results
We observed a depletion of plasma total fatty acids and cholesterol, but an increase in certain free fatty acids with the observed decline in sphingolipids, phosphocholines, triglycerides and cholesterol esters probably driven by enhanced fatty acid oxidation in AML cells. Arachidonic acid and precursors were elevated in AML, particularly in patients with high bone marrow (BM) or peripheral blasts and unfavorable prognostic risk. PGF2α was also elevated, in patients with low BM or peripheral blasts and with a favorable prognostic risk. A broad panoply of lipid classes is altered in AML plasma, pointing to disturbances of several lipid metabolic interconversions, in particular in relation to blast cell counts and prognostic risk.
Conclusions
These data indicate potential roles played by lipids in AML heterogeneity and disease outcome.
General significance
Enhanced catabolism of several lipid classes increases prognostic risk while plasma PGF2α may be a marker for reduced prognostic risk in AML.
Abbreviations: AML, acute myeloid leukemia; MUFA, monounsaturated fatty acid; CML, chronic myelogenous leukemia; ALL, acute lymphoblastic leukemia; FAO, fatty acid oxidation; CPT1a, carnitine palmitate transferase 1a; SCD1, stearoyl CoA desaturase 1; FAB, French-American-British classification; GCMS, gas chromatography–mass spectrometry; FAME, fatty acid methyl ester; PCA, principal components analysis; PLS-DA, projection to latent structures-discriminant analysis; OPLS-DA, orthogonal PLS-DA; UPLC-ESI-QTOFMS, ultraperformance liquid chromatography-electrospray ionization-quadrupole time-of-flight mass spectrometry; ESI +, electrospray ionization positive mode; ESI-, electrospray ionization negative mode; PUFA, polyunsaturated fatty acid; FLC-QqLIT-MS, fast liquid chromatography-quadrupole linear ion-trap mass spectrometry; MRM, multiple reactions monitoring; LPE, lysophosphatidylethanolamine; MG, monoacylglycerol; DG, diacylglycerol; TG, triacylglycerol (triglyceride); FAA, fatty acid amide; POEA, palmitoleoyl ethanolamide; LPC, lysophosphatidylcholine; PC, phosphatidylcholine; PE, phosphatidylethanolamine; SM, sphingomyelin; Cer, ceramide; CE, cholesterol ester; CoQ10, coenzyme Q10; DGLA, dihomo-γ-linoleic acid; AA, arachidonic acid; 8,9-DHET, 8,9-dihydroxy-5Z,11Z,14Z-eicosatrienoic acid; EPA, eicosapentaenoic acid (20:5;5Z,8Z,11Z,14Z,17Z); 12-HEPE, 12-hydroxy-5Z,8Z,10E,14Z,17Z-eicosapentaenoic acid; 12-LOX, 12-lipoxygenase; PGE2, prostaglandin E2; PGF2α, prostaglandin F2α; PGF1α, prostaglandin 1α; TxB2, thromboxane B2; TxA2, thromboxane A2; mPGES-1, microsomal prostaglandin E synthase-1; 2OG, 2-oxoglutarate; 2HG, (R)-2-hydroxyglutarate; DIC, disseminated intravascular coagulation; PGH2, prostaglandin H2
Keywords: Acute myeloid leukemia, Lipidomics, Fatty acids, Eicosanoids, Blast cell number, Prognostic risk
Graphical abstract
The AML lipidomic landscape.
Highlights
-
•
Many lipids are depleted in AML plasma likely due to induced fatty acid oxidation
-
•
Plasma arachidonic acid and precursors correlated with unfavorable prognostic risk
-
•
Plasma PGF2α at diagnosis correlated with favorable prognostic risk
-
•
Many metabolic interconversions perturbed in relation to blast cell numbers
-
•
AML heterogeneity related to plasma lipidome disturbances
1. Introduction
Acute myeloid leukemia (AML) is a fatal disease with a heterogeneous genomic and cytogenetic profile. While traditional cell signaling cascades have attracted considerable research interest in an attempt to understand the pathogenesis of AML and to optimize AML therapy [1], little is known regarding the role of lipid mediators in AML cell proliferation or in disease prognosis. However, the study of lipid profiles and lipid metabolism in myeloid leukemias such as AML has a long history. In the 1960s, the lipid content of various normal and abnormal leukocytes was reported, in which total cholesterol, for example, was significantly lower in AML cells than in normal leukocytes [2]. In a single chronic myelogenous leukemia (CML), total lymphatic cell lipid was almost five-times lower than from corresponding healthy cells. In addition, the pattern of ether-linked neutral glycerides also differed considerably between CML and healthy cells [3]. Subsequently, the lipid composition of AML myeloblasts and immature bone marrow myeloid cells from healthy persons was compared with that of normal mature human neutrophils. There was a decreased total cholesterol and cholesterol/phospholipid ratio, with an increased percentage of unsaturated fatty acids, when compared with normal mature neutrophils [4]. Recently, AML patients were reported to display lower HDL cholesterol than either healthy controls or ALL patients, with lower total cholesterol and LDL cholesterol seen only in male AML patients [5]. It thus appeared that AML cells might display an increased lipid catabolism. A later study showed that pharmacological inhibition of fatty acid oxidation (FAO) retarded proliferation of AML cells cultured on a feeder layer of mesenchymal stromal cells. The authors proposed that the shift to nonoxidative fatty acid metabolism, such as generation of ceramide, may decrease cell survival [6]. Accordingly, FAO may represent a biochemical characteristic of AML proliferation and this was borne out by a study of the proliferation of AML blasts from 23 patients that was inhibited by incubation with the non-β-oxidizable fatty acid tetradecylthioacetic acid [7]. FAO has recently been recognized as a key component of cancer cells [8].
In order to attempt to understand better the extent of perturbation of the plasma lipidome in AML, we have conducted a mass spectrometry-based lipidomic investigation of AML patients and healthy blood donor controls. Additionally, we have analyzed certain clinical disease features at the time of AML diagnosis in relation to eicosanoid lipid mediators and their metabolic precursors, to comprehend what role these may play in the heterogeneity of this disease.
2. Materials and methods
2.1. Patients and samples
2.2. Gas chromatography–mass spectrometry fatty acid profiling
Profiles of plasma total (free and esterified) fatty acids were determined for 20 AML patients and 20 controls by gas chromatography–mass spectrometry (GCMS) after conversion to their corresponding fatty acid methyl esters (FAMEs) as we have previously described [12].
2.3. Ultraperformance liquid chromatography-electrospray ionization-quadrupole time-of-flight mass spectrometry untargeted lipidomics
An untargeted lipidomic investigation of plasma from 20 AML patients and 20 controls was conducted using ultraperformance liquid chromatography-electrospray ionization-quadrupole time-of-flight mass spectrometry (UPLC-ESI-QTOFMS) by a modification of our published method [13]. Data were collected in continuum mode and the raw chromatographic data were imported into Progenesis QI 2.1 software (Nonlinear Dynamics, Newcastle-upon-Tyne, UK) for visualization of chromatograms as ion intensity maps, chromatogram alignment, peak picking, deconvolution and normalization. In ESI+ mode, the following adducts were considered when solving empirical formulae from accurate mass determinations: [M + H]+, [M + Na]+, [M + NH4]+, [2M + H]+, and [M + H-H2O]+. In ESI- mode, the following adducts were considered: [M-H]- and [M + Cl]-. When two or more different adducts were aligned, a neutral mass could be determined. Both up- and down-regulated AML plasma lipids were identified in Progenesis QI by searching various databases, including HMDB, ChemSpider, and Lipid Maps and reported on the basis of their fold change from control plasma and their statistical significance on the basis of ANOVA. Matching to database entries was made on the basis of accurate mass, isotope similarity and retention time.
2.4. Determination of plasma eicosanoids by fast liquid chromatography-quadrupole linear ion-trap mass spectrometry
Patient and control plasmas were analyzed in Leipzig for their concentration of seven polyunsaturated fatty acids (PUFAs) and 94 eicosanoids using fast liquid chromatography-quadrupole linear ion-trap mass spectrometry. The assay involved analysis of analytes and deuterated internal standards detected by 123 multiple reaction monitoring transitions using a 5500 QTrap mass spectrometer (AB Sciex, Darmstadt, Germany) operating in ESI mode and with quantitation performed against deuterated internal standards as described [14]. The specific multiple reaction monitoring transitions employed for 38 eicosanoids and 10 deuterated internal standards, together with their retention times, are given in Supplemental Table 1.
Table 1.
Characteristics of AML patients.
| Patient | Sex | Age at diagnosis | FAB | Molecular diagnosis | Cytogenetics | Prognostic risk stratification |
|---|---|---|---|---|---|---|
| 001 | f | 81 | M1 | NPM1mut; FLT3-ITD | Normal | Intermediate |
| 002 | m | 75 | M5 | normal | Complex abnormalities | Unfavorable |
| 003 | f | 70 | M1 | NPM1mut | Normal | Favorable |
| 005 | m | 72 | M6 | normal | Normal | Intermediate |
| 007 | m | 42 | M4 | normal | Monosomy 7 | Unfavorable |
| 009 | m | 70 | M0 | normal | Complex abnormalities | Unfavorable |
| 011 | m | 54 | M1 | normal | Trisomies 8, 19 | Unfavorable |
| 015 | m | 59 | M5 | NPM1mut | Normal | Favorable |
| 017 | m | 55 | M0 | normal | Normal | Intermediate |
| 022 | m | 68 | M0 | NPM1mut; FLT3-ITD | Normal | Intermediate |
| 027 | m | 59 | M6 | normal | Normal | Intermediate |
| 033 | m | 67 | M4 | normal | Normal | Intermediate |
| 040 | m | 67 | M4 | normal | Normal | Intermediate |
| 045 | f | 65 | M1 | normal | Complex abnormalities | Unfavorable |
| 046 | f | 62 | M2 | NPM1mut | Normal | Favorable |
| 050 | m | 49 | M0 | normal | Trisomy 11 | Intermediate |
| 051 | m | 72 | M2 | CEPBA | Normal | Favorable |
| 052 | m | 64 | M2 | CEPBA | Normal | Favorable |
| 056 | m | 62 | M1 | NPM1mut | Normal | Favorable |
| 058 | m | 66 | M1 | FLT3-ITD | Complex abnormalities | Unfavorable |
2.5. Statistics
Univariate statistical analysis was performed using GraphPad Prism 6.07 (GraphPad Software, Inc., La Jolla, CA). Means are expressed ± S.D. and p values are all two-sided. Group differences were analyzed nonparametrically using Kruskal-Wallis for three or more data sets or the Mann-Whitney U test for two data sets. In order to minimize false discovery, p values derived from multiple comparisons were subjected to Dunn's correction. These conservative measures were adopted to minimize the likelihood that lipid biomarkers for AML arose stochastically. Multivariate data analysis was conducted using SIMCA 14 (MKS Umetrics AB, Malmö, Sweden).
3. Results
3.1. Targeted fatty acid lipidomics by GCMS
Fig. 1.

Multivariate data analysis for GCMS lipidomics in AML and control plasmas. A, Unsupervised PCA scores plot. Red and green symbols represent AML and control plasma samples, respectively. B, Supervised PLS-DA. Red and green symbols represent AML and control plasma samples, respectively. C, Validation of the PLS-DA model using the leave-one-out procedure whereby one-seventh of the data is removed, randomized, returned, and the PLS-DA model rebuilt. One hundred such permutations were used. R2 is the correlation (purple symbols) and Q2 is the predictability (orange symbols). The data are not over-fitted because the permuted values for R2 and Q2 fall below 0.3 and zero, respectively, and therefore the PLS-DA model is valid. D, Orthogonal PLS-DA loadings S-plot showing lipid molecules that are upregulated (upper right quadrant) and downregulated in AML plasma (lower left quadrant). 1 = 18:2n-6, 2 = cholesterol, 3 = 18:0, 4 = 20:4n-6, 5 = 20:3n-6, 6 = 20:2n-6, 7 = 20:5n-3.
3.2. Untargeted lipidomics by UPLC-ESI-QTOFMS
UPLC-ESI-QTOFMS was used to conduct an untargeted lipidomic investigation [13] of 20 AML and 20 control plasmas. A total of 50 lipid molecules from several classes were found to be highly statistically significantly altered in the plasma of AML patients relative to healthy controls. The mass errors for their assignments averaged 0.9 ppm. Of these lipid molecules, 12 were elevated in AML plasma and comprised five free fatty acids (palmitic, palmitoleic, oleic, linoleic and arachidonic acids; 1.5- to 5.2-fold increase), two fatty acid amides (oleamide and palmitoleoyl ethanolamide; ∞-fold increase), two lysophosphatidylethanolamines (LPE(18:0) and LPE(20:2); 2.6- to 2.8-fold increase), and three glycerolipids, comprising a monoacylglycerol (MG(18:0)), a diacylglycerol (DG(34:1)) and a triacylglycerol (triglyceride; TG(56:5)) that were elevated 1.3- to 2.7-fold (Supplemental Table 2). The greatest statistical and biological finding was the appearance of two fatty acid amides (FAAs), oleamide and palmitoleoyl ethanolamide (POEA), in AML plasma, both of which can be considered to be endocannabinoids that act on CB1 and CB2 receptors [18], [19], with elevated plasma POEA reported to be associated with certain anxiety states [19] and oleamide being an important regulator of sleep via the CB1 receptor [20].
The 38 diminished lipid molecules in AML patient plasma are also shown in Supplemental Table 2. They fell into six lipid categories, comprising two lysophospholipids (LPC(20:2) and LPE(18:2); lowered 2.8- to 3.2-fold), four triglycerides (TG(51:8), TG(52:8), TG(53:8), and TG(53:9); lowered 2.1- to 2.7-fold), 13 phospholipids (PC(32:0), PC(36:2), two PC(36:3)s, PC(36:4), PC(36:5), PC(38:3), PC(40:7), PC(40:8), PC(P-36:4), PE(40:6), PE(44:10), and PS(36:2); lowered 1.4- to 29.5-fold), 14 sphingolipids (SM(18:1/12:0), SM(18:1/14:0), SM(18:1/20:0), two SM(18:1/22:1)s, SM(18:1/23:0), SM(18:1/24:1), SM(18:0/12:0), SM(18:0/22:0), SM(18:0/22:1), SM(18:0/24:0), two SM(18:0/24:1)s, and Cer(18:1/24:1); lowered 1.3- to 4.8-fold), four cholesterol esters (cholesterol 3-O-sulfate, CE(18:2), CE(18:3), and CE(20:3); lowered 1.4- to 2.1-fold), and coenzyme Q10 (CoQ10; lowered 1.6-fold). This fall in individual plasma triglycerides, phospholipids and cholesterol esters probably contributed to the attenuation of free and esterified individual plasma fatty acids and cholesterol observed above.
Selected lipids were tested for gender differences. No statistically significant differences between males and females, neither for the AML cases nor the controls, were found for these plasma lipids.
3.3. Targeted PUFA and eicosanoid lipidomics by FLC-QqLIT-MS
The targeted determination of PUFAs and their eicosanoid metabolites was conducted using FLC-QqLIT-MS. The MRM procedures used (Supplemental Table 1) permitted determination of plasma concentrations of PUFAs in the ng/ml range and eicosanoids in the pg/ml range [14]. This group of lipids comprises many important bioactive lipid mediators and their precursors. Because AML is a heterogeneous malignancy, these lipidomic findings have been expressed in relation to the severity of disease, in particular, number of bone marrow (BM) blasts, number of peripheral blasts, and prognostic risk stratification [11]. The current lipidomic study was of insufficient size to analyze the findings in relation to the major Class I—V gene mutations that co-operate in AML leukemogenesis [11]. As shown in Supplemental Fig. 4A, the % BM blasts were divided into intermediate (5–65%) and high (> 80%). Similarly, % peripheral blasts were divided into low (≤ 10%) and high (> 10%) (Supplemental Fig. 5A). Finally risk stratification was based upon a cytogenetic primary evaluation with genetic mutations providing secondary information [11], [21] (Table 1).
Fig. 3.

Lipid metabolic pathways significantly altered in relation to bone marrow (BM) blasts at diagnosis. Blue bars = plasma metabolite level in controls, orange bars = plasma metabolite level in AML patients with intermediate (5–65%) BM blasts, red bars = plasma metabolite level in AML patients with high (> 80%) BM blasts. Error bars represent SD. For distribution of BM blasts see S3A. * means p < 0.05; ** means p < 0.01; *** means p < 0.001 for differences from controls. # means p < 0.05 for differences from intermediate BM blasts. Enzymes involved are 1, fatty acid elongase; 2, Δ5-desaturase; 3, COX-1 and COX-2; 4, PGE synthase; 5, carbonyl reductase 1; 6, 15-hydroxyprostaglandin dehydrogenase; 7, prostacyclin synthase; 8, spontaneous reaction; 9, spontaneous reaction; 10, prostaglandin D synthase; 11, spontaneous reaction; 12, 5-lipoxygenase; 13, arachidonic acid 11-oxidoreductase; 14, 15-lipoxygenase.
Fig. 4.

Lipid metabolic pathways significantly altered in relation to peripheral blasts at diagnosis. Blue bars = plasma metabolite level in controls, orange bars = plasma metabolite level in AML patients with intermediate (≤ 10%) peripheral blasts, red bars = plasma metabolite level in AML patients with high (> 10%) peripheral blasts. Error bars represent SD. For distribution of BM blasts see Supplemental Fig. 5A. * means p < 0.05; ** means p < 0.01; *** means p < 0.001 for differences from controls. For key to enzymes, see Fig. 3.
Fig. 5.

Lipid metabolic pathways significantly altered in relation to risk stratification at diagnosis. Blue bars = plasma metabolite level in controls; green, orange and red bars = good, intermediate and bad risk, respectively. Error bars represent SD. * means p < 0.05; ** means p < 0.01; *** means p < 0.001 for differences from controls. # means p < 0.05 for differences from good risk patients. For key to enzymes, see Fig. 3.
Finally, in contrast to the AA canonical eicosanoid cascade, leading to the metabolites discussed above, data from the parallel pathway starting with eicosapentaenoic acid (EPA; 20:5n-3) was also collected (Supplemental Fig. 11C). Plasma concentrations of free EPA in controls and AML patients were similar (Supplemental Fig. 11A), in contrast to total (free and esterified) EPA (Fig. 2H). EPA is converted to thromboxane A3 (TxA3) by COX-1/COX-2 and CYP5A1, and then spontaneously hydrolyses to TxB3 [1]. This EPA metabolite was not detected in AML patient plasma, relative to control plasma (Supplemental Fig. 11B). Since COX-1/COX-2 clearly operate in AML, evidenced by AA metabolites in AML plasma, it must be assumed that CYP5A1 is deficient in AML. CYP5A1 is found in platelets [25] and, since the AML patients were thrombocytopenic, this may explain this finding.
Fig. 2.

Univariate data analysis for GCMS lipidomics in AML and control plasmas. Red and green symbols represent AML and control plasma samples, respectively. Vertical dotted lines are median values, with the same color code. p-Values derive from the Mann-Whitney U test. Values are relative concentrations (peak area/internal standard peak area) measured by GCMS. A, palmitic acid (16:0); B, stearic acid (18:0); C, oleic acid (18:1n-9); D, linoleic acid (18:2n-6); E, eicosadienoic acid (20:2n-6); F, eicosatrienoic acid (20:3n-6); G, arachidonic acid (20:4n-6); H, eicosapentaenoic acid (20:5n-3); I, lignoceric acid (24:0); J, cholesterol.
Selected lipids were tested for gender differences. No statistically significant differences between males and females, neither for the AML cases nor the controls, were found for the following plasma eicosanoids: DHGLA [20:3(8Z,11Z,14Z)], arachidonic acid [20:4(5Z, 8Z,11Z,14Z)], EPA [20:5(5Z, 8Z,11Z,14Z,17Z)], 8,9-DHET, 11-dehydro-TxB2, PGE2/8-isoPGE2, 12-HEPE, 11β-PGE2, 15-keto-PGF2α, 15-keto-PGE2, 8-iso-15-keto-PGF2α, PGF1α, and PGF2α.
3.4. Summary description of the AML lipidome
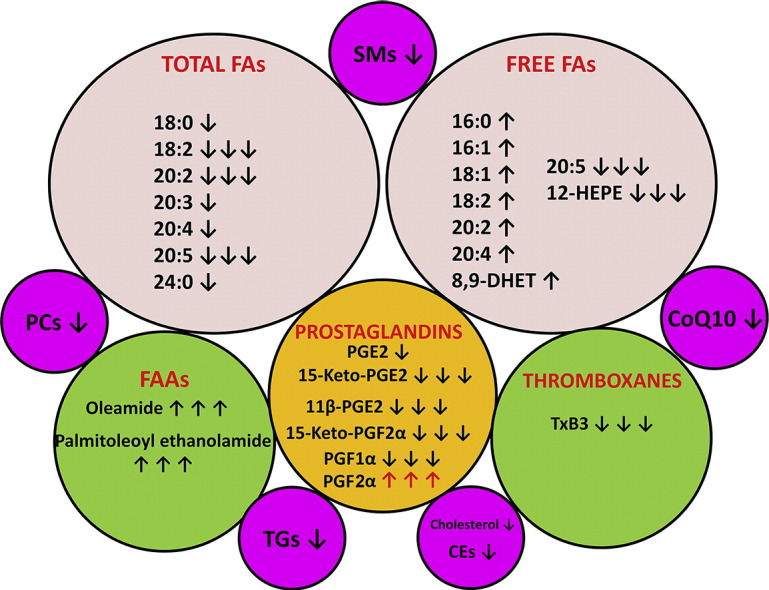
Fig. 6.

The AML lipidomic landscape. Ten lipid pools are shown. ↑ means lipid upregulated in AML plasma relative to control plasma. ↑↑↑ means lipid massively upregulated in AML plasma, not detected in control plasma. ↓ means lipid downregulated in AML plasma. ↓↓↓ means lipid massively downregulated in AML plasma or not detected in AML plasma. For lipid names see text.
Two FAAs, that are considered to act as endocannabinoids, displayed an unquantifiable increase in AML plasma, since they were undetectable in control plasmas (Supplemental Table 2). Finally, CoQ10, which is a key component of the mitochondrial electron transport chain that generates ATP by aerobic respiration and also a component of serum lipoproteins [30], was 40% reduced in AML plasma (p = 5.5 × 10− 11).
Thus, the landscape of the AML plasma lipidome is one involving a global reprogramming of lipid biochemistry that involves many of the major classes of lipids, including highly potent lipid mediators. This metabolic rewiring displays no gender differences. The implications of these findings for AML patients will be hitherto discussed.
4. Discussion
The concept that cancer cells engage in major metabolic reprogramming is borne out by this lipidomic study of AML plasma. A major depletion of total fatty acids and cholesterol was observed. This is remarkable given that the AML patients were older than the controls with a greater proportion of males. Had it been possible to obtain truly age- and sex-matched controls, it is very likely that these differences would have been even greater. These observation are consistent with earlier reports [2], [3], [4], [5] that lead to the conclusion that AML cells may display elevated lipid catabolic rates, including FAO, a process now recognized as a key component of cancer cells [8]. CT2 (SLC22A16) is a plasma membrane carnitine transporter that is over expressed in AML cells relative to normal hematopoietic cells from healthy subjects. Carnitine is required for the transport of fatty acids into the mitochondrion for FAO [31]. Knockdown of CT2 with shRNA reduced the growth and viability of AML cells, suggesting that CT2 and therefore FAO, may be a target for AML treatment [31]. The observations reported here of massively reduced total fatty acid plasma concentrations supports the essential role of FAO in AML. Because FAO feeds the Krebs cycle with abundant acetyl-CoA, increased amounts of citrate will be generated, which is the starting point for de novo fatty acid synthesis for new membrane components [32]. This represents a “futile metabolic cycle” of fatty acid synthesis and breakdown, suggested to be a hallmark of cancer cell metabolism [8]. Breaking this cycle has been seen as a means to develop novel therapeutic strategies against AML, for example, by blocking the rate limiting enzyme of FAO, carnitine palmitoyl transferase 1a (CPT1a), with the inhibitor ST1326 [33]. Clearly, understanding in vivo lipid homeostasis in AML patients may assist in the development of new drug targets and therapies.
The observed decline in sphingolipids, phosphocholines, triglycerides and cholesterol esters in AML plasma relative to controls may be driven by the enhanced rates of FAO in AML cells. Further work will be required to examine the discrete mechanisms involved in these phenomena within AML blasts, but this falls beyond the scope of this project. However, it is of interest to note that, although the AML patients were significantly older, with more males, than the blood donor controls recruited, they, counterintuitively, had lower plasma lipids, including total cholesterol and total fatty acids. As stated earlier, had the two groups been age- and sex-matched, the plasma lipid differences would have been expected to be even greater than reported here.
Two fatty acyl amides (FAAs), oleamide and POEA, were detected in AML plasma but were not detected in any control plasma. Oleamide is a sleep regulator via the CB1 receptor [20] and POEA has been implicated in anxiety disorders seen in abstinent cocaine addicts [19]. The appearance of these endocannabinoid-like FAAs in AML plasma may be responsible for some of the symptoms experienced by AML patients, such as lethargy and loss of appetite. Moreover, it has been reported that both pediatric [34] and adult [35] AML patients can develop hyperglycemia. The endocannabinoid system, which can also promote hyperglycemia [36], [37] warrants further investigation in AML.
The reduction in CoQ10 plasma concentration in AML may be of relevance. Chemical inhibition of CoQ10 in AML HL-60 cells led to increased cellular levels of superoxide [30]. It is unclear the extent to which plasma CoQ10 concentrations reflect cellular levels and to what degree CoQ10 protects against apoptosis of AML cells as it does for cells in culture [38]. Little literature exists for CoQ10 in AML.
The finding reported here that are most relevant to the biology of AML are the differential levels in AML and control plasma of the measured PUFAs and their eicosanoid metabolites. Although AA is principally produced by the action of sPLA2 and cPLA2 on plasma membrane phospholipids [39], which release preformed AA into the cytosol, it may also be synthesized de novo from 18:2n-6 by the action of elongases and desaturases [40]. Both AA and its precursors 18:3n-6 and 20:3n-6 were elevated in AML plasma. The association of prominent AA concentrations in AML plasma particularly with high BM blasts, high peripheral blasts and both intermediate and unfavorable prognostic risk patients may reflect the importance of the AA cascade in AML. Interestingly, AML cells express a reduced capacity for stimulated release of AA that is unrelated to cPLA2 [41]. This may switch AA production towards de novo synthesis, as observed here. AA is the precursor of many biological mediators, including prostaglandins, prostacyclin, thromboxanes, leukotrienes and hepoxilins. Various members of these groups were determined in this study. Of potential importance was the observation that PGF2α was elevated, in particular in relation to AML with low BM and peripheral blast and with a favorable prognostic risk. Early studies reported that, at low concentration, PGF2α had stimulatory effects upon cultured mouse leukemia lymphoblasts, but at higher concentrations, PGF2α was inhibitory [42]. In addition, mouse myeloid leukemia cells were inhibited by PGF2α from differentiating into macrophages and granulocytes on stimulation with various inducers [43]. These observations would be consistent with the findings reported here. Evidence was obtained that 15-hydroxyprostaglandin dehydrogenase (NAD+) (15-PGDH; EC 1.1.1.141) activity was downregulated in AML, due to the massively reduced plasma levels of 15-keto-PGE2, which was detected in only 1/20 AML plasma samples. HL-60 cells have been reported to possess this metabolic activity with respect to PGE2 and 15-HETE [44]. Both of these activities in vivo were observed in this study to be diminished in AML patients. HPGD that encodes 15-PGDH has been proposed to act as a tumor suppressor gene that is lost in pituitary adenomas [45]. The metabolic data presented here point to the loss or impairment of this potential tumor suppressor pathway in AML. Finally, PGJ2 is formed rapidly and spontaneously from PGD2 in mast cells and has antiproliferative potency [23], [46]. AML plasma had markedly reduced concentrations of PGJ2, particularly in high BM blast, high peripheral blast and unfavorable prognosis risk patients. The extent to which this prostaglandin and its metabolite Δ12-PGJ2 contribute to the clinical picture in certain AML patients is currently unknown.
Despite the profound alteration in the plasma lipidome in AML, a number of important and related questions remain. First, is the changed lipid profile in AML contributing to the pathobiology or is AML itself contributing to the altered plasma lipidome? The answer to this may come from the observation that our patient group, albeit small, was highly genetically heterogeneous. Moreover, some patients had a high proportion and others a low proportion of blasts. Yet, many of the reported lipidomic changes were universal to AML, suggesting that even a small number of blasts could disturb the plasma lipidome. In contrast, other lipid alterations, particularly among the eicosanoids, were related to disease severity and prognostic risk. Here, the disease may have contributed to the distorted lipid metabolism.
Second, to what extent do AML blast cells produce the observed lipid metabolites or lipid metabolizing enzymes? This is partly answered by the foregoing, in that certain eicosanoids, in particular, were elevated in patients with the greatest proportion of blasts. Future investigations will be required to provide definitive answers through a comparison of the AML blast lipidome with that of leukocytes from healthy subjects, where early data already exist [2], [3], [4].
Finally, as stated above, disturbances of lipid homeostasis in leukemias were first observed from the lowered lipid content of leukemic cells [2], [3], [4]. The data presented here are from lipidomic analyses of AML and control plasma. The question remains, are these intracellular and extracellular lipid perturbations related and, if so, by what mechanisms? The relatively recent recognition that membrane ABC transporters were able to transport natural lipids and lipid analogues [47] has led to further investigation of cellular lipid export by ABC transporters. For example, cholesterol was reported to be transported out of cells by ABCA1 and that mutations in the ABCA1 gene were responsible for Tangier disease [47], [48], [49]. Furthermore, activity of the multidrug transporter P-glycoprotein (ABCB1) has been shown to be stimulated by the presence of cholesterol either extracellularly [50] or in the membrane [51] and ABCB1 actively redistributes cholesterol from the cytoplasmic side to the exoplasmic side of the membrane [51]. Evidence has been presented for the cellular export of other lipid entities by a range of ABC transporters [52]. With regard to AML, cell culture experiments demonstrated that AML cells are able to upregulate through epigenetic mechanisms the expression of a number of transporter proteins [53]. The mRNA expression for ABCA5, ABCB6, ABCC1, and ABCC3 has recently been reported to be upregulated in cells from 233 AML patients [54]. It is therefore likely that perturbed plasma lipid profiles in AML are related to intracellular changes in circulating AML cells through mechanisms involving multiple ABC transporters.
In summary, this multiplatform lipidomic investigation of AML has uncovered a number of metabolic pathways that were both up- and down-regulated in AML, many of which were previously unknown in AML. Several of these dysregulated pathways point towards potential contributing mechanisms in AML pathogenesis. Moreover, some pathways mapped to the most severe clinical disease features, including high BM and peripheral blasts and unfavorable prognostic risks, determined at diagnosis by cytogenetic and genetic analyses. This first lipidomic study of AML provides a springboard for several new directions of research into AML and may provide opportunities to identify novel druggable targets for the treatment of this leukemia that currently has only a 26% 5-year survival rate.
5. Conclusion
Patients with AML have a significant number of altered plasma lipids at diagnosis. Components of the lipidome affected include total cholesterol and fatty acids, fatty acid amides, glycerolipids, phospholipids, sphingolipids, cholesterol esters, coenzyme Q10, together with many eicosanoids. In particular, arachidonic acid precursors and eicosanoid metabolites are positively related to disease severity and prognostic outlook and therefore may be drivers of the disease. Of all lipids determined in this study, plasma PGF2α at the time of diagnosis may be a marker for reduced disease severity and prognostic risk. Understanding perturbations in the AML plasma lipidome may lead to new targets for drug therapy of AML.
Transparency document
Transparency document.
Acknowledgments
DB and JRI wish to thank Kjetil Johnsen of Johnsen Consulting and Services, Geneva for GCMS instrument maintenance and training.
Footnotes
The transparency document associated with this article can be found, in online version.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbacli.2017.03.002.
Appendix A. Supplementary data
Supplementary material
References
- 1.Ruvolo P.P., Qiu Y., Coombes K.R., Zhang N., Neeley E.S., Ruvolo V.R., Hail N., Jr., Borthakur G., Konopleva M., Andreeff M., Kornblau S.M. Phosphorylation of GSK3alpha/beta correlates with activation of AKT and is prognostic for poor overall survival in acute myeloid leukemia patients. BBA Clin. 2015;4:59–68. doi: 10.1016/j.bbacli.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gottfried E.L. Lipids of human leukocytes: relation to celltype. J. Lipid Res. 1967;8:321–327. [PubMed] [Google Scholar]
- 3.Snyder F., Wood R. Alkyl and alk-1-enyl ethers of glycerol in lipids from normal and neoplastic human tissues. Cancer Res. 1969;29:251–257. [PubMed] [Google Scholar]
- 4.Klock J.C., Pieprzyk J.K. Cholesterol, phospholipids, and fatty acids of normal immature neutrophils: comparison with acute myeloblastic leukemia cells and normal neutrophils. J. Lipid Res. 1979;20:908–911. [PubMed] [Google Scholar]
- 5.Usman H., Rashid R., Ameer F., Iqbal A., Zaid M., Hasnain S., Kalbacher H., Zaidi N. Revisiting the dyslipidemia associated with acute leukemia. Clin. Chim. Acta. 2015;444:43–49. doi: 10.1016/j.cca.2015.01.038. [DOI] [PubMed] [Google Scholar]
- 6.I. Samudio, R. Harmancey, M. Fiegl, H. Kantarjian, M. Konopleva, B. Korchin, K. Kaluarachchi, W. Bornmann, S. Duvvuri, H. Taegtmeyer, M. Andreeff, Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction, J. Clin. Invest., 120 (2010) 142–156. [DOI] [PMC free article] [PubMed]
- 7.Tronstad K.J., Bruserud O., Berge K., Berge R.K. Antiproliferative effects of a non-beta-oxidizable fatty acid, tetradecylthioacetic acid, in native human acute myelogenous leukemia blast cultures. Leukemia. 2002;16:2292–2301. doi: 10.1038/sj.leu.2402698. [DOI] [PubMed] [Google Scholar]
- 8.Carracedo A., Cantley L.C., Pandolfi P.P. Cancer metabolism: fatty acid oxidation in the limelight. Nat. Rev. Cancer. 2013;13:227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett J.M., Catovsky D., Daniel M.T., Flandrin G., Galton D.A., Gralnick H.R., Sultan C. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br. J. Haematol. 1976;33:451–458. doi: 10.1111/j.1365-2141.1976.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 10.Bennett J.M., Catovsky D., Daniel M.T., Flandrin G., Galton D.A., Gralnick H.R., Sultan C. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann. Intern. Med. 1985;103:620–625. doi: 10.7326/0003-4819-103-4-620. [DOI] [PubMed] [Google Scholar]
- 11.Kansal R. Acute myeloid leukemia in the era of precision medicine: recent advances in diagnostic classification and risk stratification. Cancer Biol. Med. 2016;13:41–54. doi: 10.28092/j.issn.2095-3941.2016.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patterson A.D., Maurhofer O., Beyoglu D., Lanz C., Krausz K.W., Pabst T., Gonzalez F.J., Dufour J.F., Idle J.R. Aberrant lipid metabolism in hepatocellular carcinoma revealed by plasma metabolomics and lipid profiling. Cancer Res. 2011;71:6590–6600. doi: 10.1158/0008-5472.CAN-11-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beyoglu D., Krausz K.W., Martin J., Maurhofer O., Dorow J., Ceglarek U., Gonzalez F.J., Dufour J.F., Idle J.R. Disruption of tumor suppressor gene Hint1 leads to remodeling of the lipid metabolic phenotype of mouse liver. J. Lipid Res. 2014;55:2309–2319. doi: 10.1194/jlr.M050682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kortz L., Dorow J., Becker S., Thiery J., Ceglarek U. Fast liquid chromatography-quadrupole linear ion trap-mass spectrometry analysis of polyunsaturated fatty acids and eicosanoids in human plasma. J. Chromatogr. B. 2013;927:209–213. doi: 10.1016/j.jchromb.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 15.Lanz C., Ledermann M., Slavik J., Idle J.R. The production and composition of rat sebum is unaffected by 3 Gy gamma radiation. Int. J. Radiat. Biol. 2011;87:360–371. doi: 10.3109/09553002.2010.537432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braiterman L.T., Watkins P.A., Moser A.B., Smith K.D. Peroxisomal very long chain fatty acid beta-oxidation activity is determined by the level of adrenoleukodystrophy protein (ALDP) expression. Mol. Genet. Metab. 1999;66:91–99. doi: 10.1006/mgme.1998.2789. [DOI] [PubMed] [Google Scholar]
- 17.Christinat N., Morin-Rivron D., Masoodi M. High-throughput quantitative lipidomics analysis of nonesterified fatty acids in human plasma. J. Proteome Res. 2016;15:2228–2235. doi: 10.1021/acs.jproteome.6b00198. [DOI] [PubMed] [Google Scholar]
- 18.Hopps J.J., Dunn W.R., Randall M.D. Enhanced vasorelaxant effects of the endocannabinoid-like mediator, oleamide, in hypertension. Eur. J. Pharmacol. 2012;684:102–107. doi: 10.1016/j.ejphar.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 19.Pavon F.J., Araos P., Pastor A., Calado M., Pedraz M., Campos-Cloute R., Ruiz J.J., Serrano A., Blanco E., Rivera P., Suarez J., Romero-Cuevas M., Pujadas M., Vergara-Moragues E., Gornemann I., Torrens M., de la Torre R., Rodriguez de Fonseca F. Evaluation of plasma-free endocannabinoids and their congeners in abstinent cocaine addicts seeking outpatient treatment: impact of psychiatric co-morbidity. Addict. Biol. 2013;18:955–969. doi: 10.1111/adb.12107. [DOI] [PubMed] [Google Scholar]
- 20.Murillo-Rodriguez E. The role of the CB1 receptor in the regulation of sleep. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2008;32:1420–1427. doi: 10.1016/j.pnpbp.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 21.Falini B., Martelli M.P. Impact of genomics in the clinical management of patients with cytogenetically normal acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2015;28:90–97. doi: 10.1016/j.beha.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Kauffman G.L., Jr., Whittle B.J. Gastric vascular actions of prostanoids and the dual effect of arachidonic acid. Am. J. Physiol. 1982;242:G582–G587. doi: 10.1152/ajpgi.1982.242.6.G582. [DOI] [PubMed] [Google Scholar]
- 23.Fukushima M. Biological activities and mechanisms of action of PGJ2 and related compounds: an update. Prostaglandins Leukot. Essent. Fatty Acids. 1992;47:1–12. doi: 10.1016/0952-3278(92)90178-l. [DOI] [PubMed] [Google Scholar]
- 24.Agins A.P., Delhagen J.E. Metabolism of prostaglandin E2 by human HL-60 leukemia cells. Agents Actions. 1987;21:400–402. doi: 10.1007/BF01966528. [DOI] [PubMed] [Google Scholar]
- 25.Nakahata N. Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol. Ther. 2008;118:18–35. doi: 10.1016/j.pharmthera.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Babcock T., Helton W.S., Espat N.J. Eicosapentaenoic acid (EPA): an antiinflammatory omega-3 fat with potential clinical applications. Nutrition. 2000;16:1116–1118. doi: 10.1016/s0899-9007(00)00392-0. [DOI] [PubMed] [Google Scholar]
- 27.Gomolka B., Siegert E., Blossey K., Schunck W.H., Rothe M., Weylandt K.H. Analysis of omega-3 and omega-6 fatty acid-derived lipid metabolite formation in human and mouse blood samples. Prostaglandins Other Lipid Mediat. 2011;94:81–87. doi: 10.1016/j.prostaglandins.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 28.Yokomizo T., Izumi T., Takahashi T., Kasama T., Kobayashi Y., Sato F., Taketani Y., Shimizu T. Enzymatic inactivation of leukotriene B4 by a novel enzyme found in the porcine kidney. Purification and properties of leukotriene B4 12-hydroxydehydrogenase. J. Biol. Chem. 1993;268:18128–18135. [PubMed] [Google Scholar]
- 29.Stenke L., Sjolinder M., Miale T.D., Lindgren J.A. Novel enzymatic abnormalities in AML and CML in blast crisis: elevated leucocyte leukotriene C4 synthase activity paralleled by deficient leukotriene biosynthesis from endogenous substrate. Br. J. Haematol. 1998;101:728–736. doi: 10.1046/j.1365-2141.1998.00752.x. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez-Aragon D., Buron M.I., Lopez-Lluch G., Herman M.D., Gomez-Diaz C., Navas P., Villalba J.M. Coenzyme Q and the regulation of intracellular steady-state levels of superoxide in HL-60 cells. Biofactors. 2005;25:31–41. doi: 10.1002/biof.5520250105. [DOI] [PubMed] [Google Scholar]
- 31.Wu Y., Hurren R., MacLean N., Gronda M., Jitkova Y., Sukhai M.A., Minden M.D., Schimmer A.D. Carnitine transporter CT2 (SLC22A16) is over-expressed in acute myeloid leukemia (AML) and target knockdown reduces growth and viability of AML cells. Apoptosis. 2015;20:1099–1108. doi: 10.1007/s10495-015-1137-x. [DOI] [PubMed] [Google Scholar]
- 32.Samudio I., Konopleva M. Targeting leukemia's “fatty tooth”. Blood. 2015;126:1874–1875. doi: 10.1182/blood-2015-08-665125. [DOI] [PubMed] [Google Scholar]
- 33.Ricciardi M.R., Mirabilii S., Allegretti M., Licchetta R., Calarco A., Torrisi M.R., Foa R., Nicolai R., Peluso G., Tafuri A. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. 2015;126:1925–1929. doi: 10.1182/blood-2014-12-617498. [DOI] [PubMed] [Google Scholar]
- 34.Banihashem A., Ghasemi A., Ghaemi N., Moazzen N., Amirabadi A. Prevalence of transient hyperglycemia and diabetes mellitus in pediatric patients with acute leukemia. Iran J Pediatr. Hematol. Oncol. 2014;4:5–10. [PMC free article] [PubMed] [Google Scholar]
- 35.Ali N.A., O'Brien J.M., Jr., Blum W., Byrd J.C., Klisovic R.B., Marcucci G., Phillips G., Marsh C.B., Lemeshow S., Grever M.R. Hyperglycemia in patients with acute myeloid leukemia is associated with increased hospital mortality. Cancer. 2007;110:96–102. doi: 10.1002/cncr.22777. [DOI] [PubMed] [Google Scholar]
- 36.Gruden G., Barutta F., Kunos G., Pacher P. 2015. Role of the Endocannabinoid System in Diabetes and Diabetic Complications. Br J Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Semmo N., Weber T., Idle J.R., Beyoglu D. Metabolomics reveals that aldose reductase activity due to AKR1B10 is upregulated in hepatitis C virus infection. J. Viral Hepat. 2015;22:617–624. doi: 10.1111/jvh.12376. [DOI] [PubMed] [Google Scholar]
- 38.Fernandez-Ayala D.J., Martin S.F., Barroso M.P., Gomez-Diaz C., Villalba J.M., Rodriguez-Aguilera J.C., Lopez-Lluch G., Navas P. Coenzyme Q protects cells against serum withdrawal-induced apoptosis by inhibition of ceramide release and caspase-3 activation. Antioxid. Redox Signal. 2000;2:263–275. doi: 10.1089/ars.2000.2.2-263. [DOI] [PubMed] [Google Scholar]
- 39.Balsinde J., Dennis E.A. Distinct roles in signal transduction for each of the phospholipase A2 enzymes present in P388D1 macrophages. J. Biol. Chem. 1996;271:6758–6765. doi: 10.1074/jbc.271.12.6758. [DOI] [PubMed] [Google Scholar]
- 40.Murff H.J., Edwards T.L. 2014. Endogenous Production of Long-Chain Polyunsaturated Fatty Acids and Metabolic Disease Risk, Curr Cardiovasc Risk Rep, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Runarsson G., Feltenmark S., Forsell P.K., Sjoberg J., Bjorkholm M., Claesson H.E. The expression of cytosolic phospholipase A2 and biosynthesis of leukotriene B4 in acute myeloid leukemia cells. Eur. J. Haematol. 2007;79:468–476. doi: 10.1111/j.1600-0609.2007.00967.x. [DOI] [PubMed] [Google Scholar]
- 42.Yang T.J., Dale J.B., Machanoff R. Effects of prostaglandins E1, E2, and F2alpha on the growth of leukaemia cells in culture. J. Cell Sci. 1976;20:199–206. doi: 10.1242/jcs.20.1.199. [DOI] [PubMed] [Google Scholar]
- 43.Takenaga K., Honma Y., Kado J., Hozumi M. Mechanisms of inhibition of mouse myeloid leukemic cell differentiation by prostaglandin F2 alpha. Gann. 1982;73:175–183. [PubMed] [Google Scholar]
- 44.Agins A.P., Zipkin R.E., Taffer I.M. Metabolism of cyclooxygenase and lipoxygenase products by 15-prostaglandin dehydrogenase from human HL-60 leukemia cells. Agents Actions. 1987;21:397–399. doi: 10.1007/BF01966527. [DOI] [PubMed] [Google Scholar]
- 45.Bai J.W., Wang Z., Gui S.B., Zhang Y.Z. Loss of 15-hydroxyprostaglandin dehydrogenase indicates a tumor suppressor role in pituitary adenomas. Oncol. Rep. 2012;28:714–720. doi: 10.3892/or.2012.1806. [DOI] [PubMed] [Google Scholar]
- 46.Haberl C., Hultner L., Flugel A., Falk M., Geuenich S., Wilmanns W., Denzlinger C. Release of prostaglandin D2 by murine mast cells: importance of metabolite formation for antiproliferative activity. Mediators Inflamm. 1998;7:79–84. doi: 10.1080/09629359891216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borst P., Zelcer N., van Helvoort A. ABC transporters in lipid transport. Biochim. Biophys. Acta. 2000;1486:128–144. doi: 10.1016/s1388-1981(00)00053-6. [DOI] [PubMed] [Google Scholar]
- 48.Bodzioch M., Orso E., Klucken J., Langmann T., Bottcher A., Diederich W., Drobnik W., Barlage S., Buchler C., Porsch-Ozcurumez M., Kaminski W.E., Hahmann H.W., Oette K., Rothe G., Aslanidis C., Lackner K.J., Schmitz G. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 49.Brooks-Wilson A., Marcil M., Clee S.M., Zhang L.H., Roomp K., van Dam M., Yu L., Brewer C., Collins J.A., Molhuizen H.O., Loubser O., Ouelette B.F., Fichter K., Ashbourne-Excoffon K.J., Sensen C.W., Scherer S., Mott S., Denis M., Martindale D., Frohlich J., Morgan K., Koop B., Pimstone S., Kastelein J.J., Genest J., Jr., Hayden M.R. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 50.Connelly-Smith L., Pattinson J., Grundy M., Shang S., Seedhouse C., Russell N., Pallis M. P-glycoprotein is downregulated in KG1a-primitive leukemia cells by LDL cholesterol deprivation and by HMG-CoA reductase inhibitors. Exp. Hematol. 2007;35:1793–1800. doi: 10.1016/j.exphem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 51.Garrigues A., Escargueil A.E., Orlowski S. The multidrug transporter, P-glycoprotein, actively mediates cholesterol redistribution in the cell membrane. Proc. Natl. Acad. Sci. U. S. A. 2002;99:10347–10352. doi: 10.1073/pnas.162366399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kruh G.D., Belinsky M.G. The MRP family of drug efflux pumps. Oncogene. 2003;22:7537–7552. doi: 10.1038/sj.onc.1206953. [DOI] [PubMed] [Google Scholar]
- 53.Hauswald S., Duque-Afonso J., Wagner M.M., Schertl F.M., Lubbert M., Peschel C., Keller U., Licht T. Histone deacetylase inhibitors induce a very broad, pleiotropic anticancer drug resistance phenotype in acute myeloid leukemia cells by modulation of multiple ABC transporter genes. Clin. Cancer Res. 2009;15:3705–3715. doi: 10.1158/1078-0432.CCR-08-2048. [DOI] [PubMed] [Google Scholar]
- 54.Varatharajan S., Abraham A., Karathedath S., Ganesan S., Lakshmi K.M., Arthur N., Srivastava V.M., George B., Srivastava A., Mathews V., Balasubramanian P. ATP-binding casette transporter expression in acute myeloid leukemia: association with in vitro cytotoxicity and prognostic markers. Pharmacogenomics. 2017;18:235–244. doi: 10.2217/pgs-2016-0150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparency document.
Supplementary material