

Abstract
To understand the interaction between Pt and surface oxygenated species in electrocatalysis, this paper correlates the electrochemistry of atomic oxygen on Pt formed in the gas phase with electrochemically generated oxygen species on a variety of single-crystal platinum surfaces. The atomic oxygen adsorbed on single-crystalline Pt electrodes, made by thermal dissociation of molecular oxygen, is used for voltammetry measurements in acidic electrolytes (HClO4 and H2SO4). The essential knowledge of coverage, binding energy, and surface construction of atomic oxygen is correlated with the charge, potential, and shape of voltammograms, respectively. The differences of the voltammograms between the oxide made by thermal dissociation of molecular oxygen and electrochemical oxidation imply that atomic oxygen is not an intermediate of the electrochemical oxidation of Pt(111). The reconstruction of (100) terrace and step and the low-potential stripping of atomic oxygen on (111) step site provide insight into the first stages of degradation of Pt-based electrocatalysts.
Platinum-based electrocatalysts are highly catalytic for the conversion cycles of carbon, nitrogen, and oxygen species. Catalyzing these redox cycles is becoming increasingly important regarding the urgent demands of industry, energy, and environment in modern society, such as the hydrogen–oxygen fuel cell, water electrolysis, CO2 reduction, nitrate reduction, ammonia production, and so on.1−5 Because electrocatalysis is usually conducted in aqueous electrolyte, the platinum electrocatalyst may be covered by some intermediate stage of Pt surface oxide, for instance, during oxidation reactions or during the oxygen reduction reaction (ORR). The long-term degradation of the catalytic activity of a Pt electrocatalyst is thought to be related to the formation of this Pt oxide.6 Thus the surface oxygenated species or surface oxide that forms on Pt has attracted great attention and has been intensively studied in the past decades.3,7,8
The large number of investigations on the oxidation of Pt can be categorized in two groups, that is, ultrahigh vacuum (UHV)-based surface science studies and electrochemical studies. In particular, surface science studies have established a detailed and systematic understanding of the oxidation of single-crystalline Pt on the atomic scale.7 On the basis of Clavilier’s pioneering work of preparing single-crystalline electrodes by flame annealing, single-crystalline Pt has also been intensively studied in electrocatalysis.9,10 However, while most surface science studies are conducted in a UHV chamber (although recent work on the oxidation of Pt(111) at higher oxygen pressure exists11), electrochemical studies are conducted in an aqueous electrolyte under ambient conditions, making these two cases of Pt surface oxidation drastically different. Importantly, the electrochemical oxidation of Pt involves H2O, whereas Pt in vacuum or gas phase is oxidized by O2, NO2, or other active oxygen species. Thus it is highly desirable to understand the differences between the oxidation at the Pt/gas interface and the Pt/electrolyte interface. Attempts have been made to correlate the structure of the oxygenated species on Pt(111)/vacuum with surface oxygen species prepared by electrochemical oxidation on Pt(111).12 Many of the species involved have also been considered as intermediates in the oxygen reduction reaction (ORR).13 Understanding the nature of the surface species and their binding energetics at the single-crystal Pt electrode surface is of paramount importance in surface electrochemistry and electrocatalysis.3 Unfortunately, these correlations are not straightforwardly verified by experiment, as there is little direct spectroscopic evidence of surface oxygen species; therefore, many correlations are actually based on (idealized) density functional theory (DFT) calculations.12−14 Remarkably, our recent in situ shell-isolated nanoparticle-enhanced Raman spectroscopic (SHINERS) measurements suggested that the electrochemical oxidation of Pt(111) yields a 2D (su)peroxide surface layer, in stark disagreement with the expectation of the formation of atomic oxygen.15 This result suggested that there may be significant differences between gas-phase and electrochemical oxidation of platinum surfaces that have hitherto not been fully appreciated. Specifically, the participation of atomic oxygen in electrochemical oxidation is under discussion.
To shed further light on this issue, we propose here to measure the electrochemical stripping of atomic oxygen on single-crystalline Pt and compare it to the voltammetric features of electrochemical oxidation. The idea is to generate (atomic) oxygen on single-crystalline Pt by bringing the Pt single crystal into contact with a molecular oxygen atmosphere (O2) at ambient temperature and pressure (generating a species that we will refer to as Pt-OGAS). The fingerprint reductive stripping voltammogram of the atomic oxygen can be used to compare to the electrochemical characteristics of electrochemically generated oxygen species (Pt-OEC) to draw conclusions regarding the nature of the oxygen species, coverage, binding energy, surface sites, surface reconstruction, and so on. In this way, the comparison of the reductive voltammograms of atomic oxygen on Pt, prepared by thermal dissociation of molecular oxygen, and electrochemical oxidation will allow us to analyze the oxygenated species in intermediate stages of electrochemical oxidation of Pt.
We prepare the Pt-OGAS electrode by contacting a flame-annealed Pt crystal with a purely O2 atmosphere. Because investigations of thermal dissociation of O2 on Pt(111) by UHV show that the coverages of O generated at 420 and 620 K are different,16 first the effect of the crystal temperature during cooling in O2 atmosphere is carefully measured and controlled to avoid deformation of the surface (see Experimental Methods section and Supporting Information for details.) After cooling in oxygen, the Pt-OGAS electrode is protected by a droplet of electrolyte-free water. The open-circuit potential of the Pt(111)-OGAS measured under this condition is ca. 1.17 V (vs RHE). To ensure the stability of the Pt(111)-OGAS upon transfer to the electrochemical cell, we set the starting potential of voltammetric scan to be 1.15 V to avoid (further) electrochemical oxidation or reduction. At the moment that the crystal contacts the electrolyte, we observed only a transient charging current, which suggests that the surface of Pt(111)-OGAS is indeed stable at 1.15 V.
Figure 1 shows the reductive stripping voltammograms of the Pt(111)-OGAS with a cooling time of 10 min in 0.1 mol/L HClO4 (red curve) and 0.1 mol/L H2SO4 electrolytes (blue curve) in comparison with the blank voltammetry of Pt(111) in sulfuric acid (black dashed line). It is clearly observed in Figure 1 that the reduction features observed between 1.0 and 0.6 V of the Pt(111)-OGAS, indicated by Area 1 (HClO4) and Area 2 (H2SO4), are significantly different from the Pt(111). We ascribe this current to the reductive stripping of OGAS. Before we discuss the nature of this reductive stripping in more detail, we note that at potentials negative to 0.4 V the voltammograms for all three electrodes overlap, which indicates that the Pt(111)-OGAS has been completely reduced. In the H2SO4 electrolyte, the voltammetric features of Pt(111)-OGAS between 0.4 and 0.6 V are identical with Pt(111), which implies similar adsorption/desorption of bisulfate (HSO4–). The small peak from the {111}-type defects at ca. 0.12 V is also observed on the Pt(111)-OGAS electrode, which indicates that the surface of Pt(111)-OGAS is clean. Area 1 in HClO4 and Area 2 in H2SO4 are assigned to the stripping OGAS (Pt(111)-OGAS + 2H+ + 2e– → Pt(111) + H2O; see the mechanistic analysis in the Supporting Information). According to the charge of the stripping charge of OGAS and HSO4–, we conclude that the coverage of OGAS is ∼0.336 (See Supporting Information), which is reasonably consistent with the measurement of 0.4 ML on Pt(111) oxidized in 0.5 Torr O2.11 We will assume that the OGAS layer formed is a layer of atomic oxygen, as assumed in the determination of the coverage, for which we will give further arguments in the remainder of the paper.
Figure 1.
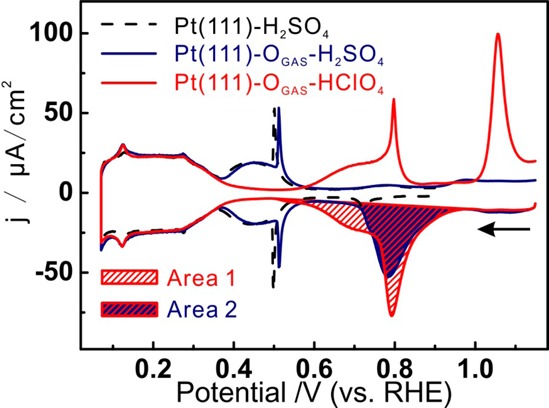
Voltammograms of Pt(111)-OGAS and Pt(111) in 0.1 mol/L HClO4 and H2SO4; the voltammetry of the Pt(111)-OGAS starts at 1.15 V with a scan rate of 50 mV/s.
Temperature-programmed desorption of atomic oxygen to O2 (TPD-O2) is a widely used technique to study the interaction between the Pt surface and atomic oxygen, giving insight into the binding energy and the coverage. We will use here stripping voltammetry as a potential-programmed desorption of atomic oxygen on Pt(hkl) surfaces. For example, in the TPD-O2 measurement of Pt(553)-OGAS generated in UHV, the desorption temperature of oxygen atoms adsorbed on the step is higher than oxygen on the terrace, which indicates a higher binding energy of atomic oxygen at step edges.17 According to the reaction equation Pt(111)-OGAS + 2H+ + 2e– → Pt(111) + H2O, the stripping potential directly describes the binding energy of the atomic oxygen versus the reference reaction (H+ + e– ⇆ 1/2 H2) in an electrochemical environment, which allows us to compare binding energies in UHV and electrochemistry.18Figure 2a compares voltammograms of Pt(553)-OGAS and Pt(553) in 0.1 mol/L HClO4 electrolyte. The standard voltammograms of the under-potential deposition hydrogen (UPD-H) region negative of 0.4 V on Pt(553) and Pt(553)-OGAS overlap, which indicates that the adsorption of atomic oxygen does not lead to the creation of more defects. The stripping peak of oxygen on Pt(553)-OGAS exhibits two peaks centered at 0.6 and 0.8 V, which we will refer to as peaks 1 and 2, respectively. Figure 2b shows a series of stripping voltammograms of Pt(s)-[n(111) × (111)]-OGAS for varying terrace width n. Note that the stripping of OGAS from the stepped surfaces overlaps with a broad feature between 0.6 and 0.8 V, which is ascribed to the reduction of OH from (111) terraces.19,20 To demonstrate the facet dependence of the stripping of atomic oxygen, the stripping peak is deconvoluted as shown in Figure 2a. The fraction of step sites to the total number of surface sites is defined as s/(s + t), as illustrated in Figure 2c. Figure 2d shows the charge density corresponding to peak 1 and the potential of peak 1 on Pt(s)-[n(111) × (111)]-OGAS as a function of the fraction of step sites. First, the charge density of peak 1 linearly depends on the step fraction, on the basis of which we assign peaks 1 and 2 to the stripping of atomic oxygen on the step and terrace sites, respectively. This assignment is also supported by DFT calculations of the binding energy of oxygen on the step site, showing that on Pt(553) step-site oxygen is ∼0.37 eV more stable than terrace-site oxygen,21 which would correspond to a 0.185 V difference in stripping potential, in good agreement with experiment. Second, the peak potential shifts positively with increasing step site fraction, which we ascribe to step–step interactions (meaning that oxygen on isolated steps is more stable).
Figure 2.
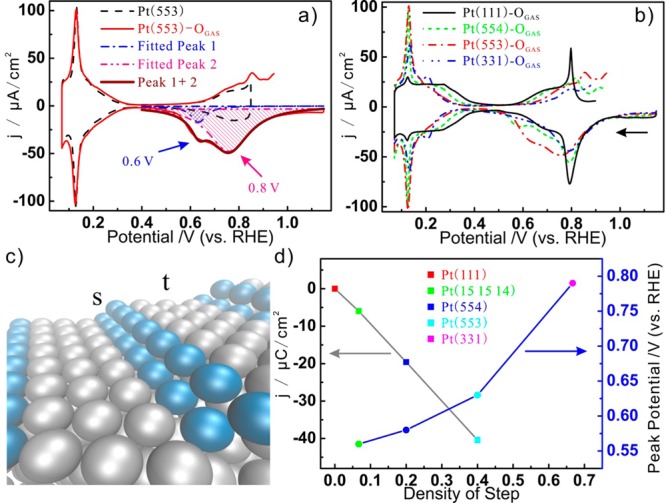
(a) Voltammograms of Pt(553)-OGAS and Pt(553).(b) Voltammograms of Pt(111)-OGAS, Pt(554)-OGAS, Pt(553)-OGAS, and Pt(331)-OGAS. (c) Illustration of step and terrace atoms on Pt(553). (d) Peak potential and integrated charge of stripping voltammograms of Pt(s)-[n(111) × (111)]-OGAS. All of the measurements were performed in 0.1 M HClO4 with a scan rate of 50 mV/s.
To further illustrate the correlation with UHV characterization, we also performed the measurements on Pt(100)-OGAS, where we note that the Pt(100)-(1 × 1) surface is usually reconstructed by the adsorption of oxygenated species.22Figure 3a shows the stripping voltammograms on Pt(100)-OGAS (red curve), where the broad peak centered at ca. 0.9 V is assigned to the stripping of oxygen. The voltammetric profile observed after oxygen stripping suggests that the adsorption of atomic oxygen indeed caused a surface reconstruction based on the following observations. First, the voltammogram is not symmetric at potentials negative of 0.5 V. In particular, the cathodic current is lower than the corresponding anodic current in the potential region between 0.35 and 0.5 V, which is assigned to the formation of OH on Pt(100).3,23 Second, it has been found that the reconstruction at room temperature is a transient complex surface structure between the Pt(100)-(1 × 1) and the reconstructed Pt(100)-(1 × 3) superstructure.24,25 Comparing Pt(100)-OGAS with the standard voltammograms of Pt(100), Pt(19 1 1), and Pt(9 1 1), the current between 0.35 and 0.45 V decreases with increasing step density, which indicates that the adsorption of oxygen on Pt(100)-(1 × 1) causes a partial deformation of the (100) terrace. Finally, on Pt(100)-OGAS there is a redox couple in the potential region between 0.6 and 0.8 V, as shown in Figure 3b. This redox couple is manifested only if the potential remains above 0.5 V, so that the surface reconstruction is not lifted by UPD-H adsorption. We ascribe this feature to OH formation on hexagonally reconstructed parts of the surface. The reconstruction of the {100} step can be also observed for Pt(533)-OGAS surface. (See the Supporting Information.)
Figure 3.
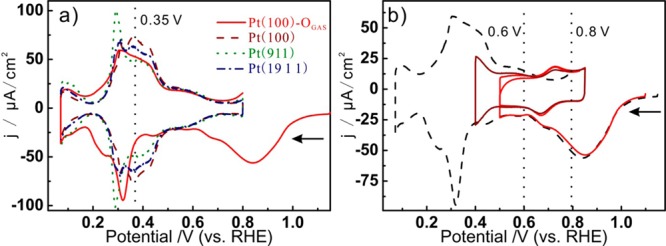
(a) Stripping voltammograms of Pt(100)-OGAS and standard voltammograms of Pt(100), Pt(19 1 1), and Pt(911). (b) Low vertex potential dependent stripping voltammogram of Pt(100)-OGAS. All measurements were performed in 0.1 M HClO4 with a scan rate of 50 mV/s.
We will now discuss the difference between the oxygen species on Pt(111) made by thermal dissociation of O2 (Pt(111)-OGAS) and the oxygen species generated by electrochemical oxidation (Pt(111)-OEC). For Pt(111)-OEC, the coverage of oxygen and the nature of the surface oxygen species on Pt depends on the applied oxidation potential. Let us first discuss the oxygen species formed at 1.15 V (Pt(111)-OEC,1.15V). It is generally agreed that the peak at 0.8 V corresponds to the formation of OHads on the Pt(111). This surface hydroxyl phase is oxidized in a peak at 1.07 V. On the basis of in situ SHINERS measurements, we recently proposed that the Pt(111)-OEC formed in this peak corresponds to a form of surface (su)peroxide on Pt(111) rather than to atomic oxygen on Pt(111), as was previously suggested.15 In Figure 4a we compare the reductive stripping voltammograms of Pt(111)-OGAS and Pt(111)-OEC,1.15V in HClO4 electrolyte. We note that the cathodic peak at ca. 1.05 V for Pt(111)-OEC is not observed on Pt(111)-OGAS, which is a first indication that Pt(111)-OEC is different from Pt(111)-OGAS. This cathodic peak is apparently associated with the anodic peak at 1.07 V, as can be observed when oxygen is stripped from Pt(111)-OGAS until 0.9 V and then scanned positively again (see Figure 4a). The potential of the anodic peak at 1.07 V shifts negatively in more alkaline electrolyte,3 which implies that it may correspond to the reduction of a negatively charged oxygen species.15 The irreversibility of the stripping of the oxygen species of Pt(111)-OEC generated at 1.07 V is not fully understood. According to Gómez-Marín and Feliu, the irreversibility is kinetic in origin and related to a slow nucleation-and-growth mechanism.26 The significant conclusion of our work here is that the oxygen species generated in Pt(111)-OGAS has no counterpart in Pt(111)-OEC, confirming our previous conclusion that the atomic oxygen is not an intermediate in the electrochemical oxidation of Pt(111).15
Figure 4.
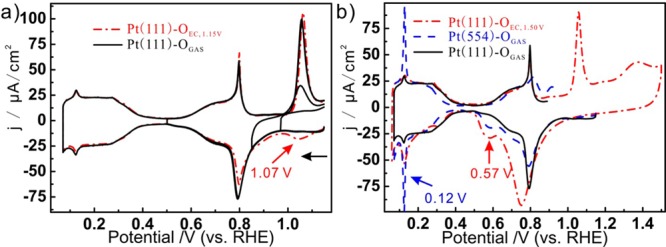
(a) Voltammograms of Pt(111)-OGAS and Pt(111)-OEC,1.15V. (b) Voltammograms of Pt(111)-OGAS, Pt(554)-OGAS, and Pt(111)-OEC,1.50 V in 0.1 mol/L HClO4 electrolyte (scan rate: 50 mV/s).
When the potential of electrochemical oxidation is scanned more positive than 1.2 V, the oxide formed is different from that negative of 1.2 V, and the well-defined Pt(111) surface is irreversibly damaged.26Figure 2b compares the stripping voltammograms of Pt(111)-OGAS, Pt(554)-OGAS, and Pt(111)-OEC,1.50V in 0.1 mol/L HClO4 electrolyte. It can be seen that the voltammograms of Pt(554)-OGAS and Pt(111)-OEC,1.50V both show peaks at 0.57 and 0.12 V, which implies the formation of adsorbed atomic oxygen and adsorbed hydrogen on (111) type step edges formed on Pt(111)-OEC,1.50V. According to the charges of 33.4 and 16.0 μC/cm2 of the peaks at 0.57 and 0.12 V, respectively, we estimate the defect density to be ca. 0.2 from the step density plot in Figure 2d. The step-edge features of Pt(111)-OEC,1.50V are broader than those of stepped surfaces, suggesting that the defects are much more random and heterogeneous than regular step edges.
In summary, we have measured the stripping voltammograms of atomic oxygen on various single-crystal Pt electrodes, as generated by the dissociation of molecular oxygen at room temperature. The coverage of atomic oxygen on Pt(111) was measured to be ca. 0.33, as concluded from the stripping charge. The stripping potential of atomic oxygen from the step-edge and terrace sites on Pt(s)-[n(111) × (111)] agrees with the expected binding energy difference; that is, the oxygen adsorbed on the {111} step type has a ca. 0.37 eV higher binding energy and is stripped at ca. 0.2 V more negative potential compared with atomic oxygen on the terrace. The stripping measurements on Pt(s)-[n(100) × (111)] surfaces indicate the reconstruction of {100} terrace and step sites caused by adsorption of oxygen, leading to a mixture of {100} terrace and {111}-type domains on the surface. The comparison between the reductive voltammograms of the oxygen on Pt(111) made by thermal dissociation of molecular oxygen and electrochemical oxidation at 1.15V suggests that electrochemical oxidation of Pt(111) does not lead to atomic oxygen state on Pt(111), consistent with our previous SHINERS measurements showing the formation of a surface (su)peroxide phase instead. When Pt(111) is oxidized electrochemically at 1.5 V, the surface disorders and leads to a reduction peak that is very similar to the stripping voltammograms of atomic oxygen from the step edges in Pt(s)-[n(111) × (111)], indicating that atomic oxygen adsorbed on {111} step sites appears to be an intermediate in electrochemical oxidation.
Experimental Methods
The semibead-like single-crystalline Pt electrodes were purchased from iCryst. The electrolyte solutions of HClO4 and H2SO4 were prepared by HClO4 (70%, Suprapur, Merck) and H2SO4 (96%, Suprapur, Merck). The ultrapure water (resistivity >18.2 MΩcm) was produced by a Milli-Q gradient A10 system. The gases of H2, O2, and Ar with a purity grade of 6.0 (99.9999%) were provided by Linde.
The Pt-OGAS was prepared in a round-bottomed flask with a volume of 250 mL containing ∼150 mL of O2-saturated water without any electrolyte. The input gas flow goes through a trap of 3 M NaOH solution before it enters the flask. To avoid explosion, H2 is switched off after the Ar flow is increased slowly and the cap of the flask is released. Next, the O2 flow is switched on. To avoid the influence from any possible reaction on the thermal couple, we used pure Ar with a same flow rate as H2/Ar in the measurement of crystal temperature.
Acknowledgments
This work was financially supported by the European Commission Horizon 2020—Research and Innovation Framework Programme (Marie Skłodowska-Curie actions Individual Fellowship awarded to Y.-F.H., no. 661145, DYNECAT).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.7b00136.
Experimental details of Pt-OGAS preparation including temperature dependence. pH dependence of stripping voltammogram and determination of OGAS coverage. Stripping voltammogram of Pt(533)-OGAS. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Canfield D. E.; Glazer A. N.; Falkowski P. G. The Evolution and Future of Earth’s Nitrogen Cycle. Science 2010, 330, 192–196. 10.1126/science.1186120. [DOI] [PubMed] [Google Scholar]
- Debe M. K. Electrocatalyst Approaches and Challenges for Automotive Fuel Cells. Nature 2012, 486, 43–51. 10.1038/nature11115. [DOI] [PubMed] [Google Scholar]
- Marković N. M.; Ross P. N. Jr Surface Science Studies of Model Fuel Cell Electrocatalysts. Surf. Sci. Rep. 2002, 45, 117–229. 10.1016/S0167-5729(01)00022-X. [DOI] [Google Scholar]
- Tian N.; Zhou Z.-Y.; Sun S.-G.; Ding Y.; Wang Z. L. Synthesis of Tetrahexahedral Platinum Nanocrystals with High-Index Facets and High Electro-Oxidation Activity. Science 2007, 316, 732–735. 10.1126/science.1140484. [DOI] [PubMed] [Google Scholar]
- Rosca V.; Duca M.; de Groot M. T.; Koper M. T. M. Nitrogen Cycle Electrocatalysis. Chem. Rev. 2009, 109, 2209–2244. 10.1021/cr8003696. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Yuan X.-Z.; Hin J. N. C.; Wang H.; Friedrich K. A.; Schulze M. A Review of Platinum-Based Catalyst Layer Degradation in Proton Exchange Membrane Fuel Cells. J. Power Sources 2009, 194, 588–600. 10.1016/j.jpowsour.2009.06.073. [DOI] [Google Scholar]
- Somorjai G. A.; Li Y.. Introduction to Surface Chemistry and Catalysis, 2nd ed.; John Wiley & Sons: Hoboken, NJ, 2010. [Google Scholar]
- Conway B. E. Electrochemical Oxide Film Formation at Noble Metals as a Surface-Chemical Process. Prog. Surf. Sci. 1995, 49, 331–452. 10.1016/0079-6816(95)00040-6. [DOI] [Google Scholar]
- Clavilier J.; Faure R.; Guinet G.; Durand R. Preparation of Monocrystalline Pt Microelectrodes and Electrochemical Study of the Plane Surfaces Cut in the Direction of the {111} and {110} Planes. J. Electroanal. Chem. Interfacial Electrochem. 1980, 107, 205–209. 10.1016/S0022-0728(79)80022-4. [DOI] [Google Scholar]
- Climent V.; Feliu J. M. Thirty Years of Platinum Single Crystal Electrochemistry. J. Solid State Electrochem. 2011, 15, 1297–1315. 10.1007/s10008-011-1372-1. [DOI] [Google Scholar]
- Miller D. J.; Öberg H.; Kaya S.; Sanchez Casalongue H.; Friebel D.; Anniyev T.; Ogasawara H.; Bluhm H.; Pettersson L. G. M.; Nilsson A. Oxidation of Pt(111) under Near-Ambient Conditions. Phys. Rev. Lett. 2011, 107 (19), 195502. 10.1103/PhysRevLett.107.195502. [DOI] [PubMed] [Google Scholar]
- Gómez-Marín A. M.; Feliu J. M. Pt(111) Surface Disorder Kinetics in Perchloric Acid Solutions and the Influence of Specific Anion Adsorption. Electrochim. Acta 2012, 82, 558–569. 10.1016/j.electacta.2012.04.066. [DOI] [Google Scholar]
- Nørskov J. K.; Rossmeisl J.; Logadottir A.; Lindqvist L.; Kitchin J. R.; Bligaard T.; Jónsson H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. 10.1021/jp047349j. [DOI] [Google Scholar]
- Tian F.; Jinnouchi R.; Anderson A. B. How Potentials of Zero Charge and Potentials for Water Oxidation to OH(ads) on Pt(111) Electrodes Vary With Coverage. J. Phys. Chem. C 2009, 113, 17484–17492. 10.1021/jp905377d. [DOI] [Google Scholar]
- Huang Y. F.; Kooyman P. J.; Koper M. T. M. Intermediate Stages of Electrochemical Oxidation of Single-Crystalline Platinum Revealed by in situ Raman spectroscopy. Nat. Commun. 2016, 7, 12440. 10.1038/ncomms12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashlakov D. L.; Juurlink L. B. F.; Koper M. T. M.; Yanson A. I. Subsurface Oxygen on Pt(111) and Its Reactivity for CO Oxidation. Catal. Lett. 2012, 142, 1–6. 10.1007/s10562-011-0730-z. [DOI] [Google Scholar]
- van der Niet M. J. T. C.; den Dunnen A.; Juurlink L. B. F.; Koper M. T. M. A Detailed TPD Study of H2O and Pre-adsorbed O on the Stepped Pt(553) Surface. Phys. Chem. Chem. Phys. 2011, 13, 1629–1638. 10.1039/C0CP01162B. [DOI] [PubMed] [Google Scholar]
- Koper M. T. M. Blank voltammetry of hexagonal surfaces of Pt-group metal electrodes: Comparison to Density Functional Theory Calculations and Ultra-high Vacuum Experiments on Water Dissociation. Electrochim. Acta 2011, 56, 10645–10651. 10.1016/j.electacta.2011.02.001. [DOI] [Google Scholar]
- Berná A.; Climent V.; Feliu J. M. New Understanding of the Nature of OH Adsorption on Pt(111) Electrodes. Electrochem. Commun. 2007, 9, 2789–2794. 10.1016/j.elecom.2007.09.018. [DOI] [Google Scholar]
- Wakisaka M.; Suzuki H.; Mitsui S.; Uchida H.; Watanabe M. Identification and Quantification of Oxygen Species Adsorbed on Pt(111) Single-Crystal and Polycrystalline Pt Electrodes by Photoelectron Spectroscopy. Langmuir 2009, 25, 1897–1900. 10.1021/la803050r. [DOI] [PubMed] [Google Scholar]
- Kolb M. J.; Calle-Vallejo F.; Juurlink L. B. F.; Koper M. T. M. Density Functional Theory Study of Adsorption of H2O, H, O, and OH on Stepped Platinum Surfaces. J. Chem. Phys. 2014, 140, 134708. 10.1063/1.4869749. [DOI] [PubMed] [Google Scholar]
- Lang B.; Légaré P.; Maire G. Adsorption Studies on the Pt (100) Surface. Surf. Sci. 1975, 47, 89–97. 10.1016/0039-6028(75)90275-7. [DOI] [Google Scholar]
- van der Niet M. J. T. C.; Garcia-Araez N.; Hernández J.; Feliu J. M.; Koper M. T. M. Water Dissociation on Well-defined Platinum Surfaces: The Electrochemical Perspective. Catal. Today 2013, 202, 105–113. 10.1016/j.cattod.2012.04.059. [DOI] [Google Scholar]
- Griffiths K.; Jackman T. E.; Davies J. A.; Norton P. R. Interaction of O2 with Pt(100). Surf. Sci. 1984, 138, 113–124. 10.1016/0039-6028(84)90499-0. [DOI] [Google Scholar]
- Kibler L. A.; Cuesta A.; Kleinert M.; Kolb D. M. In-situ STM characterisation of the Surface Morphology of Platinum Single Crystal Electrodes as a Function of Their Preparation. J. Electroanal. Chem. 2000, 484, 73–82. 10.1016/S0022-0728(00)00065-6. [DOI] [Google Scholar]
- Gómez-Marín A. M.; Clavilier J.; Feliu J. M. Sequential Pt(111) Oxide Formation in Perchloric Acid: an Electrochemical Study of Surface Species Inter-conversion. J. Electroanal. Chem. 2013, 688, 360–370. 10.1016/j.jelechem.2012.07.016. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.