

Abstract
Neurofibrillary tangles (NFTs), composed of hyperphosphorylated tau, are a key pathologic feature of Alzheimer’s disease (AD). Tau phosphorylation is under the control of multiple kinases and phosphatases, including Fyn. Previously, our group found an association between two regulatory single nucleotide polymorphisms (SNPs) in the FYN gene with increased tau levels in the CSF. In this study, we hypothesized that Fyn expression in the brain is influenced by AD status and genetic content. We found that Fyn protein, but not mRNA, levels were increased in AD patients compared to cognitively normal controls and are associated with regulatory region SNPs. Additionally, expression of the FYN 3’ UTR can decrease expression in multiple cell lines, suggesting this regulatory region plays an important role in FYN expression. Taken together, these data suggest that FYN expression is regulated according to AD status and regulatory region haplotype, and genetic variants may be instrumental in the development of NFTs in AD and other tauopathies.
Keywords: Fyn, Alzheimer’s disease, SNP, Cerebellum, Hippocampus, Reporter assay
1. Introduction
Alzheimer’s disease (AD) is a debilitating neurological disorder which accounts for over 50% of dementia cases worldwide, affecting more than 44 million people (L. M. Bekris, Yu, Bird, & Tsuang, 2010; Iqbal, Liu, & Gong, 2015). The two major pathologic hallmarks of AD are the presence of amyloid-beta (Aβ) plaques and neurofibrillary tangles (NFTs), the latter comprised of hyperphosphorylated tau protein (Allen et al., 2014). Tau protein, the product of the MAPT gene, associates with and stabilizes microtubules for proper axonal transport (Mandelkow, 2004; Sato-Harada, Okabe, Umeyama, Kanai, & Hirokawa, 1996). Tau is a dynamically structured protein, with multiple phosphorylation sites impacting the protein’s ability to bind microtubules (Huang, Wu, & Zhou, 2015; Sato-Harada et al., 1996). The tau protein has at least 85 putative phosphorylation sites, and multiple proteins are involved in the phosphorylation and dephosphorylation of tau, including GSK3β, Fyn, CDK5, PP2A, and many others (Lynn M. Bekris, Millard, et al., 2012; Huang et al., 2015; Jayapalan & Natarajan, 2013; Liao, Yang, Weng, Kuo, & Chang, 2015; Shanley et al., 2015; Sontag, Nunbhakdi-Craig, White, Halpain, & Sontag, 2012; Tenreiro, Eckermann, & Outeiro, 2014; Xia, Li, & Götz, 2015). Notably, hyperphosphorylation of tau is a common feature of neurodegenerative tauopathies, and results in impaired axonal transport, NFT formation, and neuronal cell death (Lynn M. Bekris, Millard, et al., 2012; Duka et al., 2013; Huang et al., 2015; Krüger & Mandelkow, 2016; Sontag et al., 2012; Tenreiro et al., 2014). Therefore, understanding the mechanisms by which tau phosphorylation is controlled is an important priority.
Fyn, a Src-family non-receptor tyrosine kinase, has prominent roles in T-cell signaling, myelination, learning and memory, neuroinflammation, apoptosis, and cell adhesion (Chin, 2005; Du, Tan, & Hou, 2012; Kaufman et al., 2015; Ko et al., 2015; Resh, 1998). Fyn also has multiple isoforms, known as FynB, FynT, and FynΔ7, which arise as a result of alternative splicing at exon 7 and demonstrate differential kinase activity and expression patterns, mainly in the immune system and brain respectively (Davidson, Chow, Fournel, & Veillette, 1992; Davidson, Viallet, & Veillette, 1994; Goldsmith, Hall, & Atkinson, 2002; C Lee et al., 1998). Importantly, Fyn has recently been implicated in AD, where inhibition of Fyn kinase activity can lead to improved memory and synaptic function in animal models (Chin, 2005; Kaufman et al., 2015; Nygaard, van Dyck, & Strittmatter, 2014; Yang et al., 2011). Notably, Fyn can phosphorylate tau on tyrosine-18, a post-translational modification found only early in development and in a subset of hyperphosphorylated tau species in the AD brain (G. Lee et al., 2004). In mouse models, Fyn has also been implicated as a downstream target of Aβ. Extracellular oligomeric Aβ can interact with prion protein (PrPC), and along with mGluR5, initiate a signal transduction cascade in the postsynaptic density leading to increased Fyn activity, increased tau-Fyn interactions, as well as Fyn-mediated phosphorylation of NMDA receptors resulting in excitotoxicity (Larson et al., 2012; Nygaard et al., 2014; Roberson et al., 2011; J W Um et al., 2012). Together, these findings demonstrate a role for Fyn phosphorylation activity in AD pathogenesis.
However, despite these findings, the link between FYN gene regulation, Fyn phosphorylation, and tau pathology remains unclear. In a previous study, we discovered two SNPs located in the FYN gene, rs7768046 and rs1621289, associated with increased total tau (t-tau) levels in AD cerebrospinal fluid (CSF) (Lynn M. Bekris, Millard, et al., 2012). Notably, rs7768046 tags the long isoform promoter region and rs1621289 is located within the 3’ UTR region of FYN, suggesting that FYN regulatory region genetic content may play a role in FYN gene regulation, which may in turn contribute to elevated CSF phosphorylated tau in AD. Others have described the FynB promoter region and the FYN 3’UTR (Gao, Howard, Ban, & Chandra, 2009; Grossman et al., 2015; Ninio-Many, Grossman, Shomron, Chuderland, & Shalgi, 2013) and increased Fyn expression has been previously described in AD (Chingli Lee et al., 2015), but little is known about how genetic variants within FYN regulatory elements might impact expression.
Therefore, the aim of this investigation was to examine the influence of genetic variation on FYN expression by measuring mRNA and protein levels and regulatory region reporter activity. Specifically, the hypothesis was that FYN expression and promoter activity are significantly influenced by genetic content. Our findings indicate that Fyn protein expression, but not mRNA, varies according to AD status and FYN 3’UTR genetic variation, and that FYN regulatory region reporter genetic content impacts expression only in certain cell types.
2. Materials and Methods
2.1 Population Description
Two regions of post-mortem brain (cerebellum (CB) and hippocampus (HP)) were obtained from post-mortem samples in the Neuropathology Core of the Alzheimer’s Disease Research Center (ADRC) at the University of Washington (UW). Use of human tissue was approved by the UW and Cleveland Clinic Foundation (CCF) Institutional Review Boards. All subjects, or their surrogates, had provided informed consent. Tissue was flash frozen at the time of autopsy and stored at –80 °C. Patients with late-onset AD (n = 21) and cognitively normal controls (n = 22) were UW ADRC volunteers as previously described (Lynn M. Bekris, Lutz, et al., 2012). Further patient information can be observed in Table 1.
Table 1.
Population Description
| Brain mRNA and protein | ||
|---|---|---|
|
|
||
| Controls | AD | |
|
|
|
|
| n= | 22 | 21 |
| % Female | 50 | 55 |
| % APOE ε4 | 18 | 68 |
| Mean Age (St.Dev) | 87 (5) | 82 (7) |
| Plaque Score | Absent - Moderate | Sparse-Frequent |
| Braak Stage | I – IV | IV – VI |
2.2 Postmortem brain DNA, RNA, and protein extraction
DNA, RNA, and protein were extracted from postmortem brain tissue using the Qiagen Allprep DNA/RNA Mini Kit (Qiagen, Valencia, CA) as previously described (Lynn M. Bekris, Lutz, et al., 2012). Soluble and insoluble protein was isolated from the column flow through using acetone precipitation according to Qiagen protocol. The protein sample was then resuspended in immunoprecipitation buffer (150 mM NaCl, 50 mM Tris-HCl (pH 6.8), 0.5% NP-40, 0.5% sodium deoxycholate, 5 mM EDTA, 50 mg/ml leupeptin, 10 mg/ml aprotinin, and 0.25 mM PMSF). FYN mRNA was measured in triplicate by quantitative real-time PCR (qRT-PCR) using the 7500 Real Time PCR System (Applied Biosystems, Foster City, CA), with β-actin (ACTB) as a loading control (Applied Biosystems). All qRT-PCR results are presented as a FYN/ACTB (ΔCT). FYN protein levels were measured by Western blot using a mouse monoclonal anti-Fyn (p59) primary antibody (Thermo Scientific, Waltham, MA) and visualized with a donkey anti-mouse secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA) and a ECL Prime Western blotting detection reagent (Sigma-Aldrich, St. Louis, MO). Band intensity was measured with ImageJ (US National Institutes of Health, rsb.info.nih.gov/ij/) (Abràmoff, Magalhães, & Ram, 2004). All Fyn protein results are presented as integrated density of Fyn/Actin. Subjects were categorized with neuritic plaque scores of absent, sparse, moderate, frequent diffuse, or frequent, and by Braak stage for NFT (Braak & Braak, 1991)(Table 1).
2.3 Immunohistochemistry
Fresh frozen human brain samples from one AD patient and one cognitively normal control were used for immunohistochemistry. Ten micron HP frozen sections were dried onto Superfrost Plus charged microscope slides (Fisher Scientific, Pittsburgh, PA) and then pretreated with Triton-X prior to the immunostaining procedure. Immunodetection was performed with anti-Fyn (p59) mouse monoclonal antibody as described in Section 2.2 (1:50, 0.004 µg/µl) and secondary goat biotinylated anti-mouse IgG (H+L) antibodies (1:200, 0.0075 µg/µl; Vector Laboratories, Burlingame, CA). The specificity of antigen detection was determined by omitting the primary antibody.
2.4 SNP selection and genotyping
Nine FYN SNPs chosen for mRNA and protein level analysis were chosen due to their association with cerebrospinal fluid (CSF) tau levels in a previous study or location within a potential regulatory region in the FYN gene (Lynn M. Bekris, Millard, et al., 2012). (see Figure 1 for visualization). SNPs used in promoter haplotype constructs (Table 2) were chosen based on their minor allele frequency (≥10% in the CEU population) and location in the constructs. Regulatory regions were chosen according to the UCSC Genome Browser GRCh37/hg19 Assembly, ENCODE, and regions previously described in literature (Rosenbloom et al., 2013, 2015).
Figure 1. The FYN gene encodes multiple isoforms.

Three representative promoters were used for analysis: a promoter resulting in transcription of a long isoform (2538 bp; located at chr6:112,192,288–112,194,825 in the UCSC Genome Browser Human Feb 2009 GRCh37/hg19 Assembly, NCBI Gene NM_002037; also known as FynB), a promoter transcribing an isoform of intermediate size (1201 bp; chr6:112,114,983–112,116,183, NCBI NM_153048; also known as FynΔ7), and a promoter producing a short isoform (1134 bp, chr6:112,041,163–112,042,296, NCBI NM_153047; also known as FynT) (Davidson et al., 1992; Goldsmith et al., 2002; Rosenbloom et al., 2015) (A) All promoters share a common 3’ UTR (1824 bp). Thirteen SNPs found in FYN regulatory regions were studied for associations with FYN expression levels in AD: long promoter SNPs rs62413757, rs72944244, and rs9481198, as well as rs7768046 (B); medium promoter SNPs rs6939256, rs11967460 and rs1409839 (C); short promoter SNP rs706895 (D); and 3’ UTR SNPs rs9387025, rs12191154, rs1621289, and rs9320374 (B–D). Of particular interest are two SNPs (in bold): rs7768046, located in the first intron downstream of the long promoter outside of our constructs but in linkage disequilibrium with other SNPs, and rs1621289, in the 3’ UTR, which have previously shown associations with increased t-tau levels in AD (Lynn M. Bekris, Millard, et al., 2012).
Table 2. Single Nucleotide Polymorphism Frequency and Analysis Summary.
The relationship between SNP and expression was evaluated using two types of analyses. First, brain mRNA (qRT-PCR) and protein (Western Blot) expression assays and second regulatory region reporter assays. SNPs with >1% rare allele frequency were evaluated. Seven SNPs were evaluated in the brain expression assays and ten were evaluated in the reporter assays. One SNP, rs1621289, showed a significant difference between alleles in the brain. Three SNPs; rs11967460 (Promoter 1), rs1409839 (Promoter 2), rs9320374 (3’UTR) in the reporter assays.
| Location | SNP | Type of Analyses | UCSC Allele Frequency | Brain | Reporter |
|---|---|---|---|---|---|
|
|
|
|
|
|
|
| FYN Intron 1 | rs7768046 | Brain Expression | A: 83.7 G: 16.3 | − | NA |
| FYN Promoter 1 (FynB) | rs62413757 | Reporter Assay | A: 2.1 G: 97.9 | NA | − |
| rs72944244 | Reporter Assay | C: 8.6 T: 91.4 | NA | − | |
| rs9481198 | Brain Expression, Reporter Assay | C: 43.8 T: 56.28 | − | − | |
| FYN Promoter 1 (FynΔ7) | rs11967460 | Reporter Assay | A: 7.3 G: 92.7 | NA | + |
| rs1409839 | Brain Expression, Reporter Assay | C: 36.6 T: 63.4 | − | + | |
| FYN Promoter 3 (FynT) | rs706895 | Brain Expression, Reporter Assay | C: 51.0 T: 49.0 | − | − |
| FYN 3'UTR | rs9387025 | Brain Expression, Reporter Assay | A: 39.1 G: 60.9 | − | − |
| rs12191154 | Brain Expression, Reporter Assay | A: 95.3 G: 4.7 | − | − | |
| rs1621289 | Brain Expression, Reporter Assay | C: 24.3 T: 75.7 | + | − | |
| rs9320374 | Reporter Assay | A: 49.9 G: 50.1 | NA | + |
Genotypes were selected according to their location within regulatory elements, or if there was not a SNP within the regulatory region that met the frequency criteria, then a regulatory region tagging SNP was selected as previously described (Lynn M. Bekris, Lutz, et al., 2012; Lynn M Bekris et al., 2008). TaqMan allelic discrimination assays (Applied Biosystems) were used to determine genotype. Plates were subject to an end-point read using a 7500 Real Time PCR System.
2.5 Generation of haplotype reporters
DNA regions of interest were amplified from genomic DNA using PCR, inserted into reporter constructs, and validated by sequencing. FYN promoter constructs were inserted into the HindIII restriction site and upstream (5’) of the luciferase gene of the pGL4.10[luc2] vector (Promega, Madison, WI) to produce promoter-only constructs. To create promoter – 3’ UTR constructs, the FYN 3’ UTR was inserted into the FseI restriction site downstream (3’) of the luciferase coding region and upstream from the poly-A. The regulatory region primer locations and reporter inserts used for cloning are represented in Figure 1 B–C and further described in Figures 3–6. The In-Fusion PCR Cloning System (Takara Clontech, Mountain View, CA) was used for all cloning procedures. After propagating recombinant DNA in E. coli host cells, reporter constructs were isolated and purified using an ion exchange column (Qiagen), and then fully sequenced to validate genetic content as previously described (Lynn M. Bekris, Lutz, et al., 2012). The following regulatory region reporter construct primer sequences were designed using the chr6:111,977,363–112,203,913; UCSC Genome Browser on Human Feb. 2009 (GRCh37/hg19) Assembly (Figure 1A) for the FYN gene. The 3 FYN promoters and the 3’UTR primers were designed according to previous reports and location of ENCODE described regulatory elements (Rosenbloom et al., 2013, 2015). The primers are listed here in the 3’ to 5’ orientation as observed in the UCSC Genome Browser and their location is noted as Figure 1A BLAT sequences:
-
Long FynB promoter (insert size: 2538):
Forward primer 3’- CGAGTGGGTTGAGCGTTACT-5’
Reverse primer 3’- TCGAGGAGCCATACTTTTAGGA-5’
-
Medium FynΔ7 promoter (insert size: 1201):
Forward primer 3’-TCACACTAACCGGGTAAGCC-5’
Reverse primer 3’-TAGCCATGTGCTCCACAGAG-5’
-
Short FynT promoter (insert size: 1134):
Forward primer 3'-TGAACACTTTTTCCCCTTCC-5’
Reverse primer 3'-CCATAGCGGTACCCAGAGC-5’
-
FYN 3’ UTR (insert size: 2014):
Forward primer 3’- TTCTTCCCCTATTTCCCAGG-5’
Reverse primer 3’- CAAACACCTGTCCTGATTGG-5’
Figure 3. FYN promoter haplotype functional analysis in human cell lines.
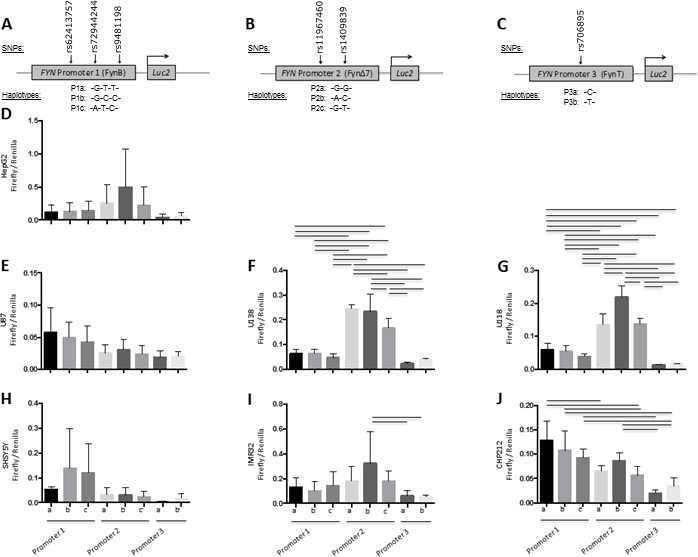
FYN promoter activity was evaluated using luciferase reporter vectors (pGL4.10[luc2]) with FYN promoter haplotypes inserted. Promoter 1 reporter constructs consisted of FynB promoter region haplotypes (A). Promoter 2 reporter constructs consisted of FynΔ7 promoter region haplotypes (B). Promoter 3 reporter constructs consisted of FynT promoter region haplotypes (C). Promoter region haplotype reporter constructs were transiently transfected into a human hepatocyte cell line (HepG2) (D), human glial cell lines (U87, U138, U118) (E–G), and human neuronal cell lines (SHSY5Y, IMR32, CHP212) (H–J). Results are presented as firefly/Renilla luciferase activity and SEM for each construct, n = 6 for each group. Black lines denote a significant difference between promoter haplotypes (p-value of < 0.05 after Bonferroni correction for multiple comparisons).
Figure 6. FYN 3’UTR haplotype functional analysis relative to FYN promoter haplotypes in human neuronal cell lines.

FYN 3’UTR activity was evaluated using luciferase reporter vectors (pGL4.10[luc2]) with both the FYN 3’UTR and FYN promoter haplotypes inserted as depicted in panels A–C. Promoter 1 plus 3’UTR reporter constructs consisted of FynB promoter region haplotype and the FYN 3’UTR (A and D, G, J). Promoter 2 plus 3’UTR reporter constructs consisted of FynΔ7 promoter region haplotypes (B and E, H, K). Promoter 3 plus 3’UTR reporter constructs consisted of FynT promoter region haplotypes (C and F, I, L). Promoter region haplotype reporter constructs and Promoter plus 3’UTR reporter constructs were transiently transfected into a human neuronal cell lines (SHSY5Y, IMR32, CHP212) (D–F). Results are presented as Firefly/Renilla luciferase activity for the promoter plus 3’UTR constructs relative to the promoter only constructs (Relative Quantification) and SEM for each construct, n = 6 for each group. Black lines denote a significant difference between reporter construct haplotypes (p-value of < 0.05 after Bonferroni correction for multiple comparisons).
2.6 Cell culture
For promoter – 3’ UTR construct experiments, six cell lines were used. Human neuroblastoma SH-SY5Y and CHP-212 cells (ATCC, Manassas, VA) were grown in 44.5% Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY) with 44.5% F12 (Gibco), 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (100 µU/ml penicillin and 100 µg/ml streptomycin). IMR-32 neuroblastoma cells and U-87, U-118, and U-138 glioblastoma cells were grown in 89% DMEM, 10% FBS, and 1% penicillin/streptomycin. All cells were grown at 37 °C in a 5% CO2 atmosphere, and passaged at a concentration of 2.5 × 104 per well into 96-well tissue culture plates 48 h before transfection.
2.7 Reporter construct assays
SH-SY5Y, CHP-212, IMR-32, U-87, U-118, and U-138 cell lines were transiently transfected for 48 h with Firefly luciferase pGL4.10[luc2] haplotype reporter constructs with Renilla luciferase pGL4.75[hRluc/CMV] constructs as a loading control in the same well using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions (Lynn M. Bekris, Lutz, et al., 2012). Firefly reporter constructs contained either one of the FYN promoter region haplotypes or a FYN promoter haplotype and the FYN 3’ UTR (Table 2, Figures 4–6). All transfection assays contained duplicate transfections within the same assay, and were performed at least six times. After 48 h, transiently transfected cells were harvested using the Dual-Glo® Luciferase Assay System (Promega) that allows for high-throughput analysis of Firefly luciferase pGL4.10[luc2] constructs in the first step. In the second step, Firefly luminescence was quenched and the Renilla luciferase (pGL4.75[hRluc/CMV]) was activated, and the internal loading control was analyzed. Luciferase reporter counts per second (CPS) were measured using an LMax II 384 luminometer (Molecular Devices, Sunnyvale, CA) and were presented as a ratio of Firefly/Renilla that represents reporter construct activity relative to the loading control (Lynn M. Bekris, Lutz, et al., 2012).
Figure 4. FYN 3’ UTR haplotype functional analysis relative to FYN promoter haplotypes in human HepG2 cells.
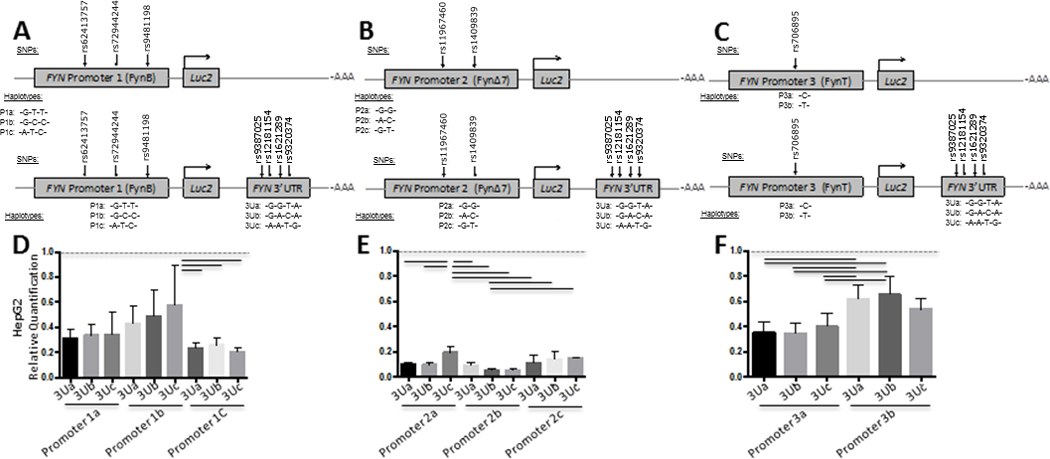
FYN 3’UTR activity was evaluated using luciferase reporter vectors (pGL4.10[luc2]) with both the FYN 3’UTR and FYN promoter haplotypes inserted as depicted in panels A–C. Promoter 1 plus 3’UTR reporter constructs consisted of FynB promoter region haplotype and the FYN 3’UTR (A and D). Promoter 2 plus 3’ UTR reporter constructs consisted of FynΔ7 promoter region haplotypes (B and E). Promoter 3 plus 3’ UTR reporter constructs consisted of FynT promoter region haplotypes (C and F). Promoter region haplotype reporter constructs and Promoter plus 3’UTR reporter constructs were transiently transfected into a human hepatocyte cell line (HepG2) (D–F). Results are presented as Firefly/Renilla luciferase activity for the promoter plus 3’UTR constructs relative to the promoter only constructs (Relative Quantification) and SEM for each construct, n = 6 for each group. Black lines denote a significant difference between reporter construct haplotypes (p-value of < 0.05 after Bonferroni correction for multiple comparisons).
2.8 Statistical analysis
Post-mortem brain FYN mRNA, Fyn protein levels, and construct luciferase activity were analyzed using SPSS Version 22 (IBM) and GraphPad Prism 6 software (La Jolla, CA). For SNP analysis, genotypes containing the minor alleles were collapsed as previously described (Matthews, Haynes, Liu, & Ott, 2008). Linear regression models or one-way ANOVA were used where mRNA expression level, protein expression level, or reporter activity were dependent variables, genotype or haplotype were independent variables, and disease status plus brain region or promoter construct were selection variables. All p-values were corrected with the Bonferroni correction for multiple comparisons.
3. Results
3.1 FYN mRNA and Protein Expression in AD Post-mortem Brain
To determine if FYN expression is higher in AD, as others have described, we first measured FYN mRNA by qRT-PCR and protein by Western blot in post-mortem brain CB and HP. There was no significant difference in FYN mRNA levels in AD compared to cognitively normal controls (Figure 2A). There was significantly higher Fyn protein in AD compared to controls in both the CB (unadjusted p-value, 0.006) and HP (unadjusted p-value, 0.002) (Figure 2B). In addition, in both AD and controls CB Fyn protein levels were significantly higher than HP (p-value, 0.003; p-value, 0.001, respectively) (Figure 2B). Representative Western blots of Fyn (59 kDa) and Actin B (42 kDa) protein in cognitively normal controls and AD patients in post-mortem CB and HP are shown (Figure 2D). Immunohistochemistry demonstrated Fyn immunopositivity in both AD and control tissue (Figure 2E–H). In both AD and control section immunostaining was robust in white matter oligodendrocyte nuclei and to a lesser extent in associated cytoplasm. In control HP neurons (CA1) similar levels of staining was observed in nuclei and cytoplasm. In AD HP, CA1, staining was observed in a more diffuse manner in atrophic neurons and unique staining was observed in plaque neurites.
Figure 2. FYN mRNA and protein expression in the human post-mortem brain.
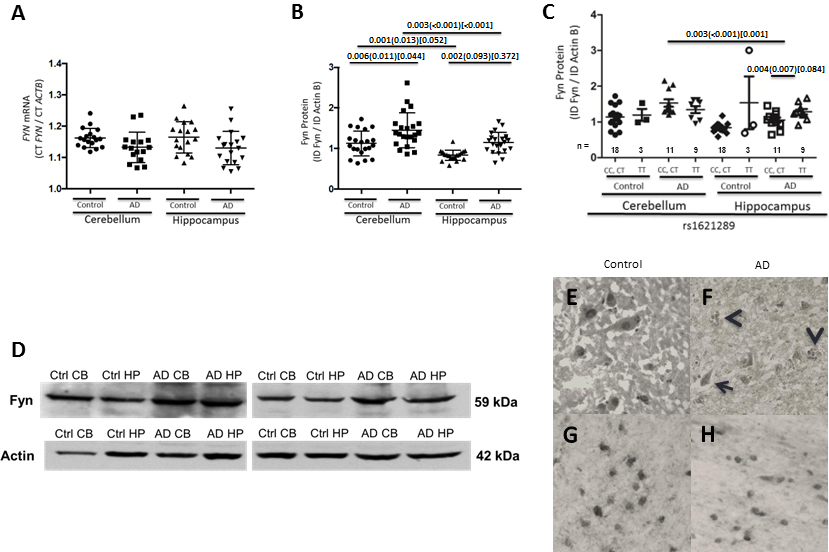
(A) FYN mRNA levels measured in the hippocampus and cerebellum in AD patients and cognitively normal controls; (B) Fyn protein levels are significantly increased in both the cerebellum and hippocampus of AD patients compared to cognitively normal controls. Data are presented as the ratio of FYN to ACTB with mean CT and SEM values indicated with error bars for mRNA levels, and the ratio of Fyn to ActB with mean integrated density and SEM values indicated with error bars for protein. n values are visible under the graphs. (C) Fyn protein levels are significantly higher in AD patient hippocampus with the 3’ UTR SNP rs1621289 TT genotype (CC, CT) compared to the C allele carriers (CC, CT). (D) Representative blots of Fyn protein levels in cognitively normal controls and AD patients in post-mortem cerebellum and hippocampus. P-values were obtained by linear regression with age, sex, and APOE ε4 status as covariates, and Bonferroni corrected for multiple comparisons. P-values are listed as unadjusted, adjusted for covariates in parentheses, and corrected for multiple comparisons after adjustment in brackets. (E–H) Post-mortem human brain Fyn immunohistochemistry. Neuronal staining in normal hippocampus (E) demonstrates nuclear and light cytoplasmic labeling, while staining in AD hippocampus (F) demonstrates plaque neurite (arrow heads) and atrophic neuronal labeling (arrow). In white matter temporal lobe) equivalent staining was observed in oligodendrocyte nuclei in both normal (G) and AD (H) brain.
3.2 Fyn Protein in AD Post-mortem Brain Stratified by Regulatory Region Genotype
Next, we evaluated Fyn protein expression in post-mortem brain tissue with respect to genetic content. Our previous study found an association between CSF t-tau levels in AD and the FYN long isoform (FynB) intron 1 SNP (rs7768046) (Lynn M. Bekris, Millard, et al., 2012). This SNP is in linkage disequilibrium with regulatory region SNPs in the FynB promoter. It was not significantly associated with Fyn protein expression. Other regulatory region SNPs were evaluated and all but one 3’UTR SNP were not significantly associated with Fyn mRNA or protein levels (data not shown). These included SNPs within the FynB promoter (rs9481198), within the FynΔ7 promoter (rs1409839), within the FynT promoter (rs706895) and within the FYN 3’ UTR (rs12191154, rs1621289). The 3’UTR rs1621289 TT genotype, compared to the CC, CT, was associated with higher Fyn protein levels in the AD HP (Figure 2C) suggesting that the FYN 3’UTR might play a role in FYN gene regulation.
3.3 FYN Promoter Haplotype Functional Analysis in Human Cell Lines
Regulatory region reporter constructs were designed to examine whether the FYN promoters are influenced by regulatory region genetic content (haplotype). FYN promoter-only constructs were made containing multiple haplotypes. The long isoform FynB promoter constructs contained three haplotypes consisting of rs62413757, rs72944244 and rs9481198 (Figure 3A). Importantly, the rs7768046 SNP analyzed previously was not included in the FYN promoter constructs, since it is in linkage disequilibrium with FynB promoter SNPs and is not located within the promoter region. For the medium FynΔ7 promoter, three haplotypes were analyzed derived from two SNPs; rs11967460 and rs1409839 (Figure 3B). The short FynT promoter constructs contained one SNP, rs706895 and thus only two alleles were analyzed (Figure 3C).
All three FYN promoter haplotypes were analyzed for differences between FYN promoter haplotype activity in multiple human cell lines, including hepatocytes (Figure 3D: HepG2), glia (Figure 3E–G: U-87, U-138, U-118) and neurons (Figure 3H–J: SH-SY5Y, IMR-32, CHP-212). FYN promoter activity was not significantly different according to genetic content in HepG2, U-87 or SH-SY5Y cell lines (Figure 3D, E, H). In 2 glial cell lines (U-138, U-118), FYN promoter 2 (FynΔ7: P2) had significantly higher activity than FYN promoter 1 (FynB: P1) or FYN promoter 3 (FynT: P3) (Figure 3F, G). In addition, FYN promoter 2 (FynΔ7) activity was significantly different according to haplotype in these two glial cell lines where haplotype P2b was significantly higher than P2a (Figure 3F, G) suggesting this promoter haplotype may be more active in glial cells than the other promoter haplotypes. Increased FYN promoter 2 (FynΔ7) activity, compared to FYN promoter 3 (FynT) was observed in neuronal cell lines (IMR-32, CHP-212), but demonstrated a smaller effect size and was only higher than FYN promoter 3 (FynT) and actually lower than FYN promoter 1 (FynB) in CHP-212 cells (Figure 3I, J).
3.4 FYN 3’UTR Haplotype Functional Analysis in Human Cell Lines
Next, since we found an association between Fyn protein levels (Figure 2C) and a 3’UTR SNP (rs1621289), FYN regulatory region reporter constructs were designed to examine whether the FYN gene regulation is influenced by FYN 3’UTR regulatory region genetic content (haplotype) (Figure 4–6). The FYN 3’UTR is shared by all 3 FYN isoforms. FYN 3’UTR containing multiple haplotypes were inserted into the FYN promoter constructs. Each promoter – 3’ UTR construct contained a different promoter haplotype as well as a different 3’ UTR haplotype consisting of rs9387025, rs12191154, rs1621289 and rs9329374 (Figures 4–6). Results are presented as FYN promoter – 3’UTR haplotype constructs relative to FYN promoter only constructs (Relative Quantification) (Figures 4–6). All FYN promoter only activity was set to one and is represented by a dotted line. Standard error bars that do not cross the dotted line indicate a significant difference in FYN promoter only activity and FYN promoter – 3’UTR haplotype constructs. Promoters 1, 2 and 3 are labeled as P1, P2 and P3 and each haplotype is labeled as a, b or c. Each 3’UTR haplotype is labeled as 3Ua, 3Ub, 3Uc (Figures 4–6A, B, C).
In HepG2 cells, all FYN promoter – 3’UTR haplotype constructs had significantly lower activity compared to FYN promoter only construct activity (Figure 4D–F) and differences between haplotypes was particularly pronounced for the FYN promoter 2 (FynΔ7) and FYN promoter 3 (FynT) (Figure 4E, F). Haplotype P1b-3Uc was higher than P1c-3Ua, 3Ub and 3Uc (Figure 4D). Haplotype P3a was lower than P3b regardless of 3’UTR haplotype (Figure 4F). Haplotype P2a-3Uc was higher than P2a-3Ub, and P2a-3Ua (Figure 4E) suggesting inhibition specific to the P2-3Uc haplotype in HepG2 cells.
In glial cells, all FYN promoter 2 – 3’UTR haplotype constructs had significantly lower activity compared to FYN promoter only activity (Figure 5E, H, K). In addition, FYN promoter 1 – 3’UTR and FYN promoter 2 – 3’UTR haplotype constructs in U-118 cells had significantly lower activity than the corresponding FYN promoter only construct (Figure 5J, L). The FYN promoter 1b – 3’UTR haplotypes, compared to the promoter only constructs and compared to the promoter 1a – 3’UTR or promoter 1c – 3’UTR was not influenced by any of the 3’UTR in U-87 and U-138 cells (Figure 4D, G). The FYN promoter 3b – 3’UTR haplotype, compared to the promoter only constructs and compared to the promoter 3a – 3’UTR haplotypes in U87 and U138 cells (Figure 5F, I). Overall, in glial cells, the P1b and P3b haplotypes showed the least inhibition regardless of 3’UTR haplotype (Figure 5).
Figure 5. FYN 3’UTR haplotype functional analysis relative to FYN promoter haplotypes in human glial cell lines.
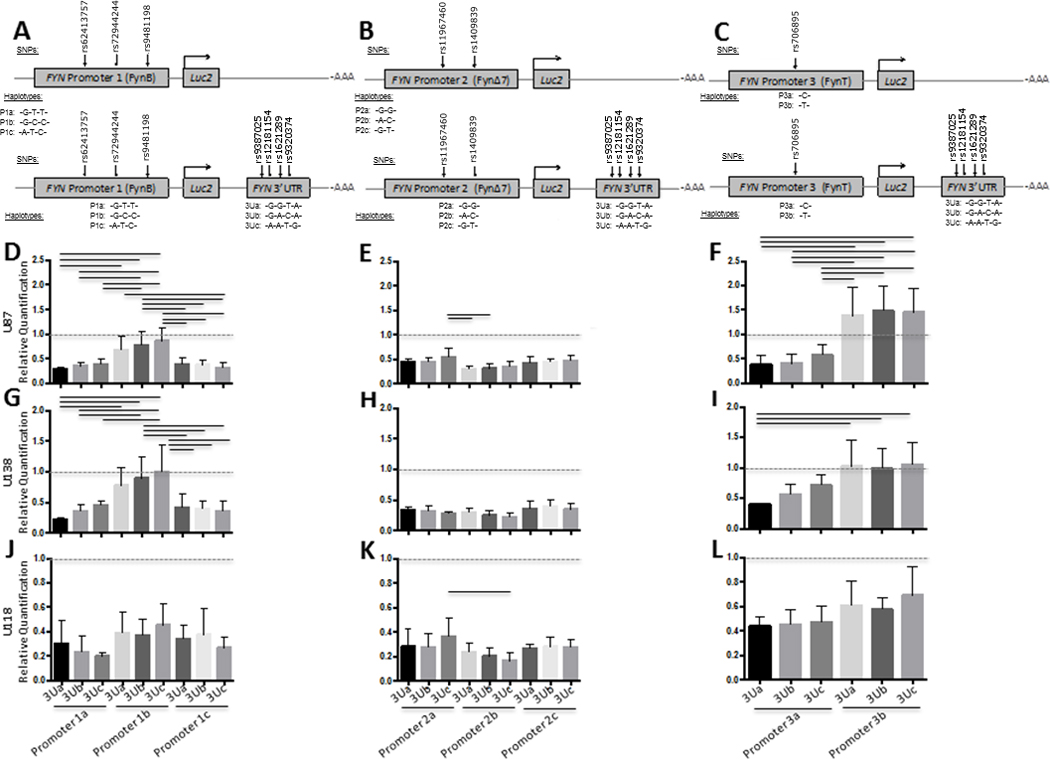
FYN 3’UTR activity was evaluated using luciferase reporter vectors (pGL4.10[luc2]) with both the FYN 3’UTR and FYN promoter haplotypes inserted as depicted in panels A-C. Promoter 1 plus 3’UTR reporter constructs consisted of FynB promoter region haplotype and the FYN 3’UTR (A and D, G, J). Promoter 2 plus 3’UTR reporter constructs consisted of FynΔ7 promoter region haplotypes (B and E, H, K). Promoter 3 plus 3’UTR reporter constructs consisted of FynT promoter region haplotypes (C and F, I, L). Promoter region haplotype reporter constructs and Promoter plus 3’UTR reporter constructs were transiently transfected into a human astroglial cell lines (U87, U138, U118) (DΔF). Results are presented as Firefly/Renilla luciferase activity for the promoter plus 3’UTR constructs relative to the promoter only constructs (Relative Quantification) and SEM for each construct, n = 6 for each group. Black lines denote a significant difference between reporter construct haplotypes (p-value of < 0.05 after Bonferroni correction for multiple comparisons).
In SH-SY5Y cells there was significant inhibition for all FYN promoter 1a – 3’UTR, FYN promoter 1c – 3’UTR FYN promoter 3a – 3’UTR haplotypes, compared to corresponding promoter only constructs, but not FYN promoter 1b -3’UTR 3Ub or FYN promoter 3b -3’UTR 3Uc (Figure 6D–F). In IMR-32 cells all FYN promoter – 3’UTR haplotypes had lower activity compared to their corresponding promoter only constructs (Figure 6G–J). There were significant differences between haplotypes in IMR-32 cells, most pronounced was the differences between P 1b -3Uc compared to the other haplotypes (Figure 6G). In CHP-212 cells all promoter 2 – 3’UTR, promoter 1a – 3’UTR, promoter 1c – 3’UTR haplotype activity, but not promoter 1b – 3’UTR 3Uc, was significantly lower compared to corresponding promoter constructs (Figure 6J, K, L) while the promoter 3b – 3’UTR haplotypes were significantly higher (Figure 6L). In IMR-32 neuronal cells haplotype P1c-3Ua was significantly less inhibited than the P1c-3Uc, and the P1b-3Uc was significantly less inhibited than the P1b-3Ub or P1b-3Ua, suggesting that in IMR-32 cells inhibition is disrupted by the 3’UTR 3Uc haplotype (Figure 6G).
4. Discussion
One of the major hallmarks of AD is the presence of NFTs, thought to be composed of hyperphosphorylated and aggregated tau protein (Boehm, 2013; Duka et al., 2013; Nisbet, Polanco, Ittner, & Götz, 2014; Xia et al., 2015). Notably, phosphorylation of tau by the Src family kinase Fyn is thought to contribute to buildup of NFTs, neuronal excitotoxicity, synaptic deficits and cellular apoptosis (Boehm, 2013; Trepanier, Jackson, & MacDonald, 2012; Xu et al., 2014; Yang et al., 2011)(Haas & Strittmatter, 2016; Larson et al., 2012; Nygaard et al., 2014; Roberson et al., 2011; J W Um et al., 2012; Ji Won Um et al., 2013).
This investigation expands upon information from our previous study which implicated two SNPs in regulatory regions of the FYN gene, rs7768046 and rs1621289, as correlates of CSF tau levels (Lynn M. Bekris, Millard, et al., 2012). Here, we tested the hypotheses that regulatory region SNPs may influence FYN expression in post-mortem brain and human cell lines. Three FYN isoforms were studied: a long isoform (FynB), an isoform of intermediate length (FynΔ7, “medium isoform”), and a short isoform (FynT) that were present in the GRCh37/hg19 assembly of the UCSC Genome Browser (Figure 1) (Rosenbloom et al., 2015). Interestingly, each of these FYN isoforms have the same 3’ UTR, suggesting that the FYN 3’ UTR may be a master regulator of expression regardless of isoform.
To examine the relationship between regulatory region SNPs on Fyn expression, we first evaluated FYN mRNA and protein levels in post-mortem brain tissue with respect to AD status and genotype. FYN mRNA levels were not significantly different in AD patients compared to cognitively normal controls (Figure 2A). In contrast, Fyn protein levels were increased in both CB and HP from AD patients compared to controls (Figure 2B). Together, these data suggest that Fyn protein levels are elevated in AD regardless of mRNA levels, highlighting the importance of Fyn regulation in the brain. Others have reported Fyn as elevated in AD brain compared to controls (Chingli Lee et al., 2015). Interestingly, Fyn has been described as expressed in glial cells and up-regulated FynT is activated in astrocytes upon treatment with amyloid peptides (Chun, Crispino, & Tocco, 2004; Chingli Lee et al., 2015). Therefore, it could be speculated that higher Fyn protein in AD compared to controls (Figure 2B) may be partly driven by a preponderance of glial versus neuronal cells in AD brain. Fyn immunostaining revealed a similar pattern of staining of oligodendroglial nuclei and cytoplasm in the white matter of both AD and controls. However, in the HP the pattern of Fyn immunostaining was different in AD compared to controls, in that we observed plaque-associated neurite staining only in AD (Figure 2D–G)(Peckham et al., 2016).
One possible explanation for a lack of FYN mRNA increase, but an increase in Fyn protein, is that AD patients may exhibit defects in the turnover of the Fyn kinase protein. Similar results have been found by others studying glutamate receptor activity in the rat retina, where improper regulation of the GluR1 AMPA receptor subunit and other associated proteins had low mRNA levels, high protein levels, and increased reactive oxygen species associated with excitotoxicity and cell death (Challenor et al., 2015). Therefore, aberrant Fyn protein expression levels could be a result of misregulation of a protein that modifies Fyn activity, such as STEP (Boehm, 2013; Kaufman et al., 2015; Nygaard et al., 2014). Another intriguing possibility is the role of stress granules, which may sequester FYN mRNA in the aging brain and in response to increased cellular stress caused by the accumulation of amyloid plaques and NFTs allow translation of FYN (Ash, Vanderweyde, Youmans, Apicco, & Wolozin, 2014; Vanderweyde et al., 2012).
Since SNPs located in key regulatory regions, such as the promoter or the 3’ UTR, can affect translational dynamics and alter protein levels. Therefore, we examined 11 SNPs in promoter and 3’UTR regulatory regions and their relationship with Fyn expression levels. Consistent with the aggregate data presented in Figure 2A, individual regulatory SNP genotype had no significant effect on FYN mRNA levels (data not shown). In contrast, Fyn protein levels were significantly elevated in AD tissue (Figure 2B), were elevated in CB compared to HP in both AD and controls (Figure 2B), and a 3’UTR SNP was associated with Fyn protein levels in AD HP (Figure 2C). However, this difference did not remain significant after correction for multiple comparisons. Taken together, these data suggest that promoter genetic content may influence Fyn expression in a brain region specific manner both in cognitively normal controls and AD patients while FYN 3’UTR genetic content may abnormally influence Fyn protein levels in AD HP. One possible explanation for this finding is that in the toxic AD microenvironment of the HP there are specific trans-acting factors abnormally regulating FYN expression and the genetic variant acts to disrupts or enhance this regulation. Alternatively, these SNPs could be surrogates for the true genetic variant culprit. Notably, other groups have demonstrated differential Fyn isoform imbalance in AD, specifically an increase of the FynT/FynB ratio in neurons presenting NFTs (Chingli Lee et al., 2015) supporting the notion that Fyn isoform regulation may be different depending on cell type or disease status.
Interestingly, when evaluating the influence of promoter genetic content on expression using a reporter assay, there was not a significant difference in reporter activity between FYN promoter haplotypes in HepG2, U-87 or SH-SY5Y cells (Figure 3D, E, H). Notably, in the U-138 and U-118 cell lines, the promoter 2 (FynΔ7) haplotypes showed significantly elevated reporter activity compared to the other haplotypes (Figure 3F, G) suggesting this promoter may be more active in glial cells compared to the other promoters. However, it is difficult to draw a meaningful parallel to what is happening in vivo given that these are immortalized cell lines. Furthermore, P2b was significantly higher P2a or P2c haplotypes in the U-118 cells (Figure 3G) and higher in U138 compared to P2c, suggesting P2b containing an rs11967460 A allele may contribute to increased Fyn expression in glial cell types. Notably, in the U-118 glial cells and the CHP-212 neuronal cells, promoter 1 (FynB) was elevated compared to promoter 3 (FynT) which is show an increase in FynT/FynB ratio in neurons, suggesting Fyn isoform imbalance in AD (Chingli Lee et al., 2015). Elevated promoter 2b reporter activity was also seen in the IMR-32 and CHP-212 cells (Figure 3I, J) but showed a different pattern in CHP-212 compared to the glial cell lines in that the promoter 2 activity was lower than promoter 1. Taken together, these data suggest that differences in FYN promoter activity are not only dependent upon cell type and FYN promoter isoform, but also promoter genetic content.
Interestingly, the promoter 2 (FynΔ7) haplotype contains rs1409839 which has potential overlap with a region of p300 histone acetyltransferase activity in HepG2 cells (Genome Browser hg19, ENCODE HepG2 P300 s, chr6:112,114,855–112,115,855) (Rosenbloom et al., 2013, 2015). Therefore, it is possible that genetic variants in this region may result in differing histone acetylation patterns, thereby changing transcriptional activation and FYN expression. Also of note, the medium promoter drives transcription of the FynΔ7 isoform lacking kinase activity, so tight control of this isoform’s expression is logical for proper Fyn activity (Goldsmith et al., 2002). Similar to the medium promoter site, the long promoter is extensively covered by histone modification sites, which could play a likely role in gene expression.
SNPs, rs12191154 and rs1621289, are both located just upstream of the shared FYN 3’ UTR in the last intron of FYN within a strong H3K4me1 region (Figure 1A) suggesting that an extended 3’region may influence expression. Therefore, we examined whether genetic content of this expanded FYN 3’UTR might differentially influence FYN expression. Interestingly, the rs1621289 TT genotype in AD HP is associated with higher levels of Fyn protein in our post-mortem brain sample (Figure 2E). Thus, if it can be speculated that high levels of Fyn are associated with an increase in p-tau/t-tau, then these results further support our previous findings that showed the rs1621289 T allele is associated with marginally, but non-significant, higher p-tau/t-tau levels in AD CSF (Lynn M. Bekris, Millard, et al., 2012). The 3’ UTR SNP rs1621289 was not associated with FYN mRNA levels in the brain (data not shown), but was associated with Fyn protein levels (Figure 2C) suggesting post-transcriptional regulation at the translational level. Previous work has demonstrated that FYN can be targeted by the microRNA (miRNA) hsa-miR-125a-3p, but a link between this miRNA and AD has yet to be described (Grossman et al., 2015; Ninio-Many et al., 2013). Transcriptional inhibition is a well-documented phenomenon where the 3’ UTR contributes to translation repression or mRNA degradation (Hughes, Gilley, Kristiansen, & Ham, 2011; Le Cam & Legraverend, 1995; Ogbourne & Antalis, 1998). Genetic variants in AD have been described as influencing miRNA binding sites. For example, SNPs in the 3’ UTR of the APP gene have been associated with disruption of APP gene regulation that leads to increased APP expression (Delay, Calon, Mathews, & Hébert, 2011). Therefore, it is also possible that miRNAs targeting the 3’ UTR could regulate Fyn activity in a similar way.
Therefore, to examine the role of 3’ UTR inhibition on FYN expression, we transiently transfected FYN promoter – 3’ UTR luciferase reporter constructs in seven cell lines (Figures 4–6). Interestingly, the FYN promoter — 3’ UTR reporter constructs inhibited reporter activity, compared to the promoter only construct activity, in most of the cell lines tested. Furthermore, the level of inhibition varied by cell type and haplotype. However, there were some notable exceptions to this inhibition (Figure 4D–E). For example, P2a – 3Uc had significantly less inhibition than the other P2a – 3Ua or P2a – 3Ub in HepG2 cells (Figure 4E) suggesting that in these cells 3’UTR specific trans-acting factor post-transcriptional regulation is disrupted by haplotype. Interestingly, the 3Uc 3’UTR haplotype differs from the other 3’UTR haplotypes by rs9320374 G allele suggesting that this region may play a role in post-transcriptional regulation in HepG2 cells (Figure 4E). In contrast, in glial cells P1b and P3b haplotypes showed a lack of inhibition regardless of 3’UTR haplotype suggesting that a promoter specific trans-acting factor is post-transcriptionally disrupted in these cells (Figure 5F, I, D, G). The P1b haplotype differs from the P1a and P1c by the rs72944244 C allele while the P3b has the rs706895 T allele suggesting that these SNP regions may play a role in P1 post-transcriptional regulation in glial cells. Furthermore, in neuronal cell lines the P1b and P3b haplotypes showed a significant lack of inhibition compared to the other promoter haplotypes (Figure 6D, G, I, J, F, L). Interestingly, there was a lack of inhibition for the P1b – 3Uc haplotype in IMR-32 and CHP-212 cells suggesting that the 3’UTR rs9320374 G allele may play a post-transcriptional role in these cells. Taken together, these data suggest that differences in 3’UTR activity may be dependent upon cell type, FYN promoter isoform and genetic content of both the promoter and the 3’UTR.
Higher FYN protein levels in the AD HP are associated with the FYN 3’UTR rs1621289 TT genotype. Notably, the FYN 3’UTR 3Ua and 3Uc haplotype contain the rs1621289 T allele; however, this allele only showed a lack of inhibition (higher expression) compared to other haplotypes in the presence of the P1b and P2a promoter haplotypes and did not show higher expression in the context of the 3Ua haplotypes regardless of promoter haplotype. These results suggest that specific cell lines may contain a miRNA or other trans-acting factor acting upon the 3’ UTR not at the rs1621289 site but rather the rs9320374 since 3Uc differs from the haplotype at this SNP by the G allele. Together, these data suggest that FYN regulation is a complex, multifactorial process under regulation by both its promoter and 3’ UTR.
In summary, our previous investigation found an association between a FYN regulatory region genetic variants and CSF tau levels. In the present investigation, these previous results are supported by our findings that expression in both the AD HP and regulatory region reporter assays are associated with FYN regulatory region genetic variation. Together, these results suggest that FYN promoter and FYN 3’ UTR genetic content can alter Fyn expression, potentially providing us with insight into misregulation of Fyn in AD. These results have important implications for AD since aberrant increased Fyn expression may lead to hyperphosphorylation of its downstream targets, such as tau.
FYN Paper Highlights.
FYN mRNA levels in the cerebellum or hippocampus of AD patients are similar to wild type
Fyn protein levels are increased in the cerebellum of AD patients
Fyn mRNA and protein expression are altered by FYN promoter and 3’ UTR SNPs
The FYN 3’ UTR can inhibit promoter activity in multiple cell lines
Acknowledgments
This work was supported by grants from the National Institutes of Health (K99/R00 AG034214 and P50 AG05136) and the Jane and Lee Seidman Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: The authors declare that they have no actual or potential conflicts of interest related to the publishing of this work.
References
- Abràmoff MD, Magalhães PJ, Ram SJ. Image processing with imageJ. Biophotonics International. 2004;11(7):36–41. http://doi.org/10.1117/1.3589100. [Google Scholar]
- Allen M, Kachadoorian M, Quicksall Z, Zou F, Chai HS, Younkin C, Ertekin-Taner N. Association of MAPT haplotypes with Alzheimer’s disease risk and MAPT brain gene expression levels. Alzheimer’s Research & Therapy. 2014;6(4):39. doi: 10.1186/alzrt268. http://doi.org/10.1186/alzrt268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash PEa, Vanderweyde TE, Youmans KL, Apicco DJ, Wolozin B. Pathological Stress Granules in Alzheimer’s Disease. Brain Research. 2014;1584:1–7. doi: 10.1016/j.brainres.2014.05.052. http://doi.org/10.1016/j.brainres.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekris LM, Lutz F, Li G, Galasko DR, Farlow MR, Quinn JF, Yu C-E. ADAM10 expression and promoter haplotype in Alzheimer’s disease. Neurobiology of Aging. 2012;33(9):2229.e1–2229.e9. doi: 10.1016/j.neurobiolaging.2012.03.013. http://doi.org/10.1016/j.neurobiolaging.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekris LM, Millard S, Lutz F, Li G, Galasko DR, Farlow MR, Peskind ER. Tau phosphorylation pathway genes and cerebrospinal fluid tau levels in Alzheimer’s disease. American Journal of Medical Genetics, Part. B: Neuropsychiatric Genetics. 2012;159(B(7)):874–883. doi: 10.1002/ajmg.b.32094. http://doi.org/10.1002/ajmg.b.32094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekris LM, Millard SP, Galloway NM, Vuletic S, Albers JJ, Li G, Yu C-E. Multiple SNPs within and surrounding the apolipoprotein E gene influence cerebrospinal fluid apolipoprotein E protein levels. Journal of Alzheimer’s Disease : JAD. 2008;13(3):255–266. doi: 10.3233/jad-2008-13303. http://doi.org/10.1016/j.bbi.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekris LM, Yu C-E, Bird TD, Tsuang DW. Review Article: Genetics of Alzheimer Disease. Journal of Geriatric Psychiatry and Neurology. 2010;23(4):213–227. doi: 10.1177/0891988710383571. http://doi.org/10.1177/0891988710383571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm J. A “danse macabre”: tau and Fyn in STEP with amyloid beta to facilitate induction of synaptic depression and excitotoxicity. European Journal of Neuroscience. 2013;37(12):1925–1930. doi: 10.1111/ejn.12251. http://doi.org/10.1111/ejn.12251. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica. 1991;82(4):239–259. doi: 10.1007/BF00308809. http://doi.org/10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Challenor M, O’Hare Doig R, Fuller P, Giacci M, Bartlett C, Wale CH, Fitzgerald M. Prolonged glutamate excitotoxicity increases GluR1 immunoreactivity but decreases mRNA of GluR1 and associated regulatory proteins in dissociated rat retinae in vitro. Biochimie. 2015;112:160–171. doi: 10.1016/j.biochi.2015.03.008. http://doi.org/10.1016/j.biochi.2015.03.008. [DOI] [PubMed] [Google Scholar]
- Chin J. Fyn Kinase Induces Synaptic and Cognitive Impairments in a Transgenic Mouse Model of Alzheimer’s Disease. Journal of Neuroscience. 2005;25(42):9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. http://doi.org/10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun JT, Crispino M, Tocco G. The dual response of protein kinase Fyn to neural trauma: Early induction in neurons and delayed induction in reactive astrocytes. Experimental Neurology. 2004;185(1):109–119. doi: 10.1016/j.expneurol.2003.09.019. http://doi.org/10.1016/j.expneurol.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Davidson D, Chow LM, Fournel M, Veillette A. Differential regulation of T cell antigen responsiveness by isoforms of the src-related tyrosine protein kinase p59fyn. The Journal of Experimental Medicine. 1992;175(6):1483–1492. doi: 10.1084/jem.175.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson D, Viallet J, Veillette a. Unique catalytic properties dictate the enhanced function of p59fynT, the hemopoietic cell-specific isoform of the Fyn tyrosine protein kinase, in T cells. Molecular and Cellular Biology. 1994;14(7):4554–4564. doi: 10.1128/mcb.14.7.4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delay C, Calon F, Mathews P, Hébert SS. Alzheimer-specific variants in the 3’UTR of Amyloid precursor protein affect microRNA function. Molecular Neurodegeneration. 2011;6(1):70. doi: 10.1186/1750-1326-6-70. http://doi.org/10.1186/1750-1326-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C-P, Tan R, Hou X-Y. Fyn Kinases Play a Critical Role in Neuronal Apoptosis Induced by Oxygen and Glucose Deprivation or Amyloid-β Peptide Treatment. CNS Neuroscience & Therapeutics. 2012;18(9):754–761. doi: 10.1111/j.1755-5949.2012.00357.x. http://doi.org/10.1111/j.1755-5949.2012.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duka V, Lee JH, Credle J, Wills J, Oaks A, Smolinsky C, Sidhu A. Identification of the Sites of Tau Hyperphosphorylation and Activation of Tau Kinases in Synucleinopathies and Alzheimer’s Diseases. PLoS ONE. 2013;8(9):1–11. doi: 10.1371/journal.pone.0075025. http://doi.org/10.1371/journal.pone.0075025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Howard A, Ban K, Chandra J. Oxidative stress promotes transcriptional up-regulation of Fyn in BCR-ABL1-expressing cells. Journal of Biological Chemistry. 2009;284(11):7114–7125. doi: 10.1074/jbc.M804801200. http://doi.org/10.1074/jbc.M804801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith JF, Hall CG, Atkinson TP. Identification of an alternatively spliced isoform of the fyn tyrosine kinase. Biochemical and Biophysical Research Communications. 2002;298:501–504. doi: 10.1016/s0006-291x(02)02510-x. http://doi.org/10.1016/S0006-291X(02)02510-X. [DOI] [PubMed] [Google Scholar]
- Grossman H, Chuderland D, Ninio-Many L, Hasky N, Kaplan-Kraicer R, Shalgi R. A novel regulatory pathway in granulosa cells, the LH/human chorionic gonadotropin-microRNA-125a-3p-Fyn pathway, is required for ovulation. The FASEB Journal. 2015;29(8):3206–3216. doi: 10.1096/fj.14-269449. http://doi.org/10.1096/fj.14-269449. [DOI] [PubMed] [Google Scholar]
- Haas LT, Strittmatter SM. Oligomers of Amyloid-beta Prevent Physiological Activation of the Cellular Prion Protein-Metabotropic Glutamate Receptor 5 Complex by Glutamate in Alzheimer′s Disease. Journal of Biological Chemistry. 2016 doi: 10.1074/jbc.M116.720664. jbc.M116.720664, http://doi.org/10.1074/jbc.M116.720664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Wu Z, Zhou B. Behind the curtain of tauopathy: a show of multiple players orchestrating tau toxicity. Cellular and Molecular Life Sciences. 2015 doi: 10.1007/s00018-015-2042-8. http://doi.org/10.1007/s00018-015-2042-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes R, Gilley J, Kristiansen M, Ham J. The MEK-ERK pathway negatively regulates bim expression through the 3 ’ UTR in sympathetic neurons. BMC Neuroscience. 2011;12(1):69. doi: 10.1186/1471-2202-12-69. http://doi.org/10.1186/1471-2202-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong C-X. Tau and neurodegenerative disease: the story so far. Nature Reviews Neurology. 2015;12(1):1–14. doi: 10.1038/nrneurol.2015.225. http://doi.org/10.1038/nrneurol.2015.225. [DOI] [PubMed] [Google Scholar]
- Jayapalan S, Natarajan J. The role of CDK5 and GSK3B kinases in hyperphosphorylation of microtubule associated protein tau (MAPT) in Alzheimer’s disease. Bioinformation. 2013;9(20):1023–1030. doi: 10.6026/97320630091023. http://doi.org/10.6026/97320630091023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev Ma, Jeng AT, Strittmatter SM. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Annals of Neurology. 2015 doi: 10.1002/ana.24394. n/a-n/a, http://doi.org/10.1002/ana.24394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HM, Lee SH, Kim KC, Joo SH, Choi WS, Shin CY. The Role of TLR4 and Fyn Interaction on Lipopolysaccharide-Stimulated PAI-1 Expression in Astrocytes. Molecular Neurobiology. 2015;52(1):8–25. doi: 10.1007/s12035-014-8837-z. http://doi.org/10.1007/s12035-014-8837-z. [DOI] [PubMed] [Google Scholar]
- Krüger L, Mandelkow EM. Tau neurotoxicity and rescue in animal models of human Tauopathies. Current Opinion in Neurobiology. 2016;36:52–58. doi: 10.1016/j.conb.2015.09.004. http://doi.org/10.1016/j.conb.2015.09.004. [DOI] [PubMed] [Google Scholar]
- Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, Lesné SE. The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2012;32(47):16857–16871a. doi: 10.1523/JNEUROSCI.1858-12.2012. http://doi.org/10.1523/JNEUROSCI.1858-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Cam A, Legraverend C. Transcriptional repression, a novel function for 3 ’ untranslated regions. European Journal of Biochemistry. 1995;231(3):620–627. [PubMed] [Google Scholar]
- Lee C, Kim MG, Jeon SH, Park DE, Park SD, Seong RH. Two species of mRNAs for the fyn proto-oncogene are produced by an alternative polyadenylation. Mol Cells. 1998;8(6):746–749. [PubMed] [Google Scholar]
- Lee C, Low CYB, Francis PT, Attems J, Wong PTH, Lai MKP, Tan MGK. An isoform-specific role of FynT tyrosine kinase in Alzheimer’s disease. Journal of Neurochemistry. 2015:637–650. doi: 10.1111/jnc.13429. http://doi.org/10.1111/jnc.13429. [DOI] [PubMed] [Google Scholar]
- Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Ksiezak-Reding H. Phosphorylation of tau by fyn: implications for Alzheimer’s disease. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2004;24(9):2304–2312. doi: 10.1523/JNEUROSCI.4162-03.2004. http://doi.org/10.1523/JNEUROSCI.4162-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Yang TT, Weng RR, Kuo C, Chang C. TTBK2: A Tau Protein Kinase beyond Tau Phosphorylation. BioMed Research International. 2015;2015:1–10. doi: 10.1155/2015/575170. http://doi.org/10.1155/2015/575170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E-M. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. The Journal of Cell Biology. 2004;167(1):99–110. doi: 10.1083/jcb.200401085. http://doi.org/10.1083/jcb.200401085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews AG, Haynes C, Liu C, Ott J. Collapsing SNP Genotypes in Case-Control Genome-Wide Association Studies Increases the Type I Error Rate and Power. Statistical Applications in Genetics and Molecular Biology. 2008;7(1):1325. doi: 10.2202/1544-6115.1325. http://doi.org/10.2202/1544-6115.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninio-Many L, Grossman H, Shomron N, Chuderland D, Shalgi R. microRNA-125a-3p reduces cell proliferation and migration by targeting Fyn. Journal of Cell Science. 2013;126(13):2867–2876. doi: 10.1242/jcs.123414. http://doi.org/10.1242/jcs.123414. [DOI] [PubMed] [Google Scholar]
- Nisbet RM, Polanco J-C, Ittner LM, Götz J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathologica. 2014;129(2):207–220. doi: 10.1007/s00401-014-1371-2. http://doi.org/10.1007/s00401-014-1371-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard HB, van Dyck CH, Strittmatter SM. Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alzheimer’s Research & Therapy. 2014;6(1):8. doi: 10.1186/alzrt238. http://doi.org/10.1186/alzrt238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogbourne S, Antalis TM. Transcriptional control and the role of silencers in transcriptional regulation in eukaryotes. Biochem J. 1998;14:1–14. doi: 10.1042/bj3310001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckham H, Giuffrida L, Wood R, Gonsalvez D, Ferner A, Kilpatrick TJ, Xiao J. Fyn is an intermediate kinase that BDNF utilizes to promote oligodendrocyte myelination. Glia. 2016;64(2):255–269. doi: 10.1002/glia.22927. http://doi.org/10.1002/glia.22927. [DOI] [PubMed] [Google Scholar]
- Resh MD. Fyn, a Src family tyrosine kinase. Int.J.Biochem.Cell Biol. 1998;30(11):1159–1162. doi: 10.1016/s1357-2725(98)00089-2. http://doi.org/10.1016/S1357-2725(98)00089-2. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Mucke L. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2011;31(2):700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. http://doi.org/10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Kent WJ. The UCSC Genome Browser database: 2015 update. Nucleic Acids Research. 2015;43(D1):D670–D681. doi: 10.1093/nar/gku1177. http://doi.org/10.1093/nar/gku1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan Ca, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Kent WJ. ENCODE Data in the UCSC Genome Browser: year 5 update. Nucleic Acids Research. 2013;41(D1):D56–D63. doi: 10.1093/nar/gks1172. http://doi.org/10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato-Harada R, Okabe S, Umeyama T, Kanai Y, Hirokawa N. Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Structure and Function. 1996;21:283–295. doi: 10.1247/csf.21.283. http://doi.org/10.1247/csf.21.283. [DOI] [PubMed] [Google Scholar]
- Shanley MR, Hawley D, Leung S, Zaidi NF, Dave R, Schlosser Ka, Liu M. LRRK2 Facilitates tau Phosphorylation through Strong Interaction with tau and cdk5. Biochemistry. 2015;54(33):5198–5208. doi: 10.1021/acs.biochem.5b00326. http://doi.org/10.1021/acs.biochem.5b00326. [DOI] [PubMed] [Google Scholar]
- Sontag JM, Nunbhakdi-Craig V, White CL, Halpain S, Sontag E. The protein phosphatase PP2A/Bα binds to the microtubule-associated proteins Tau and MAP2 at a motif also recognized by the kinase Fyn: Implications for tauopathies. Journal of Biological Chemistry. 2012;287(18):14984–14993. doi: 10.1074/jbc.M111.338681. http://doi.org/10.1074/jbc.M111.338681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenreiro S, Eckermann K, Outeiro TF. Protein phosphorylation in neurodegeneration: friend or foe? Frontiers in Molecular Neuroscience. 2014 May;7:1–30. doi: 10.3389/fnmol.2014.00042. http://doi.org/10.3389/fnmol.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepanier CH, Jackson MF, MacDonald JF. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS Journal. 2012;279(1):12–19. doi: 10.1111/j.1742-4658.2011.08391.x. http://doi.org/10.1111/j.1742-4658.2011.08391.x. [DOI] [PubMed] [Google Scholar]
- Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Strittmatter SM. Metabotropic Glutamate Receptor 5 Is a Coreceptor for Alzheimer Aβ Oligomer Bound to Cellular Prion Protein. Neuron. 2013;79(5):887–902. doi: 10.1016/j.neuron.2013.06.036. http://doi.org/10.1016/j.neuron.2013.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Strittmatter SM. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012;15(9):1227–1235. doi: 10.1038/nn.3178. http://doi.org/10.1038/nn.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro a, Ikezu T, Wolozin B. Contrasting Pathology of the Stress Granule Proteins TIA-1 and G3BP in Tauopathies. Journal of Neuroscience. 2012;32(24):8270–8283. doi: 10.1523/JNEUROSCI.1592-12.2012. http://doi.org/10.1523/JNEUROSCI.1592-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia D, Li C, Götz J. Pseudophosphorylation of Tau at distinct epitopes or the presence of the P301L mutation targets the microtubule-associated protein Tau to dendritic spines. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2015;1852(5):913–924. doi: 10.1016/j.bbadis.2014.12.017. http://doi.org/10.1016/j.bbadis.2014.12.017. [DOI] [PubMed] [Google Scholar]
- Xu J, Chatterjee M, Baguley TD, Brouillette J, Kurup P, Ghosh D, Lombroso PJ. Inhibitor of the Tyrosine Phosphatase STEP Reverses Cognitive Deficits in a Mouse Model of Alzheimer’s Disease. PLoS Biology. 2014;12(8):e1001923. doi: 10.1371/journal.pbio.1001923. http://doi.org/10.1371/journal.pbio.1001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Belrose J, Trepanier CH, Lei G, Jackson MF, MacDonald JF. Fyn, a potential target for alzheimer’s disease. Journal of Alzheimer’s Disease. 2011;27(2):243–252. doi: 10.3233/JAD-2011-110353. http://doi.org/10.3233/JAD-2011-110353. [DOI] [PubMed] [Google Scholar]