

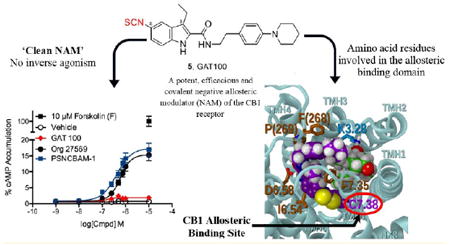
Abstract
One of the most abundant G-protein coupled receptors (GPCRs) in brain, the cannabinoid 1 receptor (CB1R), is a tractable therapeutic target for treating diverse psychobehavioral and somatic disorders. Adverse on-target effects associated with small-molecule CB1R orthosteric agonists and inverse agonists/antagonists have plagued their translational potential. Allosteric CB1R modulators offer a potentially safer modality through which CB1R signaling may be directed for therapeutic benefit. Rational design of candidate, druglike CB1R allosteric modulators requires greater understanding of the architecture of the CB1R allosteric endodomain(s) and the capacity of CB1R allosteric ligands to tune the receptor’s information output. We have recently reported the synthesis of a focused library of rationally designed, covalent analogues of Org27569 and PSNCBAM-1, two prototypic CB1R negative allosteric modulators (NAMs). Among the novel, pharmacologically active CB1R NAMs reported, the isothiocyanate GAT100 emerged as the lead by virtue of its exceptional potency in the [35S]GTPγS and β-arrestin signaling assays and its ability to label CB1R as a covalent allosteric probe with significantly reduced inverse agonism in the [35S]GTPγS assay as compared to Org27569. We report here a comprehensive functional profiling of GAT100 across an array of important downstream cell-signaling pathways and analysis of its potential orthosteric probe-dependence and signaling bias. The results demonstrate that GAT100 is a NAM of the orthosteric CB1R agonist CP55,940 and the endocannabinoids 2-arachidonoylglycerol and anandamide for β-arrestin1 recruitment, PLCβ3 and ERK1/2 phosphorylation, cAMP accumulation, and CB1R internalization in HEK293A cells overexpressing CB1R and in Neuro2a and STHdhQ7/Q7 cells endogenously expressing CB1R. Distinctively, GAT100 was a more potent and efficacious CB1R NAM than Org27569 and PSNCBAM-1 in all signaling assays and did not exhibit the inverse agonism associated with Org27569 and PSNCBAM-1. Computational docking studies implicate C7.38(382) as a key feature of GAT100 ligand-binding motif. These data help inform the engineering of newer-generation, druggable CB1R allosteric modulators and demonstrate the utility of GAT100 as a covalent probe for mapping structure–function correlates characteristic of the druggable CB1R allosteric space.
Keywords: Allosteric covalent probe, allosteric site, biased signaling, binding domain, cannabinoid 1 receptor, cysteine, functional selectivity, G protein-coupled receptors, homology modeling, isothiocyanate, ligand bias, ligand-binding motif, negative allosteric modulator, 7-transmembrane receptor, signal transduction, therapeutics discovery
Graphical abstract
The ubiquitous mammalian endocannabinoid biosignaling system includes the cannabinoid 1 and (CB1R) and 2 (CB2R) G-protein coupled receptors (GPCRs), their endogenous agonist ligands (endocannabinoids) anandamide (AEA) and 2-arachidonoylglycerol (2-AG), and enzymes responsible for endocannabinoid synthesis and inactivation.1,2 As the mostexpressed GPCR in brain, CB1R’s principal neurobiological function is to transduce endocannabinoid signaling and thereby inhibit neurotransmitter release.3 On the other hand, CB2R is mainly expressed in peripheral tissues and to a lesser extent in CNS and has been pursued for treating pain and inflammation. 4-10 In the periphery, CB1R activity helps regulate metabolic processes including substrate storage and mobilization. Dysregulation of CB1R-mediated signaling is implicated in the etiology of several disorders, making CB1R an attractive target of small-molecule pharmacotherapeutics for controlling pain and nausea and treating diseases including obesity, type 2 diabetes, glaucoma, and substance abuse.11,12
Structurally diverse CB1R orthosteric ligands have been identified with varying chemotypes, potencies, and selectivities.12-19 These include agonists such as the natural phytocannabinoid ∆9- tetrahydrocannabinol and the synthetic agentCP55,940 ([2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol])] and the antagonist/inverse agonist SR141716A ([5-(4-chlorophenyl)-1-(2,4-dichloro-phenyl)-4- methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide]). These and other CB1R orthosteric agonists and antagonists/inverse agonists can exert therapeutic effects in preclinical disease models and, as demonstrated to a more limited extent, in humans.20 However, typical orthosteric CB1R agonists carry risks of abuse potential, euphoria, anxiety, panic, and impaired cognition and motor function.14 CB1R orthosteric antagonists/inverse agonists are associated with serious negative psychobehavioral (e.g., depression, social aversion, suicidal ideation) and somatic (e.g., gastrointestinal) effects, making for an unacceptable adverseevent profile that has been attributed to their inverse-agonist property.21,22
Nonselective (de)activation of downstream CB1R signaling pathways by typical CB1R orthosteric ligands is believed to contribute to the unwanted side effects that have impeded their therapeutic application as drugs. Although CB1R preferentially couples to Gαi/o-type G proteins, it may also interact with Gαs or Gαq.23,24 The CB1R-mediated signaling network involves diverse G protein-dependent (e.g., adenylate cyclase) and -independent (e.g., β-arrestin) pathways and modulation of complex communication networks such as mitogen-activated protein kinase (MAPK) cascades.25-27 The variegated nature of the CB1R-associated signaling network is indicative of multiple, finely controlled information-transducing mechanisms that could be selectively exploited for therapeutic gain with the potential of reduced adverse-event risk as compared to typical orthosteric CB1R agonists antagonists/inverse agonists. Mounting evidence over the past decade indicates that CB1R contains (an) allosteric binding site(s) topographically distinct from the receptor’s orthosteric site that can engage endogenous and synthetic ligands.28-32 Allosteric-based drugs represent a pharmacological approach to therapeutic CB1R modulation that could offer distinct translational advantages over ligands that bind at the receptor’s orthosteric site, as implied by the extended ternary complex model of GPCR multistate structure–function dynamics.33,34 This model posits that engagement of an allosteric modulator by a GPCR alters the receptor’s affinity for an orthosteric ligand and/or the orthosteric ligand’s efficacy by inducing cooperative conformational transitions in the receptor that affect both orthosteric-ligand (dis)association rates and downstream functional effects.33,34 In this manner, an allosteric modulator may stabilize a GPCR in conformations not readily attainable with orthosteric ligands so as to “fine-tune” the pharmacological activity of the orthosteric ligand and elicit unique, therapeutically advantageous downstream signaling patterns. The enhanced selectivity, reduced inter-receptor promiscuity, and higher-resolution control of information transmission characteristic of GPCR allosteric modulators are also believed to contribute to an improved safety profile as compared to typical orthosteric ligands.33,34
The two classical CB1R allosteric ligands, Org2756928 (5-chloro-3-ethyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole- 2-carboxamide) and PSNCBAM-129 (1-(4-chlorophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridine-2-yl)phenyl)urea) (Figure 1), each behave as both a negative allosteric modulator (NAM) of CP55,940 potency and efficacy in G-protein mediated signaling and a positive allosteric modulator (PAM) of CP55,940 binding affinity. Additionally, few endogenous ligands (Lipoxin A4, pregnenolone, and pepcans; see Figure 1 for representative structures) have also been shown to act as CB1R allosteric modulators. 28,31,32,35 Org27569 and PSNCBAM-1 displace the CB1R orthosteric antagonist/inverse agonist SR141716A in competitive binding assays, suggesting that these three ligands may partially share a binding region.28,29,36,37 Although Org27569 and PSNCBAM-1 have been intensively studied over the past decade, their therapeutic application has been hampered by the lack of suitably liganded CB1R crystal structures for atomic-level structural characterization and, perhaps most critically, their inverse-agonist property which—by extension from CB1R orthosteric antagonists/inverse agonists—could predispose to unacceptable adverse events.29,38,39 To inform rational drug design aimed at therapeutic CB1R allosteric modulation and delineate the optimal structural and pharmacophore requirements thereof, it thus becomes necessary to expand the currently limited repertoire of CB1R allosteric ligands and detail their receptor–interaction profile and effects on CB1R structure and function.
Figure 1.
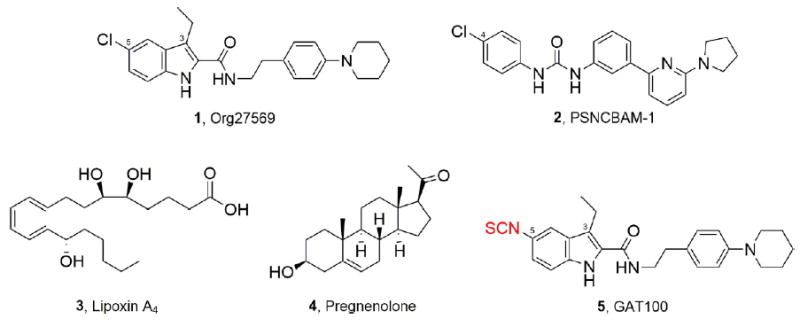
Representative allosteric modulators of the CB1 receptor and the “first-ever” CB1R allosteric site covalent probe.
Although homology modeling and mutation studies have offered some insight into the Org27569 and PSNCBAM-1 binding domains at CB1R, inherent limitations associated with these techniques do not allow direct experimental delineation of intermolecular interactions between NAMs and functional, wild-type CB1R and the consequent effect of NAM engagement on CB1R signaling and trafficking.37,40 Purpose-designed, pharmacologically active orthosteric ligands bearing functionalities reactive with specific GPCR amino acid(s) are increasingly being utilized as covalent molecular probes for GPCR activity profiling and orthosteric binding-site mapping.41,42 For instance, this approach has been successfully integrated with mutational, mass spectrometry, and in silico modeling studies in an experimental paradigm called ligand-assisted protein structure (LAPS) to provide unique information on the interaction of various CB1R orthosteric ligands with biologically active receptor.41,43-47
In an effort to extend the application of covalent probes for experimental mapping of the CB1R druggable allosteric space, we recently detailed the rational design and synthesis of novel Org27569 and PSNCBAM-1 analogues as the first-reported designer CB1R allosteric probes.48 In contrast to those two prototypical noncovalent CB1R allosteric ligands, the pharmacologically active derivatives in our library, by virtue of their electrophilic or photoactivatable moiety, are capable of reacting covalently and in a chemically defined, amino acid residuespecific manner within the allosteric ligand-binding domain of functional, wild-type CB1R, thus serving as covalent molecular probes. Among those CB1R designer allosteric probes, GAT100 (Figure 1), featuring an electrophilic NCS functionality at the C-5 position, emerged as the most potent ligand in the two functional assays (GTPγS and β-arrestin) evaluated. GAT100 retained the paradoxical effects of parent compound Org27569, behaving as a PAM of CP55,940 binding, but with improved potency over Org27569.48 Distinctively, in the GTPγS assay, GAT100 did not exhibit the inverse agonism that is a feature of both Org2756928,36,37 and PSNCBAM-1.29,36 Since the association between inverse agonism at CB1R and undesirable psychological and somatic side effects has limited the therapeutic application of orthosteric CB1R antagonists/inverse agonists, GAT100’s lack of inverse-agonist property holds therapeutic promise and underscores the importance of the C-5 site for enhancing Org27569’s druggability.48 The translational implications of those data are further emphasized by the resurgence of interest in the therapeutic opportunities afforded by targeted covalent receptor ligands, especially with respect to simplifying dosing regimens, widening therapeutic margins, and increasing potency, duration, and specificity.49-51
These considerations make it essential to characterize the signaling repertoire of GAT100 at CB1R as compared to that of Org27569 and PSNCBAM-1 in the same cellular systems and analyze the potential of GAT100 to display orthosteric-probe dependence [i.e., the effect of different orthosteric ligands (“orthosteric probes”) on the potency and efficacy of a given allosteric ligand] and functional selectivity/ligand bias (i.e., ligand-specific selective modulation of the activity of select signal transduction pathways over others through a given receptor).33,34 For this purpose, we report here a comprehensive in vitro evaluation of GAT100 downstream signaling in cell systems heterologously or endogenously expressing CB1R. To evaluate the orthosteric-probe dependence of GAT100, we chose the high-affinity, potent synthetic cannabinoid CP55,940 and the endocannabinoids 2-AG and AEA. Pharmacological profiling encompassed quantification of CB1R-mediated, G protein-independent β-arrestin1 recruitment, Gαq-dependent phospholipase Cβ3 (PLCβ3) activation, Gαi/o-dependent extracellular signal-regulated kinase (ERK) activation and cAMP production, and CB1R internalization.
The data demonstrate that GAT100 is a more potent NAM of CB1R signaling than Org27569 or PSNCBAM-1 and, notably, lacks inverse-agonist activity compared to Org27569 and PSNCBAM-1 in the cAMP assay. In radioligand equilibriumbinding assays with hCB1R-transfectedCHOcells, GAT100 acted as a NAMof CB1R antagonist/inverse agonist ([3H]SR141716A) binding and was more potent than Org27569. In silico molecular modeling studies suggest that C7.38(382) is a key feature of GAT100’s ligand-binding motif as the most likely amino acid reactive with the probe’s isothiocyanate moiety. The data constitute the first comprehensive characterization of the functional consequences of CB1R modulation by a potent covalent CB1R allosteric NAM (GAT100) and provide initial insight into that ligand’s binding motif and receptor-interaction profile.
RESULTS AND DISCUSSION
CB1R-Mediated β-Arrestin1 Recruitment
A bioluminescence resonance energy transfer (BRET) assay was used to evaluate the effect of GAT100 on β-arrestin1 recruitment by CB1R as compared to that of the two prototypic CB1R NAMs, Org27569 and PSNCBAM-1. Three cell systems were used: HEK293A cells overexpressing human CB1R (hCB1R) (Figure 2, panels A, D, andG) and twomurine cells,Neuro2a neuroblastoma stem cells (Figure 2, panels B, E, and H) and differentiated STHdhQ7/Q7 striatal progenitor cells (Figure 2, panels C, F, and I), endogenously expressing CB1R. Cells were treated with 1 nM to 10 μM 2-AG (Figure 2, panels A–C), AEA (Figure 2, panels D–F), or CP55,940 (Figure 2, panels G–I) with or without 1 μM GAT100, Org27569, or PSNCBAM-1 for 30 min before quantifying the assay endpoint, BRET efficiency of energy transfer (BRETEff) between hCB1R green fluorescent protein 2 (GFP2) and β-arrestin1-Renilla luciferase (Rluc), as a sensitive measure of β-arrestin1 recruitment. GAT100 treatment reduced the potency and decreased themaximum possible effect (i.e., efficacy) (Emax) of all CB1R orthosteric ligands on CB1R-mediated β-arrestin1 recruitment to a greater extent than vehicle, Org27569, and PSNCBAM-1 in all cell types studied (Figure 2). The decrease in Emax observed in the presence of Org27569 or PSNCBAM-1 is presented in Table 1. In the absence of orthosteric agonists, neither GAT100, Org27569, nor PSNCBAM-1 (1 nM to 10 μM) evidenced inverse agonism toward β-arrestin1 recruitment in HEK293A (Figure 2, panel J), Neuro2a (Figure 2, panel K), and STHdhQ7/Q7 cells (Figure 2, panel L). Inhibition of orthosteric agonist-mediated β-arrestin1 recruitment by Org27569, PSNCBAM-1, and GAT100 was observed here, as has been shown elsewhere.29,48,52,53 Although Org27569 has been reported to promote the recruitment of β-arrestin1 in immunohistochemical colocalization assays,54 β-arrestin1 recruitment was not observed here using the BRET assay.
Figure 2.
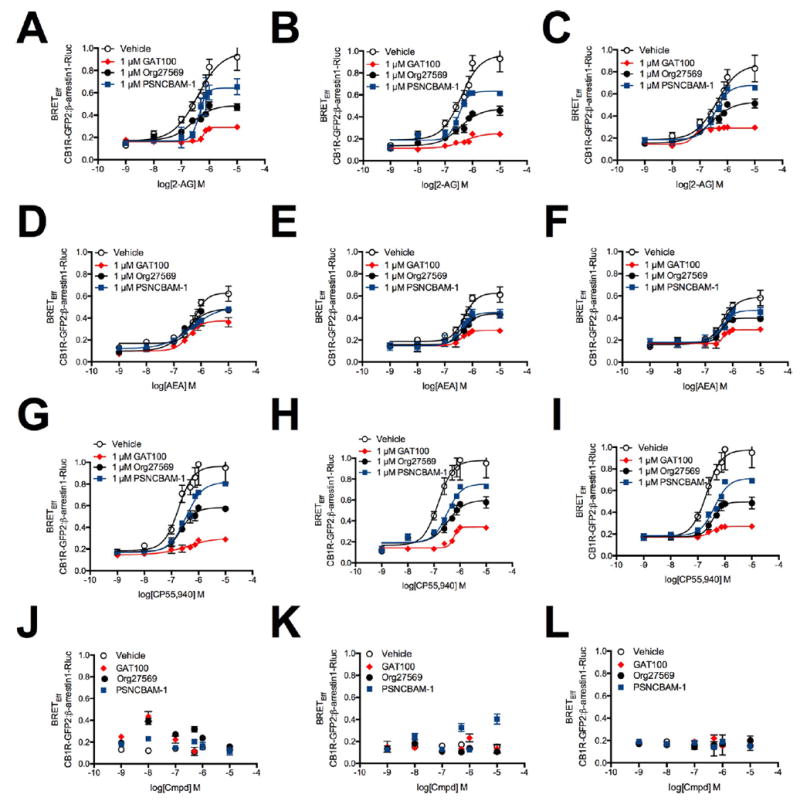
BRETEff between β-arrestin1-Rluc and CB1R-GFP2 in the presence of 1 nM to 10 μM 2-AG, AEA, or CP55,940. HEK293A (A,D,G), Neuro2a (B,E,H), and STHdhQ7/Q7 (C,F,I) cells were transfected with β;-arrestin1-Rluc- and CB1R-GFP2-containing plasmids and BRET2 was measured 30 min after treatment with 2-AG (A–C), AEA (D–F), or CP55,940 (G–I) ± 1 μM GAT100, Org27569, or PSNCBAM-1. (J–L) HEK293A (J), Neuro2a (K), and STHdhQ7/Q7 (L) cells were transfected with β-arrestin1-Rluc- and CB1R-GFP2-containing plasmids, and BRET2 was measured 30 min after treatment with 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1 alone. Concentration–response curves were fit using the allosteric modulator shift nonlinear regression models. N = 4.
Table 1.
BRETEff between β-Arrestin1-Rluc and CB1-GFP2 in the Presence of GAT100, Org27569, and PSNCBAM-1a
| HEK293Ab | ||||||
| 2-AG
|
AEA
|
CP55,940
|
||||
| IC50 (nM) | Emax | IC50 (nM) | Emax | IC50 (nM) | Emax | |
|
| ||||||
| GAT100 | 54.0 (39.8–79.5) | 0.29 ± 0.01 | 48.6 (29.2–123) | 0.37 ± 0.03 | 24.9 (0.59–104) | 0.30 ± 0.04 |
| Org27569 | 85.1 (79.3–94.7)* | 0.49 ± 0.03* | 447 (216–636)*† | 0.49 ± 0.02** | 240 (175–333)*† | 0.58 ± 0.03*** |
| PSNCBAM-1 | 162 (143–183)* | 0.64 ± 0.06* | 263 (255–294)*† | 0.50 ± 0.04** | 209 (193–234)*† | 0.81 ± 0.10*** |
|
| ||||||
| Neuro2ab | ||||||
| 2-AG
|
AEA
|
CP55,940
|
||||
| IC50 (nM) | Emax | IC50 (nM) | Emax | IC50 (nM) | Emax | |
|
| ||||||
| GAT100 | 28.8 (6.4–34.1) | 0.25 ± 0.03 | 57.5 (46.0–62.6) | 0.29 ± 0.02 | 33.9 (1.40–99.7) | 0.34 ± 0.02 |
| Org27569 | 31.6 (23.9–44.1) | 0.47 ± 0.03* | 138 (63.7–202)*† | 0.44 ± 0.02*** | 263 (165–420)*† | 0.60 ± 0.05*** |
| PSNCBAM-1 | 74.1 (57.1–93.2)* | 0.63 ± 0.03* | 355 (203–520)*† | 0.45 ± 0.03*** | 316 (240–424)*† | 0.75 ± 0.08*** |
|
| ||||||
| STHdhQ7/Q7b | ||||||
| 2-AG
|
AEA
|
CP55,940
|
||||
| IC50 (nM) | Emax | IC50 (nM) | Emax | IC50 (nM) | Emax | |
|
| ||||||
| GAT100 | 17.4 (2.90–29.6) | 0.29 ± 0.01 | 20.0 (0.33–12.9) | 0.29 ± 0.02 | 58.9 (4.90–109) | 0.27 ± 0.02 |
| Org27569 | 123 (89.8–160)* | 0.53 ± 0.03* | 514 (369–752)*† | 0.40 ± 0.02*** | 288 (195–419)*† | 0.50 ± 0.03*** |
| PSNCBAM-1 | 355 (123–593)* | 0.68 ± 0.03* | 510 (374.3–751)*† | 0.47 ± 0.02*** | 398 (318–489)*† | 0.71 ± 0.02*** |
IC50 (nM) determined using nonlinear regression analysis; Emax (BRETEff), determined using nonlinear regression analysis (Emax 2-AG alone 0.97 ± 0.01, AEA alone 0.63 ± 0.04, CP55,940 alone 0.97 ± 0.02). Data are mean with 95% CI (IC50) and mean ± SEM (Emax).
P < 0.05,
P < 0.01,
P < 0.001 compared to GAT100 within cell type and orthosteric agonist treatment;
To quantify the concentration-dependence of test compounds for β-arrestin1 recruitment, BRETEff between hCB1R-GFP2 and β-arrestin1-Rluc was then measured in HEK293A (Figure 3, panels A, D, and G), Neuro2a (Figure 3, panels B, E, and H), and STHdhQ7/Q7 cells (Figure 3, panels C, F, and I) treated with 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1 and 500 nM 2-AG (Figure 3, panels A–C), AEA (Figure 3, panels D–F), or CP55,940 (Figure 3, panels G–I) for 30 min. As summarized in Table 1, GAT100 was a more potent NAM of CB1R-mediatedarrestin1 recruitment than either Org27569 or PSNCBAM-1 in all cell types tested and with all orthosteric ligands. No difference in potency (IC50) or efficacy (Emax) was observed in cells treated with GAT100 and 2-AG, AEA, or CP55,940. In contrast, Org27569 and PSNCBAM-1 displayed probe dependence in all cell types, since both were more potent and efficacious NAMs in 2-AG-treated cells as compared to AEA- and CP55,940-treated cells (Table 1).
Figure 3.
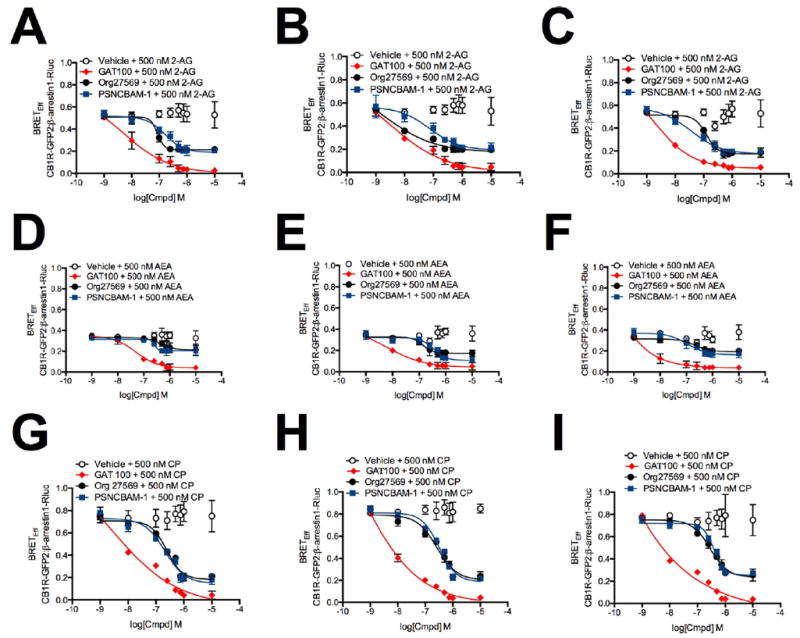
BRETEff between β-arrestin1-Rluc and CB1R-GFP2 in the presence of 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1. HEK293A (A,D,G), Neuro2a (B,E,H), and STHdhQ7/Q7 (C,F,I) cells were transfected with β-arrestin1-Rluc- and CB1R-GFP2-containing plasmids, and BRET2 was measured 30 min after treatment with GAT100, Org27569, or PSNCBAM-1 + 500 nM 2-AG (A–C), AEA (D–F), or CP55,940 (G–I). Concentration–response curves were fit using the allosteric modulator shift nonlinear regression models. N = 4.
CB1R-Mediated PLCβ3 Phosphorylation
We previously reported that the Gαq effector PLCβ3 is phosphorylated via CB1R following treatment of STHdhQ7/Q7 cells with 2-AG, AEA, or CP55,940.55 We have now measured CB1R-dependent PLCβ3 phosphorylation in HEK293A cells transiently transfected with hCB1R-GFP2-expressing plasmid (Figure 4, panels A, D, and G) and in Neuro2a (Figure 4, panels B, E, and H), and STHdhQ7/Q7 cells (Figure 4, panels C, F, and I) expressing endogenous CB1R upon cell treatment with 1 nM to 10 μM 2-AG (Figure 4, panels A–C), AEA (Figure 4, panels D–F), or CP55,940 (Figure 4, panels G–I) with or without 1 μM GAT100, Org27569, or PSNCBAM-1 for 10 min. GAT100 treatment reduced the potency (EC50) and efficacy (Emax) of agonist-dependent PLCβ3 phosphorylation to a greater extent than vehicle, Org27569, and PSNCBAM-1 in all cell types and with all orthosteric ligands (Figure 4). The decrease in Emax observed in the presence of Org27569 or PSNCBAM-1 is presented in Table 2. To assess possible intrinsic activity associated with these CB1R allosteric modulators, PLCβ3 phosphorylation was also measured in HEK293A (Figure 4, panel J), Neuro2a (Figure 4, panel K), and STHdhQ7/Q7 cells (Figure 4, panel L) treated with 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1 for 10 min. GAT100, Org27569, and PSNCBAM-1 had no effect on basal PLCβ3 phosphorylation in the absence of orthosteric agonists, indicating a lack of intrinsic activity in this assay (Figure 4, panels J–L).
Figure 4.
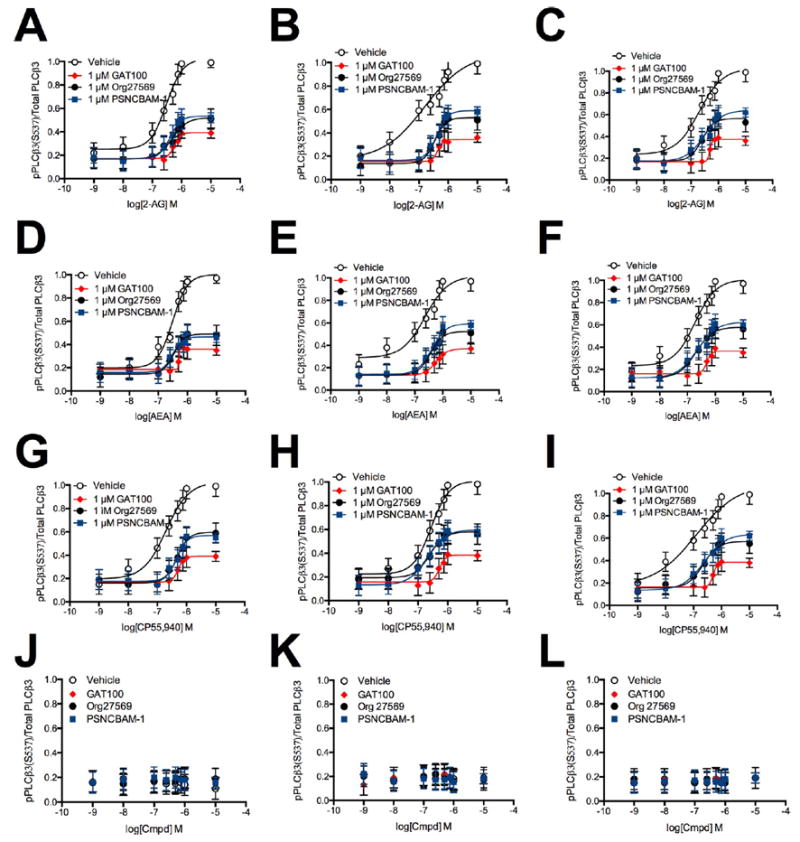
PLCβ3 phosphorylation in the presence of 1 nM to 10 μM 2-AG, AEA, or CP55,940. PLCβ3 phosphorylation was measured in HEK293A (A,D,G), Neuro2a (B,E,H), and STHdhQ7/Q7 (C,F,I) cells 10 min after treatment with 2-AG (A–C), AEA (D–F), or CP55,940 (G–I) ± 1 μM GAT100, Org27569, or PSNCBAM-1. (J–L) PLCβ3 phosphorylation was measured in HEK293A (J), Neuro2a (K), and STHdhQ7/Q7 (L) cells 10 min after treatment with 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1 alone. Concentration–response curves were fit using the allosteric modulator shift nonlinear regression models. N = 4.
Table 2.
PLCβ3 Phosphorylation in the Presence of GAT100, Org27569, and PSNCBAM-1a
| HEK293Ab | ||||||
| 2-AG
|
AEA
|
CP55,940
|
||||
| IC50 (nM) | Emax | IC50 (nM) | Emax | IC50 (nM) | Emax | |
|
| ||||||
| GAT100 | 56.1 (44.0–62.6) | 0.39 ± 0.02 | 50.2 (23.9–65.3) | 0.36 ± 0.01 | 76.6 (60.8–103)† | 0.39 ± 0.01 |
| Org27569 | 105 (85.8–123)* | 0.52 ± 0.02* | 138 (124–156)*† | 0.49 ± 0.02** | 151 (125–184)*† | 0.60 ± 0.03** |
| PSNCBAM-1 | 166 (106–239)* | 0.53 ± 0.02* | 95.5 (90.7–101)*† | 0.47 ± 0.02** | 174 (121–247)* | 0.57 ± 0.02** |
|
| ||||||
| Neuro2ab | ||||||
| 2-AG
|
AEA
|
CP55,940
|
||||
| IC50 (nM) | Emax | IC50 (nM) | Emax | IC50 (nM) | Emax | |
|
| ||||||
| GAT100 | 50.1 (33.4–79.2) | 0.34 ± 0.02 | 60.3 (28.1–131) | 0.37 ± 0.02 | 49.0 (25.2–93.4) | 0.39 ± 0.02 |
| Org27569 | 551 (519–615)* | 0.53 ± 0.02*** | 324 (219–501)*† | 0.52 ± 0.02**†† | 148 (98.0–247)* | 0.58 ± 0.02**†† |
| PSNCBAM-1 | 595 (514–610)* | 0.59 ± 0.02** | 282 (213–381)* | 0.59 ± 0.02** | 123 (104–149)*† | 0.60 ± 0.02** |
|
| ||||||
| STHdhQ7/Q7b | ||||||
| 2-AG
|
AEA
|
CP55,940
|
||||
| IC50 (nM) | Emax | IC50 (nM) | Emax | IC50 (nM) | Emax | |
|
| ||||||
| GAT100 | 52.7 (39.8–63.1) | 0.37 ± 0.01 | 57.4 (35.9–66.1) | 0.37 ± 0.01 | 71.0 (67.2–79.7)† | 0.38 ± 0.01 |
| Org27569 | 148 (98.9–216)* | 0.56 ± 0.02** | 151 (129–181)* | 0.59 ± 0.02*** | 141 (124–161)* | 0.57 ± 0.02** |
| PSNCBAM-1 | 138 (107–160)* | 0.64 ± 0.04** | 191 (172–214)*† | 0.63 ± 0.02** | 190 (158–228)*† | 0.64 ± 0.02** |
IC50 (nM) determined using nonlinear regression analysis; Emax pPLCβ3/Total PLCβ3 In-cell western determined using nonlinear regression analysis (Emax 2-AG alone 1.04 ± 0.02, AEA alone 1.01 ± 0.05, CP55,940 alone 1.04 ± 0.05). Data are mean with 95% CI (IC50) and mean ± SEM (Emax).
P < 0.05,
P < 0.01,
P < 0.001 compared to GAT100 within cell type and orthosteric agonist treatment;
P < 0.05,
To obtain comparative potency (IC50) values for the effect of these allosteric ligands on CB1R-mediated PLCβ3 phosphorylation, we used HEK293A (Figure 5, panels A, D, and G), Neuro2a (Figure 5, panels B, E, and H), and STHdhQ7/Q7 cells (Figure. 5, panels C, F, and I) treated with 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1 and 500 nM 2-AG (Figure 5, panels A–C), AEA (Figure 5, panels D–F), or CP55,940 (Figure 5, panels G–I) for 10 min. GAT100 was a more potent NAM of CB1R-dependent PLCβ3 phosphorylation than either Org27569 or PSNCBAM-1 in all cell types and with all orthosteric ligands tested (Table 2). GAT100 displayed marginal orthosteric-probe dependence in CP55,940-treated HEK293A and STHdhQ7/Q7 cells, being less potent than in 2-AGor AEA-treated cells (Table 2). In contrast, Org27569 displayed probe-dependence in HEK293A and Neuro2a cells as a more potent NAM in 2-AG-treated HEK293A cells and in 2-AG- and CP55,940-treated Neuro2a cells (Table 2). PSNCBAM-1 was a more potent NAM in AEA-treated HEK293A cells, CP55,940- treated Neuro2a cells, and 2-AG-treated STHdhQ7/Q7 cells as compared to other orthosteric ligands (Table 2). No orthostericprobe dependence in efficacy (Emax) was observed among cells treated with GAT100, Org27569, or PSNCBAM-1 and 2-AG, AEA, or CP55,940 (Table 2).
Figure 5.
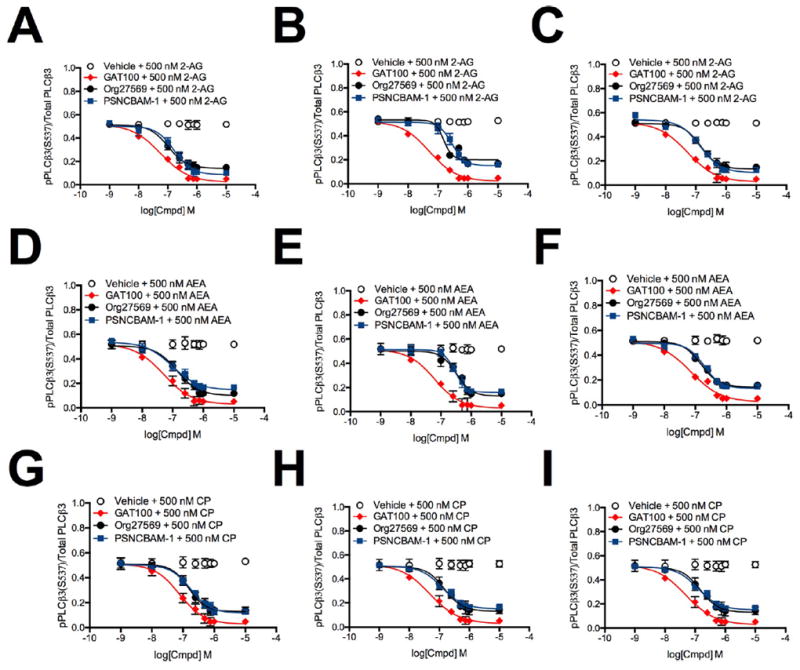
PLCβ3 phosphorylation in the presence of 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1. PLCβ3 phosphorylation was measured in HEK293A (A,D,G), Neuro2a (B,E,H) and STHdhQ7/Q7 (C,F,I) cells 10 min after treatment with GAT100, Org27569, or PSNCBAM-1 + 500 nM 2-AG (A–C), AEA (D–F), or CP55,940 (G–I). Concentration-response curves were fit using the allosteric modulator shift nonlinear regression models. N = 4.
CB1R-Mediated ERK1/2 Phosphorylation
Rapid, transient ERK1/2 phosphorylation of the MAPK family member, ERK1/2, is a hallmark of CB1R activation via Gαi/o.56 Consequently, we evaluated CB1R-dependent ERK1/2 phosphorylation in HEK293A cells transiently transfected with hCB1R-GFP2-expressing plasmid (Figure 6, panels A, D, and G) and in Neuro2a (Figure 6, panels B, E, and H) and STHdhQ7/Q7 cells (Figure 6, panels C, F, and I) expressing endogenous CB1R in response to treatment with 1 nM to 10 μM2-AG (Figure 6, panels A–C),AEA (Figure 6, panelsD–F), or CP55,940 (Figure 6, panels G–I) with or without 1 μMGAT100, Org27569, or PSNCBAM-1 for 10 min. GAT100 reduced the potency and efficacy of orthosteric agonist-induced ERK1/2 phosphorylation to a greater extent than vehicle, Org27569, and PSNCBAM-1 in all cell types and with all orthosteric ligands tested (Figure 6, Table 3).
Figure 6.
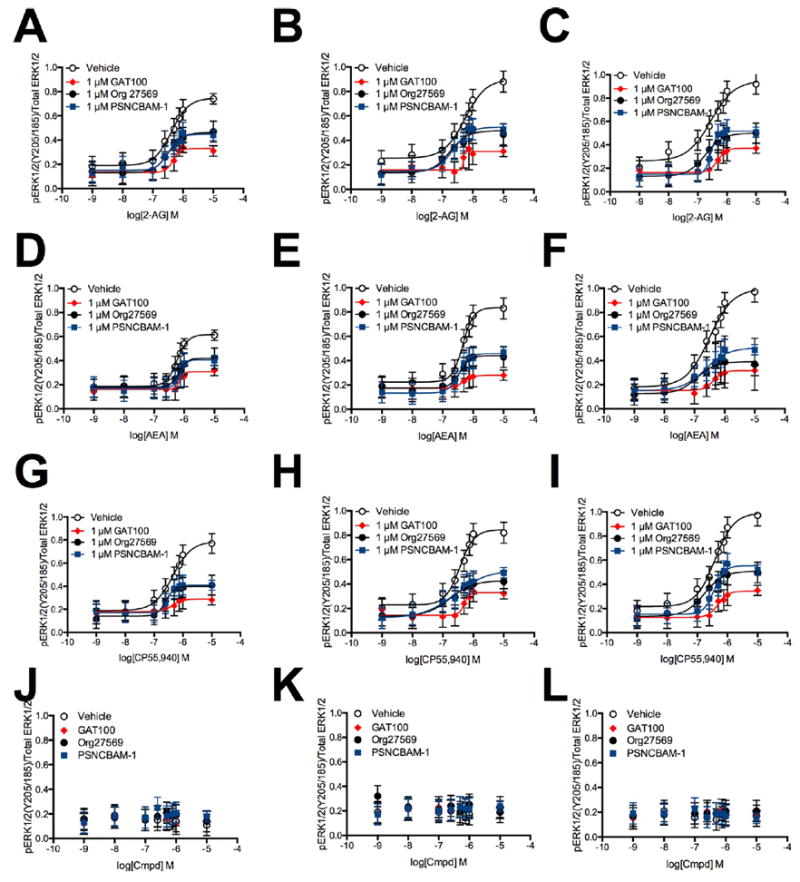
ERK1/2 phosphorylation in the presence of 1 nM to 10 μM 2-AG, AEA, or CP55,940. ERK1/2 phosphorylation was measured in HEK293A (A,D,G), Neuro2a (B,E,H), and STHdhQ7/Q7 (C,F,I) cells 10 min after treatment with 2-AG (A–C), AEA (D–F), or CP55,940 (G–I) ± 1 μM GAT100, Org27569, or PSNCBAM-1. (J–L) ERK1/2 phosphorylation was measured in HEK293A (J), Neuro2a (K), and STHdhQ7/Q7 (L) cells 10 min after treatment with 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1 alone. Concentration–response curves were fit using the allosteric modulator shift nonlinear regression models. N = 4.
Table 3.
ERK1/2 Phosphorylation in the Presence of GAT100, Org27569, and PSNCBAM-1a
| HEK293Ab | ||||||
| 2-AG
|
AEA
|
CP55,940
|
||||
| IC50 | Emax | IC50 | Emax | IC50 | Emax | |
|
| ||||||
| GAT100 | 69.4 (50.2–76.5) | 0.33 ± 0.02 | 56.6 (29.3–71.6) | 0.31 ± 0.02 | 84.1 (78.3–96.1)† | 0.29 ± 0.02 |
| Org27569 | 126 (106–149)* | 0.46 ± 0.02*** | 324 (236–447)*† | 0.42 ± 0.02* | 309 (200–461)*† | 0.40 ± 0.01** |
| PSNCBAM-1 | 135 (105–162)* | 0.45 ± 0.02*** | 302 (230-393)*† | 0.41 ± 0.02* | 240 (164–357)*† | 0.41 ± 0.01** |
|
| ||||||
| Neuro2ab | ||||||
| 2-AG
|
AEA
|
CP55,940
|
||||
| IC50 | Emax | IC50 | Emax | IC50 | Emax | |
|
| ||||||
| GAT100 | 57.5 (29.1–112) | 0.31 ± 0.02 | 57.5 (32.9–72.1) | 0.28 ± 0.02 | 47.9 (24.4–94.8) | 0.33 ± 0.02 |
| Org27569 | 339 (227–512)* | 0.48 ± 0.03** | 189 (147–217)*† | 0.44 ± 0.02*** | 219 (141–337)*† | 0.43 ± 0.02** |
| PSNCBAM-1 | 229 (180–267)* | 0.51 ± 0.02** | 115 (75.0–177)*† | 0.46 ± 0.02** | 302 (284–339)*† | 0.51 ± 0.03** |
|
| ||||||
| STHdhQ7/Q7b | ||||||
| 2-AG
|
AEAM
|
CP55,940
|
||||
| IC50 | Emax | IC50 | Emax | IC50 | Emax | |
|
| ||||||
| GAT100 | 57.9 (28.4–75.1) | 0.37 ± 0.02 | 51.3 (19.6–136) | 0.32 ± 0.02 | 52.3 (21.8–74.2) | 0.34 ± 0.02 |
| Org27569 | 138 (78.6–242)* | 0.50 ± 0.02* | 295 (259–356)*† | 0.39 ± 0.03* | 81.3 (75.4–98.1)*† | 0.51 ± 0.02** |
| PSNCBAM-1 | 155 (93.3–252)* | 0.52 ± 0.01** | 170 (97.3–297)* | 0.51 ± 0.02*** | 153 (114–192)* | 0.55 ± 0.03** |
IC50 (nM) determined using nonlinear regression analysis; Emax pERK1/2/Total ERK1/2 In-cell western determined using nonlinear regression analysis (Emax 2-AG alone 0.87 ± 0.06, AEA alone 0.81 ± 0.11, CP55,940 alone 0.86 ± 0.07). Data are mean with 95% CI (IC50) and mean ± SEM (Emax)
P < 0.05,
P < 0.01,
P < 0.001 compared to GAT100 within cell type and orthosteric agonist treatment;
P < 0.05, compared to 2-AG within cell type and NAM treatment as determined by one-way ANOVA followed by Dunnett’s multiple comparison (Emax) or nonoverlapping 95% CI (IC50). Data are from Figures 6 (Emax) and 7 (IC50). N. = 4.
We next determined the effect of allosteric ligands on CB1Rdependent ERK1/2 phosphorylation. For this purpose, ERK1/2 phosphorylation was measured in HEK293A (Figure 6, panel J), Neuro2a (Figure 6, panel K), and STHdhQ7/Q7 cells (Figure 6, panel L) treated with 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1 for 10 min.GAT100, Org27569, and PSNCBAM-1 had no effect on basal ERK1/2 phosphorylation in the absence of orthosteric agonists (Figure 6, panels J–L). ERK1/2 phosphorylation was then measured in HEK293A (Figure 7, panels A, D, and G), Neuro2a (Figure 7, panels B, E, and H), and STHdhQ7/Q7 cells (Figure 7, panels C, F, and I) treated with 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1 and with 500 nM 2-AG (Figure 7, panels A–C), AEA (Figure 7, panels D–F), or CP55,940 (Figure 7, panels G–I) for 10 min. GAT100 was a more potent NAM of CB1R-dependent ERK1/2 phosphorylation than either Org27569 or PSNCBAM-1 in all cell types and with all orthosteric ligands tested (Table 3). GAT100 only displayed probe-dependence in CP55,940-treated HEK293A cells, where GAT100 was less potent compared to 2-AG- or AEA-treated cells (Table 3). In contrast, Org27569 was most potent in the presence of 2-AG in HEK293A cells, AEA in Neuro2a cells, and CP55,940 in STHdhQ7/Q7 cells (Table 3). PSNCBAM-1 was most potent in the presence of 2-AG in HEK293A cells and AEA in Neuro2a cells, but did not display orthosteric-probe dependence in STHdhQ7/Q7 cells (Table 3). No probe dependence was observed in terms of efficacy for any of the three allosteric ligands examined (Emax). Inhibition of orthosteric agonist-mediated Gαi/o signaling by Org27569, PSNCBAM-1, and GAT100 was observed here, as has been shown elsewhere.29,48,52,53 In contrast, Org27569 has been reported to promote the ERK1/2 phosphorylation via β-arrestin1 in the absence of orthosteric ligand in Western blot experiments,54 Org27569-dependent ERK1/2 phosphorylation was not observed in the in-cell Western assay used here.
Figure 7.
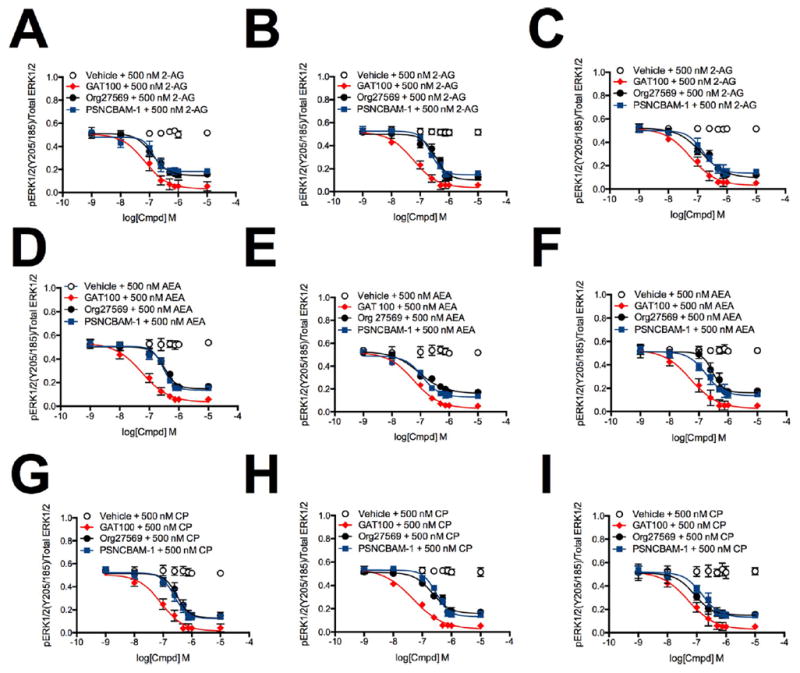
ERK1/2 phosphorylation in the presence of 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1. ERK1/2 phosphorylation was measured in HEK293A (A,D,G), Neuro2a (B,E,H), and STHdhQ7/Q7 (C,F,I) cells 10 min after treatment with GAT100, Org27569, or PSNCBAM-1 + 500 nM 2-AG (A–C), AEA (D–F), or CP55,940 (G–I). Concentration-response curves were fit using the allosteric modulator shift nonlinear regression models. N = 4.
CB1R-Mediated Adenylate Cyclase Activity
Cellular adenylate cyclase activity has been used extensively to index Gαi-dependent signal modulation by CB1R allosteric ligands.27 We used a luciferase-based reporter assay to measure indirectly CB1R-dependent modulation of forskolin-stimulated adenylate cyclase activity (i.e., modulation of cAMP accumulation) in HEK-CRE cells transiently transfected with hCB1R-GFP2- expressing plasmid and treated with 10 μM forskolin and 1 nM to 10 μM 2-AG (Figure 8, panel A), AEA (Figure 8, panel B), or CP55,940 (Figure 8, panel C) with or without 1 μM GAT100, Org27569, or PSNCBAM-1 for 4 h. Relative to orthosteric ligand alone, GAT100 shifted the orthosteric ligand-cAMP concentration–response curves rightward to a greater extent than Org27569 or PSNCBAM-1 in the presence of 2-AG, AEA, or CP55,940 (Figure 8, panels A–C). In HEK-CRE cells treated with 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1 for 4 h, Org27569 and PSNCBAM-1 alone, but not GAT100, increased cAMP levels significantly in the absence of orthosteric agonists (Figure 8, panel D). Therefore, Org27569 and PSNCBAM-1, but not GAT100, demonstrated measurable inverse agonist activity in the cAMP assay, as has been shown previously for Org27569 and PSNCBAM-1.29,36,52,53
Figure 8.
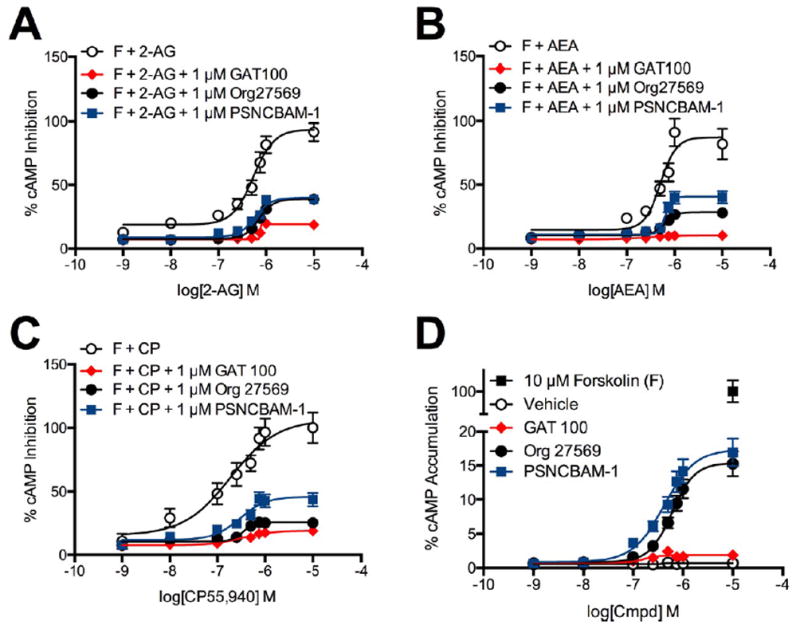
cAMP inhibition in the presence of 1 nM to 10 μM 2-AG, AEA, or CP55,940. cAMP levels were measured in HEK-CRE cells 4 h after treatment with 10 μM forskolin (F) + 2-AG (A), AEA (B), or CP55,940 (C) ± 1 μM GAT100, Org27569, or PSNCBAM-1. (D) cAMP levels were measured in HEK-CRE cells 4 h after treatment with 10 μM forskolin (F) + 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1 alone. Concentration–response curves were fit using the allosteric modulator shift nonlinear regression models. N = 4.
Accumulation of cAMP was also measured in HEK-CRE cells treated with 10 μM forskolin in combination with 1 nMto 10 μM GAT100, Org27569, or PSNCBAM-1 and with 500 nM 2-AG (Figure 9, panel A), AEA (Figure 9, panel B), or CP55,940 (Figure 9, panel C) for 4 h. In the absence of orthosteric agonist, GAT100 did not affect cAMP accumulation, whereas Org27569 and PSNCBAM-1 both increased cellular cAMP in a concentration-dependent manner (Figure 9). These results support the literature evidence that Org27569 and PSNCBAM-1 possess inverse-agonist activity,28,29,37 whereas GAT100 does not.48,57 Furthermore, GAT100 was the most potent and efficacious inhibitor of cAMP accumulation in the HEK-CRE cells treated with orthosteric agonist (Figure 9). The data in Figure 9 also demonstrate that GAT100, Org27569, and PSNCBAM-1 did not display probe dependence in HEK-CRE cells with respect to potency (IC50) or efficacy (Emax) in the cAMP assay. The data presented in Figures 8 and 9 demonstrate that GAT100 did not display inverse agonist activity at Gαi-dependent signal pathways.
Figure 9.
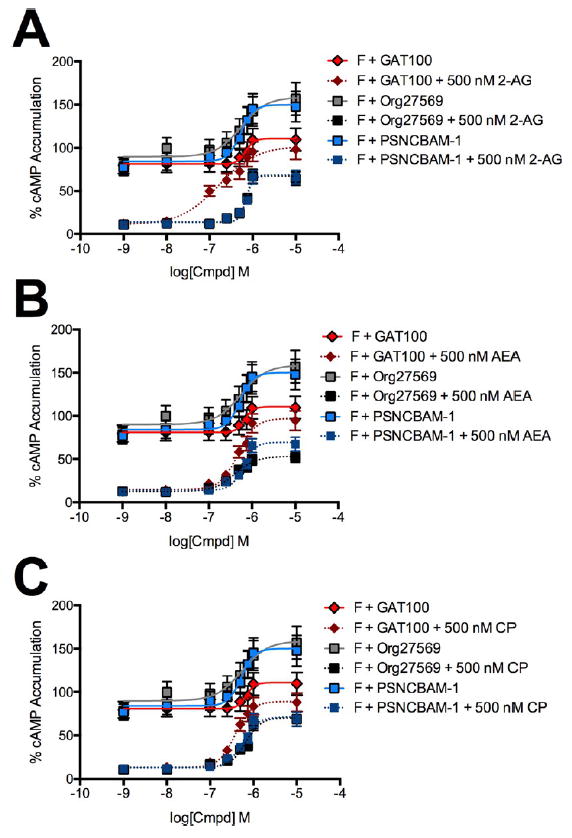
cAMP accumulation in the presence of 1 nM to 10 μM GAT100, Org27569, or PSNCBAM-1. cAMP levels were measured in HEK-CRE cells 4 h after treatment with 10 μM forskolin (F) + GAT100, Org27569, or PSNCBAM-1 + 500 nM 2-AG (A), AEA (B), or CP55,940 (C). Concentration–response curves were fit using the allosteric modulator shift nonlinear regression models. N = 4.
CP55,940-Dependent CB1R Internalization
As with other GPCRs, CB1R internalization is recognized as a temporal and spatial modulator of CB1R-mediated signaling.58,59 Accordingly, we determined the effect of allosteric ligands on CP55,940-induced CB1R internalization in STHdhQ7/Q7 cells using on- and in-cell Western analysis. CP55,940 alone resulted in sustained CB1R internalization beginning at 30 min after treatment inception (Figure 10). GAT100, Org27569, or PSNCBAM-1 reversed this effect, increasing the fraction of CB1R at the plasma membrane. GAT100 elicited a more rapid reversal of CB1R internalization than Org27569 or PSNCBAM-1.
Figure 10.
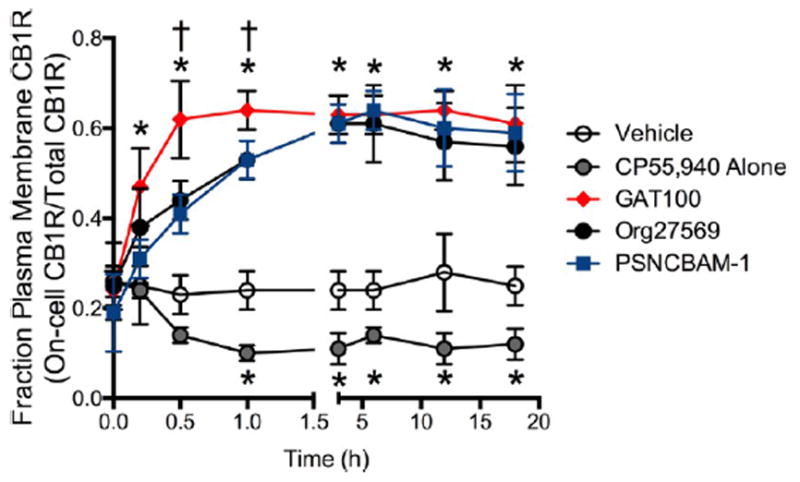
CB1R internalization in the presence of 1 μM GAT100, Org27569, or PSNCBAM-1. Fraction of CB1R at the plasma membrane was determined in STHdhQ7/Q7 cells following treatment with 1 μM GAT100, Org27569, or PSNCBAM-1 + 500 nM CP55,940. *P < 0.01 compared to vehicle within time point, †P < 0.01 compared to Org27569 or PSNCBAM-1 within time point. Data are mean ± SEM N = 4.
Summary of Potency and Efficacy for GAT100, Org27569, and PSNCBAM-1
Table 5 summarizes the signaling profile of GAT100 across the activity assays, cell systems, and orthosteric ligands utilized along with the parallel data for Org27569 and PSNCBAM-1. All three allosteric modulators profiled in this study were active in each of the signaling assays conducted. The potencies observed for Org27569 and PSNCBAM-1 were comparable to published values for arrestin recruitment and G protein-dependent [35S]GTPγS binding conducted with cellular CB1R overexpression and isolatedmembrane systems.28,29,36 However, in the presence of CP55,940 or the endocannabinoids 2-AG or AEA, GAT100 was more potent, and efficacious NAM than Org27569 and PSNCBAM-1.
Table 5.
Summary of Data for GAT100, Org27569, and PSNCBAM-1a
| pathway ligand biasbe | ||||||||
|---|---|---|---|---|---|---|---|---|
|
β-arrestin1
|
PLCβ3
|
ERK
|
cAMP
|
|||||
| IC50 (nM) | Emax | IC50 (nM) | Emax | IC50 (nM) | Emax | IC50 (nM) | Emax | |
| GAT100 | 35.8 (4.40–51.1) | 0.30 ± 0.01 | 60.3 (32.5–95.6) | 0.38 ± 0.01 | 59.4 (55.7–63.1)† | 0.32 ± 0.01 | 741(534–937)† | 0.16 ± 0.04†† |
| Org27569 | 236 (229–341)* | 0.50 ± 0.02*** | 206 (141–228)* | 0.55 ± 0.01** | 213 (178–249)* | 0.45 ± 0.01*** | 940 (858–1099)* | 0.31 ± 0.04***† |
| PSNCBAM-1 | 293 (247–318)* | 0.63 ± 0.04*** | 217 (152–261)* | 0.58 ± 0.02*** | 200 (177–224)* | 0.58 ± 0.02*** | 936 (858–982)* | 0.42 ± 0.03***† |
| probe dependencece | ||||||
| 2-AG
|
AEA
|
CP55,940
|
||||
| IC50 (nM) | Emax | IC50 (nM) | Emax | IC50 (nM) | Emax | |
|
| ||||||
| GAT100 | 57.3 (43.1–111) | 0.31 ± 0.02 | 55.5 (11.9–104) | 0.36 ± 0.03 | 55.4 (14.1–147) | 0.33 ± 0.02 |
| Org27569 | 169 (114–219)* | 0.49 ± 0.02*** | 280 (185–378)* | 0.45 ± 0.03** | 205 (183–252)* | 0.50 ± 0.04*** |
| PSNCBAM-1 | 223 (202-248)* | 0.56 ± 0.03*** | 253 (178–263)* | 0.49 ± 0.03*** | 234 (210–287)* | 0.60 ± 0.04*** |
|
| ||||||
| cell dependencede | ||||||
| HEK293A
|
Neuro2a
|
STHdhQ7/Q7
|
||||
| IC50 (nM) | Emax | IC50 (nM) | Emax | IC50 (nM) | Emax | |
|
| ||||||
| GAT100 | 52.4 (34.8–70.9) | 0.34 ± 0.01 | 39.8 (17.0–48.9) | 0.32 ± 0.01 | 40.7 (7.38–84.0) | 0.34 ± 0.02 |
| Org27569 | 204 (174–262)* | 0.49 ± 0.02*** | 191 (154–219)* | 0.50 ± 0.02** | 186 (162–209)* | 0.51 ± 0.02*** |
| PSNCBAM-1 | 191 (174–215)* | 0.53 ± 0.04*** | 229 (208–267)* | 0.57 ± 0.03** | 182 (159–217)* | 0.59 ± 0.03*** |
IC50 (nM) determined using nonlinear regression analysis; Emax determined using nonlinear regression analysis. Data are mean with 95% CI (IC50) or mean ± SEM (Emax).
Pathway ligand bias data were calculated as the mean of data for each pathway (Tables 1-4) for GAT100, Org27569, or PSNCBAM-1. cAMP Emax divided by 100 in order to compare to other data sets.
Probe dependence data were calculated as the mean of data for each orthosteric ligand (Tables 1-4) for GAT100, Org27569, or PSNCBAM-1.
Cell dependence data were calculated as the mean of data for each cell type (Tables 1-3) for GAT100, Org27569, or PSNCBAM-1.
P < 0.05,
P < 0.01,
P < 0.001 compared to GAT100 within column for “pathway ligand bias”, “probe dependence”, and “cell dependence” as determined by one-way ANOVA followed by Dunnett’s multiple comparison.
P < 0.05, compared to β-arrestin1 within “pathway ligand bias” as determined by nonoverlapping 95% CI. N = 4.
To determine whether GAT100 displayed functional selectivity, the NAM activity of GAT100 was characterized at several distinct pathways: β-arrestin1 (G protein-independent), PLCβ3 (Gαq-dependent), and ERK1/2 and adenylate cyclase (cAMP) (Gαi/o-dependent). GAT100 was a more potent inhibitor of β-arrestin1 recruitment compared to its inhibitory potency in the ERK1/2, and cAMP assays. GAT100, Org27569, and PSNCBAM-1 were generally more efficacious inhibitors of adenylate cyclase-mediated pathways as compared to their efficacy in the β-arrestin1, PLCβ3, and ERK1/2 assays.
Potential signaling bias was further analyzed using the global nonlinear regression operational model of Black and Leff (eq 1).60 Concentration—response data from Figures 3, 5, 7, and 9 were fit to that operational model in order to calculate the transduction coefficients [log R(τ/KA)] of each allosteric modulator and its relative activity (∆log R) as compared to GAT100 in the presence of 2-AG (eq 2) ( Table S1). GAT100 in the presence of 500 nM 2-AG was chosen as the reference ligand because it was the most potent and efficacious compound in all assays used; all other agents were compared to the transduction coefficient (log R) of GAT100. Relative activity values were then used to calculate a bias factor (∆∆log R) (eq 3) for each signaling pathway relative to β-arrestin1 (Table 6). Within this model, GAT100 displayed signaling bias for β-arrestin1 compared to PLCβ3 (in the presence of AEA in Neuro2a and STHdhQ7/Q7 cells) and ERK1/2 (in the presence of AEA and CP55,940 in Neuro2a cells). Org27569 displayed signaling bias against β-arrestin1 compared to PLCβ3 (in the presence of AEA in HEK293A cells) and cAMP (in the presence of AEA in HEK293A cells). PSNCBAM-1 displayed signaling bias against β-arrestin1 compared to PLCβ3 (in the presence of AEA and CP55,940 in HEK293A cells) and ERK1/2 (in the presence of AEA in HEK293A cells). The Black and Leff operational model has been used to study CB1R agonist bias.52,61 This study is the first to apply the Black and Leff operational model to CB1R NAMs.
Table 6.
Bias Factors for GAT100, Org27569, and PSNCBAM-1a
|
β-arrestin1/pPLCβ3
|
β-arrestin1/pERK1/2
|
β-arrestin1/cAMP
|
||||||
|---|---|---|---|---|---|---|---|---|
| cell model | orthosteric ligand | allosteric ligand | ΔΔlog R | BF | ΔΔlog R | BF | ΔΔlog R | BF |
| HEK293A | 2-AG | GAT100 | 0.0 | 1.0 | 0.0 ± 0.1 | 1.0 | 0.0 ± 0.1 | 1.0 |
| Org27569 | –1.0 ± 0.9 | 0.1 | –1.1 ± 0.1 | 0.1 | –0.6 ± 0.8 | 0.2 | ||
| PSNCBAM-1 | –1.3 ± 0.7 | 0.1 | –0.7 ± 0.2 | 0.2 | –0.2 ± 0.5 | 0.6 | ||
| AEA | GAT100 | –1.2 ± 0.8 | 0.1 | –0.6 ± 0.1 | 0.3 | –0.1 ± 0.1 | 0.8 | |
| Org27569 | –1.6 ± 0.6* | 0.03 | 1.2 ± 0.8 | 15.8 | –1.2 ± 0.6* | 0.1 | ||
| PSNCBAM-1 | –1.7 ± 0.2* | 0.02 | –1.2 ± 0.2* | 0.1 | 0.7 ± 0.7 | 5.0 | ||
| CP55,940 | GAT100 | –0.2 ± 0.1 | 0.6 | –0.2 ± 0.1 | 0.6 | 0.1 ± 0.1 | 1.3 | |
| Org27569 | –0.3 ± 0.1 | 0.5 | 0.6 ± 0.4 | 4.0 | 0.0 ± 0.6 | 1.0 | ||
| PSNCBAM-1 | –1.5 ± 0.5* | 0.03 | –0.7 ± 0.6 | 0.2 | –1.1 ± 0.6 | 0.1 | ||
| Neuro2a | 2-AG | GAT100 | 0.0 ± 0.1 | 1.0 | 0.0 ± 0.1 | 1.0 | ||
| Org27569 | –0.2 ± 0.2 | 0.6 | 0.5 ± 0.2 | 3.2 | ||||
| PSNCBAM-1 | 1.0 ± 0.5 | 10.0 | 0.5 ± 0.4 | 3.2 | ||||
| AEA | GAT100 | 1.2 ± 0.1* | 15.8 | 0.9 ± 0.1* | 7.9 | |||
| Org27569 | 0.6 ± 0.2 | 4.0 | 0.2 ± 0.2 | 1.6 | ||||
| PSNCBAM-1 | 0.7 ± 0.7 | 5.0 | –0.1 ± 0.1 | 0.8 | ||||
| CP55,940 | GAT100 | 0.5 ± 0.2 | 3.2 | 1.0 ± 0.1* | 10.0 | |||
| Org27569 | 0.6 ± 0.4 | 4.0 | 0.5 ± 0.9 | 3.2 | ||||
| PSNCBAM-1 | –0.5 ± 0.2 | 0.3 | –0.1 ± 0.9 | 0.8 | ||||
| STHdhQ7/Q7 | 2-AG | GAT100 | 0.0 ± 0.1 | 1.0 | 0.0 ± 0.1 | 1.0 | ||
| Org27569 | –0.6 ± 0.7 | 0.3 | –0.3 ± 0.6 | 0.5 | ||||
| PSNCBAM-1 | 0.6 ± 0.2 | 4.0 | 0.0 ± 0.3 | 1.0 | ||||
| AEA | GAT100 | 0.9 ± 0.1* | 7.9 | 0.1 ± 0.1 | 1.3 | |||
| Org27569 | –0.7 ± 0.6 | 0.2 | 0.1 ± 0.9 | 1.3 | ||||
| PSNCBAM-1 | 0.5 ± 0.5 | 3.2 | 0.2 ± 0.2 | 1.6 | ||||
| CP55,940 | GAT100 | 0.7 ± 0.1* | 5.0 | –0.2 ± 0.1 | 0.6 | |||
| Org27569 | –0.1 ± 0.6 | 0.8 | –0.5 ± 0.3 | 0.3 | ||||
| PSNCBAM-1 | 0.4 ± 0.2 | 2.5 | 0.1 ± 0.3 | 1.3 | ||||
∆∆log R and bias factor (BF) determined using operational model analysis as described in Methods (eqs 1–3). Calculations are from data presented in Figures 3 (β-arrestin1), 5 (pPLCβ3), 7 (pERK1/2), and 9 (cAMP) in cells treated with 500 nM orthosteric agonist +1 nM - 10 μM allosteric modulator. ∆∆log R for GAT100 compared with Org27569 and PSNCBAM-1.
P < 0.01 compared to 500 nM 2-AG + GAT100 within cell type as determined by one-way ANOVA followed by Dunnett’s multiple comparison. Data are mean ± SEM from at least four independent experiments. N = 4.
The present study is the first evaluation of Org27569 and PSCNBAM-1 for potential PLCβ3-related pathway bias.36,52,54,62 Previous observations have been made that Org27569 is biased against both Gαi/o and β-arrestin signaling.52,54 Such differences among studies likley reflect the well-recognized difficulty in extrapolating ligand bias across different assay systems/experimental conditions, especially when comparing ligands.63,64
An important advantage of using the operational model to estimate the relative activity and ligand bias is that this model negates the effects of differences in receptor density.65 Therefore, differences in bias between cell types are not the result of changes in agonist potency or efficacy.65,66
Recognition of CP55,940 as a standard reference synthetic cannabinoid and 2-AG and AEA as the principal endocannabinoids justified their use in this study to determine if GAT100 displayed orthosteric-probe dependence.1 When data from different cell models and assays were pooled to calculate arithmetic means, GAT100, Org27569, and PSNCBAM-1 did not display significant orthosteric-probe dependence among CP55,940, 2-AG, and AEA (Table 5). Baillie et al.36 reported that the Org27569 and PSNCBAM-1 both showed probedependence as more potent modulators of CP55,940 activity as compared to WIN 55,212-2. Khajehali et al.52 observed that Org27569 fully inhibits CP55,940, only weakly inhibits 2-AG, and does not inhibit AEA activity. Neither of these cited studies examined PSNCBAM-1 probe-dependence with the endocannabinoids 2-AG or AEA.
Furthermore, GAT100, Org27569, and PSNCBAM-1 did not display cell-dependence among HEK293A, Neuro2a, and STHdhQ7/Q7 cells (Table 5). The lack of orthosteric-probe and cell-dependence suggests that all threeCB1R allostericmodulators could be active in distinct biological environments.
Notably, GAT100 could be differentiated pharmacologically from both Org27569 and PSNCBAM-1 according to therapeutically relevant characteristics. As compared to those prototypic CB1R allosteric ligands across all cell types and information pathways examined, GAT100 was a more potent and efficacious NAM of endocannabinoid activity (Table 5) and was devoid of the appreciable inverse-agonist activity48 we and others have observed to be a hallmark of both Org27569 and PSNCBAM-1.28,29,36 By directly evaluating the downstream signaling profiles of these three CB1R NAMs in parallel across three distinct cell systems with both overexpressed and native CB1R and assessing multiple G-protein-dependent and -independent signaling pathways, we have countered the possibility that the GAT100 signaling pattern described and the pharmacological differences uncovered between GAT100 vs Org27569 and PSNCBAM-1 reflect assay or cell-context dependent factors rather than the multidimensional information network of CB1R itself.63,64
Effect of GAT100 on [3H]SR141716A Equilibrium Binding by hCB1R
GAT100, Org27569, and PSNCBAM-1 increase CP55,940 equilibrium binding by CB1R, GAT100 being the most potent and efficacious enhancer.28,29,48 Both Org27569 and PSNCBAM-1 can displace the CB1R antagonist/inverse agonist SR141716A, suggesting that the binding domains of these three ligands may overlap.37 These combined data invited examination of the effect of GAT100 on equilibrium binding of SR141716A in order to gain some insight into the CB1R GAT100 binding domain. At 1 and 10 μM, both GAT100 and Org27569 reduced [3H]SR141716A specific binding to membranes obtained from CHO cells transfected with hCB1R in a concentration-related manner (Figure 11). Unlike Org27569, GAT100 did not completely abrogate [3H]SR141716A binding at a concentration of 10 μM. GAT100 displayed ~6-fold greater potency (EC50 = 409.8 nM) than Org27569 (EC50 = 2522 nM) as an inhibitor of [3H]SR141716A binding.
Figure 11.
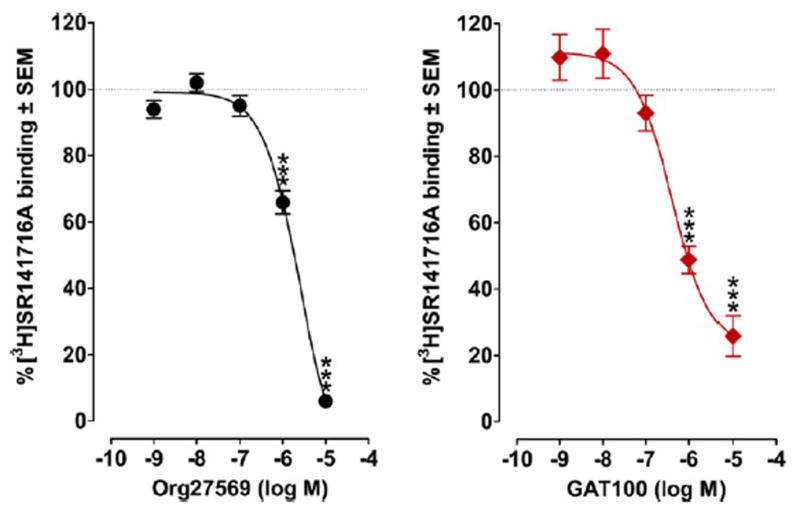
Effects of GAT100 and Org27569 on the binding of [3H]SR141716A (2 nM) to membranes obtained from CHO cells transfected with human CB1R receptors. Symbols represent mean percentage changes in [3H]SR141716A binding values ± SEM (n = 6). The mean Emax values of GAT100 and Org27569, with their 95% confidence limits shown in brackets, are 22.59% (8.95 and 36.22%) and −17.57% (−4.54 and −30.61%), respectively. The corresponding EC50 values, again with 95% con3dence limits shown in brackets, are 409.8 nM (185.2 and 906.6 nM) and 2522 nM (1700 and 3741 nM). Asterisks indicate mean values that are significantly different from 100% (***P < 0.001 via Student’s one sample t test).
Computational Modeling of the Interaction Profile between GAT100 and Intermediate-State hCB1R (hCB1R**)
The absence of a reported CB1R crystal structure led us to apply computational methods for gaining insight into the GAT100 binding site within hCB1R and potential GAT100–amino acid interactions critical to the allosteric ligand’s functional pharmacology. Four considerations guided this effort: (a) Ligand binding to CB1R and other class A GPCRs is widely accepted to occur within transmembrane helices (TMHs) and extracellular loops (ECLs) with lipophilic ligands predisposed to accessing the TMH bundle via the membrane lipid bilayer.43,67 (b) GAT100’s reactive isothiocyanate functionality is a conservative modification of the parent Org27569 structure at the critical C-5 position such that Org27569 can be used as direct comparator to GAT100 (Figure 1).48 (c) Under physiological incubation/reaction conditions as used in our GAT100-CB1R studies (i.e., aqueous milieu and physiological pH), the nucleophilic thiol moiety of cysteine residues renders them by far the most reactive nucleophilic amino acid toward various electrophiles, including isothiocyanates,68-70 making cysteine the most likely participant in a nucleophilic addition reaction with the isothiocyanate GAT100. Indeed, under these conditions, isothiocyanates have been recognized as “sulfhydryl-reactive affinity ligands,” and as such useful probes “to investigate ligandreceptor interactions.”69 The avidity and facile reactivity of designer isothiocyanate probes for GPCR cysteine residues has been previously exploited by us and others to help map critical ligand-interaction domains.41,43-45 Given that GAT100 is an isothiocyanate, the above context—in addition to prior in silico and experimental descriptions of CB1R next discussed—provides a sound rationale for focusing the modeling studies on cysteine and no other nucleophilic amino acids. (d) Of the 13 total hCB1R cysteine residues, two ECL cysteines (C257 and C264) are essential to hCB1R high-level expression and function by virtue of the disulfide bridge between them that would preclude their reaction with GAT100.43 Six other hCB1R cysteines may likewise be eliminated as potential covalent attachment sites for GAT100 because of their location outside of hCB1R TMHs. The five remaining hCB1R cysteine residues [C1.55(139), C4.47(238), C6.47(355), C7.38(382), and C7.42(386)] are located in hCB1R TMHs. C1.55(139) and C4.47(238) are on the intracellular side of the CB1R 7-TMH bundle, and face outward toward bilayer lipid,71,72 rendering thempotential targets for GAT100 molecules in the inner leaflet of the bilayer. In the CB1R inactive state, C7.42(386) is oriented toward the binding site crevice, whereas C6.47(355) and C7.38(382) are oriented toward lipid with C6.47(355) deeper in the lipid bilayer, making these latter two residues potential targets for GAT100 in the outer bilayer leaflet.
We used molecular dynamics (MD) simulation to identify the CB1R TMH cysteine residue most likely to react with GAT100. For this purpose, a model was employed representing a receptor conformation (hCB1R**) in a lipid bilayer intermediate between inactive (R) and active (R*) states that is promoted by Org27569 in the presence of CP55,940 and that preferentially binds agonist but cannot signal in G protein-mediated pathways, as elaborated elsewhere.37 Given that GAT100 is a direct Org27569 analogue, we first analyzedMDprofiles for Org27569-hCB1R** in a 1-palmitoyl-2-oleoyl-phosphatidylcholine (POPC) bilayer across fourteen Org27569 trajectories. As illustrated by the MD snapshot in Figure 12, Org27569 contoured at its Van der Waals radii is horizontal to the POPC membrane acyl chains and sits two turns down from the extracellular TMH termini. In this figure, the phosphorus atoms of the phospholipid head groups are shown contoured at their Van der Waals radii, but the rest of the lipid, including acyl chains, have been turned off for clarity. The Org27569 indole ring is located at the level of C7.38(382) and is higher in the bilayer than either C7.42(386) or C6.47(355). Over both 1 μs MD trajectories, only C1.55(139) and C7.38(382) achieved a distance of less than 5 Å from the Org27569 chlorine atom, a proximity making them the most likely cysteine residues to participate in a nucleophilic-addition reaction with GAT100. C1.55(139) is part of a cholesterol binding site73 formed by 44% of Class A GPCRs at the intracellular end of the receptor in the TMH1-2-3-4. Although CB1R does not have the exact sequence motif for this binding site, recent covalent labeling studies identified this region as a possible site for Org27569 interaction with CB1.74 However, binding at this site does not explain the positive allosteric effects that Org27569 has upon CP55,940 binding, negative allosteric effects upon CP55,940 signaling and partial displacement of SR141716A binding rendering C1.55(139) an unlikely candidate for interaction withGAT100.28,74
Figure 12.
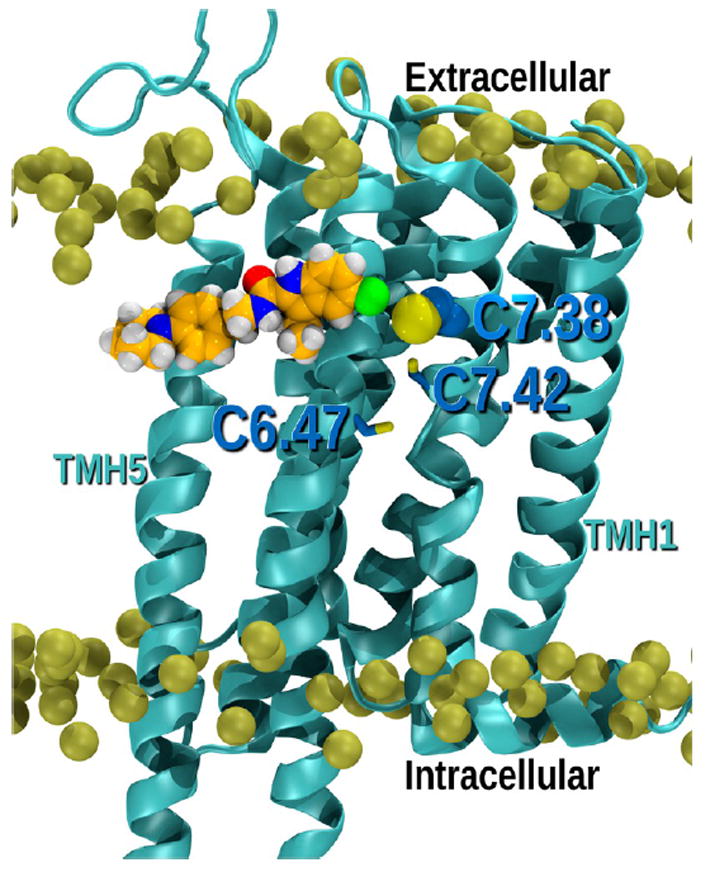
Figure illustrating the position of ORG27569 in the lipid bilayer relative to the cluster of CYS residues in the TMH6-TMH7 region. It is clear here that the ORG27569 indole ring is located at the level of C7.38(382) and is higher in the bilayer than Cys7.42(386) and Cys6.47(355).
Previous studies have identified CB1R C6.47(355) as the site of CB1R covalent labeling by analogues of classical cannabinoids43,44 and endocannabinoids46 derivatized with reactive isothiocyanate functionalities. However, ourMDanalysis indicates that Org27569 resides, on average, higher in the lipid bilayer from C6.47(355), nearer to C7.38(382) (Figure 12). These aggregate data with parent compound Org27569 led us to focus subsequent modeling studies on covalent GAT100-C7.38(382) interaction in the outer leaflet of the membrane.
GAT100 Covalent Labeling at C7.38(382)
Modeling studies revealed that if GAT100 labels C7.38(382), it can enter the hCB1R** bundle above CP55,940 such that it can interact with CP55,940, while simultaneously interacting with binding site residues, including I6.54, D6.58, F7.35, P(269), and F(268). CP55,940 retains all of its interactions in hCB1R** including the crucial hydrogen bond with K3.28.37 Figure 13 shows GAT100 (purple) covalently attached to C7.38(382) interacting with CP55,940 (green) in the hCB1R** binding site. The conformational energy expense for GAT100 to assume this position in the binding pocket was 1.00 kcal/mol. Figure S2 shows an overlay of the global minimum energy conformer of Org27569 to the conformation of GAT100. It is clear here that the two compounds overlay very well. The interaction energy between GAT100 and CP55,940 in this dock was calculated to be −2.08 kcal/mol. The total interaction energy for GAT100 in Figure 13 is −41.2 kcal/mol. The interaction energies with individual receptor residues are given in Table S2. CP55,940 in this dock (Figure 13) has a total interaction energy of −61.8 kcal/mol. The CP55,940 interaction energies with individual receptor residues are provided in Table S3.
Figure 13.
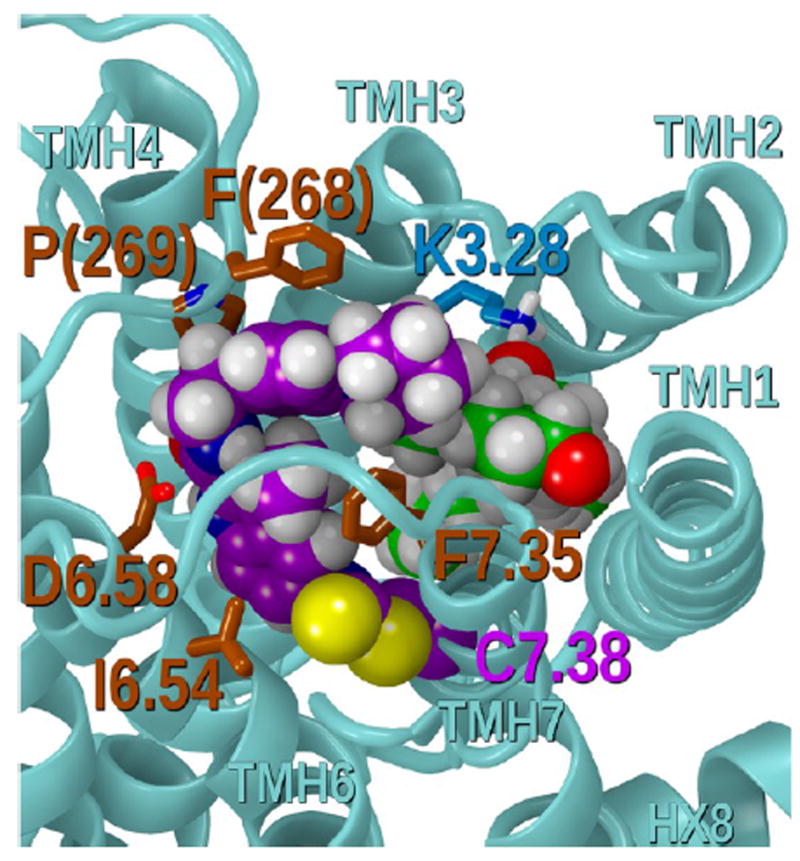
Figure showing GAT100 (purple) covalently attached to C7.38(382) interacting with CP55940 (green) in the hCB1R** binding site. Modeling studies revealed that if GAT100 labels C7.38(382), it can enter the hCB1R** bundle above CP55940 such that it can interact with CP55940, while simultaneously interacting with binding site residues, including I6.54, D6.58, F7.35, P(269), and F(268). CP55940 (green) retains all of its interactions in hCB1R** including the crucial hydrogen bond with K3.28.
GAT100 as Positive Allosteric Modulator of CP55,940 Binding
Tables S2 and S3 show that CP55,940 has a favorable interaction energy (−2.08 kcal/mol) with GAT100. In addition, GAT100 is located above CP55,940, blocking CP55,940 exit from the binding pocket. Both effects will increase CP55,940 binding affinity. Thus, GAT100 functions here as a positive allosteric modulator of CP55,940 binding.
GAT100 as a Negative Allosteric Modulator of CP55,940 Signaling
Class A GPCR activation is accompanied by TMH conformational changes, as well as changes in IC and EC loop conformations. We have previously reported that, upon receptor activation, an important ionic interaction forms between TMH2 and the EC-3 loop (specifically, an interaction forms between residues D2.63(176) and K373);75 this ionic interaction is necessary for signal transduction and promotes a conformation of the EC-3 loop that is pulled over the top (extracellular face) of the receptor (see Figure 11A in ref 37). We have shown previously that Org27569 can impact CP55,940 signaling by interrupting this EC-3 loop movement.37 Figure 14 illustrates that when GAT100 is bound in the hCB1R** binding pocket, it sits high enough to prevent the D2.63(176)/K373 interaction. In this way, GAT100 can prevent CP55,940 signaling.
Figure 14.
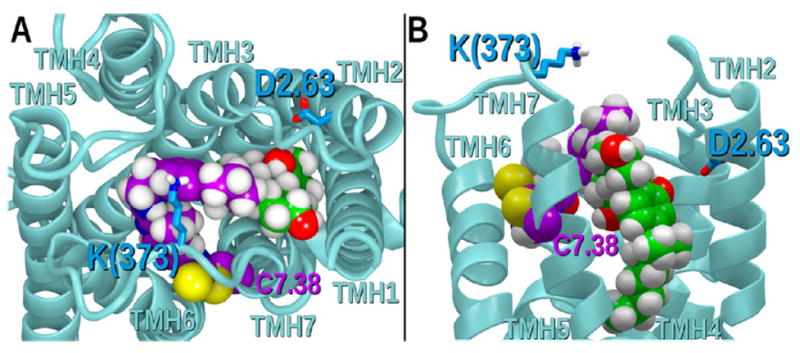
Figure showing a (A) top view and (B) side view of GAT100 bound in the hCB1R** binding pocket. Here GAT100 sits high enough to prevent the D2.63176/K373 interaction that is necessary for receptor activation. In this way, GAT100 (purple) can prevent CP55940 (green) signaling.
GAT100 Displacement of SR141716A
Figure 15A provides an extracellular view of SR141716A (orange) docked in hCB1R**.76 Figure 15B (view from extracellular) and C (view from lipid bilayer) shows that when GAT100 (purple) is docked in hCB1R**, it partially overlaps the SR141716A binding site. This overlap would account for the partial displacement of SR141716A binding by GAT100.
Figure 15.
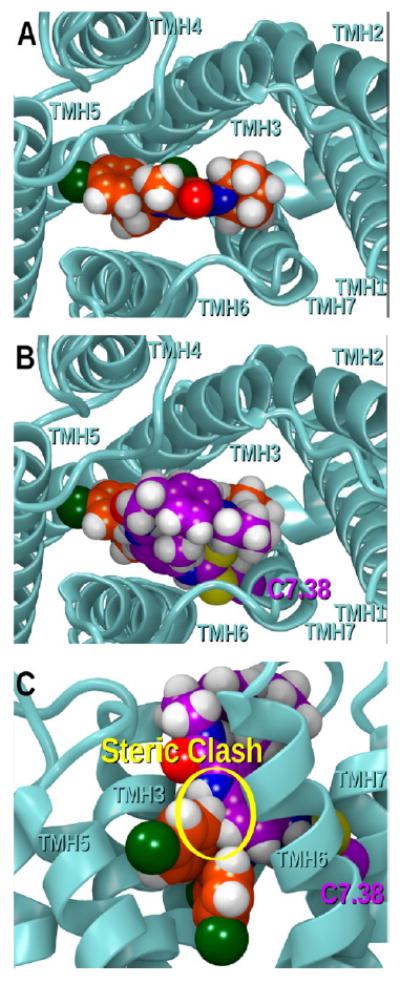
(A) Extracellular view of SR141716A (orange) docked in hCB1R**. Panels (B) (view from extracellular) and (C) (view from lipid bilayer) show that when GAT100 (purple) is docked in hCB1R**, it partially overlaps the SR141716A binding site. This overlap would account for the partial displacement of SR141716A binding by GAT100.
It remains for future studies to determine whether GAT100’s superior potency and efficacy; its G-protein-independent, β-arrestin1-mediated signaling bias; and its freedom from significant inverse-agonist activity engender (pre)clinical therapeutic advantages in vivo. In this regard, it is noteworthy that the on-target specificity and selectivity associated with GPCR covalent ligands have been considered to hold promise of improved drug safety, tolerance, and efficacy,77 and arrestinbiased GPCR ligands can promote salutary effects and simultaneously dampen deleterious ones in vivo.64 Arguably of greatest translational relevance, the propensity of Org27569 and PSNCBAM-1 to interfere with constitutive CB1R signaling through their inverse-agonist action is not shared by GAT100, making it tempting to speculate that GAT100 might have less potential to induce the adverse events associated with typical orthosteric CB1R antagonists/inverse agonists and suggesting more generally that the binding sites for NAM and inverse agonist activity can be separated. By extension, NAMs such as GAT100 might represent a tenable route toward resolving the beneficial and deleterious routes of CB1R information output and reducing the side-effect risk of modulating/attenuating CB1R signaling for therapeutic exploitation. Furthermore, GAT100’s more rapid reversal of agonist-induced CB1R internalization may beneficially attenuate net receptor desensitization, such as that induced by increased orthosteric ligand binding to the receptor. The unique pharmacological distinctions between GAT100 and parent NAM Org27569 in our signaling assays suggest that (non)covalent GAT100 derivatives with alternative modifications at the C-5 site are worthy of synthesis and cell-based profiling. Such derivatives could be purposedesigned to react with amino acids other than cysteine and validated as probes for more global experimental identification of critical ligand-interaction sites within the allosteric hCB1R binding domain(s).41
Conclusion
Biologically active allosteric ligands capable of fine-tuning GPCR information output are being increasingly sought for their potential advantages over orthosterically directed drugs, specifically in terms of improved on-target potency, efficacy, and safety.78,79 In this regard, allosteric modulation of the principal GPCR inmammalian brain, CB1R, has gained particularly intense interest as a therapeutic modality from the unacceptable adverseevent risk associated with typical orthosteric CB1R agonists and antagonists/inverse agonists that has plagued their development as drugs. For those GPCRs such as CB1R for which crystal structures remain elusive, molecular probes have assumed increasing importance for interrogating receptor ligand-binding domains and elucidating the functional consequences of ligand engagement in order to guide rational drug design.42 The availability of the first, purpose-designed CB1R covalent allosteric probe and Org27569 analogue, GAT100, has enabled initial application of such an experimental approach to this well-recognized drug target.
The present work constitutes the detailing of GAT100’s downstream signaling profile and the application of computational methods to gain insight into the structure–function correlates of GAT100 engagement by CB1R. Our ligand-docking and in silico analysis of the GAT100-hCB1R** interaction profile identifies C7.38(382) as the hCB1R** amino acid with which GAT100 is most likely to participate in a covalent interaction by virtue of its isothiocyanate moiety. The availability of GAT100 as a functional, allosteric-site covalent probe enables additional experimental studies aimed at further characterization of CB1R allosteric ligand-binding motifs and the pharmacology of CB1R functional modulation by such a ligand. Determination of the effect of targeted CB1R mutations on GAT100-CB1R complex formation and the application of peptide-level tandem mass spectrometry to define directly the allosteric-site amino acid(s) reacting with this covalent ligand are also made possible. GAT100 may be used to stabilize allosteric CB1R conformations to aid their crystallization as prerequisite to atomic-level X-ray structure analysis of the liganded receptor. Proof-of-concept animal studies aimed at defining the effects of GAT100 in vivo and interrogating the extent to which GAT100-sensitive signaling pathways identified herein are responsible for those effects could help in realizing the therapeutic promise and purported translational advantages of covalent allosteric GPCR modulators.
METHODS
Compounds
2-AG, AEA, and CP55,940 were purchased from Tocris Bioscience (Bristol, U.K.). Org27569, PSNCBAM-1, and GAT100 were synthesized in the laboratory of Dr. Ganesh A Thakur (Northeastern University, Boston, MA). All compounds were dissolved in DMSO (final concentration, 0.1% in culture media for all cell-based assays) and added directly to the media at concentrations and times indicated.
Amino Acid Descriptor
The Ballesteros–Weinstein formalism is used to designate the loci of specific amino acids as derived from the hCB1R sequence numbers.37,80 In this system, the most highly conserved amino acid residue in eachTMH is assigned a locant of 0.5. This number is preceded by the helix number followed in parentheses by the sequence number. All other residues in a TMH are numbered relative to that residue.
Cell Culture
HEK293A cells were a generous gift from Dr. Denis J. Dupré. (Dalhousie University, Nova Scotia, Canada) and were originally obtained from the American Type Culture Collection (ATCC, Manaassas, VA). HEK293A were transfected with 400 ng of the CB1-GFP2-expressing plasmid described below using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen, Burlington, ON, Canada). HEK293A cells were maintained at 37 °C, 5% CO2-95% air in Dulbecco’s minimal essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 104 U/mL penicillin (Pen)/streptomycin (Strep). HEK293A Cignal Lenti CRE (HEK-CRE) reporter cells were a generous gift by Dr. Christopher J. Sinal (Dalhousie University, Nova Scotia, Canada). HEK-CRE cells stably express the firefly luciferase gene driven by tandem repeat elements of the cAMP transcriptional response element (Qiagen, Toronto, Ontario, Canada). Thus, the luciferase signal is directly proportional to cAMP/protein kinase A (PKA) pathway activity. HEK-CRE cells were maintained at 37 °C, 5%CO2-95% air in DMEM supplemented with 10% FBS, 104 U/mL Pen/Strep, and 200 μg/mL puromycin. Neuro2a mouse neuroblastoma stem cells were purchased from ATCC and maintained 37 °C, 5% CO2-95% air in Eagle’s minimal essential medium (MEM) supplemented with 10% FBS and 104 U/mL Pen/Strep.81 STHdhQ7/Q7 cells are derived from the conditionally immortalized striatal progenitor cells of embryonic day 14 C57BlJ/6 mice (Coriell Institute, Camden,NJ) and were maintained at 33 °C, 5%CO2-95% air inDMEMsupplemented with 10% FBS, 2mML-glutamine, 104 U/mL Pen/Strep, and 400 μg/mL Geneticin.82 Cells were serum-deprived for 24 h prior to experiments to promote differentiation.55,82
Plasmids
Human CB1-green fluorescent protein2 (GFP2) C-terminal fusion protein was generated using the pGFP2-N3 (PerkinElmer, Waltham, MA) plasmid, as described previously.83 Human arrestin2 (β-arrestin1)-Renilla luciferase (Rluc) C-terminal fusion protein was generated using the pcDNA3.1 plasmid and provided by Dr. Denis J. Dupré. (Dalhousie University, NS, Canada). The GFP2-Rluc fusion construct and Rluc plasmids have also been described.83
CHO Cells
Chinese hamster ovary (CHO) cells transfected with cDNA encoding human cannabinoid CB1 receptors were maintained at 37 °C in Dulbecco’s modified Eagle’s medium nutrient mixture F-12 HAM, supplemented with 1 mM l-glutamine, 10% fetal bovine serum, 0.6% Pen/Strep, and G418 (400 mg/mL). All cells were exposed to 5% CO2 in their media, and were passaged twice a week using nonenzymatic cell dissociation solution. For membrane preparation, cells were removed from flasks by scraping, centrifuged, and then frozen as a pellet at −20 °C until required. Before use in radioligand binding assays, cells were defrosted, diluted in Tris buffer (50 mM Tris–HCl and 50 mM Tris–base), and homogenized with a 1 mL hand-held homogenizer.
Bioluminescence Resonance Energy Transfer2 (BRET2)
Direct interactions between CB1 and β-arrestin1 were quantified via BRET2.84 Cells were grown in a 6-well plate and transfected with CB1-GFP2 and β-arrestin1-Rluc using Lipofectamine 2000, according to the manufacturer’s instructions (Invitrogen) and as previously described.55 At 48 h post-transfection, cells were washed twice with cold 0.1 M PBS and suspended in BRET buffer [0.1 M PBS supplemented with glucose (1 mg/mL), benzamidine (10 mg/mL), leupeptin (5 mg/mL), and a trypsin inhibitor (5 mg/mL)]. Cells were dispensed into white 96-well plates (10 000 cells/well) and treated as indicated (PerkinElmer). Coelenterazine 400a substrate (50 μM; Biotium, Hayward, CA) was added and light emissions were measured at 460 nm (Rluc) and 510 nm (GFP2) using a Luminoskan Ascent plate reader (Thermo Scientific, Waltham, MA), with an integration time of 10 s and a photomultiplier tube voltage of 1200 V. BRET efficiency (BRETEff) was determined using previously described methods55,83 such that Rluc alone was used to calculate BRETMIN and the Rluc-GFP2 fusion protein was used to calculate BRETMAX.
On- and In-Cell Westerns
Cells were fixed for 10 min at room temperature with 4% paraformaldehyde and washed three times with 0.1 M PBS for 5 min each. Cells were incubated with blocking solution [0.1 M PBS, 5% normal goat serum, and 0.3% TritonX-100 (in-cell Western only), in dH2O] for 1 h at room temperature. Cells were incubated with primary antibody solutions directed against N-CB1 (1:500) (Cayman Chemical Company, Ann Arbor, MI, Cat. No. 101500), pERK1/2(Tyr205/185) (1:200), ERK1/2 (1:200), pPLCβ3- (S537) (1:500), or PLCβ3 (1:1000) (Santa Cruz Biotechnology) diluted in antibody dilution buffer [0.1MPBS, 1% (w/v) BSA, in dH2O] overnight at 4 °C. Cells were washed three times with 0.1 M PBS for 5 min each. Cells were incubated in IRCW700dye or IRCW800dye (1:500; Rockland Immunochemicals) and washed again three times with 0.1 M PBS for 5min each.On- and in-cell Western analyses were then conducted using the Odyssey Imaging system and software (version 3.0; Li-Cor). For on-cellWestern analyses, this protocol was used to detect CB1 on the plasma membrane and cells were then used in the same procedure again for in-cellWesterns to detect total CB1. In this way, the fraction of CB1 at the plasma membrane was calculated.55 A CB1 blocking peptide control was used for all assays where CB1 levels were measured (Cayman Chemical Company).
cAMP Luciferase Reporter Assay
HEK-CRE cells were transfected with CB1-GFP2. At 48 h post-transfection, cells were washed twice with cold 0.1 M PBS and suspended in BRET buffer. Cells were dispensed into 96-well plates (10 000 cells/well) and treated as indicated (PerkinElmer). Media was aspirated from cells and cells were lysed with passive lysis buffer for 20 min at room temperature (Promega, Oakville, ON, Canada). A volume of 20 μL of cell lysate was mixed with luciferase assay reagent (50 μM; Promega, Oakville, ON, Canada), and light emissions were measured at 405 nm using a Luminoskan Ascent plate reader (Thermo Scientific, Waltham, MA), with an integration time of 10 s and a photomultiplier tube voltage of 1200 V. Data are presented as % cAMP inhibition (relative to the Emax for the orthosteric ligand used in the presence of 10 μM forskolin) or % cAMP accumulation (relative to cAMP levels in the presence of 10 μM forskolin alone).
Radioligand Displacement Assays
The assays were carried out with [3H]SR141716A and Tris binding buffer (50 mM Tris–HCl, 50 mM Tris–base, 0.1% BSA, pH 7.4), total assay volume 500 μL, using the filtration procedure described previously.36,85 Binding was initiated by the addition of transfected human CB1 CHO cell membranes (50 μg protein per well). All assays were performed at 37 ΰC for 60 min before termination by the addition of ice-cold Tris binding buffer, followed by vacuum filtration using a 24-well sampling manifold (Brandel Cell Harvester; Brandel Inc., Gaithersburg, MD) and Brandel GF/B filters that had been soaked in wash buffer at 4 °C for at least 24 h. Each reaction well was washed six times with a 1.2 mL aliquot of Tris-binding buffer. The filters were oven-dried for 60 min and then placed in 3 mL of scintillation fluid (Ultima Gold XR, PerkinElmer, Seer Green, Buckinghamshire, U.K.). Radioactivity was quantified by liquid scintillation spectrometry. Specific binding was defined as the difference between the binding that occurred in the presence and absence of 1 μM unlabeled SR141716A. The concentration of [3H]SR141716A used in our displacement assays was 2 nM. The compounds under investigation were stored as stock solutions of 10 mM in DMSO, with the vehicle concentration in all assay wells being 0.1% DMSO.
Most results were calculated as percentage changes from a basal level (100%) of [3H]SR141716A binding. Prism 5.0 (GraphPad, San Diego, CA) was used to construct sigmoidal log concentration–response curves, and to calculate values of EC50, Emax, SEM, and 95% confidence intervals. Some mean values were compared using Student’s one sample t test. P values < 0.05 were considered to be significant.
Statistical Analyses
Concentration–response curves were fit to nonlinear regression with variable slope (four parameter) model, Allosteric Modulator EC50 Shift model, or global nonlinear regression using the operational model60,65,66 (eq 1) in Prism v. 6.0 (GraphPad Software Inc., San Diego, CA). The global nonlinear regression model was used to estimate the transduction coefficient [log R(τ/KA)], change in transducer coefficient relative to the reference ligand (∆log R), and bias factor (∆∆log R), as indicated. In eq 1, E is the response, Emax is the maximal response, [A] is ligand concentration, n is transducer slope, τ is agonist efficacy, and KA is the agonist’s affinity for the receptor.65 In order to obtain a global least-squares fit of the data to the operational model, all parameters except log R and ∆log R were shared between all data sets using 2-AG, AEA, or CP55,940 as the orthosteric ligand within a cell type; log KA was constrained to be greater than −15, and log τ was constrained to be less than 10.66,86 Relative activity (∆log R) was calculated in Prism as the difference between transduction coefficients [log R(τ/KA)] values for two ligands, a “test” ligand and a reference ligand (here GAT100 in the presence of 500 nM 2-AG) as measured between sample-matched replicates65 (eq 2). In eq 3, bias factor (i.e., log bias, ∆∆log R) is the difference between response 1 (R1) and response 2 (R2).65 All calculations of ∆∆log R are reported using β-arrestin1 response as R1. Statistical analyses were conducted by one- or two-way analysis of variance (ANOVA), as indicated, using GraphPad (version 5.0, Prism). Post hoc analyses were performed using Bonferroni’s or Tukey’s tests, as indicated. Homogeneity of variance was confirmed using Bartlett’s test. The level of significance was set to P < 0.001, < 0.01, or < 0.05, as indicated, and all results are reported as the mean ± the standard error of the mean (SEM) from at least four independent experiments.
| (1) |
| (2) |
| (3) |
Equilibration of the CB1R and Construction of the Org27569/CB1 Simulation Cell
The CB1 receptor, truncated at SER88/GLY417 for the N-terminus/C-terminus, respectively, was aligned with the S1P1 structure from the OPMdatabase.87 This resulted in the transmembrane region being centered at the middle of the 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) lipid bilayer and the amphipathic helix 8 oriented parallel to the plane of the membrane at approximately the lipid/water interface. Prior to the addition of ligands, CB1 was simulated for 335 ns in the POPC lipid bilayer. The model membrane simulation cell was constructed using the method described in Grossfield et al.88 The CHARMM22 protein force field89 and the CHARMM 36 lipid force field90 were used in this study. Charge neutrality was enforced with addition of chloride counterions and an overall ionic strength of 0.1 M was obtained by adding NaCl.
Initial Minimization
To relieve poor initial contacts, 2500 steps of steepest descent minimization were performed using CHARMM,91,92 with all heavy atoms of the protein fixed. This was followed by a series of restrained minimizations using NAMD.93 In this slow release phase, the CB1 heavy atoms were restrained to their initial positions and starting with a force constant of k = 5.0 kcal/mol/Å2, 500 steps of conjugate gradient minimization were performed. The force constant was subsequently reduced to 2.5, 1.0, 0.5, 0.25, 0.1, and finally 0.05 kcal/mol/Å2, with 500 steps of minimization at each step. Finally, 20,000 steps of restraint free minimization were executed. Molecular dynamics was then performed on the fully minimized system.
Details of Molecular Dynamics Simulations
For all production runs, the GPU accelerated Particle Mesh Ewald (PME) AMBER12 package was utilized.92,94 Long range electrostatics were included using PME with an 8 Å cutoff and default values for the charge grid spacing (chosen to be approximately 1 Å) and cubic B-spline. The NPTensemble was used to maintain temperature (T = 300 K, Langevin dynamics with a collision frequency of 5 ps−1) and pressure (P = 1.0 bar, using the weak coupling Berendsen pressure control95 with pressure relaxation time of 8 ps). High frequency bonds to hydrogen were restrained using the Shake method allowing the use of a 2 fs integration time step.
Addition of Org27569 to the Simulation
An initial MD simulation run was used to relax the hCB1R in an explicit, fully hydrated lipid bilayer. Upon equilibration of the receptor, taken as a leveling of the RMSD of the TMH of hCB1R (Figure S1), a snapshot was taken at 70 ns, and the hCB1R conformation was extracted. During the course of this and all subsequent trajectories with Org27569 molecules added, the hCB1R ionic lock (R3.50-D6.30 salt bridge) was maintained. A frame (at 70 ns) was extracted, and the simulation cell was rebuilt with the inclusion of fourteen Org27569s. The ligands were placed as follows: Seven extracellular (defined by the last four turns of the TMHs) locations were defined in a circle relative to the center of the TMHs at a radial distance such that at least one phospholipid could be placed between the receptor and a given Org27569 molecule. The seven locations were randomized on this circle and the center of mass (COM) of an Org27569 was placed at this site. The orientation and location of the Org27569 molecules was such that the indole moiety of Org27569 was approximately at the lipid/water interface. Each molecule was rotated by a random amount. If contacts between the Org27569 and CB1R ensued, the ligand was moved radially away from the receptor until the contacts were relieved. Similar placement was performed for the 7 intracellular Org27569 molecules. Parameters for Org27569 were initially obtained via the ParamChem server.96 Parameters were developed as described (see http://mackerell.umaryland.edu/ff_dev.shtml) for any initial torsion or bond angle with high penalties. The final system contained 90,921 atoms including 177 POPC molecules, fourteen Org27569 molecules, the protein, ions, and 20,300 solvating water molecules. Minimization and molecular dynamics were performed as described above for the CB1/POPC system. Two independent trajectories were generated, differing only in their initial placement of lipids, ions, and waters. Each was simulated for at least 1 μs.
Ligand Conformational Search
Complete conformational analyses performed for GAT100 and CP55,940 here. Local energy minima were identified by rotation of a subject torsion angle through 360° in 45° increments (8-fold search), followed by semiempirical AM1 energy minimization of each rotamer generated. The geometries of the resulting unique conformers were optimized with ab initio Hartree–Fock calculations at the 6-31G* level as encoded in Spartan ’08 (Wave function, Inc., Irvine, CA). To calculate the energy difference between the global minimum energy conformer of GAT100 or CP55,940 and their final docked conformations, the single point energy of each was obtained using the all atom OPLS3 force field in Macromodel 10.9 (Schrödinger Inc., Portland, OR) and the difference was calculated.
Docking of Covalently Attached GAT100 into the hCB1R**/CP55,940 Complex
GAT100 was covalently attached to C7.38(382) and docked into the Org27569 hCB1R** binding site previously identified by Glide docking studies.37 hCB1R** represents an intermediate CB1R conformation that can bind agonists, yet does not signal in G protein-mediated pathways.37 In this model, the EC3 loop/TMH2 salt bridge at K(373)/D2.63 had to be broken to allow GAT100 to bind in the presence of CP55,940. The energy of the ligand(s)–hCB1R** complex, including loop regions, was minimized using the OPLS3 force field in Macromodel 10.9 (Schrödinger LLC). The first stage consisted of Polak–Ribier conjugate gradient minimization using a distance dependent dielectric function with a base constant of 2. No harmonic constraints were placed on the side chains, but 250 kcal/mol fixed atom constraints were applied to hold all the backbone atoms in place. The termini and loops were not allowed to move during this stage. The minimization was continued until the bundle reached the 0.01 kcal/mol/Å2 gradient. An 8.0 Å nonbonded cutoff (updated every 10 steps), a 20.0 Å electrostatic cutoff, and a 4.0 Å hydrogen bond cutoff were used in each stage of the calculation. In the second stage, loops were relaxed via a Polak–Ribier conjugate gradient minimization until a 0.01 kcal/mol/Å2 gradient was reached. The loop and termini regions were left free, while the transmembrane regions and ligands were not allowed to move. An 8.0 Å extended nonbonded cutoff (updated every 10 steps), 20.0 Å electrostatic cutoff, and 4.0 Å hydrogen bond cutoff were used in this calculation, and the generalized Born/surface area (GB/SA) continuum solvation model for water available in Macromodel was employed.
Assessment of Pairwise Interaction and Total Energies
Interaction energies between each bound ligand and residue in the complex were calculated using Macromodel, as described.75
Supplementary Material
Table 4.
cAMP Inhibition in the Presence of GAT100, Org27569, and PSNCBAM-1a
| HEK-CREb | ||||||
|---|---|---|---|---|---|---|
| 2-AG
|
AEA
|
CP55,940
|
||||
| IC50 (nM) | Emax | IC50 (nM) | Emax | IC50 (nM) | Emax | |
| agonist alone | 338 (203–516) | 93.6 ± 5.47 | 508 (358–719) | 87.2 ± 7.36 | 587 (474–678) | 106.3 ± 11.4 |
| GAT100 | 644 (575–761)ˆ | 19.2 ± 0.95ˆˆˆ | 774 (735–805)†ˆ | 10.4 ± 1.05ˆˆˆ | 834 (786-952)† ˆ | 19.2 ± 1.62ˆˆˆ |
| Org27569 | 970 (836–1180)*ˆ | 39.0 ± 1.40***ˆˆˆ | 882 (825–961)* ˆ | 28.4 ± 2.07***ˆˆˆ | 969 (829–998)* ˆ | 25.7 ± 1.64*ˆˆˆ |
| PSNCBAM-1 | 978 (916–1223)*ˆ | 40.0 ± 2.13*** ˆˆˆ | 870 (849–919)* ˆ | 40.7 ± 2.45***ˆˆˆ | 962 (842–997)*ˆ | 45.7 ± 3.93***ˆˆˆ |
IC50 (nM) determined using nonlinear regression analysis; Emax (% cAMP inhibition) determined using nonlinear regression analysis. Data are mean with 95% CI (IC50) and mean ± SEM (Emax).
P < 0.05,
P < 0.01,
P < 0.001compared to GAT100 within orthosteric agonist treatment;
P < 0.05, compared to 2-AG within NAM treatment,
P < 0.05,
P < 0.01,
Acknowledgments
We thank Dr. Brian D. Hudson for his independent review of the data.
Funding
This work was supported by a Canadian Institutes of Health Research (CIHR) operating grant (MOP-97768) to M.E.M.K., a Bridge Funding Grant from Dalhousie University to E.M.D.-W., and National Institutes of Health Grants DA027113 and EY024717 to G.A.T., and DA03934 and DA021358 to P.H.R. R.B.L. is supported by studentships from CIHR, the Huntington Society of Canada, Killam Trusts, and the Nova Scotia Health Research Foundation.
Abbreviations
- 2-AG
2-arachidonylglycerol
- AEA
anandamide
- ANOVA
analysis of variance
- BRET
bioluminescence resonance energy transfer
- CB1
type 1 cannabinoid receptor
- CB2
type 2 cannabinoid receptor
- CP55,940
2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol
- GFP2
green fluorescent protein 2
- HEK-CRE
human embryonic kidney 293A Cignal Lenti cAMP response element cells
- Rluc
Renilla luciferase
- LAPS
ligand-assisted protein structure
- NAM
negative allosteric modulator
- Org27569
5-chloro-3-ethyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxa-mide
- PAM
positive allosteric modulator
- pregnenolone
1-((3S,8S,9S,10R,13S,14S,17R)-3-hydroxy-10,12,13-trimethyl-2,3,4,7,\8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl) ethan-1-one
- PSNCBAM-1
1-(4-chlorophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridine-2-yl) phenyl)urea
- SAR
structure–activity relationship
- GAT100
3-ethyl-5-isothiocyanato-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide
- TMH
transmembrane helix
- NCS
isothiocyanate
- hCB1R**
human cannabinoid receptor 1 active state
- IC
intracellular
- EC
extracellular
- [35S]GTPγS
guanosine 5’-O-(3-[35S]thio)-triphosphate
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
Footnotes
Author Contributions
Designed research: R.B.L., D.P.H, D.L., P.H.R., R.G.P., M.K.M., E.M.D.-W., and G.A.T. Synthesized GAT100, Org27569, and PSNCBAM-1: A.R.K. and P.M.K. Conducted experiments: R.B.L., D.P.H., D.L., and L.A.S. Analyzed data and literature and wrote the manuscript: R.B.L., A.R.K, D.P.H., P.H.R., D.R.J, R.G.P, M.E.M.K., E.M.D.-W., and G.A.T.
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.6b00041.
Transduction coefficients and relative activity for GAT100, Org27569, and PSNCBAM-1 (Table S1); ligand/receptor interaction energies for GAT100** (Table S2); ligand/receptor interaction energies for CP55,940** (Table S3); RMSD of the transmembrane residues of CB1 relative to the initial model in the equilibration run (Figure S1); overlay of the Org27569 global minimum energy conformation (cyan) and the docked conformation of GAT100 (purple) illustrated in Figure 13 (Figure S2) (PDF)
References
- 1.Pertwee RG, Howlett AC, Haas D, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB(1) and CB(2) Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elphick MR. The evolution and comparative neurobiology of endocannabinoid signalling. Philos Trans R Soc, B. 2012;367:3201–3215. doi: 10.1098/rstb.2011.0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 5.Rahn EJ, Deng L, Thakur GA, Vemuri K, Zvonok AM, Lai YY, Makriyannis A, Hohmann AG. Prophylactic cannabinoid administration blocks the development of paclitaxelinduced neuropathic nociception during analgesic treatment and following cessation of drug delivery. Mol Pain. 2014;10:27. doi: 10.1186/1744-8069-10-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng L, Guindon J, Vemuri VK, Thakur GA, White FA, Makriyannis A, Hohmann AG. The maintenance of cisplatin- and paclitaxel-induced mechanical and cold allodynia is suppressed by cannabinoid CB(2) receptor activation and independent of CXCR4 signaling in models of chemotherapy-induced peripheral neuropathy. Mol Pain. 2012;8:71. doi: 10.1186/1744-8069-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilkerson JL, Gentry KR, Dengler EC, Wallace JA, Kerwin AA, Armijo LM, Kuhn MN, Thakur GA, Makriyannis A, Milligan ED. Intrathecal cannabilactone CB(2)R agonist, AM1710, controls pathological pain and restores basal cytokine levels. Pain. 2012;153:1091–1106. doi: 10.1016/j.pain.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahn EJ, Thakur GA, Wood JA, Zvonok AM, Makriyannis A, Hohmann AG. Pharmacological characterization of AM1710, a putative cannabinoid CB2 agonist from the cannabilactone class: antinociception without central nervous system side-effects. Pharmacol, Biochem Behav. 2011;98:493–502. doi: 10.1016/j.pbb.2011.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilkerson JL, Gentry KR, Dengler EC, Wallace JA, Kerwin AA, Kuhn MN, Zvonok AM, Thakur GA, Makriyannis A, Milligan ED. Immunofluorescent spectral analysis reveals the intrathecal cannabinoid agonist, AM1241, produces spinal anti-inflammatory cytokine responses in neuropathic rats exhibiting relief from allodynia. Brain Behav. 2012;2:155–177. doi: 10.1002/brb3.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fichna J, Bawa M, Thakur GA, Tichkule R, Makriyannis A, McCafferty DM, Sharkey KA, Storr M. Cannabinoids Alleviate Experimentally Induced Intestinal Inflammation by Acting at Central and Peripheral Receptors. PLoS One. 2014;9:e109115. doi: 10.1371/journal.pone.0109115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pacher P, Kunos G. Modulating the endocannabinoid system in human health and disease–successes and failures. FEBS J. 2013;280:1918–1943. doi: 10.1111/febs.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thakur GA, Tichkule R, Bajaj S, Makriyannis A. Latest advances in cannabinoid receptor agonists. Expert Opin Ther Pat. 2009;19:1647–1673. doi: 10.1517/13543770903436505. [DOI] [PubMed] [Google Scholar]
- 13.Palmer SL, Thakur GA, Makriyannis A. Cannabinergic ligands. Chem Phys Lipids. 2002;121:3–19. doi: 10.1016/s0009-3084(02)00143-3. [DOI] [PubMed] [Google Scholar]
- 14.Pertwee RG. Emerging strategies for exploiting cannabinoid receptor agonists as medicines. Br J Pharmacol. 2009;156:397–411. doi: 10.1111/j.1476-5381.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dixon DD, Sethumadhavan D, Benneche T, Banaag AR, Tius MA, Thakur GA, Bowman A, Wood JT, Makriyannis A. Heteroadamantyl cannabinoids. J Med Chem. 2010;53:5656–5666. doi: 10.1021/jm100390h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thakur GA, Palmer SL, Harrington PE, Stergiades IA, Tius MA, Makriyannis A. Enantiomeric resolution of a novel chiral cannabinoid receptor ligand. J Biochem Biophys Methods. 2002;54:415–422. doi: 10.1016/s0165-022x(02)00144-6. [DOI] [PubMed] [Google Scholar]
- 17.Sharma R, Nikas SP, Paronis CA, Wood JT, Halikhedkar A, Guo JJ, Thakur GA, Kulkarni S, Benchama O, Raghav JG, Gifford RS, Jarbe TU, Bergman J, Makriyannis A. Controlled-deactivation cannabinergic ligands. J Med Chem. 2013;56:10142–10157. doi: 10.1021/jm4016075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thakur GA, Bajaj S, Paronis C, Peng Y, Bowman AL, Barak LS, Caron MG, Parrish D, Deschamps JR, Makriyannis A. Novel adamantyl cannabinoids as CB1 receptor probes. J Med Chem. 2013;56:3904–3921. doi: 10.1021/jm4000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dixon DD, Tius MA, Thakur GA, Zhou H, Bowman AL, Shukla VG, Peng Y, Makriyannis A. C3-heteroaroyl cannabinoids as photolabeling ligands for the CB2 cannabinoid receptor. Bioorg Med Chem Lett. 2012;22:5322–5325. doi: 10.1016/j.bmcl.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sink KS, Segovia KN, Nunes EJ, Collins LE, Vemuri VK, Thakur G, Makriyannis A, Salamone JD. Intracerebroventricular administration of cannabinoid CB1 receptor antagonists AM251 and AM4113 fails to alter food-reinforced behavior in rats. Psychopharmacology (Berl) 2009;206:223–232. doi: 10.1007/s00213-009-1602-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janero DR. Cannabinoid-1 receptor (CB1R) blockers as medicines: beyond obesity and cardiometabolic disorders to substance abuse/drug addiction with CB1R neutral antagonists. Expert Opin Emerging Drugs. 2012;17:17–29. doi: 10.1517/14728214.2012.660916. [DOI] [PubMed] [Google Scholar]
- 22.Kangas BD, Delatte MS, Vemuri VK, Thakur GA, Nikas SP, Subramanian KV, Shukla VG, Makriyannis A, Bergman J. Cannabinoid discrimination and antagonism by CB(1) neutral and inverse agonist antagonists. J Pharmacol Exp Ther. 2013;344:561–567. doi: 10.1124/jpet.112.201962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN55,212–2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A. 2005;102:19144–19149. doi: 10.1073/pnas.0509588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turu G, Hunyady L. Signal transduction of the CB1 cannabinoid receptor. J Mol Endocrinol. 2010;44:75–85. doi: 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- 26.Smith TH, Sim-Selley LJ, Selley DE. Cannabinoid CB1 receptor-interacting proteins: novel targets for central nervous system drug discovery? Br J Pharmacol. 2010;160:454–466. doi: 10.1111/j.1476-5381.2010.00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Howlett AC, Blume LC, Dalton GD. CB(1) cannabinoid receptors and their associated proteins. Curr Med Chem. 2010;17:1382–1393. doi: 10.2174/092986710790980023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G, Westwood P, Marrs J, Thomson F, Cowley P, Christopoulos A, Pertwee RG, Ross RA. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- 29.Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, Reynet C, Wong Kai In P. PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol. 2007;152:805–814. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pamplona FA, Ferreira J, Menezes de Lima O, Jr, Duarte FS, Bento AF, Forner S, Villarinho JG, Bellocchio L, Wotjak CT, Lerner R, Monory K, Lutz B, Canetti C, Matias I, Calixto JB, Marsicano G, Guimaraes MZ, Takahashi RN. Antiinflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proc Natl Acad Sci U S A. 2012;109:21134–21139. doi: 10.1073/pnas.1202906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vallee M, Vitiello S, Bellocchio L, Hebert-Chatelain E, Monlezun S, Martin-Garcia E, Kasanetz F, Baillie GL, Panin F, Cathala A, Roullot-Lacarriere V, Fabre S, Hurst DP, Lynch DL, Shore DM, Deroche-Gamonet V, Spampinato U, Revest JM, Maldonado R, Reggio PH, Ross RA, Marsicano G, Piazza PV. Pregnenolone can protect the brain from cannabis intoxication. Science. 2014;343:94–98. doi: 10.1126/science.1243985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bauer M, Chicca A, Tamborrini M, Eisen D, Lerner R, Lutz B, Poetz O, Pluschke G, Gertsch J. Identification and quantification of a new family of peptide endocannabinoids (Pepcans) showing negative allosteric modulation at CB1 receptors. J Biol Chem. 2012;287:36944–36967. doi: 10.1074/jbc.M112.382481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discovery. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- 35.Pertwee RG. Endocannabinoids and Their Pharmacological Actions. Handb Exp Pharmacol. 2015;231:1–37. doi: 10.1007/978-3-319-20825-1_1. [DOI] [PubMed] [Google Scholar]
- 36.Baillie GL, Horswill JG, Anavi-Goffer S, Reggio PH, Bolognini D, Abood ME, McAllister S, Strange PG, Stephens GJ, Pertwee RG, Ross RA. CB(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Mol Pharmacol. 2013;83:322–338. doi: 10.1124/mol.112.080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shore DM, Baillie GL, Hurst DH, Navas F, 3rd, Seltzman HH, Marcu JP, Abood ME, Ross RA, Reggio PH. Allosteric modulation of a cannabinoid G protein-coupled receptor: binding site elucidation and relationship to G protein signaling. J Biol Chem. 2014;289:5828–5845. doi: 10.1074/jbc.M113.478495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gamage TF, Ignatowska-Jankowska BM, Wiley JL, Abdelrahman M, Trembleau L, Greig IR, Thakur GA, Tichkule R, Poklis J, Ross RA, Pertwee RG, Lichtman AH. Invivo pharmacological evaluation of the CB1-receptor allosteric modulator Org-27569. Behav Pharmacol. 2014;25:182–185. doi: 10.1097/FBP.0000000000000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding Y, Qiu Y, Jing L, Thorn DA, Zhang Y, Li JX. Behavioral effects of the cannabinoid CB1 receptor allosteric modulator ORG27569 in rats. Pharmacol Res Perspect. 2014;2:e00069. doi: 10.1002/prp2.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fay JF, Farrens DL. A key agonist-induced conformational change in the cannabinoid receptor CB1 is blocked by the allosteric ligand Org 27569. J Biol Chem. 2012;287:33873–33882. doi: 10.1074/jbc.M112.352328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weichert D, Gmeiner P. Covalent Molecular Probes for Class A G Protein-Coupled Receptors: Advances and Applications. ACS Chem Biol. 2015;10:1376–1386. doi: 10.1021/acschembio.5b00070. [DOI] [PubMed] [Google Scholar]
- 42.Schreiber SL, Kotz JD, Li M, Aube J, Austin CP, Reed JC, Rosen H, White EL, Sklar LA, Lindsley CW, Alexander BR, Bittker JA, Clemons PA, de Souza A, Foley MA, Palmer M, Shamji AF, Wawer MJ, McManus O, Wu M, Zou B, Yu H, Golden JE, Schoenen FJ, Simeonov A, Jadhav A, Jackson MR, Pinkerton AB, Chung TD, Griffin PR, Cravatt BF, Hodder PS, Roush WR, Roberts E, Chung DH, Jonsson CB, Noah JW, Severson WE, Ananthan S, Edwards B, Oprea TI, Conn PJ, Hopkins CR, Wood MR, Stauffer SR, Emmitte KA. Advancing biological understanding and therapeutics discovery with small-molecule probes. Cell. 2015;161:1252–1265. doi: 10.1016/j.cell.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Picone RP, Khanolkar AD, Xu W, Ayotte LA, Thakur GA, Hurst DP, Abood ME, Reggio PH, Fournier DJ, Makriyannis A. (–)-7’-Isothiocyanato-11-hydroxy-1’,1’-dimethylheptylhexahydrocannabinol (AM841), a high-affinity electrophilic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor. Mol Pharmacol. 2005;68:1623–1635. doi: 10.1124/mol.105.014407. [DOI] [PubMed] [Google Scholar]
- 44.Pei Y, Mercier RW, Anday JK, Thakur GA, Zvonok AM, Hurst D, Reggio PH, Janero DR, Makriyannis A. Ligand-binding architecture of human CB2 cannabinoid receptor: evidence for receptor subtype-specific binding motif and modeling GPCR activation. Chem Biol. 2008;15:1207–1219. doi: 10.1016/j.chembiol.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szymanski DW, Papanastasiou M, Melchior K, Zvonok N, Mercier RW, Janero DR, Thakur GA, Cha S, Wu B, Karger B, Makriyannis A. Mass spectrometry-based proteomics of human cannabinoid receptor 2: covalent cysteine 6.47(257)-ligand interaction affording megagonist receptor activation. J Proteome Res. 2011;10:4789–4798. doi: 10.1021/pr2005583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janero DR, Yaddanapudi S, Zvonok N, Subramanian KV, Shukla VG, Stahl E, Zhou L, Hurst D, Wager-Miller J, Bohn LM, Reggio PH, Mackie K, Makriyannis A. Molecularinteraction and signaling profiles of AM3677, a novel covalent agonist selective for the cannabinoid 1 receptor. ACS Chem Neurosci. 2015;6(8):1400–1410. doi: 10.1021/acschemneuro.5b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kapur A, Samaniego P, Thakur GA, Makriyannis A, Abood ME. Mapping the structural requirements in the CB1 cannabinoid receptor transmembrane helix II for signal transduction. J Pharmacol Exp Ther. 2008;325:341–348. doi: 10.1124/jpet.107.133256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kulkarni PM, Kulkarni AR, Korde A, Tichkule RB, Laprairie RB, Denovan-Wright EM, Zhou H, Janero DR, Zvonok N, Makriyannis A, Cascio MG, Pertwee RG, Thakur GA. Novel electrophilic and photoaffinity covalent probes for mapping the cannabinoid 1 receptor allosteric site(s) J Med Chem. 2016;59(1):44–60. doi: 10.1021/acs.jmedchem.5b01303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discovery. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 50.Kalgutkar AS, Dalvie DK. Drug discovery for a new generation of covalent drugs. Expert Opin Drug Discovery. 2012;7:561–581. doi: 10.1517/17460441.2012.688744. [DOI] [PubMed] [Google Scholar]
- 51.Nussinov R, Tsai CJ. The design of covalent allosteric drugs. Annu Rev Pharmacol Toxicol. 2015;55:249–267. doi: 10.1146/annurev-pharmtox-010814-124401. [DOI] [PubMed] [Google Scholar]
- 52.Khajehali E, Malone DT, Glass M, Sexton PM, Christopoulos A, Leach K. Biased agonism and biased allosteric modulation at the CB1 cannabinoid receptor. Mol Pharmacol. 2015;88:368–379. doi: 10.1124/mol.115.099192. [DOI] [PubMed] [Google Scholar]
- 53.Cawston EE, Redmond WJ, Breen CM, Grimsey NL, Connor M, Glass M. Real-time characterization of cannabinoid receptor 1 (CB1) allosteric modulators reveals novel mechanism of action. Br J Pharmacol. 2013;170:893–907. doi: 10.1111/bph.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahn KH, Mahmoud MM, Shim JY, Kendall DA. Distinct roles of beta-arrestin 1 and beta-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1) J Biol Chem. 2013;288:9790–9800. doi: 10.1074/jbc.M112.438804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laprairie RB, Bagher AM, Kelly ME, Dupre DJ, Denovan-Wright EM. Type 1 cannabinoid receptor ligands display functional selectivity in a cell culture model of striatal medium spiny projection neurons. J Biol Chem. 2014;289:24845–24862. doi: 10.1074/jbc.M114.557025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daigle TL, Kearn CS, Mackie K. Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacology. 2008;54:36–44. doi: 10.1016/j.neuropharm.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abood ME. Allosteric modulators: a side door. J Med Chem. 2016;59:42–43. doi: 10.1021/acs.jmedchem.5b01824. [DOI] [PubMed] [Google Scholar]
- 58.Coutts AA, Anavi-Goffer S, Ross RA, MacEwan DJ, Mackie K, Pertwee RG, Irving AJ. Agonist-induced internalization and trafficking of cannabinoid CB1 receptors in hippocampal neurons. J Neurosci. 2001;21:2425–2433. doi: 10.1523/JNEUROSCI.21-07-02425.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- 60.Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc London, Ser B. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- 61.Laprairie RB, Bagher AM, Kelly ME, Denovan-Wright EM. Biased type 1 cannabinoid receptor signaling influences neuronal viability in a cell culture model of Huntington disease. Mol Pharmacol. 2016;89:364–375. doi: 10.1124/mol.115.101980. [DOI] [PubMed] [Google Scholar]
- 62.Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J Biol Chem. 2012;287:12070–12082. doi: 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rankovic Z, Brust TF, Bohn LM. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett. 2016;26:241–250. doi: 10.1016/j.bmcl.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luttrell LM, Maudsley S, Bohn LM. Fulfilling the promise of “biased” G protein-coupled receptor agonism. Mol Pharmacol. 2015;88:579–588. doi: 10.1124/mol.115.099630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- 66.Ehlert FJ, Suga H, Griffin MT. Quantifying agonist activity at G protein-coupled receptors. J Visualized Exp. 2011:e3179. doi: 10.3791/3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hurst DP, Grossfield A, Lynch DL, Feller S, Romo TD, Gawrisch K, Pitman MC, Reggio PH. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J Biol Chem. 2010;285:17954–17964. doi: 10.1074/jbc.M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Doorn JA, Petersen DR. Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem Res Toxicol. 2002;15:1445–1450. doi: 10.1021/tx025590o. [DOI] [PubMed] [Google Scholar]
- 69.Tahtaoui C, Balestre MN, Klotz P, Rognan D, Barberis C, Mouillac B, Hibert M. Identification of the binding sites of the SR49059 nonpeptide antagonist into the V1a vasopressin receptor using sulfydryl-reactive ligands and cysteine mutants as chemical sensors. J Biol Chem. 2003;278:40010–40019. doi: 10.1074/jbc.M301128200. [DOI] [PubMed] [Google Scholar]
- 70.Wu X, Zhou QH, Xu K. Are isothiocyanates potential anti-cancer drugs? Acta Pharmacol Sin. 2009;30:501–512. doi: 10.1038/aps.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bramblett RD, Panu AM, Ballesteros JA, Reggio PH. Construction of a 3D model of the cannabinoid CB1 receptor: determination of helix ends and helix orientation. Life Sci. 1995;56:1971–1982. doi: 10.1016/0024-3205(95)00178-9. [DOI] [PubMed] [Google Scholar]
- 72.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 73.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stornaiuolo M, Bruno A, Botta L, Regina GL, Cosconati S, Silvestri R, Marinelli L, Novellino E. Endogenous vs Exogenous Allosteric Modulators in GPCRs: A dispute for shuttling CB1 among different membrane microenvironments. Sci Rep. 2015;5:15453. doi: 10.1038/srep15453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marcu J, Shore DM, Kapur A, Trznadel M, Makriyannis A, Reggio PH, Abood ME. Novel insights into CB1 cannabinoid receptor signaling: a key interaction identified between the extracellular-3 loop and transmembrane helix 2. J Pharmacol Exp Ther. 2013;345:189–197. doi: 10.1124/jpet.112.201046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hurst D, Umejiego U, Lynch D, Seltzman H, Hyatt S, Roche M, McAllister S, Fleischer D, Kapur A, Abood M, Shi S, Jones J, Lewis D, Reggio P. Biarylpyrazole inverse agonists at the cannabinoid CB1 receptor: importance of the C-3 carboxamide oxygen/lysine3.28(192) interaction. J Med Chem. 2006;49:5969–5987. doi: 10.1021/jm060446b. [DOI] [PubMed] [Google Scholar]
- 77.Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci. 2014;35:308–316. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 78.Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discovery. 2013;12:630–644. doi: 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- 79.Gentry PR, Sexton PM, Christopoulos A. Novel allosteric modulators of G protein-coupled receptors. J Biol Chem. 2015;290:19478–19488. doi: 10.1074/jbc.R115.662759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In: Sealfon SC, Conn PM, editors. Methods in Neurosciences. Academic Press; San Diego, CA: 1995. pp. 366–428. [Google Scholar]
- 81.Blume LC, Bass CE, Childers SR, Dalton GD, Roberts DC, Richardson JM, Xiao R, Selley DE, Howlett AC. Striatal CB1 and D2 receptors regulate expression of each other, CRIP1A and delta opioid systems. J Neurochem. 2013;124:808–820. doi: 10.1111/jnc.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 83.Bagher AM, Laprairie RB, Kelly ME, Denovan-Wright EM. Co-expression of the human cannabinoid receptor coding region splice variants (hCB(1)) affects the function of hCB(1) receptor complexes. Eur J Pharmacol. 2013;721:341–354. doi: 10.1016/j.ejphar.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 84.James JR, Oliveira MI, Carmo AM, Iaboni A, Davis SJ. A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat Methods. 2006;3:1001–1006. doi: 10.1038/nmeth978. [DOI] [PubMed] [Google Scholar]
- 85.Ross RA, Gibson TM, Stevenson LA, Saha B, Crocker P, Razdan RK, Pertwee RG. Structural determinants of the partial agonist-inverse agonist properties of 6’-azidohex-2’-yne-delta8-tetrahydrocannabinol at cannabinoid receptors. Br J Pharmacol. 1999;128:735–743. doi: 10.1038/sj.bjp.0702836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Griffin MT, Figueroa KW, Liller S, Ehlert FJ. Estimation of agonist activity at G protein-coupled receptors: analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. J Pharmacol Exp Ther. 2007;321:1193–1207. doi: 10.1124/jpet.107.120857. [DOI] [PubMed] [Google Scholar]
- 87.Lomize AL, Pogozheva ID, Lomize MA, Mosberg HI. Positioning of proteins in membranes: a computational approach. Protein Sci. 2006;15:1318–1333. doi: 10.1110/ps.062126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grossfield A, Feller SE, Pitman MC. A role for direct interactions in the modulation of rhodopsin by omega-3 polyunsaturated lipids. Proc Natl Acad Sci U S A. 2006;103:4888–4893. doi: 10.1073/pnas.0508352103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, III, Roux B, Schlenkrich M, Smith JC, Stote R, RStraub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-hydrogen empirical potential for molecular modeling and dynamics studies of proteins using the CHARMM22 force field. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 90.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 92.Case DA, Darden TA, Cheatham ITE, Simmerling CL, Wang J, Duke RE. AMBER12 (version 12.3) University of California San Francisco; San Francisco: 2012. [Google Scholar]
- 93.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Salomon-Ferrer R, Goetz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER - Part II: Particle Mesh Ewald. J Chem Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 95.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 96.Vanommeslaeghe K, Raman EP, MacKerell AD., Jr Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J Chem Inf Model. 2012;52:3155–3168. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.