

Abstract
Introduction
Epidemiologic studies have demonstrated an association between diabetes and dementia. Insulin signaling within the brain, in particular within the hypothalamus regulates carbohydrate, lipid, and branched chain amino acid (BCAA) metabolism in peripheral organs such as the liver and adipose tissue. We hypothesized that cerebral amyloidosis impairs central nervous system control of metabolism through disruption of insulin signaling in the hypothalamus, which dysregulates glucose and BCAA homeostasis resulting in increased susceptibility to diabetes.
Methods
We examined whether APP/PS1 mice exhibit increased susceptibility to aging or high-fat diet (HFD)-induced metabolic impairment using metabolic phenotyping and insulin-signaling studies.
Results
APP/PS1 mice were more susceptible to high-fat feeding and aging-induced metabolic dysregulation including disrupted BCAA homeostasis and exhibited impaired hypothalamic insulin signaling.
Discussion
Our data suggest that AD pathology increases susceptibility to diabetes due to impaired hypothalamic insulin signaling, and that plasma BCAA levels could serve as a biomarker of hypothalamic insulin action in patients with AD.
Keywords: Diabetes, Alzheimer disease, Insulin signaling, Branched chain amino acids, Glucose
1. Introduction
Alzheimer’s disease (AD) is the most common progressive neurodegenerative disorder causing dementia in the elderly. Type 2 diabetes (T2D) is a complex metabolic disorder that is characterized by glucotoxicity and lipotoxicity and a pathognomonic defect in T2D is insulin resistance (IR). There have been a number of epidemiologic and clinical studies demonstrating that T2D/IR is associated with an increased risk of AD and vice versa [1–4]. The link between T2D/IR and AD is also supported by animal studies demonstrating that interventions to induce a prediabetic state such as crossing AD mice with leptin receptor deficient (db/db) mice [5] or environmental challenges such as high-fat feeding [6] exacerbate AD pathology.
The prevailing paradigm for this association is that T2D/IR increases the risk of AD through direct effects of neuronal insulin signaling as well as through indirect effects of a glucotoxic and lipotoxic microenvironment and/or detrimental effects of diabetes on the vasculature [3]. In addition to genetic susceptibility, environmental factors, in particular over-nutrition, increase the predisposition to T2D and the risk for dementia [7]. Of note, aging is a risk factor for both AD and T2D, the latter through a worsening of insulin sensitivity.
AD and T2D have been shown to share several pathophysiological features, including impaired cognitive function, oxidative and inflammatory stress, vascular dysfunction, and amyloid accumulation further illustrating the common pathophysiological themes underlying these aging-related diseases [2,8]. It is commonly believed that T2D/IR is a risk factor for AD primarily based on (1) the idea that hyperinsulinemia promotes competition of insulin degrading enzyme between degrading amyloid or insulin [9,10] and (2) the notion that a failure of insulin to inhibit glycogen synthase kinase 3 (GSK-3) results in GSK3 overactivity [11] and promotes tau hyperphosphorylation and amyloid production [12]. Although this association has been commonly interpreted as T2D/IR being a predisposing factor for AD, the reverse hypothesis—i.e., that AD causes T2D/IR—has not been adequately investigated.
Brain insulin signaling, particularly in the hypothalamus, controls several metabolic pathways in peripheral organs. Hypothalamic insulin signaling controls food intake [13], hepatic glucose production [14], adipose tissue lipolysis, and de novo lipogenesis [15] and branched-chain amino acids (BCAAs) catabolism [16]. Animal models have established that the experimental induction of brain insulin resistance can induce whole body insulin resistance, that is, a pre-diabetic state illustrating the importance of hypothalamic insulin action in metabolic homeostasis.
The role of hypothalamic insulin signaling is much more difficult to study in humans, but the fact that intranasal insulin, which appears to deliver insulin directly to the central nervous system (CNS) and may bypass decreased insulin transport from the circulation via the blood brain barrier to the brain improves memory in patients with AD [17,18] supports the concept that enhancement of brain insulin signaling is beneficial in AD. There is also a report demonstrating that insulin signaling is reduced in cerebral cortex slices derived postmortem from AD patients [19]—including AD patients without any previous clinical evidence for T2D. Hence, we speculated that AD pathology might lead to hypothalamic insulin resistance, which, in turn, increases the susceptibility to T2D/IR.
BCAA levels have been observed to be elevated in obese and diabetic individuals and appear to be one of the earliest harbingers for the future risk of diabetes. As brain insulin signaling is a regulator of hepatic BCAA catabolism [16], these data suggest that impaired brain insulin action is one of the earliest pathogenic defects in T2D. In turn, increasing plasma BCAA levels affects cognitive function such as spatial memory [20,21], possibly by reducing the expression of nerve growth factor in the hippocampus of rats [22] or via overstimulation of N-methyl-D-aspartate (NMDA) receptors [23]. Of note, mouse models with elevated BCAA levels displayed hippocampus-selective retromer deficiency which has been implicated in late onset AD [24]. The retromer is also involved in sorting of SorCS1, a gene implicated in both T2D and AD [25]. These data suggest that impaired brain insulin signaling in AD could result in elevated plasma BCAA levels that may be an attendant to, or a driver of, AD progression.
One popular mouse model of AD is the APP/PS1 mouse that carries the APP Swedish mutation at K670 N/M671 L and the presenilin-1Δexon9 mutation (APP/PS1) causing amyloid deposits by 6 and 7 months, and abundant plaque formation by 9 months of age. Studies assessing glucose metabolism in APP/PS1 mice have described similar glucose tolerance in mice that were young and fed a standard diet [5,26]. Other studies, however, have reported impaired glucose tolerance in young APP/PS1 mice that was exacerbated with high fat feeding [27]. Some evidence suggests that impaired brain insulin signaling precedes AD pathology in these mice [28]. However, the authors of that report only assessed the nonstimulated phosphorylation state of signaling molecules such as Akt and GSK 3. It is important to point out that these signaling molecules also participate in other signaling pathways such as cytokine signaling and it is impossible to ascribe a decrease in the basal phosphorylation state to decreased insulin signaling versus that of other signaling pathways. The formal way to study insulin signaling is to acutely induce insulin signaling through an insulin bolus and then compare the insulin-induced phosphorylation state of downstream molecules such as Akt and GSK 3 which requires comparison to the basal phosphorylation state in nonstimulated animals. Only then can the degree of activation of downstream molecules be specifically attributed to insulin signaling. Others have also demonstrated impaired insulin signaling in brains from APP/PS1 mice [27]; however, whole brain lysates were used for these earlier studies, and therefore, it is unclear whether insulin signaling is impaired specifically in the hypothalamus. In the present study, we have hypothesized that AD (i.e., amyloid pathology) in APP/PS1 mice impairs hypothalamic insulin signaling, disrupting the CNS control of glucose, lipid, and BCAA homeostasis and thereby increasing the susceptibility to diabetes.
2. Methods
2.1. Animals
Male APP/PS1 mice (5–6 and 11–12 months old) were used for these experiments as explained in Supplementary Information.
2.2. Glucose and insulin tolerance testing
Mice were fasted for either 5 hours or overnight and acclimated to the testing room with access to water. Animals were injected intraperitoneally (i.p) with a 1–2 g/kg glucose solution (Sigma Aldrich, St. Louis, MO) at the age of 5 to 11 months. The insulin tolerance test was performed with 0.75, 1, or 2 U/kg insulin (Humulin R, Lilly & Co., Indianapolis, IN) solution. Blood samples obtained from tail vein bleeds were used to measure blood glucose immediately before and after the glucose or insulin bolus as detailed in Supplementary Material.
2.3. Fasting-refeeding protocol
Mice were fasted for 24 hours and then refed via gavage with 300 μL (0.33 kcal) of an Ensure (Abbott Laboratories, USA) solution containing 1.1 kcal/mL with 20% of calories from fat, 13% carbohydrate, and 18% protein. Blood samples were collected after fasting and 1 and 2 hours after refeeding to measure blood glucose, insulin, triglycerides (TG), nonesterified fatty acids (NEFA), and branched chain amino acids (BCAA).
2.4. Insulin signaling studies
Insulin signaling studies were performed immediately after the insulin tolerance test by injecting mice at 90 min intraperitoneally with 100 mU of insulin in 300 μL of 5% glucose solution after which organs were harvested as detailed in the Supplementary Material.
2.5. Plasma insulin, lipids, and BCAA determinations
Plasma insulin, lipids, and BCAA levels were determined as described in Supplementary Material.
2.6. Statistics
Data were analyzed with either a two-tailed student t test or analysis of variance using GraphPad Prism (GraphPad Software, CA, USA) when appropriate. Values are displayed as group means ± SEM. A P < .05 was determined a priori as the threshold for statistically significant differences.
3. Results
Type 2 diabetes in humans is thought to emerge from a confluence of factors increasing susceptibility to insulin resistance such as genetic predisposition and environmental challenges such as excessive caloric intake due to high-fat diets (HFDs) for example. We therefore probed whether APP/PS1 mice were more susceptible to high-fat diet-induced and/or aging-induced metabolic dysregulation. To this end, we studied hypothalamic insulin signaling, glucose, lipid, and BCAA homeostasis in 5-month-old mice which approximately equates to 30-year-old humans where T2D/IR incidence is low and 11-month-old mice which approximately equates to 55-year-old humans where T2D/IR incidence peaks, before and after high fat feeding were 60% of all calories are provided in the form of fat (see experimental scheme in Fig. 1A and B).
Fig. 1.
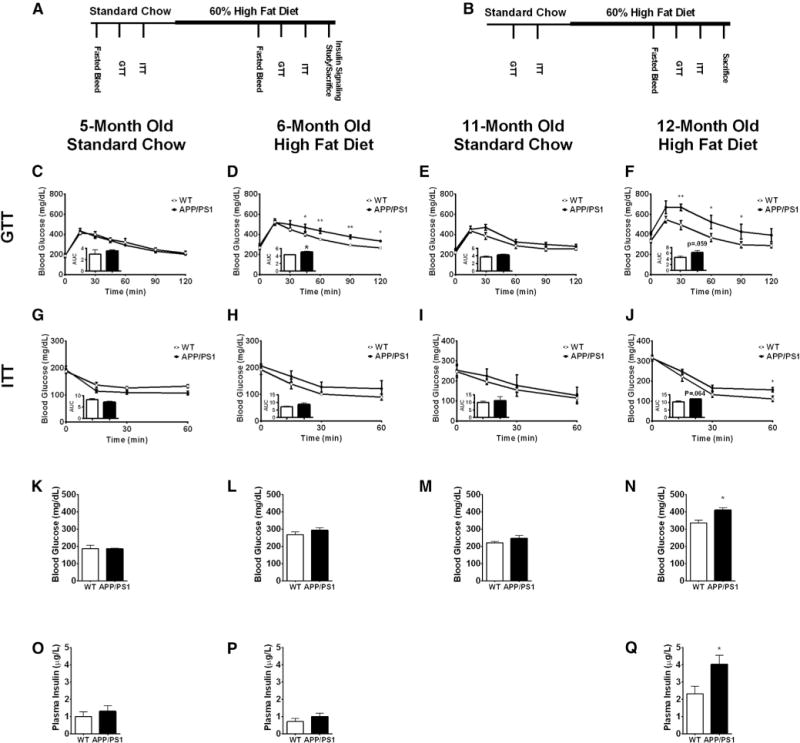
APP/PS1 mice are more susceptible to high-fat diet (HFD)–induced glucose intolerance, which is exacerbated with age. Experimental scheme for the two cohorts of (A) young (5 months) and (B) old (11 months) mice. Glucose tolerance test for young mice fed a standard chow (C) and a HFD (D) and old mice fed a standard chow (E) and a HFD (F). Insulin tolerance test for young mice fed a standard chow (G) and a HFD (H) and old mice fed a standard chow (I) and a HFD (J). Fasting blood glucose concentration for young mice fed a standard chow (K) and during high-fat feeding (L) and old mice fed a standard chow (M) and during high-fat feeding (N). Fasting insulin concentration for young mice fed a standard chow (O) and during high-fat feeding (P) and old mice during high-fat feeding (Q). Displayed are averages ± SEM. Statistical significance is defined as *P < .05, **P < .01. Wt n = 5 and APP/PS1 n = 4.
We dynamically assessed glucose homeostasis by performing a glucose tolerance test (GTT) through the administration of an i.p glucose bolus to 5- and 11-month old mice before and after high-fat feeding. When fed a standard chow diet, 5-month-old APP/PS1 mice exhibited similar glucose excursions to Wt littermates (Fig. 1C). However, when these animals were challenged with a HFD for 4 weeks, APP/PS1 mice became glucose intolerant as evident from the significantly higher glucose excursion and area under the curve during the GTT (Fig. 1D). We also studied glucose tolerance in 11-month-old mice to determine whether aging, a well-known risk factor for both T2D and AD, differentially affects glucose homeostasis in APP/PS1 mice. When fed a standard chow, old APP/PS1 mice exhibited similar blood glucose excursions to Wt mice as reflected by the comparable areas under the curve (Fig. 1E). After high fat feeding, however, old APP/PS1 mice were markedly more glucose intolerant than Wt littermates (Fig. 1F).
To determine whether the observed glucose intolerance in APP/PS1 mice is due to impaired insulin action, we performed an insulin tolerance test (ITT) by administering an i.p. insulin bolus to 5- and 11-month-old mice before and after high-fat feeding. Five-month-old APP/PS1 mice demonstrated similar decreases in blood glucose during the ITT when fed a standard chow (Fig. 1G) and after 4 weeks of high-fat feeding (Fig. 1H) suggesting that insulin sensitivity was still maintained in young APP/PS1 mice. Similarly, 11-month-old APP/PS1 mice fed a standard chow diet exhibited comparable insulin tolerance, albeit with a trend to perform worse in the APP/PS1 mice (Fig. 1I). However, when fed a HFD for 6-weeks, old APP/PS1 mice exhibited worse insulin tolerance which became statistically significant at time point 60 minutes and was also reflected in the larger area under the curve (Fig. 1J), although it did not reach statistical significance (P = .064). Taken together, these findings suggest that HFD challenge and aging unveil a metabolic phenotype in APP/PS1 mice. This was also reflected in the fasting glucose levels (Fig. 1K–N) and the fasting insulin levels (Fig. 1O–Q) that were significantly worse in the aged and HFD fed APP/PS1 mice compared to the Wt littermates.
We next probed the ability of APP1/PS1 mice to maintain metabolic homeostasis when transitioning from the fasted to the fed state, which is commonly impaired in the pre-diabetic and diabetic state and is a sign of metabolic inflexibility. We compared 5-month-old overnight fasted mice to mice refed after an overnight fast in their ability to regulate postprandial glucose, insulin, plasma lipids, and BCAAs (see experimental scheme in Fig. 2A). After refeeding, APP/PS1 mice exhibited similar glucose excursions to that of Wt mice (Fig. 2B) which is in line with the GTT results indicating that young APP/PS1 mice are able to maintain glucose homeostasis. Of note, postprandial insulin levels remained elevated in APP/PS1 mice 2 hours after refeeding (Fig. 2C), which is indicative of insulin resistance, although the glucose excursions were not different. APP/PS1 mice also exhibited higher triglyceride excursions after refeeding (Fig. 2F), whereas postprandial plasma glycerol and NEFA levels were not different (Fig. 2D and 2E). Taken together, these observations further support the notion that young APP/PS1 exhibit a very discrete metabolic phenotype that is exacerbated by nutritional challenges and/or aging.
Fig. 2.
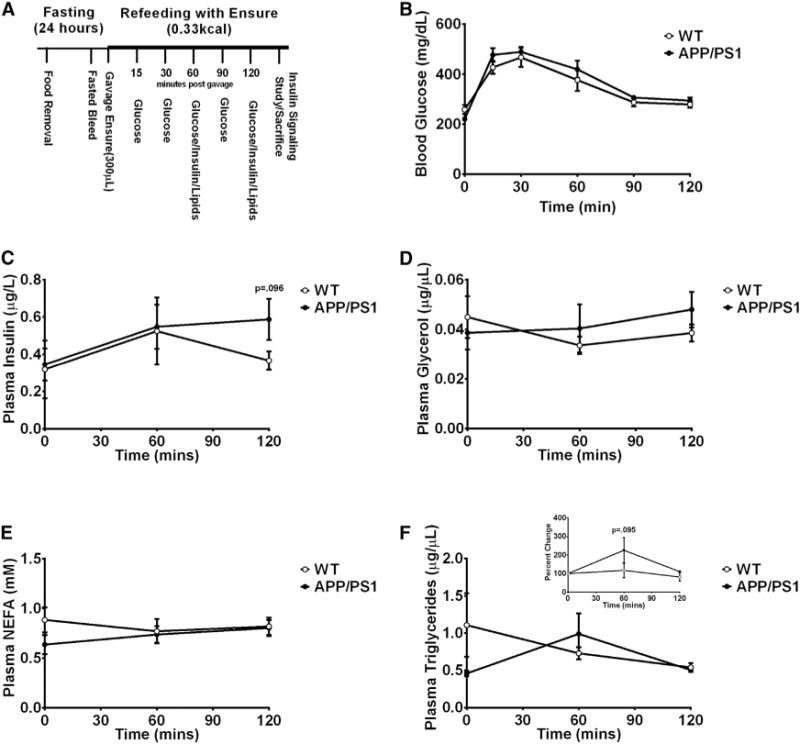
APP/PS1 mice exhibit impaired postprandial insulin and triglyceride levels. Experimental scheme for the fasting and refeeding protocol (A). Glucose excursion before and after refeeding with an Ensure solution (B). Plasma insulin (C), glycerol (D), non-esterified free fatty acids (E), and triglycerides (F) concentrations during the fasting-refeeding protocol. Displayed are averages ± SEM. Wt n = 6 and APP/PS1 n = 6.
The metabolic impairment observed in APP/PS1 mice could be due to altered energy homeostasis. To address this possibility, we measured food intake and body weight from 5- and 11-month-old mice before and/or after high-fat feeding. At the age of 5 months, there was no difference in average caloric intake between APP/PS1 and Wt mice (Fig. 3A). Although the total calories consumed increased during high-fat feeding, there was no difference in caloric intake between genotypes (Fig. 3B). APP/PS1 mice aged 11 months fed a HFD exhibited a tendency toward greater caloric intake (Fig. 3C), but this was not statistically different (P =.081). Body weight was not different between 5-month old APP/PS1 and Wt mice when fed a regular chow (Fig. 3D) or a HFD (Fig. 3E) which is in line with similar caloric intake in these mice. Similarly, 11-month-old APP/PS1 mice did not differ in body weight from Wt mice when fed a standard chow (Fig. 3F) or a HFD (Fig. 3G). Furthermore, we measured body composition in old APP/PS1 as these mice showed a tendency toward higher caloric intake and found that fat mass and lean mass were also not different before or after high-fat feeding (Supplementary Fig. 1). Thus, food intake or adiposity fails to explain the observed metabolic impairment in APP/PS1 mice.
Fig. 3.
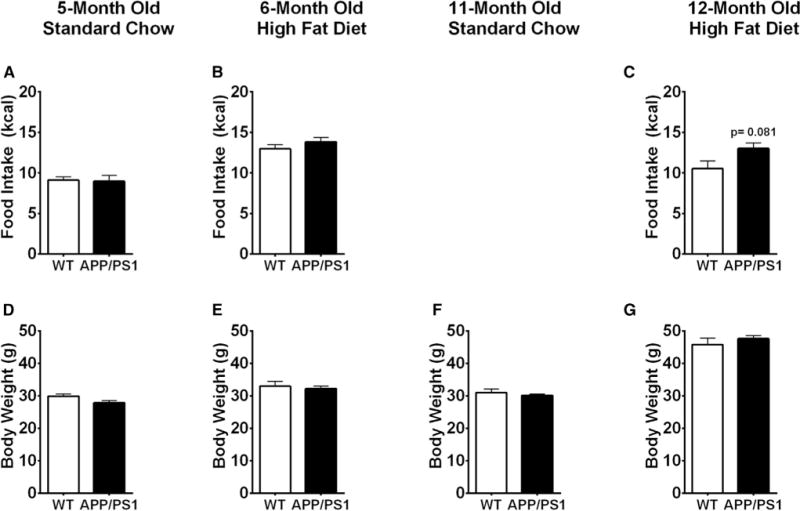
Food intake and body weight were not different between APP/PS1 and Wt mice. Averaged daily caloric intake for young mice fed a standard chow (A) and during high-fat feeding (B) and for old mice during high-fat feeding (C). Averaged body weight for young mice fed a standard chow (D) and HFD (E) and old mice fed a standard chow (F) and a HFD (G). Displayed are averages ± SEM. Wt n = 5 and APP/PS1 n = 4.
Altered lipid homeostasis can give rise to lipotoxicity, a key feature of the metabolic syndrome and T2D. We tested whether APP/PS1 mice exhibit alterations in circulating lipids by measuring fasting plasma glycerol, NEFA and triglycerides (TG) levels. Plasma glycerol levels were unaltered in 5-month-old APP/PS1 mice fed a standard chow (Fig. 4A), after high fat feeding (Fig. 4B) or after high-fat feeding in old APP/PS1 mice (Fig. 4C). Plasma NEFA levels were also not different in 5-month old mice fed a standard chow (Fig. 4D), after high-fat feeding (Fig. 4E) or after high-fat feeding in old APP/PS1 mice (Fig. 4F). Similar to glycerol and NEFA, plasma triglycerides were also not different between any of the groups (Fig. 4G–I). Thus, APP/PS1 mice are still able to maintain lipid homeostasis.
Fig. 4.
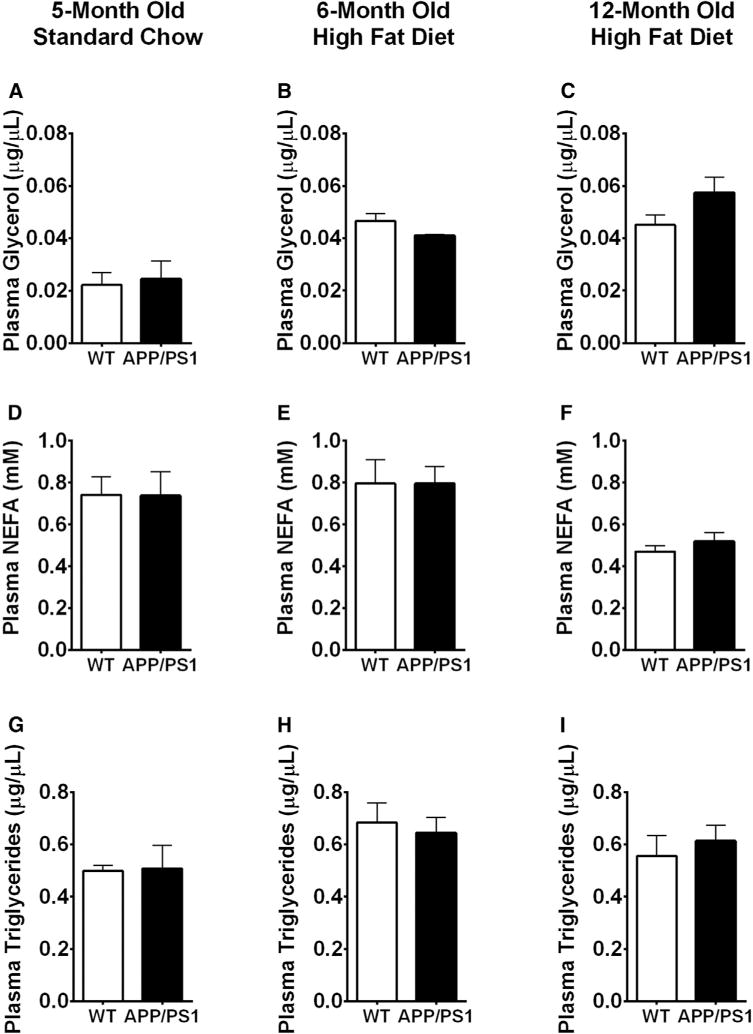
Plasma lipid parameters were not different between APP/PS1 and Wt mice. Plasma glycerol concentration for young mice fed a standard chow (A) and high-fat diet (HFD) (B) and old mice fed a HFD (C). Plasma nonesterified free fatty acids for young mice fed a standard chow (D) and high fat diet (E) and old mice fed a HFD (F). Plasma triglycerides concentrations for young mice fed a standard chow (G) and HFD (H) and old mice fed a HFD (I). Displayed are averages ± SEM. Wt n = 5 and APP/PS1 n = 4.
Hypothalamic insulin signaling is an important regulator of CNS control of nutrient partitioning and a disruption of hypothalamic insulin signaling could account for the dysmetabolic phenotype of the APP/PS1 mice. Hence we probed hypothalamic insulin signaling in 5-month old APP/PS1 mice after fasting-refeeding (standard chow) or high fat feeding, by administering an i.p. bolus of insulin and harvesting the hypothalami 15 minutes later. Western blot analysis of hypothalamic protein extracts revealed that APP/PS1 mice had decreased induction of GSK-3 and AKT phosphorylation over basal activation (Fig. 5A) and hence exhibited impaired hypothalamic insulin signaling. Thus, hypothalamic insulin resistance likely contributes to the impaired glucose homeostasis observed in these mice.
Fig. 5.
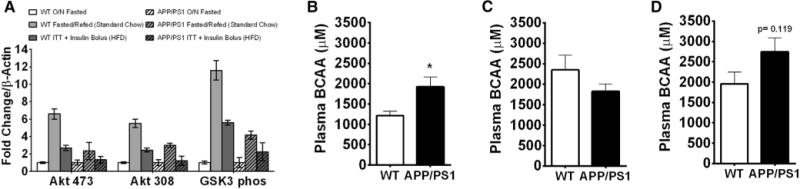
APP/PS1 mice exhibit hypothalamic insulin resistance and impaired BCAAs homeostasis. Western blot analysis of hypothalamic protein extracts from 5- to 6-month-old mice after the fasting-refeeding protocol (standard chow) and after high-fat feeding for 4 weeks (ITT plus insulin bolus) (A). Plasma BCAA concentration for young mice fed a standard chow diet (B) and after high-fat feeding (C) and old mice after high-fat feeding (D). Displayed are averages ± SEM. significance is defined as *P < .05. Wt n = 5 and APP/PS1 n = 4. Abbreviation: ITT, insulin tolerance test.
Plasma levels of branch chain amino acids (BCAAs) are commonly elevated in obese, prediabetic, and/or diabetic humans, rise very early in the course of diabetes development and are a sensitive predictor for the future risk for diabetes [29]. Our laboratory has recently demonstrated that brain insulin action induces BCAA catabolism, whereas inhibition of hypothalamic insulin signaling results in higher levels of circulating BCAAs [16]. Because hypothalamic insulin signaling was reduced in APP/PS1 mice, we assessed BCAA homeostasis. Indeed, 5-month-old APP/PS1 mice fed a regular chow diet exhibited significantly higher levels of circulating BCAAs (Fig. 5B). High-fat feeding induces hypothalamic insulin resistance, and hence, BCAA levels were higher in Wt mice compared to standard chow fed Wt mice. However, BCAA levels were not further elevated by high-fat feeding (Fig. 5C). Although BCAA levels trended to be higher in aged APP/PS1 mice, this did not reach significance (Fig. 5D) possibly due to hypothalamic insulin resistance preceding the high-fat feeding and/or the more pronounced systemic hyperinsulinemia of the APP/PS1 mice.
There are several molecular pathways that can impair insulin signaling within the CNS. Accumulation of misfolded proteins induces the unfolded protein response leading to endoplasmic reticulum (ER) stress which has been observed in the frontal cortex of AD patients [30,31] and in the brains of rodent models of T2D [32,33]. Of note, ER stress may also play an important role in hypothalamic dysfunction in obesity and diabetes [34]. Chronic and/or excessive ER stress also contributes to the activation of autophagy pathways [35] and inflammation pathways [36], and brain inflammation is implicated in both AD [37] and diabetes [38].
To determine whether APP/PS1 mice displayed an altered cortical ER stress response, we dissected the prefrontal cortex and measured mRNA expression of genes involved in ER stress including C/EBP homologous protein (CHOP), Binding immunoglobulin protein (BiP/GRP-78), and activating transcription factors (ATF), ATF3, ATF4, and ATF6. We found that 12-month-old APP/PS1 mice fed a HFD tended toward higher expression levels of (1) the ER molecular chaperone Bip/GRP-78 (a protein that plays a key role in initiating the unfolded protein response [39]); (2) the DNA damage induced transcript CHOP; (3) the CHOP-regulated protein ATF3 [40]; and (4) ATF4, a protein that is essential for the induction of genes involved in autophagy [41] (Fig. 6A). These observations suggest that 12-month-old APP/PS1 mice display a molecular signature consistent with the development of a state of increased ER stress and autophagy in the frontal cortex.
Fig. 6.
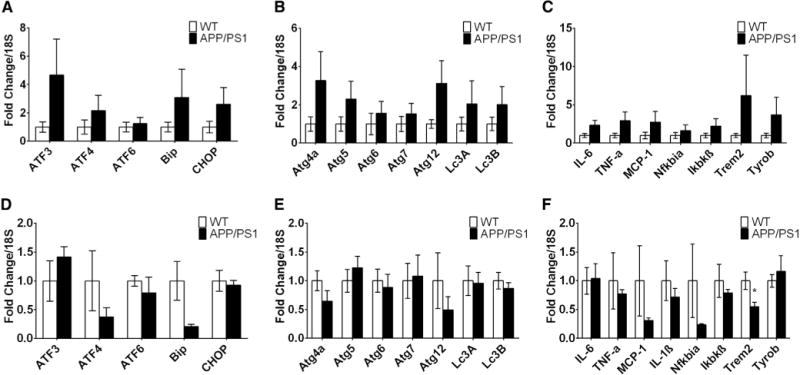
Old APP/PS1 mice exhibit greater expression of genes involved in ER stress, autophagy, and inflammation in the frontal cortex after high-fat feeding. Expression of genes involved in ER stress, autophagy, and inflammation pathways from frontal cortex samples of old (A, B, and C, respectively) and young (D, E, and F, respectively) mice fed a high-fat diet. Displayed are averages ± SEM. Wt n = 5 and APP/PS1 n = 3–4.
As the prefrontal cortex of old APP/PS1 mice exhibited elevated mRNA levels of genes involved in ER stress and in autophagy as demonstrated by the higher expression of ATF4, we measured expression levels of other genes involved in autophagy such as the autophagy-related protein (Atg) 4a, Atg5, Atg6, Atg 7, Atg12 and light chain 3 (LC3A and LCB) that are important cytoplasmic effectors of SIRT1-driven autophagy. SIRT1 is a member of the sirtuin family of deacetylases that regulates life span and nutritional state-dependent autophagy [42]. Consistent with the increased expression of ATF4, mRNA levels for all autophagy-related genes trended toward elevation in old APP/PS1 mice (Fig. 6B), suggestive of increased cellular degradation, but these differences were not statistically significant.
Based on the observation that 12-month-old APP/PS1 mice show increased expression of genes involved in ER stress and autophagy, we assessed the levels of several proinflammatory cytokines and found that old APP/PS1 mice tended to have increased mRNA expression of IL-1β, TNF-α, MCP-1, and SOCS3 (a negative regulator of cytokine signaling) and displayed a higher expression of IKBKβ which is essential for NF-κβ inflammatory signaling (Fig. 6C). Of note, APP/PS1 mice also tended to exhibit elevated expression of Trem2 and Tyrobp which play key roles in microglial-mediated immune response in postmortem brains of AD patients [43]. Thus, aged APP/PS1 mice fed a HFD tended to have higher expression of genes involved in ER signaling, autophagy, and inflammation in the prefrontal cortex, potentially exacerbating AD pathology, impairing insulin signaling, and inducing metabolic pathology.
To test whether the observed induction of ER stress, inflammation, and autophagy pathways in 12-month old high fat diet-fed APP/PS1 mice was likely to be a cause or a consequence of the AD neuropathology, we analyzed prefrontal cortex samples from 6-month-old APP/PS1 mice fed a HFD because young APP/PS1 mice are asymptomatic and as of yet have no obvious AD neuropathology [44]. In contrast to old mice, young APP/PS1 mice tended to have lower expression of genes involved in ER stress (Fig. 6D), autophagy (Fig. 6E), and inflammation (Fig. 6F) in the prefrontal cortex. Hence, it appears more likely that ER stress, autophagy, and inflammation in the brain result from AD neuropathology progression and that activation of these pathways is unlikely to be a driver of AD-associated metabolic dysregulation.
4. Discussion
Our aging societies are faced with an increased prevalence of both T2D/IR and AD. Aging is clearly a risk factor for both of these conditions and AD increases the prevalence for T2D/IR and vice versa. Here, we tested the hypothesis that in a commonly used mouse model of AD, the APP/PS1 mice, AD pathology can induce brain insulin resistance which in turn disrupts metabolic homeostasis and increases susceptibility to T2D/IR. Our data demonstrate that HFD feeding and/or aging unveils a metabolic phenotype of the APP/PS1 mice and hence increased susceptibility to diabetes. This is likely due to impaired hypothalamic insulin signaling in APP/PS1 mice which is also reflected in impaired BCAA homeostasis.
When fed a standard chow, APP/PS1 and Wt mice exhibited similar metabolic control except for very discrete abnormalities in APP/PS1 mice. Although APP/PS1 mice exhibited normal glucose excursions during the fasting-refeeding protocol, these mice were unable to maintain postprandial plasma triglyceride and insulin levels suggesting a predisposition to metabolic dysregulation. Indeed, after a high fat diet metabolic challenge, both young and old APP/PS1 mice demonstrated impaired glucose homeostasis which was more pronounced in the old cohort indicating that aging exacerbates the metabolic phenotype of AD mice. The metabolic impairment seen in APP/PS1 mice was not due to greater food intake, body weight, or adiposity as all these measures were not different from Wt controls. Hypothalamic insulin signaling is critical for the regulation of hepatic glucose production [45] and BCAA catabolism [16]. To the best of our knowledge, this study is the first to report that APP/PS1 mice exhibit hypothalamic insulin resistance, which we speculate accounts for the impaired glucose and BCAA homeostasis. Of note, hypothalamic insulin resistance in APP/PS1 mice was observed even before a HFD challenge (Fig. 5A) when the metabolic impairment in these mice still was very discrete. In line with the notion that hypothalamic insulin resistance accounts for impaired BCAA homeostasis, APP/PS1 mice exhibited significantly higher circulating BCAA levels.
High-fat feeding impaired BCAA homeostasis in Wt mice, whereas it did not further exacerbated this in APP/PS1 mice. One may speculate that in the presence of preexisting hypothalamic insulin resistance of the APP/PS1 mice, high-fat feeding does not further worsen BCAA homeostasis. It is important to note that while BCAA levels are consistently higher in insulin resistant humans, insulin resistance in rodents is not always associated with increased plasma BCAA levels. For example, BCAAs are increased in genetic forms of obesity, but not HFD fed mice, possibly due to the hyperinsulinemia which is able to maintain some metabolic parameters within the normal range such as BCAAs and triglycerides, but not others such as glucose [46]. Hence, the compensatory hyperinsulinemia is able to maintain nutrient homeostasis in younger, but not in older animals which is in line with our finding that BCAA levels are slightly increased after high fat feeding in old APP/PS1 mice.
Central nervous system inflammation is considered a primary driver of brain insulin resistance in AD [47]. Likewise, ER stress and autophagy have been demonstrated to impair insulin signaling within the CNS, at least in several studies of mouse model of diabetes [48]. As we used the hypothalami for immunoblotting in our insulin signaling studies, we were unable to assess ER stress and autophagy via mRNA analysis in the hypothalamus. Hence, we assessed ER stress, autophagy, and inflammation in the frontal cortex, a region well known to be affected in AD patients and APP/PS1 mice and to correlate with AD neuropathology [49]. We observed that 12-month-old APP/PS1 mice fed a HFD exhibited a molecular signature consistent with a state of increased ER stress, autophagy, and inflammation (Fig. 6A–C). Yet, if indeed ER stress, autophagy, and/or inflammation are primary drivers of impaired CNS control of metabolism, one would expect these pathways to be activated before metabolic regulation is impaired and/or before AD pathology is manifested. However, when we assessed 6-month-old APP/PS1 mice where a discrete metabolic impairment is already present, we failed to observe elevated mRNA levels of genes indicative of ER stress, autophagy, and/or inflammation. Thus, our data suggest that these pathways are more likely to be activated in response to AD pathology and hence are likely not primary drivers of the metabolic impairment.
Future studies should test whether brain insulin action is impaired by combining glucose clamp studies and isotope tracer dilution techniques with intracerebroventricular insulin infusion. This protocol will permit to test whether the amyloid pathology of APP/PS1 mice has any impact on the ability of brain insulin signaling to suppress hepatic glucose production and adipose lipolysis [15]. We propose that the brain amyloid pathology causes this disturbance in hypothalamic insulin signaling, and that the glucose intolerance (at least in this AD model) occurs as a result of impaired hypothalamic insulin action given its important role in controlling hepatic glucose metabolism [14]. Furthermore, as BCAAs can directly affect energy homeostasis by activating hypothalamic mammalian Target of Rapamycin (mTOR) signaling which in turn can induce or worsen hypothalamic insulin resistance [50], it would be informative to assess whether elevated BCAAs in AD mice contribute to insulin resistance by inducing hypothalamic mTOR signaling.
In summary, this study shows that APP/PS1 mice exhibit an increased susceptibility to metabolic impairment due to high-fat feeding and/or aging. This increased susceptibility to diabetes is likely due to hypothalamic insulin resistance. Furthermore, the hypothalamic insulin resistance of the APP/PS1 mice was reflected in impaired BCAA homeostasis. This observation may be of clinical relevance as it suggests that plasma BCAAs may serve as a marker for impaired brain insulin action in patients with AD.
Supplementary Material
RESEARCH IN CONTEXT.
Systemic review: To advance our understanding of the relationship between Alzheimer’s disease (AD) and diabetes, we performed an extensive Pubmed literature search to identify studies of animal models of AD that examined metabolism and insulin signaling.
Interpretation: Our studies provide evidence that AD increases the susceptibility to aging and/or HFD induced insulin resistance. As insulin action within the hypothalamus controls systemic metabolism, our finding of impaired hypothalamic insulin signaling could account for the link between AD and diabetes. Furthermore, we identified plasma branch chain amino acids (BCAA) as a potential marker of impaired hypothalamic insulin action in a mouse model of AD.
Future directions: Studies should elucidate the molecular mechanism through which AD impairs neuronal insulin signaling. Is hypothalamic insulin action, that is, the ability of brain insulin to regulate systemic metabolism impaired in patients with AD and are plasma BCAA levels a biomarker of impaired brain insulin action in patients with AD?
Acknowledgments
This study was supported by NIH grants DK074873, DK083568, DK082724, 3R01AA023416 (C.B.), ADA 1-12-BS-38 (M.E.E.), U01 AG046170 (mPI S.G., M.E.E.; Eric E. Schadt, corresponding PI), R34 AG049649 (mPI S.G. and Joel T Dudley, corresponding PI), R01 NS075685 (S.G.), RRD MERIT I01RX000684 (S.G.), BLRD MERIT I01BX000348 (S.G.), P50 AG005138 to Mary Sano (S.G.), and Cure Alzheimer’s Fund Research Consortium (S.G.).
Footnotes
Supplementary data
Supplementary data related to this article can be found online at http://dx.doi.org/10.1016/j.jalz.2016.01.008.
References
- 1.Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–81. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 2.Haan MN. Therapy Insight: type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat Clin Pract Neurol. 2006;2:159–66. doi: 10.1038/ncpneuro0124. [DOI] [PubMed] [Google Scholar]
- 3.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–42. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 4.Xu WL, Qiu CX, Wahlin A, Winblad B, Fratiglioni L. Diabetes mellitus and risk of dementia in the Kungsholmen project: a 6-year follow-up study. Neurology. 2004;63:1181–6. doi: 10.1212/01.wnl.0000140291.86406.d1. [DOI] [PubMed] [Google Scholar]
- 5.Jiménez-Palomares M, Ramos-Rodríguez JJ, López-Acosta JF, Pacheco-Herrero M, Lechuga-Sancho AM, Perdomo G, et al. Increased Aβ production prompts the onset of glucose intolerance and insulin resistance. Am J Physiol Endocrinol Metab. 2012;302:E1373–80. doi: 10.1152/ajpendo.00500.2011. [DOI] [PubMed] [Google Scholar]
- 6.Ramos-Rodriguez JJ, Ortiz-Barajas O, Gamero-Carrasco C, de la Rosa PR, Infante-Garcia C, Zopeque-Garcia N, et al. Prediabetes-induced vascular alterations exacerbate central pathology in APPswe/PS1dE9 mice. Psychoneuroendocrinology. 2014;48:123–35. doi: 10.1016/j.psyneuen.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 7.Kalmijn S, Launer LJ, Ott A, Witteman JC, Hofman A, Breteler MM. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol. 1997;42:776–82. doi: 10.1002/ana.410420514. [DOI] [PubMed] [Google Scholar]
- 8.Ristow M. Neurodegenerative disorders associated with diabetes mellitus. J Mol Med (Berl) 2004;82:510–29. doi: 10.1007/s00109-004-0552-1. [DOI] [PubMed] [Google Scholar]
- 9.Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162–7. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu WQ, Folstein MF. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: review and hypothesis. Neurobiol Aging. 2006;27:190–8. doi: 10.1016/j.neurobiolaging.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 11.Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes. 2000;49:263–71. doi: 10.2337/diabetes.49.2.263. [DOI] [PubMed] [Google Scholar]
- 12.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J Neurochem. 2008;104:1433–9. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porte D, Jr, Baskin DG, Schwartz MW. Insulin signaling in the central nervous system: a critical role in metabolic homeostasis and disease from C. elegans to humans. Diabetes. 2005;54:1264–76. doi: 10.2337/diabetes.54.5.1264. [DOI] [PubMed] [Google Scholar]
- 14.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. 2002;8:1376–82. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- 15.Scherer T, O’Hare J, Diggs-Andrews K, Schweiger M, Cheng B, Lindtner C, et al. Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab. 2011;13:183–94. doi: 10.1016/j.cmet.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shin AC, Fasshauer M, Filatova N, Grundell LA, Zielinski E, Zhou JY, et al. Brain insulin lowers circulating BCAA levels by inducing hepatic BCAA catabolism. Cell Metab. 2014;20:898–909. doi: 10.1016/j.cmet.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freiherr J, Hallschmid M, Frey WH, 2nd, Brunner YF, Chapman CD, Holscher C, et al. Intranasal insulin as a treatment for Alzheimer’s disease: a review of basic research and clinical evidence. CNS Drugs. 2013;27:505–14. doi: 10.1007/s40263-013-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holscher C. First clinical data of the neuroprotective effects of nasal insulin application in patients with Alzheimer’s disease. Alzheimers Dement. 2014;10:S33–7. doi: 10.1016/j.jalz.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 19.Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–38. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scaini G, Comim CM, Oliveira GM, Pasquali MA, Quevedo J, Gelain DP, et al. Chronic administration of branched-chain amino acids impairs spatial memory and increases brain-derived neurotrophic factor in a rat model. J Inherit Metab Dis. 2012;36:721–30. doi: 10.1007/s10545-012-9549-z. [DOI] [PubMed] [Google Scholar]
- 21.Fischer W, Wictorin K, Bjorklund A, Williams LR, Varon S, Gage FH. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature. 1987;329:65–8. doi: 10.1038/329065a0. [DOI] [PubMed] [Google Scholar]
- 22.Scaini G, Mello-Santos LM, Furlanetto CB, Jeremias IC, Mina F, Schuck PF, et al. Acute and chronic administration of the branched-chain amino acids decreases nerve growth factor in rat hippocampus. Mol Neurobiol. 2013;48:581–9. doi: 10.1007/s12035-013-8447-1. [DOI] [PubMed] [Google Scholar]
- 23.Contrusciere V, Paradisi S, Matteucci A, Malchiodi-Albedi F. Branched-chain amino acids induce neurotoxicity in rat cortical cultures. Neurotox Res. 2010;17:392–8. doi: 10.1007/s12640-009-9115-0. [DOI] [PubMed] [Google Scholar]
- 24.Morabito MV, Berman DE, Schneider RT, Zhang Y, Leibel RL, Small SA. Hyperleucinemia causes hippocampal retromer deficiency linking diabetes to Alzheimer’s disease. Neurobiol Dis. 2014;65:188–92. doi: 10.1016/j.nbd.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lane RF, Raines SM, Steele JW, Ehrlich ME, Lah JA, Small SA, et al. Diabetes-associated SorCS1 regulates Alzheimer’s amyloid-beta metabolism: evidence for involvement of SorL1 and the retromer complex. J Neurosci. 2010;30:13110–5. doi: 10.1523/JNEUROSCI.3872-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hiltunen M, Khandelwal VK, Yaluri N, Tiilikainen T, Tusa M, Koivisto H, et al. Contribution of genetic and dietary insulin resistance to Alzheimer phenotype in APP/PS1 transgenic mice. J Cell Mol Med. 2012;16:1206–22. doi: 10.1111/j.1582-4934.2011.01384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mody N, Agouni A, McIlroy GD, Platt B, Delibegovic M. Susceptibility to diet-induced obesity and glucose intolerance in the APP (SWE)/PSEN1 (A246E) mouse model of Alzheimer’s disease is associated with increased brain levels of protein tyrosine phosphatase 1B (PTP1B) and retinol-binding protein 4 (RBP4), and basal phosphorylation of S6 ribosomal protein. Diabetologia. 2011;54:2143–51. doi: 10.1007/s00125-011-2160-2. [DOI] [PubMed] [Google Scholar]
- 28.Chua LM, Lim ML, Chong PR, Hu ZP, Cheung NS, Wong BS. Impaired neuronal insulin signaling precedes Abeta42 accumulation in female AbetaPPsw/PS1DeltaE9 mice. J Alzheimers Dis. 2012;29:783–91. doi: 10.3233/JAD-2012-111880. [DOI] [PubMed] [Google Scholar]
- 29.Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17:448–53. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reinhardt S, Schuck F, Gr€osgen S, Riemenschneider M, Hartmann T, Postina R, et al. Unfolded protein response signaling by transcription factor XBP-1 regulates ADAM10 and is affected in Alzheimer’s disease. FASEB J. 2014;28:978–97. doi: 10.1096/fj.13-234864. [DOI] [PubMed] [Google Scholar]
- 31.Soejima N, Ohyagi Y, Nakamura N, Himeno E, Iinuma KM, Sakae N, et al. Intracellular accumulation of toxic turn amyloid-beta is associated with endoplasmic reticulum stress in Alzheimer’s disease. Curr Alzheimer Res. 2013;10:11–20. [PubMed] [Google Scholar]
- 32.Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 2011;108:2939–44. doi: 10.1073/pnas.1006875108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang L, Zhai Y-Q, Xu L-L, Qiao C, Sun X-L, Ding J-H, et al. Metabolic inflammation exacerbates dopaminergic neuronal degeneration in response to acute MPTP challenge in type 2 diabetes mice. Exp Neurol. 2014;251:22–9. doi: 10.1016/j.expneurol.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14:1576–82. doi: 10.1038/sj.cdd.4402200. [DOI] [PubMed] [Google Scholar]
- 36.Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci. 2014;8:213. doi: 10.3389/fncel.2014.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 38.Muriach M, Flores-Bellver M, Romero FJ, Barcia JM. Diabetes and the brain: oxidative stress, inflammation, and autophagy. Oxid Med Cell Longev. 2014;2014:102158. doi: 10.1155/2014/102158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakka V, Prakash-babu P, Vemuganti R. Crosstalk between endoplasmic reticulum stress, oxidative stress, and autophagy: potential therapeutic targets for acute CNS injuries. Mol Neurobiol. 2014;53:532–44. doi: 10.1007/s12035-014-9029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen BP, Wolfgang CD, Hai T. Analysis of ATF3, a transcription factor induced by physiological stresses and modulated by gadd153/Chop10. Mol Cell Biol. 1996;16:1157–68. doi: 10.1128/mcb.16.3.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.B’chir W, Maurin A-C, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–99. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–9. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–20. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, et al. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol. 2002;173:183–95. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- 45.Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci. 2002;5:566–72. doi: 10.1038/nn0602-861. [DOI] [PubMed] [Google Scholar]
- 46.Kadota Y, Toyoda T, Kitaura Y, Adams SH, Shimomura Y. Regulation of hepatic branched-chain alpha-ketoacid dehydrogenase complex in rats fed a high-fat diet. Obes Res Clin Pract. 2013;7:e439–44. doi: 10.1016/j.orcp.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clarke JR, Lyra ESNM, Figueiredo CP, Frozza RL, Ledo JH, Beckman D, et al. Alzheimer-associated Abeta oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol Med. 2015;7:190–210. doi: 10.15252/emmm.201404183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lopez-Gonzalez I, Schluter A, Aso E, Garcia-Esparcia P, Ansoleaga B, F LL, et al. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol. 2015;74:319–44. doi: 10.1097/NEN.0000000000000176. [DOI] [PubMed] [Google Scholar]
- 50.Ono H, Pocai A, Wang Y, Sakoda H, Asano T, Backer JM, et al. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J Clin Invest. 2008;118:2959–68. doi: 10.1172/JCI34277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.