

ABSTRACT
One of the hallmarks of cancer is the ability of tumor cells to invade surrounding tissues and metastasize. During metastasis, cancer cells degrade the extracellular matrix, which acts as a physical barrier, by developing specialized actin-rich membrane protrusion structures called invadopodia. The formation of invadopodia is regulated by Rho GTPases, a family of proteins that regulates the actin cytoskeleton. Here, we describe a novel role for RhoG in the regulation of invadopodia disassembly in human breast cancer cells. Our results show that RhoG and Rac1 have independent and opposite roles in the regulation of invadopodia dynamics. We also show that SGEF (also known as ARHGEF26) is the exchange factor responsible for the activation of RhoG during invadopodia disassembly. When the expression of either RhoG or SGEF is silenced, invadopodia are more stable and have a longer lifetime than in control cells. Our findings also demonstrate that RhoG and SGEF modulate the phosphorylation of paxillin, which plays a key role during invadopodia disassembly. In summary, we have identified a novel signaling pathway involving SGEF, RhoG and paxillin phosphorylation, which functions in the regulation of invadopodia disassembly in breast cancer cells.
KEY WORDS: RhoG, Invadopodia, SGEF, Guanine-nucleotide exchange factors, Src, Paxillin, Rac1
Highlighted Article: We describe a novel signaling pathway involving SGEF, RhoG and paxillin phosphorylation, which functions in the regulation of invadopodia disassembly in breast cancer cells.
INTRODUCTION
Rho GTPases control many aspects of cell behavior ranging from the regulation of cytoskeletal organization, cell motility and cell polarity, to nuclear gene expression and control of cell growth (Hodge and Ridley, 2016). Rho proteins cycle between an active (GTP-bound) and an inactive (GDP-bound) state. The activation of Rho proteins involves the exchange of GDP for GTP, which is catalyzed by specific guanine-nucleotide-exchange factors (GEFs). Once activated, Rho GTPases interact with a wide variety of downstream effectors to modulate their activity and/or localization. The hydrolysis of GTP to GDP, a reaction that is stimulated by GTPase-activating proteins (GAPs), inactivates the GTPases and terminates the signal. With more than 80 Rho GEFs, 70 Rho GAPs and over 100 effectors, cells regulate the activity of Rho proteins through multiple pathways, thus acting as key nodes for signal integration and dissemination (Bustelo et al., 2007; Rossman et al., 2005; Tcherkezian and Lamarche-Vane, 2007).
RhoG, a Rho protein related to Rac, has been associated with cell migration, neurite outgrowth, microtubule dynamics, micropinocytosis, bacterial uptake, phagocytosis and leukocyte trans-endothelial migration (deBakker et al., 2004; Ellerbroek et al., 2004; Jackson et al., 2015; Katoh et al., 2006, 2000; van Buul et al., 2007). Recent studies have revealed that RhoG plays a role in tumor cell invasion and may contribute to the formation of invadopodia (Hiramoto-Yamaki et al., 2010; Kwiatkowska et al., 2012). Invadopodia are actin-rich adhesive structures that form in the ventral surface of cancer cells and allow them to degrade the extracellular matrix (ECM) (Gimona et al., 2008). Formation of invadopodia involves a series of steps that include the disassembly of focal adhesions and stress fibers, and the relocalization of several of their components into the newly formed invadopodia (Hoshino et al., 2012; Oikawa et al., 2008). Invadopodia assembly starts with the formation of actin- and cortactin-rich puncta, followed by the recruitment of adhesion proteins, such as vinculin and paxillin, and proteinases that allow ECM degradation (Hoshino et al., 2013). Even though many of the molecular components required for invadopodia formation have been identified, the signaling pathways that regulate these events are still poorly understood (Linder et al., 2011). Invadopodia formation is controlled by the integrated activity of several GTPases, including Cdc42, RhoA, RhoC and Rac1 (Spuul et al., 2014). Cdc42 promotes invadopodia formation in almost every system tested (Ayala et al., 2009; Di Martino et al., 2014; Moreau et al., 2006, 2003; Nakahara et al., 2003; Tatin et al., 2006). However, generalized conclusions cannot be drawn from studies of other Rho GTPases, since their activities can both inhibit or promote invadopodia formation depending on the experimental conditions or cell type used (Spuul et al., 2014).
In this study, we describe a novel function for RhoG in the regulation of invadopodia dynamics in human breast cancer cells. Our findings describe a signaling pathway involving SGEF and RhoG that regulates invadopodia disassembly independently of Rac1 through regulation of paxillin phosphorylation.
RESULTS
RhoG is a negative regulator of invadopodia formation
We have previously shown that the breast cancer cell line SUM159 forms invadopodia when treated with phorbol esters such phorbol 12,13-dibutyrate (PDBu) and 12-O-tetradecanoylphorbol-13-acetate (PMA), and that the formation of these structures correlates with their metastatic potential (Goicoechea et al., 2009). To determine the requirement of RhoG in invadopodia formation, we generated stable SUM159 cell lines in which RhoG expression was silenced using lentivirally encoded shRNA. We used two different RhoG-specific shRNAs (shRNA#1 and shRNA#4) to rule out off-target effects. A stable cell line expressing a non-targeting shRNA was used as control (CTRL). Fig. 1A shows that RhoG silencing was efficient, especially for shRNA #4. SUM159 cells are able to form invadopodia spontaneously, so we first looked at the effect of RhoG depletion in untreated cells. To identify invadopodia structures, we stained the cells with the invadopodia markers cortactin and actin. We found that silencing RhoG induced a significant increase in the number of spontaneous invadopodia when compared to CTRL cells, from 7% in CTRL cells to 14% and 23% in RhoG shRNA#1 and shRNA#4 cells, respectively (Fig. 1B, untreated; Fig. S1A). We next looked at the effect of RhoG depletion in cells treated with phorbol esters. In PDBu-treated cells, the number of cells with invadopodia increased significantly in RhoG-knockdown (KD) cells, from 35% in CTRL cells to 64% and 81% in RhoG shRNA#1 and shRNA#4 cells respectively (Fig. 1B,C). Similarly, the percentage of cells with invadopodia increased from 31% in CTRL cells to 79% of cells in PMA-treated RhoG KD cells (Fig. 1B; Fig. S1B). Based on these results, and unless otherwise indicated, we used shRNA#4 for the rest of these studies (referred to as RhoG KD). We next performed a rescue experiment by re-expressing shRNA-resistant Myc-RhoG in RhoG KD cells (Rescue). Myc–RhoG expresses at levels that are comparable to endogenous RhoG levels in RhoG KD cells, and restores the cells ability to form invadopodia to control levels (Fig. 1D,E). We also characterized the effects of silencing RhoG on invadopodia formation in two additional human breast cancer cell lines, invasive MDA-MB-231, commonly used as a model to study invadopodia (Fig. S2A,B), and non-invasive MCF7 cells (Fig. S2C,D). In both cell lines, silencing RhoG significantly increased the number of cells with invadopodia, which suggest the effects observed are not cell line specific. To investigate the role of RhoG in more detail, we overexpressed either Myc-tagged wild-type (RhoG wt) or constitutively active RhoG (RhoG Q61L) in SUM159 cells (Fig. 1F) and tested the ability of the cells to form invadopodia (Fig. 1G). Overexpression of either RhoG wt or RhoG Q61L significantly inhibited the formation of invadopodia (Fig. 1H). As expected, the Q61L mutant, which is locked in an active conformation had a greater inhibitory effect than the wild type. Taken together, our results suggest a novel role for RhoG as a negative regulator of invadopodia formation in breast cancer cells.
Fig. 1.

RhoG negatively regulates invadopodia formation in SUM159 cells. (A) Cell lysates from SUM159 cells stably expressing non-targeting (CTRL) or RhoG-specific shRNAs (shRNA#1 and shRNA #4) were analyzed by western blotting and probed for RhoG and Rac1, and for tubulin as a loading control. (B) Quantification of cortactin- and actin-containing invadopodia in CTRL and RhoG KD cells. Results are expressed as the percentage of cells with invadopodia. Data are mean±s.e.m. for at least three independent experiments. Cells were either untreated, or treated with PDBu or PMA. (C) CTRL or RhoG KD SUM159 cells were treated with PDBu for 30 min and stained with anti-cortactin antibody (red), Alexa-Fluor-488–phalloidin (green) and Hoechst 33342 (blue). Arrowheads indicate representative invadopodia. (D) Cell lysates from CTRL, RhoG KD and RhoG KD cells expressing shRNA resistant Myc–RhoG (rescue) were immunoblotted with anti-RhoG and -Myc antibodies. Tubulin was used as a loading control. (E) Quantification of cortactin- and actin-containing invadopodia in CTRL, RhoG KD and rescue cells. Data are mean±s.e.m. of three independent experiments. (F) Cell lysates from CTRL, Myc–RhoG wt (RhoG wt) or Myc–RhoG Q61L (RhoG Q61L) cells were immunoblotted with anti-RhoG and -Myc antibodies. Tubulin was used as a loading control. (G) SUM159 cells transiently transfected with Myc-tagged wild-type (RhoG wt) or constitutively active RhoG (RhoG Q61L) were treated with PDBu for 30 min and stained with anti-cortactin antibody (red), anti-Myc antibody (green), Alexa-Fluor-647–phalloidin (magenta) and Hoechst 33342 (blue). Arrowheads indicate transfected cells and arrows indicate representative invadopodia. (H) Invadopodia were quantified in CTRL (non-transfected cells), and RhoG wt and RhoG Q61L transfected cells. Results are representative of three independent experiments in which at least 100 cells per experiment were counted. Data are mean±s.e.m. (error bars). Scale bars: 10 µm. *P<0.05; **P<0.01; ***P<0.001; ns, not significant.
Enhanced invadopodia formation after RhoG depletion requires Src activity
Phorbol esters mimic diacylglycerol (DAG), which activates protein kinase C (PKC) family members. The role and positioning of PKC in these pathways remains largely unknown, but it involves the function of Src family kinases (Hai et al., 2002; Tatin et al., 2006). Because both FAK and Src activities have been shown to be upregulated to promote invadopodia formation, and both play critical roles in cell invasion (Murphy and Courtneidge, 2011), we wanted to determine whether RhoG modulates invadopodia formation through a FAK- and/or Src-dependent pathway (FAK is also known as PTK2). Western blot analysis showed that there was no difference in the expression level of endogenous FAK and Src between CTRL and RhoG KD cells (Fig. 2A,B). However, the activity of both increased by ∼2-fold in the absence of RhoG (Fig. 2A,B). We next assayed the ability of CTRL and RhoG KD cells to form invadopodia when either Src or FAK were inhibited. Our results show that inhibition of Src with PP2 significantly inhibited invadopodia formation in both CTRL and RhoG KD cells (Fig. 2C,D), whereas inhibition of FAK with PF-573228 had no effect (Fig. 2E,F). These findings suggest that the increase in Src activity in RhoG KD cells may contribute to the increase observed in the number of cells with invadopodia, and that RhoG may function downstream of Src to regulate invadopodia formation in SUM159 cells.
Fig. 2.

Src activity is necessary for increased invadopodia formation after RhoG KD. Western blot analysis shows increased levels of phospho-Src (Src p418) (A) and phospho-FAK (FAK p397) (B) in RhoG-depleted cells. Data are mean±s.e.m. of three independent experiments. (C–F) CTRL and RhoG KD cells were cultured in the presence of vehicle control, 5 μM PP2 (C,D) or 5 μM PF-573228 (E,F) for 30 min followed by a 30 min incubation with PDBu. The efficiency of the inhibitors was tested by immunoblotting for phospho-Src (Src p418) (C) or phospho-FAK (FAK p397) (E). Quantifications in D and F show the percentage of cells with invadopodia, as determined by cortactin and actin staining. Data are mean±s.e.m. of three independent experiments. *P<0.05; **P<0.01; ***P<0.001; ns, not significant.
SGEF functions upstream of RhoG to regulate invadopodia formation
Several GEFs have been described to stimulate nucleotide exchange on RhoG (Bellanger et al., 2000; Blangy et al., 2000; Bustelo et al., 2007; D'Angelo et al., 2007; Damoulakis et al., 2014; Ellerbroek et al., 2004; Krishna Subbaiah et al., 2012; May et al., 2002; Wennerberg et al., 2002). To identify the GEF that regulates RhoG during invadopodia formation, we performed a candidate-based shRNA screen. We stably silenced the best characterized RhoG-specific GEFs, including SGEF (also known as ARHGEF26), ephexin 4 (also known as ARHGEF16), PLEKHG6 and Trio in SUM159 cells, using specific shRNAs. We tested two independent shRNAs for each of the GEFs and compared them to cells expressing a non-targeting shRNA (CTRL). The KD efficiency was verified either by quantitative real-time PCR (qRT-PCR; Fig. 3A; Fig. S3A) or western blotting (Fig. S3B). Cells were treated with PDBu, and invadopodia structures were identified in cells stained for cortactin and actin (Fig. 3B; Fig. S3C). Our results show that the number of cells with invadopodia increased significantly when SGEF was silenced (from 42% in CTRL cells, to 70% and 85% upon treatment with SGEF shRNA#4 and shRNA#5, respectively, Fig. 3C), whereas silencing PLEKHG6, ephexin 4 or Trio had no significant effect on invadopodia numbers (Fig. S3D). The phenotype observed in SGEF KD cells was similar to that observed in RhoG KD cells, both in terms of the number of cells that formed invadopodia and their appearance (Figs 3B and 1C). Based on these results, we used shRNA#5 for the rest of these studies (referred to as SGEF KD). To determine whether SGEF function was mediated through RhoG we attempted to rescue the effects of RhoG KD on invadopodia by expressing exogenous Myc–SGEF in RhoG KD cells. Our results showed that Myc–SEGF expression could not rescue the RhoG KD phenotype (Fig. 3D,E), suggesting that the role of SGEF in invadopodia is mostly mediated by RhoG. Consistent with the results showed in Fig. 1H, overexpression of Myc–SGEF wt, significantly inhibited the formation of invadopodia (Fig. 3F,G). In contrast, overexpression of a catalytically inactive mutant of SGEF (Myc–SGEF 446/621, which has E446A and N621A mutations) (Ellerbroek et al., 2004) had no effect (Fig. 3F,G), which demonstrates that SGEF exchange activity, and thus RhoG activation, is required. Only 5% of the cells transfected with Myc–SGEF wt formed invadopodia, whereas cells transfected with the Myc–SGEF 446/621 show invadopodia levels comparable to those of non-transfected cells (∼30%) (Fig. 3F). Interestingly, Myc–SGEF 446/621 localized to invadopodia (Fig. 3G). We also observed Myc–SGEF wt at invadopodia, although at a much lower frequency (∼1.25% of Myc–SGEF wt transfected cells) (Fig. S3E). This suggests that SGEF could localize transiently to invadopodia and be released to the cytoplasm after the exchange reaction is completed. In the absence of catalytic activity, SGEF may not be efficiently released and would thus be found at invadopodia.
Fig. 3.

SGEF regulates invadopodia formation. (A) Cell lysates from SUM159 cells stably expressing non-targeting (CTRL) or SGEF-specific shRNAs (shRNA#4 and shRNA #5) were analyzed by qRT-PCR for expression of SGEF. (B) CTRL and SGEF KD SUM159 cells were treated with PDBu for 30 min and stained with anti-cortactin antibody (red), Alexa-Fluor-488–phalloidin (green) and Hoechst 33342 (blue). Arrowheads indicate representative invadopodia. (C) Quantification of cortactin- and actin-containing invadopodia in CTRL and SGEF KD cells expressed as the percentage of cells with invadopodia. Data are mean±s.e.m. of four independent experiments. (D) Cell lysates from CTRL, RhoG KD and RhoG KD cells expressing Myc–SGEF (rescue Myc–SGEF) were immunoblotted with anti-RhoG and -Myc antibodies. Tubulin was used as a loading control. (E) Quantification of cortactin- and actin-containing invadopodia in CTRL, RhoG KD and rescue Myc–SGEF cells. Data are mean±s.e.m. of three independent experiments in which at least 200 cells per experiment were counted. (F) Quantification of cortactin- and actin-containing invadopodia in SUM159 cells transiently transfected with either Myc-tagged wild-type SGEF (SGEF wt) or catalytically inactive SGEF (SGEF 446/621). Data are mean± s.e.m. of at least three independent experiments in which at least 200 cells per experiment were counted. (G) Representative images of cells transfected with either Myc-tagged wild-type SGEF (SGEF wt) or catalytically inactive SGEF (SGEF 446/621). Cells were stained with anti-cortactin antibody (red), anti-Myc antibody (green), Alexa-Fluor-647–phalloidin (magenta) and Hoechst 33342 (blue). Arrows indicate transfected cells and arrowheads indicate representative invadopodia. (H–J) CTRL (H) and SGEF KD (I) cells were treated with PDBu for the indicated times. Active RhoG was precipitated from total lysates using GST–ELMO and immunoblotted with RhoG antibody. (J) For quantification, active RhoG levels were normalized to total RhoG levels. Data are mean±s.e.m. of at least three independent experiments. Scale bars: 10 µm. *P<0.05; **P<0.01; ***P<0.001.
Our results suggest that RhoG activity needs tight regulation during invadopodia formation, so we next investigated the kinetics of RhoG activation at different time points following PDBu exposure. We measured RhoG activity by using GST–ELMO to pulldown GTP-RhoG (van Buul et al., 2007). Our results show a rapid and transient decrease of RhoG activity in the first 5 min of PDBu treatment followed by a peak of activation at 15 min (Fig. 3H,J). We obtained similar kinetics of RhoG activation after treatment of cells with PMA (Fig. S4A,B). To test whether SGEF plays a role in the regulation of RhoG activation during PDBu stimulation, we measured RhoG activity in SGEF KD cells at different times following PDBu addition. Interestingly, the peak of RhoG activation observed at 15 min was completely abrogated by SGEF KD (Fig. 3I,J). In contrast, the activity of RhoG in the absence of PDBu was not significantly affected in the absence of SGEF. Therefore, our results show that SGEF specifically mediates the PDBu-induced activation of RhoG that occurs at 15 min.
Enhanced invadopodia formation in RhoG- and SGEF-depleted cells is not sufficient for invasion
In order to determine whether the invadopodia that formed following RhoG depletion retained their capacity to degrade the ECM, we plated CTRL and RhoG KD cells on Oregon Green 488-conjugated gelatin-coated coverslips and assessed invadopodia activity by quantifying the area of degradation (visible as dark on the green background) (Artym et al., 2009; Chen et al., 1985; Martin et al., 2012). Our results show that the number of cells that form invadopodia on gelatin matrix is also increased when RhoG is knocked down (CTRL=35%; RhoG KD=71%, Fig. 4A). RhoG-depleted cells also exhibited larger areas of matrix degradation compared to CTRL cells, which showed discrete areas of degradative puncta (Fig. 4B). Quantification showed a ∼9-fold increase in degradation area in RhoG KD cells relative to CTRL cells (Fig. 4C). These results demonstrate that invadopodia are still functional in the absence of RhoG. We obtained similar results, when we silenced RhoG in MDA-MB-231 cells (Fig. S2E,F). We then assayed the ability of serum-starved CTRL, RhoG KD and RhoG KD cells expressing a Myc-tagged shRNA-resistant version of RhoG (Rescue) to invade through Matrigel-coated membranes (Fig. 4D). Our results show that, even though there are more cells with invadopodia in RhoG KD cells than in CTRL cells, their ability to invade through Matrigel was significantly impaired. The ability to invade was partially restored by re-expressing Myc–RhoG (Fig. 4D). As observed in RhoG KD cells, SGEF KD cells also displayed a lower invasive capacity compared to CTRL cells (Fig. 4E). These results suggest that enhanced formation of invadopodia and increased degradation capacity in RhoG- and SGEF-deficient cells are not sufficient to enhance invasiveness. It is possible that the invasion defect observed results from the inability of cells to migrate in the absence of RhoG, since RhoG has been previously shown to play a role during cell migration (Hiramoto-Yamaki et al., 2010; Katoh et al., 2006). Supporting this, our results show that RhoG KD also impairs cell migration in SUM159 cells (Fig. S2G). In summary, our results suggest that proper coordination of matrix degradation and migration is required for efficient invasion.
Fig. 4.

Silencing RhoG increases matrix degradation but not invasion. (A) CTRL and RhoG KD cells were cultured on Oregon Green 488-conjugated gelatin and stained with anti-cortactin antibody (red). Invadopodia quantification in CTRL and RhoG KD cells is expressed as the percentage of cells with invadopodia. Data are mean±s.e.m. of three independent experiments. (B) Representative images showing matrix degradation in CTRL and RhoG KD cells. Scale bars: 20 µm. (C) Area of matrix degraded per cell area. Data are mean±s.e.m. of three independent experiments. (D) Invasion assay in CTRL, RhoG KD and RhoG KD/Myc-RhoG rescue cells. Representative images showing cells that have invaded across a Matrigel-coated transwell membrane are presented. Results are representative of at least four individual experiments. (E) Invasion assay of CTRL and SGEF KD cells. Representative images are showing on the left. Results are representative of at least four individual experiments. *P<0.05; **P<0.01.
Rac1 stimulates invadopodia formation
RhoG is a key upstream regulator of Rac1 in migrating cells (Elfenbein et al., 2009; Hiramoto et al., 2006; Katoh et al., 2006; Katoh and Negishi, 2003). However, RhoG has also been shown to function independently of Rac1 (Samson et al., 2010; Wennerberg et al., 2002). To determine whether the role of RhoG in invadopodia depends on Rac1, we first generated stable cell lines in which Rac1 expression was silenced using lentivirally encoded shRNA. Since knockdown of Rac1 was not efficient for any of the four shRNAs tested (not shown), we generated Rac1-knockout cells (Rac1 KO) using the double nicking RNA-guided Cas9 nucleases from the microbial CRISPR/Cas system. After isolation of single cell colonies, gene knockout efficiency was assayed by western blotting (Fig. 5A). Remarkably, invadopodia structures were completely absent in Rac1 KO cells (Fig. 5B). PDBu-treated Rac1 KO cells showed a very consistent morphology, with actin and cortactin concentrated at defined regions of the leading edge in small lamellipodia-like structures (Fig. 5B). Quantitative analysis in the three single cell colonies analyzed showed that Rac1 KO completely blocked the ability of cells to form invadopodia as compared to CTRL cells (Fig. 5C; Movies 1 and 2). Re-expression of Myc-tagged Rac1 in KO cells (rescue) restored the ability of the cells to form invadopodia to CTRL levels (Fig. 5D, Movie 3). To explore the role of Rac1 in more detail, we overexpressed Myc–Rac1 in SUM159 cells (Rac1 OE) and tested the cells for their ability to form invadopodia. As expected from the Rac1 KO results, cells overexpressing Rac1 formed more invadopodia than CTRL cells (Fig. 5E). We also attempted to rescue RhoG KD cells with Myc-tagged Rac1 (RhoG KD+Myc–Rac1). The rationale for this experiment was that if Rac1 is being activated downstream of RhoG, an excess of Rac1 would be able to rescue RhoG deficiency. Fig. 5F shows that ectopic expression of Rac1 could not rescue the RhoG KD invadopodia phenotype. In contrast, RhoG KD Myc–Rac1 rescue cells formed even more invadopodia than RhoG KD cells suggesting an additive effect. Taken together, these results demonstrated that Rac1 and RhoG function independently and play opposing roles in the regulation of invadopodia formation in breast cancer cells.
Fig. 5.

Rac1 is necessary for invadopodia formation in SUM159 cells. (A) Cells lysates from CTRL and Rac1 KO SUM159 cells were analyzed by western blotting and probed for Rac1 and tubulin, as a loading control. (B) CTRL and Rac1 KO cells were treated with PDBu for 30 min and stained with anti-cortactin antibody (red), Alexa-Fluor-488–phalloidin (green) and Hoechst 33342 (blue). Arrowheads indicate representative invadopodia. Scale bars: 10 µm. (C) Quantification of cortactin- and actin-containing invadopodia in CTRL and Rac1 KO cell lines expressed as percentage of number of cells with invadopodia. Data are mean±s.e.m. of at least three independent experiments in which at least 200 cells per experiment were counted. (D) Cell lysates from CTRL, Rac1 KO and Rac1 KO cells expressing Myc–Rac1 (rescue) were immunoblotted with anti-Rac1 and -Myc antibodies. Tubulin was used as a loading control (left panel). Quantification of cortactin- and actin-containing invadopodia in CTRL, Rac1 KO and rescue cells (right panel). Data are mean±s.e.m. of at least three independent experiments in which at least 200 cells per experiment were counted. (E) Cell lysates from CTRL and cells expressing Myc–Rac1 (Rac1 OE) were immunoblotted with anti-Rac1 and -Myc antibodies. Tubulin was used as a loading control (left panel). Quantification of cortactin- and actin-containing invadopodia in CTRL cells and cells expressing Myc-Rac1 (right panel). Data are mean±s.e.m. of at least three independent experiments in which at least 200 cells per experiment were counted. (F) Cell lysates from CTRL, RhoG KD and RhoG KD cells expressing Myc–Rac1 (RhoG KD+Myc-Rac1) were immunoblotted with anti-RhoG and -Myc antibodies. Tubulin was used as a loading control (left panel). Quantification of cortactin- and actin-containing invadopodia in CTRL, RhoG KD and RhoG KD+Myc-Rac1 cells (right panel). Data are mean±s.e.m. of at least three independent experiments in which at least 200 cells per experiment were counted. *P<0.05; **P<0.01; ***P<0.001.
RhoG is involved in invadopodia dynamics
To further characterize the differences in invadopodia formation between CTRL and RhoG KD cells, we analyzed invadopodia formation over time by fixing and staining SUM159 cells at different times after PDBu addition. Surprisingly, at early time points, the number of cells with invadopodia was almost identical between CTRL and RhoG KD cells, peaking at ∼70–80% (Fig. 6A). In CTRL cells, the number of cells with invadopodia decreased rapidly to ∼40% at 30 min, and remained stable even after 3 h. In contrast, in the absence of RhoG, the number of cells with invadopodia did not decrease as much, and remained stable over time at ∼70% (Fig. 6A). These results suggest that, in CTRL cells, following a rapid burst of initial invadopodia assembly in response to PDBu, invadopodia start to disassemble and the percentage of cells showing invadopodia stabilizes, probably reflecting an equilibrium between assembly and disassembly. In contrast, the percentage of cells with invadopodia remains significantly higher in RhoG KD cells, which may suggest that invadopodia are more stable or longer lived in the absence of RhoG.
Fig. 6.

Silencing RhoG or SGEF increases invadopodia lifetime. (A) CTRL or RhoG KD SUM159 cells were treated with PDBu for the indicated times and stained for cortactin and actin as invadopodia markers. Invadopodia were quantified and expressed as the percentage of cells with invadopodia. Data are mean± s.e.m. of three independent experiments in which at least 200 cells were analyzed per condition. (B) Representative time series of invadopodia formation in CTRL and RhoG KD cells. SUM159 cells expressing mCherry–cortactin were imaged for 120 min at 15 s intervals following the addition of PDBu. White arrows point to an invadopodia cluster forming in the nuclear region. Scale bars: 20 µm. (C) Invadopodia lifetime increases when RhoG and SGEF expression are silenced. Re-expression of RhoG and SGEF in RhoG KD and SGEF KD cells respectively restores the lifetime to the levels of CTRL cells. Results are representative of three independent experiments in which at least five cells were analyzed per condition. The box represents the 25–75th percentiles, and the median is indicated. The whiskers show the 5th to 95th percentiles. *P<0.05; **P<0.01.
To investigate the impact of RhoG and SGEF on invadopodia lifetime, we analyzed the dynamics of invadopodia by time-lapse confocal microscopy in SUM159 cells expressing mCherry–cortactin after PDBu treatment (Fig. 6B). In CTRL cells, invadopodia were short-lived and motile with an average lifetime of ∼10 min (Fig. 6B,C; Movie 1). However, when either RhoG or SGEF were silenced, the lifetime of invadopodia increased significantly to over 30 min, and they often persisted in a single location for time periods >1 h (Fig. 6B,C; Movies 4 and 5). When RhoG or SGEF were re-expressed in RhoG KD or SGEF KD cells, respectively, invadopodia lifetime returned to control levels (Fig. 6C; Movies 6 and 7). These results demonstrate the importance of RhoG and SGEF in the regulation of invadopodia dynamics, most likely during disassembly.
Paxillin has previously been found to be a component of invadopodia in other systems, and its tyrosine phosphorylation has been shown to play a role in invadopodia disassembly (Badowski et al., 2008). To test whether RhoG and SGEF regulate paxillin tyrosine phosphorylation, we first examined the levels of phosphorylated (phospho)-paxillin (PXN p118) and total paxillin (PXN) in CTRL, RhoG KD and SGEF KD cells by western blotting using both total and phospho-specific paxillin antibodies. Quantification of immunoblots showed that paxillin tyrosine-phosphorylation decreased ∼2.5- and ∼1.8-fold in RhoG KD and SGEF KD cells respectively compared to CTRL cells (Fig. 7A). Similar effects were also observed in MDA-MB-231 and MCF7 cells (Fig. S4C,D) with a ∼1.5- and ∼1.7-fold decrease, respectively. We also investigated the distribution of phospho-paxillin (PXN p118, red) and total paxillin (PXN, green) at invadopodia by immunofluorescence in CTRL and RhoG KD SUM159 cells. Our results show that, even though total paxillin was present at invadopodia in both CTRL and RhoG KD cells, phospho-paxillin accumulated in invadopodia structures only in CTRL cells, and was virtually absent in RhoG KD invadopodia (Fig. 7B). Line-scan analysis through invadopodial structures confirmed these results, revealing a significant decrease of phospho-paxillin fluorescence intensity on RhoG KD invadopodia (PXN p118, red line) (Fig. 7C). In addition, the average ratio of phosphorylated to total paxillin fluorescence intensity in invadopodia was two times higher in CTRL cells than in RhoG KD cells (Fig. 7D). These results show that SGEF and RhoG regulate the levels of paxillin phosphorylation, which is critical for invadopodia disassembly.
Fig. 7.

RhoG knockdown decreases paxillin phosphorylation at invadopodia. (A) Cell lysates from CTRL, RhoG KD and SGEF KD SUM159 cells were analyzed by western blotting and probed for phospho-paxillin (PXN p118), total paxillin (PXN) and tubulin as a loading control (left panel). The quantification represents the average of at least three independent experiments. Data are mean±s.e.m. (right panel). (B) CTRL and RhoG KD SUM159 cells were treated with PDBu for 30 min, fixed and stained with Alexa-Fluor-647–phalloidin (magenta), and for PXN (green) and PXN p118 (red). Scale bars: 10 µm. (C) Graphs indicate fluorescent intensity in arbitrary units (A.U.) of PXN p118 (red) with respect to PXN (green) and F-actin (blue) over the indicated line scan in (B). (D) Total fluorescence intensity for PXN and PXN p118 in individual invadopodia from CTRL and RhoG KD cells were measurements of at least 50 invadopodia per condition. Data are mean±s.e.m. (E) CTRL and RhoG KD cells were cultured in the presence of vehicle control, 5 μM PP2 or 5 μM PF-573228 for 30 min. Cell lysates were analyzed by western blotting and probed for total paxillin (PXN) and phosho-paxillin (PXN p118). (F) Quantification of PXN p118 to PXN ratio in cells treated with vehicle, PP2 and PF-573228. Results represent the mean±s.e.m. of at least three independent experiments. *P<0.05, **P<0.01.
Both Src and FAK have been associated with paxillin phosphorylation (Bellis et al., 1995; Schaller and Parsons, 1995). However, our results show that even though the phosphorylation of both kinases increased when RhoG is silenced (Fig. 2A,B), paxillin phosphorylation levels are significantly decreased (Fig. 7A), which suggests that the effect of RhoG on paxillin phosphorylation may be independent of Src and FAK. To determine whether Src and/or FAK are involved in RhoG-mediated regulation of paxillin phosphorylation, we analyzed the levels of phospho-paxillin in CTRL or RhoG KD cells in the presence or absence of Src or FAK inhibitors. Our results show that treating CTRL or RhoG KD with a FAK inhibitor did not have a significant effect in phospho-paxillin levels when compared to non-treated cells. In contrast, inhibition of Src family kinases promoted a decrease in phospho-paxillin levels in both CTRL and RhoG KD cells (Fig. 7E,F). These results suggests there are two pools of phospho-paxillin in the cells, one that is regulated by RhoG independently of Src, and the other is dependent on Src and independent of RhoG.
DISCUSSION
Cells assemble invadopodia during cell invasion, which involves a dramatic rearrangement of the actin cytoskeleton, a process that is regulated by the integrated activity of several GTPases (Spuul et al., 2014). While RhoG is known to promote migration and invasion and has been recently linked to invadopodia, the regulation and role of RhoG during invadopodia formation are still unknown. The data reported here describe a novel function for RhoG as a regulator of invadopodia disassembly in human breast cancer cells. We showed that when RhoG expression is silenced, invadopodia are more stable and live longer, whereas its overexpression has the opposite effect. We also identified SGEF as the exchange factor that regulates RhoG activation during invadopodia disassembly. Silencing SGEF, but not other RhoG-specific GEFs, phenocopied the results obtained in RhoG KD cells with a significant increase in the number of cells that form invadopodia. SGEF has been shown to direct actin cytoskeleton remodeling mainly at the dorsal surface of the cells, where it promotes the formation of dorsal ruffles (Ellerbroek et al., 2004; Patel and Galán, 2006; van Buul et al., 2007). These actin-rich dorsal structures share several of its molecular components with invadopodia and podosomes, as well as with focal adhesions, suggesting that some of the signaling pathways controlling their assembly may also be conserved (Buccione et al., 2004). In addition, recent studies have shown that SGEF may play a role during invasion in human papillomavirus-mediated cervical cancer, prostate cancer and glioblastoma (Fortin Ensign et al., 2013; Krishna Subbaiah et al., 2012; Wang et al., 2013). The molecular mechanisms by which SGEF contributes to these processes are unclear. Our studies suggest that SGEF activity is tightly regulated during the lifetime of invadopodia and that it is recruited to invadopodia where it activates RhoG during invadopodia disassembly.
RhoG can directly influence the activity of Rac1 by forming a ternary complex with its effector protein ELMO and the Rac1 exchange factor DOCK180 (also known as DOCK1) (Brugnera et al., 2002; Katoh and Negishi, 2003). However, RhoG can also signal independently of Rac1 (Gauthier-Rouviere et al., 1998; Samson et al., 2010; Wennerberg et al., 2002). Our results demonstrate that Rac1 and RhoG play opposing roles in the regulation of invadopodia dynamics, with Rac1 being essential for invadopodia formation whereas RhoG is involved in their disassembly. Rac1 has been recently shown, together with Trio and Pak1, to function in invadopodia disassembly in rat mammary adenocarcinoma cells (Moshfegh et al., 2014). In that study, silencing the expression of Rac1 or Trio was also shown to promote an increase in invadopodia lifetime, without affecting invadopodia numbers (Moshfegh et al., 2014). Even though our results contradict those of Moshfegh and colleagues (,2014), they are in agreement with several reports which show Rac1 is required for invadopodia and podosome formation in different cell lines (Furmaniak-Kazmierczak et al., 2007; Harper et al., 2010; Lin et al., 2014; Nascimento et al., 2011; Pignatelli et al., 2012; Wheeler et al., 2006). Moreover, our results show that silencing SGEF or RhoG affects both invadopodia numbers and lifetime, whereas silencing Trio has no significant effect on the number of cells that form invadopodia. Our results also differ with recent studies that analyzed the role for RhoG in invadopodia. In glioblastoma cells, depletion of RhoG inhibits invadopodia formation (Kwiatkowska et al., 2012), whereas in rat breast cancer cells it appears to have no significant effect (Moshfegh et al., 2014). The differences between these studies and the results reported here may not be necessarily contradictory. In the studies by Kwiatkowska et al., invadopodia formation was determined by measuring matrix degradation area (Kwiatkowska et al., 2012), whereas in the studies by Moshfegh et al. invadopodia were followed directly in live rat breast cancer cells following EGF stimulation (Moshfegh et al., 2014). Since matrix degradation does not necessarily correlate with invadopodia number, it is difficult to compare these studies. Alternatively, these distinct effects might reflect differences specific to the cell types used or the experimental conditions, as already reported for other GTPases (Spuul et al., 2014).
Little is known regarding the extracellular signals that regulate RhoG activity. In this study, we show that there is a rapid and transient decrease of RhoG activity in the first 5 min after PDBu and PMA treatment followed by a peak of activation at 15 min (invadopodia formation peaks at 10 min after PDBu treatment). Phorbol esters stimulate the formation of podosomes and invadopodia by activating PKCα, which lies upstream of Src (Gatesman et al., 2004; Hai et al., 2002; Tatin et al., 2006) but the mechanisms that connect them with SGEF and RhoG regulation are not known and are the focus of our future studies.
Efficient turnover of invadopodia is critical for effective cell invasion (Chan et al., 2009). It has been reported that RhoG and SGEF promote cell migration and invasion in several cell lines including glioblastoma cells, MDA-MB-231 breast cancer cells, PC-3 prostate cancer cells and HeLa cells (Chatterjee et al., 2011; Fortin Ensign et al., 2013; Hiramoto-Yamaki et al., 2010; Katoh et al., 2006; Krishna Subbaiah et al., 2012; Kwiatkowska et al., 2012). Here, we also show that RhoG- and SGEF-deficient SUM159 cells show impaired invasion, despite showing enhanced invadopodia formation and ECM degradation. These results are consistent with the notion that the ability of cancer cells to form invadopodia alone is not sufficient for invasion to occur (Chan et al., 2009). This uncoupling between invadopodia formation and invasion has been described before, and may originate from the fact that several of the proteins involved in invadopodia formation also play a role during adhesion and migration. Silencing the expression of other proteins, including FAK, laminin-332 and ezrin, has also been shown to induce an increase in invadopodia while simultaneously decreasing their invasive capacity (Chan et al., 2009; Hoskin et al., 2015; Liu et al., 2010). It is possible that the invasion defect observed is related to impaired migration. Migration is also inhibited in RhoG KD cells, as has been shown previously by others (Hiramoto-Yamaki et al., 2010; Katoh et al., 2006). Our results and those of others suggest that RhoG may be working in parallel to regulate migration through Rac1 and invadopodia disassembly independently of Rac1 (Hiramoto et al., 2006; Katoh et al., 2006).
Despite the significant advances in the characterization of the very early stages of invadopodia assembly and the role of adhesion proteins in promoting invadopodia maturation (Beaty and Condeelis, 2014), our understanding of the mechanisms involved in invadopodia disassembly is limited. A report by Badowski and colleagues describes a role for paxillin in invadopodia disassembly (Badowski et al., 2008). Paxillin tyrosine phosphorylation (Y31 and Y118) promotes ERK protein activation, which activates calpain and stimulates disassembly at the inner rim of the invadopodia ring in Rous sarcoma virus (RSV)-transformed cells (Badowski et al., 2008). These results are in agreement with a large body of literature describing the role of paxillin phosphorylation in the regulation of focal adhesion disassembly and cell migration (Brown and Turner, 2004; Nakamura et al., 2000; Petit et al., 2000; Vindis et al., 2004; Webb et al., 2004; Zaidel-Bar et al., 2007). Supporting these observations, we found a decrease in paxillin phosphorylation and an increase in invadopodia lifetime in both RhoG and SGEF KD cells, suggesting a role in invadopodia disassembly. Surprisingly, even though paxillin phosphorylation at Y31 and Y118 is mostly associated with FAK and Src kinases (Brown and Turner, 2004), we found that Src and FAK activities are not correlated with RhoG-mediated regulation of paxillin phosphorylation. Instead, we detected two pools of phospho-paxillin, one that is regulated by RhoG independently of Src, and the other is dependent on Src and independent of RhoG. FAK, on the other hand, had no effect on RhoG-mediated phosphorylation of paxillin. Taken together, our results suggest that RhoG promotes paxillin phosphorylation through a yet to be identified kinase. Alternatively, RhoG may be mediating the effect on paxillin by inhibiting a phosphatase, such as SHP-2 or PTP-PEST (also known as PTPN11 and PTPN12, respectively), which have been shown to modulate paxillin phosphorylation (Cote et al., 1999; Jamieson et al., 2005; Mañes et al., 1999; Shen et al., 2000, 1998).
In our model, RhoG and Rac1 have independent and opposite roles in the regulation of invadopodia dynamics (Fig. 8). RhoG activity needs to be downregulated for invadopodia to assemble, whereas Rac1 activity is absolutely essential. We propose that a yet to-be-identified RhoG-specific GAP downregulates RhoG during invadopodia assembly. During invadopodia disassembly, SGEF is targeted to invadopodia, where it activates RhoG. Active RhoG then promotes the phosphorylation of the adaptor protein paxillin, which stimulates the disassembly of invadopodia. Taken together, our observations suggest that RhoG promotes invadopodia turnover as the cell protrudes and prepares for tissue invasion. Our findings provide novel insights into the mechanisms of RhoG signaling in breast cancer invasion.
Fig. 8.
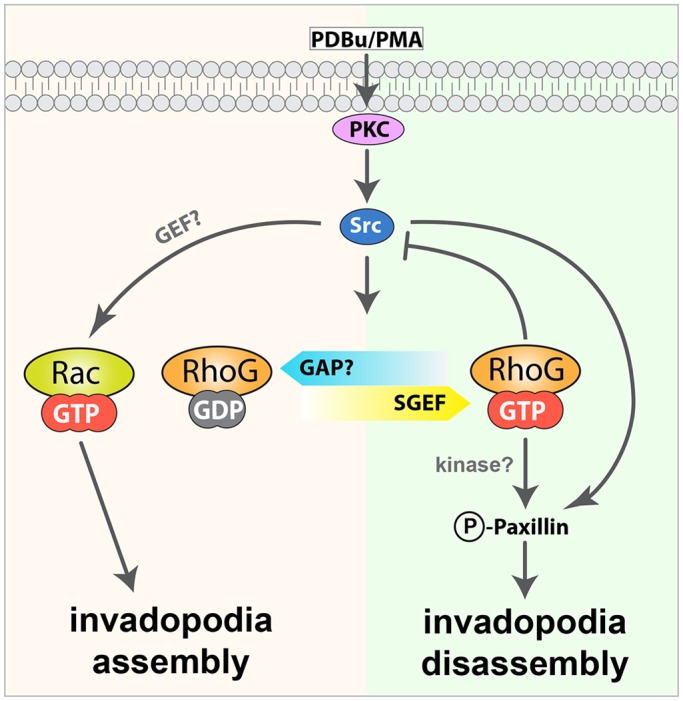
Model of RhoG function in invadopodia. Phorbol esters, including PDBu and PMA, stimulate PKC, which induces the formation of invadopodia in a Src-dependent fashion. Assembly of invadopodia requires Rac1 activity. In contrast, RhoG activity needs to be downregulated for invadopodia to form. During disassembly, SGEF is recruited to invadopodia, where it activates RhoG, which promotes the phosphorylation of paxillin.
MATERIALS AND METHODS
Cell lines
Three breast cancer cell lines were used: MCF7, MDA-MB-231 and SUM159. The cell lines were a gift from Carol Otey (UNC-Chapel Hill, NC). MCF7 and MDA-MB-231 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (GIBCO, Grand Island, NY) containing 10% fetal bovine serum (FBS) and antibiotics (penicillin-streptomycin). SUM159 cells were cultured in Ham's F12 with 5% calf serum, 5 μg/ml insulin, 1 μg/ml hydrocortisone and antibiotics. All cell lines were grown at 37°C and 5% CO2. All experiments were conducted with early passage cells that were passaged no more than 15 times. Mycoplasma was tested regularly by staining with Hoechst 33342 (83218, AnaSpec Inc., San Jose, CA).
Reagents
Antibodies against the following proteins were used: RhoG (sc-26484), Trio (sc-6060), cortactin (sc-11408) and Myc (9E10, sc-131) (Santa Cruz, Santa Cruz, CA); tubulin (T9028, Sigma, St. Louis, MO); pY118-paxillin (2541), total FAK (13009), pY397 FAK (8556), total Src (2108) and pY418 Src (2101) (Cell Signaling, Danvers, MA); total paxillin (610051) and Rac1 (610650; BD Biosciences, San Jose, CA). The antibody dilutions used are listed in Table S1. Secondary antibodies were: Alexa Fluor 488 and Alexa Fluor 594-conjugated anti-mouse-IgG and anti-rabbit-IgG secondary antibodies (A11008, A11001, A11005 and R37117) and Alexa Fluor-488 (A12379) and Alexa Fluor-647 (A22287) conjugated to phalloidin (Life Technologies, Carlsbad, CA), and horseradish peroxidase (HRP)-conjugated anti-mouse-IgG, anti-rabbit-IgG and anti-goat-IgG secondary antibodies (715-035-151, 711-035-152 and 705-035-147; Jackson Immunoresearch, West Grove, PA). PP2 (529573, Calbiochem, San Diego, CA). Phorbol-12,13-dibutyrate (PDBu) (P1269), Phorbol 12-myristate 13-acetate (PMA) (P8139) and PF-573228 (PZ0117) were from Sigma, St Louis, MO.
Transfections, immunofluorescence and treatments
Transfection of SUM159 cells was performed using Lipofectamine 2000 (Life Technologies, Carlsbad, CA). For immunofluorescence, MCF7, MDA-MB-231 and SUM159 cells grown on coverslips were fixed in 3.7% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS and then incubated with primary antibody for 1 h at room temperature. Primary antibodies were detected with Alexa Fluor 488- and Alexa Fluor 568-conjugated anti-mouse-IgG or anti-rabbit-IgG antibodies. Images were acquired on an Olympus IX81 inverted microscope using a PlanApo N 60×1.42 NA oil objective lens and a XM10 camera (Olympus, Tokyo, Japan). Image processing and quantitative analysis was performed using ImageJ. Invadopodia formation was induced by the addition of 1 μM PDBu or 7.5 μM PMA. To inhibit Src, cells were treated with PP2 (5 µM) or DMSO diluents for 30 min before treatment with PDBu. For FAK inhibition, we incubated cells with 5 μM PF-573228 for 30 min before PDBU treatment. Live imaging was performed with a Leica SP8 confocal microscope using a PlApo CS2 N 63×1.4 NA objective (Leica, Wetzlar, Germany), and equipped with an environmental chamber that controls temperature, CO2 and humidity (Tokai Hit, Fujinomiya, Japan).
Cell lysis and immunoblotting
Cells cultured on 100 mm tissue culture dishes were rinsed with PBS and then scraped into a lysis buffer containing 50 mM Tris-HCl pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100 and EZBlock protease inhibitor cocktail (BioVision, Mipitas, CA). The supernatant was collected after centrifugation at 16,800 g for 10 min. For immunoblotting, lysates were boiled in 2× Laemmli buffer, and 20 μg of protein were resolved by SDS-PAGE. The proteins were transferred onto PVDF and immunoblotted with the indicated antibodies. Immunocomplexes were visualized using the Immobilon Western Millipore Chemiluminescence HRP substrate (Millipore, Billerica, MA).
RhoG activity assay
Active RhoG pulldown experiments were performed as described previously (van Buul et al., 2007). Briefly, SUM159 cells were lysed in 50 mM Tris-HCl pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100, and EZBlock protease inhibitor cocktail. After clearing the lysates by centrifugation at 14,000 g for 5 min, the protein concentrations of the supernatants were determined, and equal amounts of total protein were incubated with 50 μg of glutathione transferase (GST)–ELMO (GST fusion protein containing the full-length RhoG effector ELMO) bound to glutathione–Sepharose beads (GE Healthcare, Pittsburgh, PA), and rotated for 30 min at 4°C. Subsequently the beads were washed four times in lysis buffer. Pull-downs and lysates were then immunoblotted for RhoG.
Gelatin degradation assay
Oregon Green 488-conjugated gelatin-coated coverslips were prepared as described previously (Martin et al., 2012). Coverslips were coated with 50 μg/ml poly-D-lysine for 15 min, washed with PBS and cross-linked with 0.5% glutaraldehyde for 15 min. Coverslips were then inverted on a 60 μl drop of 1 mg/ml Oregon Green 488-conjugated gelatin (Molecular Probes, ThermoFisher) for 20 min. After washing with PBS, coverslips were quenched with 5 mg/ml sodium borohydride for 5 min followed by washes with PBS. Finally, they were transferred into complete growth medium for 1 h before use. Cells were seeded and cultured on cross-linked gelatin for 16 h and then fixed for immunofluorescence studies. For each experimental condition, 25 images were taken in a random fashion. To quantify the gelatin degradation activity of invadopodia, we calculated the degradation area observed in images using ImageJ software and normalized the measurements to the total cell area in each image. Results are expressed as the percentage of the cell area that was degraded. At least 100 cells per experiment were analyzed.
Migration and invasion assays
Migration assays were carried out using 24-well non-coated Transwell plates (Corning, Lowel, MA) and invasion was analyzed using BD BioCoat growth-factor-reduced Matrigel Invasion Chambers (BD Biosciences, Bedford, MA). After 2 h of serum starvation, cells (1.5×104) were added to the upper chamber. The bottom chamber was filled with medium containing 10% FBS. Cells were allowed to migrate or invade for 16 h. Cells at the upper side of the membrane were removed using a Q-tip. Cells on the bottom surface were fixed and stained using Diff-Quick (IHC World, LLC, Woodstock, MD). Cells were counted from at least four individual experiments performed in triplicates.
DNA constructs
Generation of eukaryotic expression vectors pCMV-myc-RhoG-wt, wild type (WT) and mutant (E446A-N621A) Myc-tagged SGEF has been previously described (Ellerbroek et al., 2004). The RhoG Q61L was generated by site-directed mutagenesis using the Quickchange Site-Directed Mutagenesis kit (Stratagene, Santa Clara, CA). mCherry–Cortactin cDNA was a gift from James Bear, UNC-Chapel Hill, NC.
Overexpression using adenoviral system
shRNA-resistant versions of human Myc–RhoG, Myc–Rac1, Myc–SGEF and mCherry–Cortactin were subcloned into pAd/CMV/V5-DEST using Gateway recombination technology (Life Technologies Carlsbad, CA). shRNA resistance in RhoG and SGEF was achieved by introducing silence mutations in at least three bases within the corresponding shRNA targeting region. Virus particles were produced using the Virapower Adenoviral Expression System (Life Technologies, Carlsbad, CA).
CRISPR/Cas9-mediated KO
The Rac1 gene was knocked out using CRIPS/Cas9 double nickase plasmids (Santa Cruz Biotechnology, Santa Cruz, CA). Briefly, cells were transfected with the plasmid mixture (2 gRNA plasmids; strand A, 5′-AGACACGATCGAGAAACTGA-3′; strand B, 5′-TTTAGTTCCCACTAGGATGA-3′), and selected with puromycin 24 h after transfection. After selection, single cell colonies were isolated by serial dilution. The efficiency of the knockout was confirmed by western blotting.
Lentiviral constructs and transduction
pLKO lentiviral non-targeting shRNA control was from Sigma (SHC016-1EA). pLKO.1 shRNAs for human RhoG (#1 TRCN0000048018, #4 TRCN0000048021), SGEF (#4 TRCN0000048291, #5 TRCN0000048292), ephexin 4 (#1 TRCN0000047503, #3 TRCN0000047507), PLEKHG6 (#1 TRCN0000128030, #4 TRCN0000148892) and Trio (#1 TRCN0000000871, #5 TRCN0000010561) were from Open Biosystems (Huntsville, AL). Lentiviruses were prepared at the Lenti-shRNA Core Facility (UNC-Chapel Hill, NC). Cells were infected with lentivirus particles overnight. The following day, the infection medium was removed and replaced with complete medium containing puromycin (2.5 µg/ml) to select for shRNA-expressing cells. Total cell lysates were subjected to western blot analysis for protein expression as described above. For some shRNAs, single cell colonies were isolated by serial dilution.
qRT-PCR
Total RNA was purified from SGEF KD, PLEKHG6 KD and Ephexin4 KD SUM159 cells using Trizol (Life Technologies, CA) and was treated with DNAse I (NEB, Ipswich, MA). Reverse transcription was carried out using the iScript cDNA Synthesis kit (BioRad, Hercules, CA) on 1 µg of total RNA. qRT-PCR was performed with equal amounts of cDNA using the Taq PCR Master Mix kit (Qiagen, Valencia, CA); primer sequences will be made available upon request.
Statistical analysis
Values calculated from at least three independent experiments were compared by a Student's t-test using GraphPad Prism (La Jolla, CA), and P<0.05 was considered statistically significant. Error bars represent the s.e.m.
Acknowledgements
We would like to thank Carol Otey (UNC-Chapel Hill, NC) for sharing the cell lines utilized in these studies and James Bear (UNC-Chapel Hill, NC) for the mCherry–cortactin cDNA.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization and Methdology, S.M.G. and R.G.-M.; Formal Analysis, R.G.-M, S.M.G., A.Z. and K.S.; Investigation, R.G.-M, S.M.G., A.Z., S.A. and K.S. Writing – Original Draft, Writing – Review and Editing and Visualization, S.M.G. and R.G.-M. Supervision, S.M.G. and R.G.-M. Project Administration, R.G-M. Funding Acquisition, S.M.G. and R.G.-M.
Funding
R.G.-M. was supported by the National Institutes of Health (1R21CA194776-01A1, 1R15CA199101-01A1 and 1R03CA197227-01A1), Ohio Cancer Research, and the deArce-Koch Memorial Endowment fund. S.M.G. was supported by the National Institutes of Health (1R03CA161136-01). Deposited in PMC for release after 12 months.
Data availability
Supplementary movies are also available from the Dryad Digital Repository (http://dx.doi.org/10.5061/dryad.q1j50).
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.195552.supplemental
References
- Artym V. V., Yamada K. M. and Mueller S. C. (2009). ECM degradation assays for analyzing local cell invasion. Methods Mol. Biol. 522, 211-219. 10.1007/978-1-59745-413-1_15 [DOI] [PubMed] [Google Scholar]
- Ayala I., Giacchetti G., Caldieri G., Attanasio F., Mariggio S., Tete S., Polishchuk R., Castronovo V. and Buccione R. (2009). Faciogenital dysplasia protein Fgd1 regulates invadopodia biogenesis and extracellular matrix degradation and is up-regulated in prostate and breast cancer. Cancer Res. 69, 747-752. 10.1158/0008-5472.CAN-08-1980 [DOI] [PubMed] [Google Scholar]
- Badowski C., Pawlak G., Grichine A., Chabadel A., Oddou C., Jurdic P., Pfaff M., Albiges-Rizo C. and Block M. R. (2008). Paxillin phosphorylation controls invadopodia/podosomes spatiotemporal organization. Mol. Biol. Cell 19, 633-645. 10.1091/mbc.E06-01-0088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty B. T. and Condeelis J. (2014). Digging a little deeper: the stages of invadopodium formation and maturation. Eur. J. Cell Biol. 93, 438-444. 10.1016/j.ejcb.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellanger J.-M., Astier C., Sardet C., Ohta Y., Stossel T. P. and Debant A. (2000). The Rac1- and RhoG-specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nat. Cell Biol. 2, 888-892. 10.1038/35046533 [DOI] [PubMed] [Google Scholar]
- Bellis S. L., Miller J. T. and Turner C. E. (1995). Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J. Biol. Chem. 270, 17437-17441. 10.1074/jbc.270.29.17437 [DOI] [PubMed] [Google Scholar]
- Blangy A., Vignal E., Schmidt S., Debant A., Gauthier-Rouviere C. and Fort P. (2000). TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of rhoG. J. Cell Sci. 113, 729-739. [DOI] [PubMed] [Google Scholar]
- Brown M. C. and Turner C. E. (2004). Paxillin: adapting to change. Physiol. Rev. 84, 1315-1339. 10.1152/physrev.00002.2004 [DOI] [PubMed] [Google Scholar]
- Brugnera E., Haney L., Grimsley C., Lu M., Walk S. F., Tosello-Trampont A. C., Macara I. G., Madhani H., Fink G. R. and Ravichandran K. S. (2002). Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat. Cell Biol. 4, 574-582. 10.1038/ncb824 [DOI] [PubMed] [Google Scholar]
- Buccione R., Orth J. D. and McNiven M. A. (2004). Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat. Rev. Mol. Cell Biol. 5, 647-657. 10.1038/nrm1436 [DOI] [PubMed] [Google Scholar]
- Bustelo X. R., Sauzeau V. and Berenjeno I. M. (2007). GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 29, 356-370. 10.1002/bies.20558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K. T., Cortesio C. L. and Huttenlocher A. (2009). FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J. Cell Biol. 185, 357-370. 10.1083/jcb.200809110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee M., Sequeira L., Jenkins-Kabaila M., Dubyk C. W., Pathak S. and van Golen K. L. (2011). Individual rac GTPases mediate aspects of prostate cancer cell and bone marrow endothelial cell interactions. J. Signal. Transduct. 2011, 541851 10.1155/2011/541851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.-T., Chen J.-M., Parsons S. J. and Parsons J. T. (1985). Local degradation of fibronectin at sites of expression of the transforming gene product pp60src. Nature 316, 156-158. 10.1038/316156a0 [DOI] [PubMed] [Google Scholar]
- Cote J.-F., Turner C. E. and Tremblay M. L. (1999). Intact LIM 3 and LIM 4 domains of paxillin are required for the association to a novel polyproline region (Pro 2) of protein-tyrosine phosphatase-PEST. J. Biol. Chem. 274, 20550-20560. 10.1074/jbc.274.29.20550 [DOI] [PubMed] [Google Scholar]
- Damoulakis G., Gambardella L., Rossman K. L., Lawson C. D., Anderson K. E., Fukui Y., Welch H. C., Der C. J., Stephens L. R. and Hawkins P. T. (2014). P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils. J. Cell Sci. 127, 2589-2600. 10.1242/jcs.153049 [DOI] [PubMed] [Google Scholar]
- D'Angelo R., Aresta S., Blangy A., Del Maestro L., Louvard D. and Arpin M. (2007). Interaction of Ezrin with the novel guanine nucleotide exchange factor PLEKHG6 promotes RhoG-dependent apical cytoskeleton rearrangements in epithelial cells. Mol. Biol. Cell 18, 4780-4793. 10.1091/mbc.E06-12-1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- deBakker C. D., Haney L. B., Kinchen J. M., Grimsley C., Lu M., Klingele D., Hsu P.-K., Chou B.-K., Cheng L.-C., Blangy A. et al. (2004). Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr. Biol. 14, 2208-2216. 10.1016/j.cub.2004.12.029 [DOI] [PubMed] [Google Scholar]
- Di Martino J., Paysan L., Gest C., Lagree V., Juin A., Saltel F. and Moreau V. (2014). Cdc42 and Tks5: a minimal and universal molecular signature for functional invadosomes. Cell Adh. Migr. 8, 280-292. 10.4161/cam.28833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfenbein A., Rhodes J. M., Meller J., Schwartz M. A., Matsuda M. and Simons M. (2009). Suppression of RhoG activity is mediated by a syndecan 4-synectin-RhoGDI1 complex and is reversed by PKCalpha in a Rac1 activation pathway. J. Cell Biol. 186, 75-83. 10.1083/jcb.200810179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellerbroek S. M., Wennerberg K., Arthur W. T., Dunty J. M., Bowman D. R., DeMali K. A., Der C. and Burridge K. (2004). SGEF, a RhoG guanine nucleotide exchange factor that stimulates macropinocytosis. Mol. Biol. Cell 15, 3309-3319. 10.1091/mbc.E04-02-0146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin Ensign S. P., Mathews I. T., Eschbacher J. M., Loftus J. C., Symons M. H. and Tran N. L. (2013). The Src homology 3 domain-containing guanine nucleotide exchange factor is overexpressed in high-grade gliomas and promotes tumor necrosis factor-like weak inducer of apoptosis-fibroblast growth factor-inducible 14-induced cell migration and invasion via tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 288, 21887-21897. 10.1074/jbc.M113.468686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furmaniak-Kazmierczak E., Crawley S. W., Carter R. L., Maurice D. H. and Cote G. P. (2007). Formation of extracellular matrix-digesting invadopodia by primary aortic smooth muscle cells. Circ. Res. 100, 1328-1336. 10.1161/CIRCRESAHA.106.147744 [DOI] [PubMed] [Google Scholar]
- Gatesman A., Walker V. G., Baisden J. M., Weed S. A. and Flynn D. C. (2004). Protein kinase C{alpha} activates c-Src and induces podosome formation via AFAP-110. Mol. Cell. Biol. 24, 7578-7597. 10.1128/MCB.24.17.7578-7597.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier-Rouviere C., Vignal E., Meriane M., Roux P., Montcourier P. and Fort P. (1998). RhoG GTPase controls a pathway that independently activates Rac1 and Cdc42Hs. Mol. Biol. Cell 9, 1379-1394. 10.1091/mbc.9.6.1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimona M., Buccione R., Courtneidge S. A. and Linder S. (2008). Assembly and biological role of podosomes and invadopodia. Curr. Opin. Cell Biol. 20, 235-241. 10.1016/j.ceb.2008.01.005 [DOI] [PubMed] [Google Scholar]
- Goicoechea S. M., Bednarski B., García-Mata R., Prentice-Dunn H., Kim H. J. and Otey C. A. (2009). Palladin contributes to invasive motility in human breast cancer cells. Oncogene 28, 587-598. 10.1038/onc.2008.408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai C.-M., Hahne P., Harrington E. O. and Gimona M. (2002). Conventional protein kinase C mediates phorbol-dibutyrate-induced cytoskeletal remodeling in a7r5 smooth muscle cells. Exp. Cell Res. 280, 64-74. 10.1006/excr.2002.5592 [DOI] [PubMed] [Google Scholar]
- Harper K., Arsenault D., Boulay-Jean S., Lauzier A., Lucien F. and Dubois C. M. (2010). Autotaxin promotes cancer invasion via the lysophosphatidic acid receptor 4: participation of the cyclic AMP/EPAC/Rac1 signaling pathway in invadopodia formation. Cancer Res. 70, 4634-4643. 10.1158/0008-5472.CAN-09-3813 [DOI] [PubMed] [Google Scholar]
- Hiramoto K., Negishi M. and Katoh H. (2006). Dock4 is regulated by RhoG and promotes Rac-dependent cell migration. Exp. Cell Res. 312, 4205-4216. 10.1016/j.yexcr.2006.09.006 [DOI] [PubMed] [Google Scholar]
- Hiramoto-Yamaki N., Takeuchi S., Ueda S., Harada K., Fujimoto S., Negishi M. and Katoh H. (2010). Ephexin4 and EphA2 mediate cell migration through a RhoG-dependent mechanism. J. Cell Biol. 190, 461-477. 10.1083/jcb.201005141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge R. G. and Ridley A. J. (2016). Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 17, 496-510. 10.1038/nrm.2016.67 [DOI] [PubMed] [Google Scholar]
- Hoshino D., Jourquin J., Emmons S. W., Miller T., Goldgof M., Costello K., Tyson D. R., Brown B., Lu Y., Prasad N. K. et al. (2012). Network analysis of the focal adhesion to invadopodia transition identifies a PI3K-PKCalpha invasive signaling axis. Sci. Signal. 5, ra66 10.1126/scisignal.2002964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino D., Branch K. M. and Weaver A. M. (2013). Signaling inputs to invadopodia and podosomes. J. Cell Sci. 126, 2979-2989. 10.1242/jcs.079475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskin V., Szeto A., Ghaffari A., Greer P. A., Cote G. P. and Elliott B. E. (2015). Ezrin regulates focal adhesion and invadopodia dynamics by altering calpain activity to promote breast cancer cell invasion. Mol. Biol. Cell 26, 3464-3479. 10.1091/mbc.E14-12-1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson B. C., Ivanova I. A. and Dagnino L. (2015). An ELMO2-RhoG-ILK network modulates microtubule dynamics. Mol. Biol. Cell 26, 2712-2725. 10.1091/mbc.E14-10-1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson J. S., Tumbarello D. A., Halle M., Brown M. C., Tremblay M. L. and Turner C. E. (2005). Paxillin is essential for PTP-PEST-dependent regulation of cell spreading and motility: a role for paxillin kinase linker. J. Cell Sci. 118, 5835-5847. 10.1242/jcs.02693 [DOI] [PubMed] [Google Scholar]
- Katoh H. and Negishi M. (2003). RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature 424, 461-464. 10.1038/nature01817 [DOI] [PubMed] [Google Scholar]
- Katoh H., Yasui H., Yamaguchi Y., Aoki J., Fujita H., Mori K. and Negishi M. (2000). Small GTPase RhoG is a key regulator for neurite outgrowth in PC12 cells. Mol. Cell. Biol. 20, 7378-7387. 10.1128/MCB.20.19.7378-7387.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh H., Hiramoto K. and Negishi M. (2006). Activation of Rac1 by RhoG regulates cell migration. J. Cell Sci. 119, 56-65. 10.1242/jcs.02720 [DOI] [PubMed] [Google Scholar]
- Krishna Subbaiah V., Massimi P., Boon S. S., Myers M. P., Sharek L., Garcia-Mata R. and Banks L. (2012). The invasive capacity of HPV transformed cells requires the hDlg-dependent enhancement of SGEF/RhoG activity. PLoS Pathog. 8, e1002543 10.1371/journal.ppat.1002543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowska A., Didier S., Fortin S., Chuang Y., White T., Berens M. E., Rushing E., Eschbacher J., Tran N. L., Chan A. et al. (2012). The small GTPase RhoG mediates glioblastoma cell invasion. Mol. Cancer 11, 65 10.1186/1476-4598-11-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.-W., Sun M.-S., Liao M.-Y., Chung C.-H., Chi Y.-H., Chiou L.-T., Yu J., Lou K.-L. and Wu H.-C. (2014). Podocalyxin-like 1 promotes invadopodia formation and metastasis through activation of Rac1/Cdc42/cortactin signaling in breast cancer cells. Carcinogenesis 35, 2425-2435. 10.1093/carcin/bgu139 [DOI] [PubMed] [Google Scholar]
- Linder S., Wiesner C. and Himmel M. (2011). Degrading devices: invadosomes in proteolytic cell invasion. Annu. Rev. Cell Dev. Biol. 27, 185-211. 10.1146/annurev-cellbio-092910-154216 [DOI] [PubMed] [Google Scholar]
- Liu S., Yamashita H., Weidow B., Weaver A. M. and Quaranta V. (2010). Laminin-332-beta1 integrin interactions negatively regulate invadopodia. J. Cell Physiol. 223, 134-142. 10.1002/jcp.22018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mañes S., Mira E., Gómez-Mouton C., Zhao Z. J., Lacalle R. A. and Martínez-A C. (1999). Concerted activity of tyrosine phosphatase SHP-2 and focal adhesion kinase in regulation of cell motility. Mol. Cell. Biol. 19, 3125-3135. 10.1128/MCB.19.4.3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K. H., Hayes K. E., Walk E. L., Ammer A. G., Markwell S. M. and Weed S. A. (2012). Quantitative measurement of invadopodia-mediated extracellular matrix proteolysis in single and multicellular contexts. J. Vis. Exp., e4119 10.3791/4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May V., Schiller M. R., Eipper B. A. and Mains R. E. (2002). Kalirin Dbl-homology guanine nucleotide exchange factor 1 domain initiates new axon outgrowths via RhoG-mediated mechanisms. J. Neurosci. 22, 6980-6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau V., Tatin F., Varon C. and Genot E. (2003). Actin can reorganize into podosomes in aortic endothelial cells, a process controlled by Cdc42 and RhoA. Mol. Cell. Biol. 23, 6809-6822. 10.1128/MCB.23.19.6809-6822.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau V., Tatin F., Varon C., Anies G., Savona-Baron C. and Genot E. (2006). Cdc42-driven podosome formation in endothelial cells. Eur. J. Cell Biol. 85, 319-325. 10.1016/j.ejcb.2005.09.009 [DOI] [PubMed] [Google Scholar]
- Moshfegh Y., Bravo-Cordero J. J., Miskolci V., Condeelis J. and Hodgson L. (2014). A Trio-Rac1-Pak1 signalling axis drives invadopodia disassembly. Nat. Cell Biol. 16, 574-586. 10.1038/ncb2972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy D. A. and Courtneidge S. A. (2011). The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 12, 413-426. 10.1038/nrm3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara H., Otani T., Sasaki T., Miura Y., Takai Y. and Kogo M. (2003). Involvement of Cdc42 and Rac small G proteins in invadopodia formation of RPMI7951 cells. Genes Cells 8, 1019-1027. 10.1111/j.1365-2443.2003.00695.x [DOI] [PubMed] [Google Scholar]
- Nakamura K., Yano H., Uchida H., Hashimoto S., Schaefer E. and Sabe H. (2000). Tyrosine phosphorylation of paxillin alpha is involved in temporospatial regulation of paxillin-containing focal adhesion formation and F-actin organization in motile cells. J. Biol. Chem. 275, 27155-27164. [DOI] [PubMed] [Google Scholar]
- Nascimento C. F., de Siqueira A. S., Pinheiro J. J. V., Freitas V. M. and Jaeger R. G. (2011). Laminin-111 derived peptides AG73 and C16 regulate invadopodia activity of a human adenoid cystic carcinoma cell line. Exp. Cell Res. 317, 2562-2572. 10.1016/j.yexcr.2011.08.022 [DOI] [PubMed] [Google Scholar]
- Oikawa T., Itoh T. and Takenawa T. (2008). Sequential signals toward podosome formation in NIH-src cells. J. Cell Biol. 182, 157-169. 10.1083/jcb.200801042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J. C. and Galán J. E. (2006). Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J. Cell Biol. 175, 453-463. 10.1083/jcb.200605144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit V., Boyer B., Lentz D., Turner C. E., Thiery J. P. and Vallés A. M. (2000). Phosphorylation of tyrosine residues 31 and 118 on paxillin regulates cell migration through an association with CRK in NBT-II cells. J. Cell Biol. 148, 957-970. 10.1083/jcb.148.5.957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli J., Tumbarello D. A., Schmidt R. P. and Turner C. E. (2012). Hic-5 promotes invadopodia formation and invasion during TGF-beta-induced epithelial-mesenchymal transition. J. Cell Biol. 197, 421-437. 10.1083/jcb.201108143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman K. L., Der C. J. and Sondek J. (2005). GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167-180. 10.1038/nrm1587 [DOI] [PubMed] [Google Scholar]
- Samson T., Welch C., Monaghan-Benson E., Hahn K. M. and Burridge K. (2010). Endogenous RhoG is rapidly activated after epidermal growth factor stimulation through multiple guanine-nucleotide exchange factors. Mol. Biol. Cell 21, 1629-1642. 10.1091/mbc.E09-09-0809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M. D. and Parsons J. T. (1995). pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol. Cell. Biol. 15, 2635-2645. 10.1128/MCB.15.5.2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Schneider G., Cloutier J.-F., Veillette A. and Schaller M. D. (1998). Direct association of protein-tyrosine phosphatase PTP-PEST with paxillin. J. Biol. Chem. 273, 6474-6481. 10.1074/jbc.273.11.6474 [DOI] [PubMed] [Google Scholar]
- Shen Y., Lyons P., Cooley M., Davidson D., Veillette A., Salgia R., Griffin J. D. and Schaller M. D. (2000). The noncatalytic domain of protein-tyrosine phosphatase-PEST targets paxillin for dephosphorylation in vivo. J. Biol. Chem. 275, 1405-1413. 10.1074/jbc.275.2.1405 [DOI] [PubMed] [Google Scholar]
- Spuul P., Ciufici P., Veillat V., Leclercq A., Daubon T., Kramer I. J. and Génot E. (2014). Importance of RhoGTPases in formation, characteristics, and functions of invadosomes. Small GTPases 5, e28195 10.4161/sgtp.28713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatin F., Varon C., Génot E. and Moreau V. (2006). A signalling cascade involving PKC, Src and Cdc42 regulates podosome assembly in cultured endothelial cells in response to phorbol ester. J. Cell Sci. 119, 769-781. 10.1242/jcs.02787 [DOI] [PubMed] [Google Scholar]
- Tcherkezian J. and Lamarche-Vane N. (2007). Current knowledge of the large RhoGAP family of proteins. Biol. Cell 99, 67-86. 10.1042/BC20060086 [DOI] [PubMed] [Google Scholar]
- van Buul J. D., Allingham M. J., Samson T., Meller J., Boulter E., García-Mata R. and Burridge K. (2007). RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J. Cell Biol. 178, 1279-1293. 10.1083/jcb.200612053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindis C., Teli T., Cerretti D. P., Turner C. E. and Huynh-Do U. (2004). EphB1-mediated cell migration requires the phosphorylation of paxillin at Tyr-31/Tyr-118. J. Biol. Chem. 279, 27965-27970. 10.1074/jbc.M401295200 [DOI] [PubMed] [Google Scholar]
- Wang H., Li S.-H., Li H., Li C., Guan K., Luo G., Yu L., Wu R.-Q., Zhang X., Wang J. et al. (2013). SGEF enhances EGFR stability through delayed EGFR trafficking from early to late endosomes. Carcinogenesis. 34, 1976-1983. 10.1093/carcin/bgt157 [DOI] [PubMed] [Google Scholar]
- Webb D. J., Donais K., Whitmore L. A., Thomas S. M., Turner C. E., Parsons J. T. and Horwitz A. F. (2004). FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6, 154-161. 10.1038/ncb1094 [DOI] [PubMed] [Google Scholar]
- Wennerberg K., Ellerbroek S. M., Liu R.-Y., Karnoub A. E., Burridge K. and Der C. J. (2002). RhoG signals in parallel with Rac1 and Cdc42. J. Biol. Chem. 277, 47810-47817. 10.1074/jbc.M203816200 [DOI] [PubMed] [Google Scholar]
- Wheeler A. P., Wells C. M., Smith S. D., Vega F. M., Henderson R. B., Tybulewicz V. L. and Ridley A. J. (2006). Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. J. Cell Sci. 119, 2749-2757. 10.1242/jcs.03024 [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R., Milo R., Kam Z. and Geiger B. (2007). A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J. Cell Sci. 120, 137-148. 10.1242/jcs.03314 [DOI] [PubMed] [Google Scholar]