

Significance
Receptor-interacting protein kinase 3 (RIP3) is involved in many important biological processes such as necroptosis, apoptosis, and inflammation. Here, using RIP3-knockout mice, we show that RIP3 is essential for in vivo thrombosis and hemostasis. Mice lacking RIP3 exhibited prolonged tail-bleeding times and reduced arterial thrombus formation. We demonstrate that RIP3, expressed in platelets and interacting with Gα13, promotes platelet aggregation, secretion, spreading on fibrinogen, and clot retraction. RIP3 inhibitor dose-dependently inhibits platelet aggregation and prevents arterial thrombus formation. Our findings indicate a role for RIP3 in promoting in vivo thrombosis and hemostasis by amplifying platelet activation. RIP3 may represent a novel therapeutic target for thrombotic diseases.
Keywords: platelets, receptor-interacting protein kinase 3, thrombin, thromboxane A2, thrombosis
Abstract
Previous studies have shown that receptor-interacting protein kinase 3 (RIP3) is involved in many important biological processes, including necroptosis, apoptosis, and inflammation. Here we show that RIP3 plays a critical role in regulating platelet functions and in vivo thrombosis and hemostasis. Tail bleeding times were significantly longer in RIP3-knockout (RIP3−/−) mice compared with their wild-type (WT) littermates. In an in vivo model of arteriole thrombosis, mice lacking RIP3 exhibited prolonged occlusion times. WT mice repopulated with RIP3−/− bone marrow-derived cells had longer occlusion times than RIP3−/− mice repopulated with WT bone marrow-derived cells, suggesting a role for RIP3-deficient platelets in arterial thrombosis. Consistent with these findings, we observed that RIP3 was expressed in both human and mice platelets. Deletion of RIP3 in mouse platelets caused a marked defect in aggregation and attenuated dense granule secretion in response to low doses of thrombin or a thromboxane A2 analog, U46619. Phosphorylation of Akt induced by U46619 or thrombin was diminished in RIP3−/− platelets. Moreover, RIP3 interacted with Gα13. Platelet spreading on fibrinogen and clot retraction were impaired in the absence of RIP3. RIP3 inhibitor dose-dependently inhibited platelet aggregation in vitro and prevented arterial thrombus formation in vivo. These data demonstrate a role for RIP3 in promoting in vivo thrombosis and hemostasis by amplifying platelet activation. RIP3 may represent a novel promising therapeutic target for thrombotic diseases.
On vessel injury, platelets are recruited by soluble agonists, such as ADP, thrombin, and thromboxane A2 (TXA2), which interact with G protein-coupled receptors (GPCRs), leading to integrin inside-out activation and granule secretion (1–4). Ligand binding to integrin triggers outside-in signaling, initiating stable adhesion, spreading, clot retraction, and a second wave of secretion, which can result in an irreversible platelet plug and serious cardiovascular thrombotic events, including myocardial infarction and stroke (1, 5–7). Despite the well-established knowledge of the mechanisms of thrombus formation, which has contributed to the development of antithrombotic drugs, the signaling cascades leading to platelet activation are not completely understood.
The receptor-interacting protein kinase (RIP) family is a group of Ser/Thr protein kinases that share a highly homologous amino terminal kinase domain but have distinctly different carboxyl termini (8, 9). The family comprises seven members, designated RIP1–7 (8, 9). RIP3 has a carboxyl terminus that contains the RIP homotypic interaction motif, which mediates its interaction with other necroptosis-related proteins (10, 11). Downstream of death receptors, Toll-like receptors, or other sensors, RIP3 activation is tightly regulated by phosphorylation (12–15) or caspase-mediated cleavage (16, 17). Once activated, RIP3 phosphorylates its substrates, leading to programmed necrosis (13–15). Many phosphoproteins associated with the cell cycle, metabolism, or other diverse functions are regulated by RIP3 (18), suggesting that RIP3 might function beyond programmed necrosis. Indeed, the roles of RIP3 in embryonic development and various disease pathologies, such as inflammation and apoptosis, have been reported (8–10). A recent report indicated that RIP3-mediated necrotic death in areas of atherosclerotic plaques accelerates systematic inflammation and results in premature death of the animal, suggesting the possible involvement of RIP3 in cardiovascular diseases (19). The role of RIP3 in thrombosis and hemostasis has not been explored previously, however.
In this study, we demonstrate that RIP3 is expressed in both human and mouse platelets and promotes hemostasis and thrombus formation in vivo. We show that RIP3 selectively amplifies thrombin- and thromboxane A2 analog (U46619)-induced platelet aggregation and dense granule secretion. We found that RIP3 interacts with Gα13, and that Akt activation, spreading on fibrinogen, and clot retraction are defective in RIP3-deficient platelets. Moreover, RIP3 inhibitor dose-dependently inhibits platelet aggregation in vitro, and prevents thrombus formation in vivo. These results demonstrate a novel role for RIP3 in amplifying platelet activation and thrombus formation.
Results
RIP3−/− Mice Display Impaired Hemostasis and Thrombosis.
To investigate the role of RIP3 in hemostasis in vivo, we examined tail bleeding times in RIP3−/− mice. The generation of RIP3−/− mice has been described previously (11). The RIP3−/− and wild-type (WT) mice did not differ significantly in terms of RBCs, WBCs, or hemoglobin concentration (Table S1). RIP3−/− mice exhibited no tendency toward spontaneous bleeding or thrombotic events over the lifespan; however, their median tail bleeding time was significantly longer than that of WT littermates (410 s vs. 156 s; P < 0.001) (Fig. 1A). Moreover, in 44.4% of the RIP3−/− mice (vs. 16.7% of WT mice), bleeding persisted during a 10-min observation period. These data indicate that RIP3 plays an important role in primary hemostasis.
Table S1.
Hematologic analysis of WT and RIP3−/− mice
| Variable | WT (n = 27) | RIP3−/− (n = 26) |
| RBC, ×1012/L, mean ± SEM | 7.935 ± 0.113 | 8.186 ± 0.185 |
| WBC, ×109/L, mean ± SEM | 3.779 ± 0.233 | 4.421 ± 0.308 |
| Hemoglobin, g/dL, mean ± SEM | 12.37 ± 0.196 | 12.57 ± 0.265 |
| MCV, fL, mean ± SEM | 51.47 ± 0.274 | 49.92 ± 0.261* |
| MPV, fL, mean ± SEM | 5.648 ± 0.029 | 5.712 ± 0.046 |
MCV, erythrocyte mean corpuscular volume; MPV, mean platelet volume. No significant difference between WT and RIP3−/− mice was observed for RBC, WBC, hemoglobin, and MPV.
P < 0.001.
Fig. 1.

The effects of RIP3 knockout on in vivo hemostasis and thrombosis. (A) Tail bleeding time determined in groups of 36 WT and 36 RIP3−/− mice. Horizontal lines indicated the median values. Statistical analysis was performed using the two-tailed Mann–Whitney test and revealed a significant difference between the two groups of animals. ***P < 0.001. (B) Representative images of FeCl3-induced mesenteric arteriole thrombosis in WT and RIP3−/− mice as recorded by real-time microscopy. Time after FeCl3-induced injury is indicated at the bottom right of each image. (C) Occlusion time of the mesenteric arteriole as measured by FeCl3 injury in WT and RIP3−/− mice. Data are mean ± SEM values of 11 WT mice and 10 RIP3−/− mice. **P = 0.0012.
We investigated the role of RIP3 in arterial thrombosis in vivo using an FeCl3-injured mouse mesenteric arteriole thrombosis model. Platelet thrombus formation was monitored in real time by fluorescence microscopy. Compared with their WT littermates, RIP3−/− mice exhibited delayed and diminished thrombus formation (Fig. 1B). Mean occlusion time was significantly longer in the RIP3−/− mice (19.9 ± 1.1 min vs. 13.5 ± 1.2 min; P = 0.0012) (Fig. 1C). These data demonstrate that RIP3 promotes arteriole thrombus formation in vivo.
RIP3 Is Expressed in Platelets and Responsible for Impaired Platelet Thrombus Formation in RIP3−/− Mice.
Platelets play key roles in thrombosis and hemostasis. Our immunoblotting data indicate that RIP3 is expressed in both human and mouse platelets (Fig. 2A). As expected, RIP3 protein is ablated in the platelets from RIP3−/− mice, with WBCs as positive controls (20). To exclude a possibility that the defects in thrombosis and hemostasis of the RIP3−/− mice results from lack of RIP3 in vascular cells, such as endothelial cells, WT and RIP3−/− mice were repopulated with RIP3−/− or WT mouse bone marrow-derived cells, respectively, via bone marrow transplantation. The expression levels of RIP3 in platelets from RIP3−/− mice repopulated with WT mouse bone marrow-derived cells were similar to those in platelets from WT mice, whereas RIP3 expression in the platelets from WT mice repopulated with RIP3−/− bone marrow-derived cells was abolished (Fig. 2B). Occlusion times were significantly longer in WT mice repopulated with RIP3−/− bone marrow compared with RIP3−/− mice repopulated with WT bone marrow (18.4 ± 1.2 min in RIP3−/−→WT mice vs. 12.5 ± 0.9 min in WT→RIP3−/− mice; P = 0.0047) (Fig. 2C). Moreover, occlusion times of WT→WT and WT→RIP3−/− mice were similar to those of WT mice, and occlusion times of RIP3−/−→RIP3−/− and RIP3−/−→WT mice were also similar to those of RIP3−/− mice (Fig. 2C). These data indicate that a lack of RIP3 in bone marrow cells, presumably in platelets, is responsible for the impaired thrombus formation in the RIP3−/− mice.
Fig. 2.

Characterization of platelets from RIP3-deficient mice. (A) Whole blood and platelets were prepared as described in SI Materials and Methods. The washed platelets and leukocytes were lysed and probed by immunoblotting with antibodies against RIP3 and CD45 (marker for leukocytes). Blotting to β-actin was used as a lane loading control. The figures are representative of three independent experiments. (B) Expression of RIP3 in platelets from RIP3−/− (WT→RIP3−/−; n = 5) and WT (RIP3−/− →WT; n = 7) mice repopulated with WT and RIP3−/− donor bone marrow- derived cells, respectively, was detected by Western blot analysis with anti-RIP3 antibody. Platelets from WT and RIP3−/− mice were used as controls, and β-actin served as a lane loading control. (C) Occlusion time of the mesenteric arteriole was measured by FeCl3 injury in WT and RIP3−/− mice repopulated with either WT or RIP3−/− donor bone marrow- derived cells. Data are mean ± SEM values. n = 5∼7. *P = 0.035; **P = 0.0047. (D) Electron microscopy analysis of WT and RIP3−/− mouse platelets. Representative images were obtained at 1,000× and 10,000×, respectively. (E) Platelet counts were detected in whole blood from WT and RIP3−/− mice (n = 26, equivalent numbers of males and females). Results are expressed as mean ± SEM. *P < 0.05. (F) Quantification of α-granules (AG) and dense granules (DG) was calculated from 20 platelets of five different fields of view for each genotype. Data are expressed as mean ± SEM. (G) Surface expression of the major glycoproteins on WT and RIP3−/− mouse platelets on membranes determined by flow cytometry with specific antibodies. Data are expressed as the mean fluorescence intensity (MFI) ± SEM of three different experiments.
In the RIP3−/− mice, platelet size and morphology were normal (Table S1 and Fig. 2D), but the number of circulating platelets was slightly elevated (Fig. 2E). The numbers of α-granules and dense granules were comparable in the two genotypes (Fig. 2F). There were no significant differences in the surface expression of the major platelet glycoproteins (GP), CD42b (GPIbα), and CD41 (αIIb subunit) between WT and RIP3−/− platelets (Fig. 2G).
Impaired Platelet Aggregation Is Induced by U46619 and Thrombin in the Absence of RIP3.
We next investigated the role of RIP3 in platelet function by examining the effect of RIP3 deficiency on agonist-induced platelet aggregation. Platelets lacking RIP3 showed a marked defect in aggregation in response to low concentrations of thrombin or a thromboxane A2 analog, U46619 (Fig. 3 A and B). However, aggregation induced by collagen (Fig. 3C) and ADP (Fig. 3D) was not obviously different between WT and RIP3−/− platelets. To exclude the possibility that the decreased platelet aggregation induced by thrombin or U46619 is due to decreased expression levels of protease-activated receptor (PAR) 3/4 and TXA2 (TP) receptors in RIP3−/− platelets, we assayed the expression levels of these receptors. We found that deletion of RIP3 did not affect the expression levels of PAR3/4 and TP in platelets (Fig. S1). These data indicate that RIP3 promotes U46619- and thrombin-induced platelet aggregation.
Fig. 3.

The role of RIP3 in platelet aggregation. Washed platelets from WT and RIP3−/− mice were stimulated with different concentrations of U46619 (A), thrombin (B), collagen (C), and ADP (D) at 37 °C under constant stirring. Platelet aggregation was monitored using a turbidimetric aggregometer. The traces are representative of three independent experiments. In the histograms of maximal platelet aggregation under the indicated conditions, values are mean ± SEM of three independent experiments. *P = 0.016; **P = 0.0071; ****P < 0.0001.
Fig. S1.

Expression levels of TP and PAR3/4 in WT and RIP3-deficient platelets. Whole blood was collected and anticoagulated with 1/7 volume of ACD. The blood was centrifuged at 200 g for 11 min to yield platelet-rich plasma (PRP). PRP was then washed and resuspended in modified Tyrode’s buffer (3 × 108/mL). The washed platelets were lysed with SDS/PAGE loading buffer, and probed by Western blot analysis with anti-mouse PAR-3/4 (Santa Cruz Biotechnology) and TP (Abcam) antibodies. Blotting to GAPDH (anti-GAPDH antibody; Santa Cruz Biotechnology, CA) was used as a loading control. The figures are representative of three independent experiments.
Impaired U46619 and Thrombin-Induced ADP Secretion Occur in the Absence of RIP3.
ADP released from dense granules is known to play an important role in sensitizing platelets to low doses of agonist and amplifying platelet aggregation (21–24). Given our finding that RIP3 deficiency impaired platelet aggregation solely when low doses of the agonists were applied, we hypothesized that RIP3 might play a role in agonist-induced secretion. To test this hypothesis, we examined agonist-induced ATP release from RIP3−/− platelets. As shown in Fig. 4A, the second wave of ATP secretion elicited by low concentrations of U46619 was abolished in RIP3−/− platelets. Similarly, low-dose thrombin-induced ATP secretion was reduced in RIP3−/− platelets (Fig. 4B). In contrast, collagen-induced dense granule secretion was not impaired in RIP3−/− platelets (Fig. S2). These results indicate that RIP3 deficiency impairs dense granule secretion induced by U46619 or thrombin.
Fig. 4.

ATP secretion of RIP3−/− platelets in response to low doses of U46619 or thrombin. (A and B) Washed platelets from WT and RIP3−/− mice were stimulated with different concentrations of U46619 (A) or thrombin (B). ATP secretion was recorded concomitantly with platelet aggregation in the presence of luciferin/luciferase reagent (80 nM). (C and D) A low concentration of ADP (0.25 μM), insufficient to induce aggregation, reversed the inhibitory effect of RIP3 deficiency on 325 nM U46619-induced (C) or 0.008 U/mL thrombin-induced (D) platelet aggregation. Trace in the figures are representative of at least three independent experiments.
Fig. S2.

RIP3 deficiency does not affect ATP secretion in platelets stimulated with collagen. Washed platelets from WT and RIP3−/− mice were stimulated with 0.5, 1.0, 1.5, and 2.0 μg/mL collagen. ATP secretion was recorded concomitantly with platelet aggregation in the presence of luciferin/luciferase reagent at 37 °C at a stirring speed of 1,000 rpm.
As shown in Fig. 3D, ADP-induced platelet aggregation was not impaired in RIP3−/− platelets, suggesting that RIP3 is not required for ADP-induced signaling. Therefore, we considered the possibility that the inhibitory effect of RIP3 deficiency on U46619- or thrombin-induced platelet aggregation results from inhibition of ADP secretion. In support of this idea, we found that the aggregation of WT platelets induced by U46619 or thrombin was reduced to a level comparable to that seen in RIP3-deficient platelets in the presence of apyrase, which hydrolyzes released ADP (Fig. S3). Moreover, the addition of a low concentration of ADP, although insufficient by itself to induce platelet aggregation, restored the reduced aggregation of RIP3−/− platelets stimulated with a low dose of U46619 or thrombin (Fig. 4 C and D). Therefore, these results indicate that RIP3 deficiency impairs U46619- and thrombin-induced ADP secretion.
Fig. S3.

Effects of apyrase on platelet aggregation in response to low doses of U46619 or thrombin. Washed platelets (3 × 108/mL) from WT and RIP3−/− mice were stimulated with 100 nM U46619 or 0.006 U/mL thrombin in the presence or absence of apyrase (1 U/mL) at 37 °C under constant stirring. Platelet aggregation was monitored using a turbidimetric aggregometer. The traces are representative of three independent experiments.
α-Granule Secretion, TXA2 Generation, Phosphatidylserine Exposure, and Integrin Activation Occur in RIP3−/− Platelets.
Granule secretion and TXA2 generation induced by various agonists play key roles in amplifying stimulation signals and enabling robust platelet activation at the site of injury. Thus, we further evaluated α-granule secretion in RIP3−/− platelets. We found that, consistent with a previous report (25), only very small amount of P-selectin exposure was detected in U46619-stimulated platelets (Fig. S4A). There was no significant difference in P-selectin exposure between WT and RIP3−/− platelets stimulated with thrombin (Fig. S4B). Moreover, no significant difference in PF4 release was observed between WT and RIP3−/− platelets stimulated with U46619 or thrombin (Fig. S5). These data suggest that RIP3 deficiency does not impair α-granule secretion.
Fig. S4.

Analysis of α-granule secretion in RIP3−/− platelets. Flow cytometry analysis of P-selectin exposure in mouse platelets stimulated with the indicated doses of U46619 (nM) (A) or thrombin (U/mL) (B). Data are expressed as mean fluoresce intensity ± SEM of three independent experiments. There was no statistically significant difference between the two groups.
Fig. S5.

PF4 release from WT and RIP3−/− mouse platelets stimulated with U46619 and thrombin. Washed WT and RIP3−/− mouse platelets were stimulated with different concentrations of U46619 (A) or thrombin (B) for 8 min at 37 °C with stirring. The platelet suspensions were centrifuged, and the supernatants were analyzed for PF4 release using the PF4 ELISA Kit (Abcam). Quantitative results are expressed as mean ± SEM of three independent experiments.
We next investigated whether RIP3 was involved in TXA2 synthesis by measuring the stable TXA2 metabolite, TXB2. We found no statistically significant difference in TXB2 generation between WT and RIP3−/− platelets stimulated with thrombin or collagen (Fig. S6). Interestingly, ADP-induced TXB2 production was markedly elevated in RIP3−/− platelets compared with in WT platelets (10.55 ± 1.97 ng/mL vs. 0.12 ± 0.05 ng/mL).
Fig. S6.

TXB2 generation in the RIP3−/− platelets. TXB2 production in WT and RIP3−/− platelets stimulated with thrombin (0.008 U/mL), ADP (2.5 μM), collagen (0.5 μg/mL), or vehicle (control). All data are expressed as mean ± SEM of three independent experiments. *P < 0.05, Student’s t test.
At the site of vascular injury, phosphatidylserine (PS) exposure on activated platelets provides a procoagulant surface to promote thrombus formation. There is a growing body of evidence suggesting that the biochemical, morphological, and functional changes underlying agonist-induced platelet procoagulant function are broadly consistent with cell necrosis (26). Therefore, we further investigated whether RIP3 deficiency impairs PS exposure in agonist-induced platelets. The results showed no obvious difference in PS exposure between WT and RIP3−/− platelets stimulated with different concentrations of thrombin or U46619 (Fig. S7).
Fig. S7.

PS exposure in mouse platelets stimulated with thrombin and U46619. Washed WT and RIP3−/− platelets were stimulated with different concentrations of thrombin (A) or U46619 (B), or with thrombin (1 U/mL) + collagen (5 μg/mL) (C) for 30 min at 37 °C. The platelets were further incubated with Annexin V-FITC for 10 min and then analyzed by flow cytometry. Results from a representative experiment stimulated with selected concentrations of thrombin and U46619 are shown.
In vitro platelet aggregation requires integrin αIIbβ3 activation. Thus, we investigated the role of RIP3 in integrin αIIbβ3 activation. We found that U46619-induced integrin αIIbβ3 activation was decreased in RIP3−/− platelets (Fig. S8A). Moreover, U46619-induced fibrinogen binding to platelets was reduced by the inhibition of PI3K-Akt signaling (2). Similarly, thrombin-induced integrin αIIbβ3 activation was decreased in RIP3−/− platelets as well (Fig. S8B). Furthermore, fibrinogen binding was reduced in the RIP3−/− platelets stimulated with thrombin (Fig. S8C).
Fig. S8.

U46619- and thrombin-induced integrin αIIbβ3 activation in RIP3−/− platelets. (A and B) Flow cytometry analysis of PE-JON/A binding to WT and RIP3−/− platelets stimulated with vehicle (control) or the indicated doses of U46619 (A) or thrombin (B). Results from a representative experiment are shown. Quantitative results are expressed as mean ± SEM of three independent experiments. *P < 0.05. (C) Flow cytometry analysis of FITC-labeled fibrinogen binding to WT and RIP3−/− platelets stimulated with thrombin (0.002 and 0.004 U/mL) or vehicle (control). Results from a representative experiment are shown. Quantitative results from three independent experiments are expressed as mean fibrinogen binding index [total bound fluorescence (thrombin)/nonspecifically bound fluorescence (vehicle)] ± SEM. ***P < 0.001.
RIP3 Regulates Akt Phosphorylation.
It has been reported that PI3K-Akt signaling plays an essential role in regulating the second wave of ADP secretion in platelets in response to U46619 (2). Because RIP3 deficiency impairs only the second wave of ADP secretion, we investigated whether RIP3 regulates dense granule secretion via PI3K-Akt signaling. Phosphorylation of Akt at Thr308, a known marker of Akt activation that lies downstream of PI3K (2, 27), was diminished from the onset of second wave of secretion in RIP3−/− platelets stimulated with U46619 (Fig. 5A). Moreover, low-dose thrombin-induced phosphorylation of Akt also was reduced from the onset of the second wave of secretion in RIP3−/− platelets (Fig. 5B). These data suggest that RIP3 is upstream of Akt regulating ADP secretion in platelets.
Fig. 5.

The role of RIP3 in Akt phosphorylation, and the interaction of RIP3 with Gα13. (A and B) Washed platelets were stimulated with 350 nM U46619 (A) or 0.006 U/mL thrombin (B) for indicated times at 37 °C, with stirring (1,000 rpm). Stimulated platelets were lysed and samples analyzed by Western blot with anti–phospho-Akt (Thr308) and anti-total Akt antibodies. (C) Washed mouse platelets were lysed and immunoprecipitated with anti-mouse RIP3 and Gα13 antibodies or IgG controls. After incubation with protein A/G plus agarose beads, the proteins were analyzed by Western blot with anti-RIP3 and anti-Gα13 antibodies. (D) The pcDNA3.1(+)- expressing GST or GST-RIP3 and the pcDNA3.1(+)-expressing His-Gα13 were cotransfected into HEK293T cells. The cells were cultured, harvested, and lysed. The lysates were centrifuged, after which the supernatants were mixed with glutathione beads and incubated at 4 °C overnight. The beads were washed, and the bead-bound proteins were analyzed by immunoblotting. The Western blot shown is representative of three independent experiments. (E) Purified proteins and BSA (4 μg/mL) were immobilized onto the wells of microtiter plates. Increasing concentrations of His-tagged human Gα13 protein were incubated with immobilized GST (●), GST-RIP3 (■), GST-RIP3-N (▲), GST-RIP3-C (▼), and BSA. The binding of Gα13 was detected by mouse anti-His antibody and HRP-conjugated goat anti-mouse antibody. The absorbance at 450 nm was measured in three independent experiments. Data are presented as mean ± SD after subtracting the binding of Gα13 to BSA (negative control). The binding curve was fitted to the following equation: Y = Bmax × x/(Kd + x), where Y is the specific binding, x is the ligand concentration, Bmax is the binding maximum, and Kd is the equilibrium dissociation constant. For some data points, the error bars are smaller than the symbols.
RIP3 Interacts with Gα13 in Platelets.
Both Gq and Gα13 are required for platelet secretion induced by thrombin and TXA2 (4, 28, 29). Because platelet activation elicited by ADP, which activates its receptors P2Y1 and P2Y12, which couple to Gq and Gi, respectively, was not reduced in the RIP3-deficient platelets, it is less likely that RIP3 is required for Gq signaling. Therefore, we hypothesized that RIP3 might be downstream of Gα13 to regulate dense granule secretion. In support of this idea, we found that RIP3 could be coimmunoprecipitated with Gα13 (Fig. 5C), but not with Gq or Gi (Fig. S9), from the lysates of platelets. Moreover, it appears that the interaction between RIP3 and Gα13 is dynamically regulated on activation (Fig. S10).
Fig. S9.

Coimmunoprecipitation of RIP3 with Gi or Gq in mouse platelets. Washed platelets (3 × 108 /mL) from WT mice were lysed with equal volumes of 2× Nonidet P-40 lysis buffer containing protease inhibitor mixture tablets on ice for 30 min. After centrifugation at 17,000 × g at 4 °C for 10 min, the supernatants were immunoprecipitated with anti-RIP3 antibody or IgG (control) overnight. After incubation with protein A/G plus-agarose beads at 4 °C for 2 h, the proteins were analyzed by Western blot analysis with anti-Gq and anti-Gi antibodies. The figures are representative of at least three independent experiments.
Fig. S10.

Interaction of Gα13 with RIP3 in platelets stimulated with thrombin. Washed platelets (3 × 108 /mL) from WT mice were stimulated with 0.006 U/mL thrombin for the indicated times at 37 °C under stirring. The platelets were lysed with equal volumes of 2× Nonidet P-40 lysis buffer containing protease inhibitor mixture tablets on ice for 30 min. After centrifugation at 17,000 × g and 4 °C for 10 min, the supernatants were immunoprecipitated with anti-Gα13 antibody or IgG (control) overnight. After incubation with Protein A/G Plus Agarose beads at 4 °C for 2 h, the proteins were analyzed by Western blot analysis with anti-RIP3 and anti-Gα13 antibodies. The figures are representative of three independent experiments.
To further characterize the specificity of the interaction between RIP3 and Gα13, we first overexpressed RIP3 and Gα13 proteins in HEK293T cells. We found that RIP3 was pulled down with Gα13 from the HEK293T cells (Fig. 5D). We then examined the interaction between purified RIP3 and Gα13 proteins by ELISA. Fig. 5E shows a specific interaction between RIP3 and Gα13. To identify the binding site in RIP3 for Gα13, we purified two fragments of RIP3, the N terminal (1–350 aa) and C terminal (351–518 aa). We found that, similar to the full-length RIP3, the C-terminal fragment interacted with Gα13 protein, suggesting that the binding site in RIP3 for Gα13 is located at the C terminal of RIP3 (Fig. 5E). To exclude the possibility of contamination with cellular proteins during purification, we further confirmed the interaction of RIP3 with Gα13 by other sources of purified RIP3 and Gα13 proteins (Fig. S11). In addition, we investigated whether the deletion of RIP3 affects the formation of G protein heterotrimer. We found no difference in the interaction of Gα13 with Gβ between RIP3-deficient and WT platelets, suggesting that the formation of Gα13 heterotrimer is not affected by RIP3 deletion (Fig. S12). Taken together, these data suggest that RIP3 regulates platelet activation by interacting with Gα13.
Fig. S11.
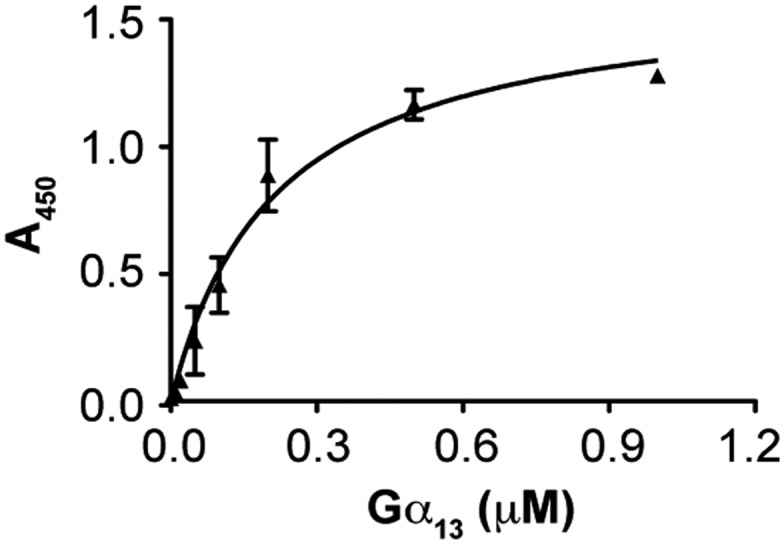
The interaction of Gα13 with RIP3. Increasing concentrations of Gα13 (His-tag; NewEast Biosciences) were incubated with immobilized RIP3 (Abcam) or BSA (control). The binding of Gα13 was assessed with mouse anti-His antibody and HRP-conjugated goat anti-mouse antibody. The absorbance at 450 nm was measured in four independent experiments, and values are presented as the mean ± SD after subtracting the binding of Gα13 to BSA (background). After the background signal had been subtracted, the binding curve was fitted to the equation Y = Bmax × x/(Kd + x), where Y is the specific binding, x is the ligand concentration, Bmax is the binding maximum, and Kd is the equilibrium dissociation constant. For some data points, the error bars are smaller than the symbols.
Fig. S12.
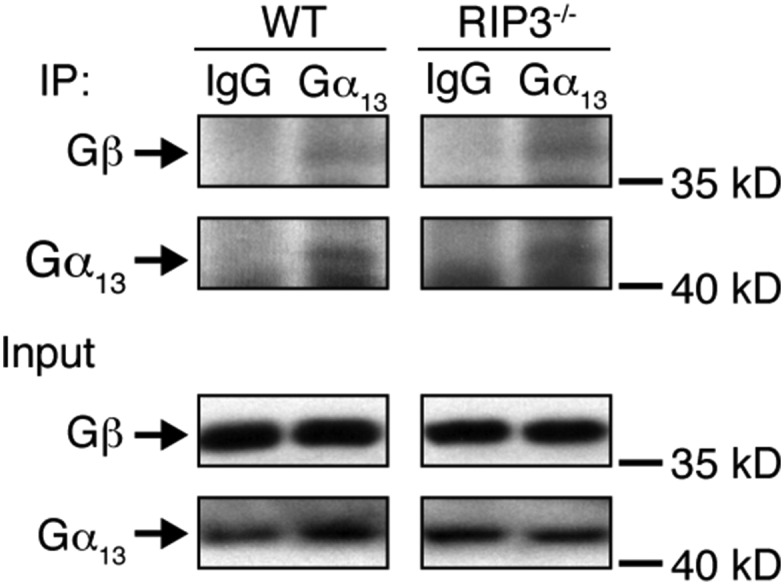
Gα13 binds to Gβ in both WT and RIP3−/− platelets. Washed WT and RIP3−/− mouse platelets (3 × 108/mL) were lysed with equal volumes of 2× Triton X-100 lysis buffer containing protease inhibitor mixture tablets on ice for 30 min. After centrifugation at 12,500 × g for 10 min at 4 °C, the supernatants were incubated with 2 μg/mL rabbit IgG and anti-Gα13 (Santa Cruz Biotechnology) antibody for 2 h at 4 °C, and then immunoprecipitated with Protein G Plus Agarose beads at 4 °C overnight. After three washes with lysis buffer, the beads were analyzed by immunoblotting. The figures are representative of three independent experiments.
RIP3 Plays a Role in Integrin Outside-In Signaling.
It is known that integrin outside-in signaling is required to induce the second wave of secretion in platelets stimulated with U46619 (2). As shown in Fig. 5 A and B, Akt phosphorylation occurred at 3 min after stimulation, corresponding to initiation of the integrin outside-in signaling. Moreover, RIP3 was found to interact with Gα13, which has been confirmed to play a key role in integrin outside-in signaling (5–7). Therefore, these findings suggest the involvement of RIP3 in integrin outside-in signaling. To test this possibility, we examined the effects of RIP3 deficiency on integrin outside-in signaling-dependent platelet spreading and clot retraction. We found markedly reduced spreading of RIP3−/− platelets on fibrinogen in the presence of thrombin compared with that of WT platelets (Fig. 6A). Furthermore, RIP3 deficiency impaired clot retraction, which requires integrin outside-in signaling (Fig. 6B). These findings suggest that RIP3 deficiency impairs integrin outside-in signaling.
Fig. 6.

RIP3 deficiency impairs integrin signaling. (A) Washed platelets were allowed to adhere and spread on fibrinogen-coated wells by thrombin or vehicle stimulation at 37 °C for 2 h. After being fixed, permeabilized, and stained, the platelets were observed with a fluorescence microscope. Images were acquired, and the spreading area of single platelets was measured using ImageJ2x software, with pixel number as the unit of size. (Left) Representative pictures. (Right) Surface areas of single platelets from 10 randomly selected fields of three different tests. ***P < 0.001. (B) Washed mouse platelets in modified Tyrode’s buffer were mixed with 150 μg/mL purified human fibrinogen. The clots were initiated by the addition of human α-thrombin (1 U/mL) and incubation at 37 °C. (Left) Representative pictures of clot retraction from three separate experiments. (Right) 2D retraction of clots was measured, and the data are expressed as retraction ratio: 1 − (final clot size/initial clot size). n = 3. Data are mean ± SEM. ***P < 0.001, Student‘s t test.
To further examine whether the integrin outside-in signaling is defective in the absence of RIP3, we assessed the phosphorylation of ERK, which is the integrin signaling-dependent activation, in RIP3−/− platelets (30, 31). We found that ERK phosphorylation was obviously reduced in RIP3−/− platelets stimulated with U46619 or thrombin (Fig. S13). Moreover, consistent with Fig. 5A, we found that the integrin antagonist RGDS peptides abolished U46619-induced Akt phosphorylation in both WT and RIP3−/− platelets (Fig. S14). These data suggest that RIP3 regulates platelet activation via the regulation of integrin outside-in signaling.
Fig. S13.

Role of RIP3 in ERK phosphorylation. Washed platelets were stimulated with 350 nM U46619 or 0.006 U/mL thrombin for the indicated times at 37 °C with stirring. The platelets were lysed and analyzed by Western blot analysis with anti–phospho-ERK1/2 and anti–total-ERK1/2 antibodies. The figures are representative of three independent experiments.
Fig. S14.

RGDS peptides abolish U46619-induced Akt phosphorylation. Washed WT and RIP3−/− mouse platelets were preincubated with RGDS (2 mM) or vehicle (−RGDS) at 37 °C for 5 min. Then 350 nM U46619 was added to platelets in an aggregometer with stirring for 5 min. The platelets were lysed and analyzed by Western blot analysis with anti–phospho-Akt (Thr308) and anti-total Akt antibodies. The Western blot shown is representative of at least three independent experiments.
RIP3 Inhibitor Blocks U46619- and Thrombin-Induced Human Platelet Aggregation and Prevents Arterial Thrombus Formation.
To investigate the role of RIP3 in human platelets, we incubated an RIP3-specific inhibitor, GSK′872 (32), with human platelets. We found that GSK′872 dose-dependently inhibited U46619- and thrombin-induced human platelet aggregation (Fig. 7 A and B), but had no effect on the aggregation of RIP3−/− platelets (Fig. S15). To further characterize the effect of RIP3 inhibitor on in vivo thrombosis, we applied the FeCl3-injured mesenteric arteriole thrombosis model using mice injected with GSK′872. Compared with vehicle-injected controls, the mice injected with GSK′872 exhibited delayed and diminished thrombus formation (Fig. 7C). Occlusion times were significantly prolonged in the mice injected with GSK′872 (29.2 ± 2.5 min in the mice injected with GSK′872 vs. 18.5 ± 0.9 min in the vehicle control mice; P < 0.01) (Fig. 7D). These data demonstrate the role of RIP3 in human platelets, and suggest the antithrombotic potential of RIP3 inhibitor.
Fig. 7.

RIP3 inhibitor GSK’872 blocks human platelet aggregation and in vivo thrombus formation. (A and B) Washed human platelets were incubated with different concentrations of GSK′872 or vehicle (DMSO) at 37 °C for 30 min, and then stimulated with U46619 (A) and thrombin (B). Platelet aggregation was monitored using a turbidimetric aggregometer. The traces are representative of 3 independent experiments. (C) Representative images of FeCl3-induced mesenteric arteriole thrombosis in C57d mice injected with GSK′872 or vehicle (DMSO) as recorded by real-time microscopy. Time after FeCl3-induced injury is indicated at the bottom right of each image. (D) The occlusion time of mesenteric arteriole injured by FeCl3 in C57d mice injected with GSK′872 or vehicle. Data are mean ± SEM values of 15 controls and 11 GSK′872-injected mice. **P < 0.01. (E) RIP3, interacting with Gα13, regulates integrin outside-in signaling.
Fig. S15.

The effects of GSK’872 on mouse platelet aggregation. Washed mouse platelets were incubated with GSK′872 (6 μM) or vehicle (DMSO) at 37 °C for 30 min, and then stimulated with U46619 and thrombin. Platelet aggregation was monitored using a turbidimetric aggregometer. The traces are representative of three independent experiments.
Discussion
In this study, we have demonstrated that RIP3 is expressed in platelets and plays important roles in arterial thrombus formation and hemostasis in vivo. We report that platelet aggregation and dense granule secretion in response to U46619 or thrombin, spreading on fibrinogen, and clot retraction are impaired in RIP3-deficient platelets, and that RIP3 antagonist inhibits platelet aggregation and prevents in vivo arterial thrombus formation.
Our data show that RIP3 deficiency selectively impairs U46619- and thrombin-induced dense granule secretion and aggregation. ADP-induced aggregation is not affected by RIP3 deletion. Because both the TXA2 receptor and the thrombin receptor couple to Gα13, we hypothesize that RIP3 may be downstream of Gα13 and mediate Gα13 signaling. In support of this hypothesis, we found that RIP3 interacts with Gα13 in platelets. Moreover, consistent with our findings, deletion of Gα13 selectively impairs low doses of thrombin- and U46619-induced platelet aggregation (4), but does not affect ADP-induced aggregation (33).
Four lines of evidence from the present study indicate that RIP3 regulates integrin outside-in signaling. First, the integrin signaling-dependent second wave of dense granule secretion and aggregation was diminished in RIP3-deficient platelets. Second, RIP3 deficiency impaired platelet spreading on fibrinogen and clot retraction. Third, ERK phosphorylation, downstream of integrin signaling (30, 31), was reduced in RIP3-deficient platelets. Fourth, RGDS, the integrin-specific antagonist, abolished the second wave of dense granule secretion and Akt phosphorylation in both WT and RIP3−/− platelets. These findings are consistent with the recent reports demonstrating that Gα13 binds to integrin αIIbβ3 and mediates integrin outside-in signaling (5–7). RIP3 was found to interact with Gα13, thereby providing a molecular basis for the involvement of RIP3 in Gα13-mediated integrin signaling (Fig. 7E).
We found that integrin activation was reduced in the RIP3-knockout platelets. Akt phosphorylation elicited by thrombin or U46619 was decreased in the absence of RIP3. These findings are consistent with previous reports indicating that deletion of Akt1 (22) or Akt2 (21), or inhibition of Akt (2), reduced thrombin- or U46619-induced fibrinogen binding. Because our data indicate impairment of dense granule secretion in RIP3 knockout mice, future work is needed to determine whether the impaired integrin activation in RIP3-deficient platelets is due to a direct role of RIP3 on integrin or results from reduced platelet secretion.
Activation of RIP3 phosphorylates its substrate, such as MLKL, leading to necrotic death and clearance of the target cells (13–15). In contrast, we demonstrate here that RIP3 promotes platelet activation and thrombus formation independent of MLKL. These findings identify RIP3 as an important regulator of platelet activation and thrombosis. In particular, the RIP3 inhibitor effectively prevented arterial thrombus formation in vivo, suggesting a novel potential antithrombotic strategy for thrombotic diseases.
In summary, our data demonstrate a critical role for RIP3 in promoting hemostasis and thrombus formation in vivo. RIP3 amplifies U46619- and thrombin-induced platelet activation by regulating integrin outside-in signaling. RIP3 antagonist inhibits human platelet aggregation in vitro and prevents arterial thrombus formation in vivo. Therefore, RIP3 may represent a signaling molecule to modulate platelet activation and a promising therapeutic target for thrombotic diseases.
Materials and Methods
Reagents, electron microscopy, measurement of TXA2 generation, flow cytometry analysis, hematologic analysis, tail bleeding time (34), in vivo thrombosis (35), irradiation and bone marrow-derived cell repopulation, platelet aggregation and secretion, assessment of fibrinogen binding, coimmunoprecipitation, plasmids and oligos, expression and purification of proteins, ELISA, transfection and protein-binding assays, platelet spreading on immobilized fibrinogen, and clot retraction are described in SI Materials and Methods.
Mice.
RIP3-deficient (RIP3−/−) mice were generated in the laboratory of Dr. Xiaodong Wang (11), and the 129/B6 RIP3−/− mice have been backcrossed to C57/B6 for 10 generations. WT control mice and RIP3-deficient mice used in this study were littermates generated from heterozygous breeding. All of the animal experiments were approved by the Ethics Committee of the First Affiliated Hospital of Soochow University.
Platelet Preparation.
Platelets from healthy volunteers were prepared as described previously (2). For studies involving human subjects, approval was obtained from the Ethics Committee of the First Affiliated Hospital of Soochow University. Written informed consent was provided by each particpant, and the studies were performed in accordance with the Declaration of Helsinki. Washed platelets from mice were prepared as described previously (2).
SI Materials and Methods
Reagents and Antibodies.
Thrombin, collagen, ADP, and Chrono-Lume were obtained from Chronolog. BSA, aspirin, pentobarbital sodium, Arg-Gly-Asp-Ser (RGDS), and Nonidet P-40 were from Sigma-Aldrich. Calcein acetoxymethyl ester (calcein-AM) was from Dojindo Molecular Technologies. U46619, LY294002, and human fibrinogen were from Calbiochem. FITC-conjugated anti-mouse P-selectin (Wug.E9), PE-conjugated anti-mouse active integrin αIIbβ3 (JON/A), and anti-mouse GPIbα antibodies were from Emfret Analytics. Mouse anti-RIP1 antibody was from BD Biosciences. RIP3 inhibitor GSK′872 was from Merck Millipore. PE-conjugated anti-mouse CD41 antibody was from BioLegend. FITC-conjugated mouse fibrinogen, anti-mouse phospho-MLKL (Ser345), anti-CD45, and anti-mouse TP antibodies, Annexin V-FITC, GST-tagged active human RIP3 full-length protein, and the PF4 ELISA Kit were from Abcam. Anti-mouse MLKL antibody was from Abgent. The TXB2 Enzyme Immunoassay (EIA) Kit was from ENZO Life Sciences. Anti-mouse RIP3 antibody was from ProSci. Anti–phospho-Akt (Thr308), anti-total Akt, anti-ERK1/2 antibody, anti–phospho-ERK1/2, anti-GAPDH, and anti-human RIP3 antibodies were from Cell Signaling Technology. Alexa Fluor 488-conjugated phalloidin was from Life Technologies. Anti-mouse P2Y12 antibody was from Alomone Labs. HRP-conjugated anti-rabbit IgG, anti-mouse PAR-3/4, Gα13, Gi and Gq, anti-6xHis, and anti-GST antibodies, Complete Protease Inhibitor Mixture tablet, Protein A/G Plus Agarose, and rabbit IgG were from Santa Cruz Biotechnology. Botrocetin was kindly provided by Dr Junling Liu, Department of Biochemistry and Molecular Cell Biology, Shanghai Jiaotong University, China. Glutathione-conjugated Sepharose beads were from GE Healthcare. Recombinant His-tagged human Gα13 full-length protein was from NewEast Biosciences.
Hematologic Analysis and Tail Bleeding Time.
Complete blood cell counts were performed with a Sysmex XP-100 Hematologic Analyzer. In the tail bleeding time experiments, 6- to 8-wk-old mice (equivalent numbers of males and females) were anesthetized with 2% pentobarbital. Tails were amputated 3 mm from the tip and immediately immersed into isotonic PBS at 37 °C. For each tail, time until bleeding had stopped (i.e., no rebleeding within 60 s), or a maximum of 600 s, was recorded.
In Vivo Thrombosis.
In the ferric chloride (FeCl3)-induced mesenteric arteriole thrombosis model experiment, platelets were isolated from donor mice and labeled with calcein-AM (5 μg/mL). Male mice were injected i.v. with calcein-labeled platelets (5 × 106/g) of matching genotype. The recipient mice were anesthetized. In inhibition experiments, GSK′872 (96 μM) or vehicle (DMSO) was retrobulbarly injected and then circulated for 30 min. The mesentery vascular bed was exteriorized, and one arteriole was chosen and visualized with an inverted fluorescent microscope (Leica Microsystems), and recorded on videotape. Thrombus formation was induced by topical application of a 3-mm2 filter paper soaked with 5% FeCl3. The vessel occlusion time was defined the time to as complete cessation of blood flow.
Irradiation and Bone Marrow-Derived Cell Repopulation.
Male WT and RIP3−/− mice (10 wk old) were lethally irradiated with 8 Gy from a cobalt-60 γ radiation source. Bone marrow-derived cells were harvested from 8-wk-old WT and RIP3−/− male mice and injected into irradiated recipient mice (1 × 107 donor cells per animal). The mice were used in experiments when blood cell counts recovered. RIP3 expression in platelets of each recipient mouse was detected by immunoblotting.
Platelet Aggregation and Secretion.
Platelet aggregation and secretion were recorded in a Chrono-Log lumi-aggregometer. Washed platelets (3 × 108 /mL) were stimulated with different agonists. Luciferin/luciferase (10 μL) was added to 240 μL of washed platelet suspension within 2 min before stimulation. In some experiments, platelets were pretreated with inhibitors at 37 °C before the addition of agonists. Platelet aggregation was monitored continuously over 5–10 min.
Electron Microscopy.
Platelets were fixed in 2.5% glutaraldehyde at 4 °C overnight and then centrifuged at 600 × g for 2 min. Platelet pellets were washed with PBS, postfixed in 1.0% osmium tetroxide for 1 h, gradiently dehydrated using acetone, and then stained with saturated uranyl acetate. The samples were infiltrated, embedded with resin, and polymerized. Finally, ultrathin sections were observed with a Hitachi H600 transmission electron microscope. For the measurement of platelet morphology and granule content, micrographs of 20 platelets from WT and RIP3−/− mice were analyzed from five different slides for each strain.
Measurement of TXA2 Generation.
TXA2 generation was assayed as its stable metabolite, TXB2, in conditions of platelet aggregation induced by aforesaid agonists. Platelet aggregation was stopped after 8 min of stimulation by adding ice-cold EDTA (1 mM) and aspirin (1 mM). Samples were centrifuged, and the supernatants were stored at −80 °C to avoid repeated freeze-thaw cycles. Levels of TXB2 were measured using the TXB2 EIA Kit following the manufacturer’s protocol.
Flow Cytometry Analysis.
Washed platelets (3 × 108/mL) were stimulated with agonists. Antibodies were added simultaneously with agonists as indicated. After incubation for 15 min at room temperature, the reaction was stopped by adding PBS, and the samples were analyzed with a Cytomics FC500 MCL flow cytometer (Beckman Coulter).
Assessment of Fibrinogen Binding.
Washed platelets were incubated with FITC-conjugated mouse fibrinogen (135 μg/mL), and thrombin or vehicle at 37 °C for 30 min. The reaction was stopped by adding PBS, and flow cytometry analysis was performed.
Immunoblotting Detection.
Washed platelets were exposed to U46619 or thrombin for different times. Platelets were then lysed and probed by immunoblotting with the antibodies indicated in the figures.
Coimmunoprecipitation.
Washed platelet suspensions (2 × 108 platelets) were lysed with equal volumes of 2 × Nonidet P-40 lysis buffer (100 mM Tris pH 7.4, 2% Nonidet P-40, 20 mM MgCl2, 300 mM NaCl, 2 mM PMSF, 2 mM NaF, and 2 mM Na3VO4) containing protease inhibitor mixture tablets on ice for 30 min. After centrifugation at 17,000 × g and 4 °C for 10 min, the supernatants were immunoprecipitated with antibodies overnight. After incubation with Protein A/G Plus Agarose beads at 4 °C for 2 h, the beads were analyzed by immunoblotting.
Plasmids and Oligos.
The GST sequence in a pGS-21a vector (Genscript) was amplified by PCR and subcloned into pcDNA3.1(+) using NheI and NotI (forward primer, 5′gggagacccaagctggctagcatgggcatgtcccctatactaggtta; reverse primer, 5′tctagactcgagcggccgcttaagagccgcctcctcccgattttggaggatggtc). The RIP3 gene in pCI-neo plasmid (Promega) and the GST sequence in the pGS-21a vector was amplified by overlap methods using the following primers: GST, forward: 5′gggagacccaagctggctagcatgggcatgtcccctatactaggtta; GST-RIP3, reverse: 5′agagccgcctcctcccgattttggaggatggtcgc; RIP3, forward: 5′tcgggaggaggcggctctatgtcgtgcgtcaagttatg; RIP3, reverse: 5′tctagactcgagcggccgcttatttcccgctatgattataccaac. The GST-RIP3 DNA sequence was subcloned into pcDNA3.1(+) using NheI and NotI. The GST-RIP3 N-terminal (1–350 aa) sequence was amplified by overlap methods using the following primers: GST, forward: 5′gggagacccaagctggctagcatgggcatgtcccctatactaggtta; GST-RIP3, reverse: 5′agagccgcctcctcccgattttggaggatggtcgc; RIP3 (1–518 aa), forward: 5′tcgggaggaggcggctctatgtcgtgcgtcaagttatg; RIP3 (1–350 aa), reverse: 5′tctagactcgagcggccgcttagtttagccactcagaaacca. The GST-RIP3-N DNA was subcloned into pcDNA3.1(+) using Nhe I and Not I. The GST-RIP3 C-terminal (351–518 aa) sequence was amplified by overlap methods using the following primers: GST, forward: 5′gggagacccaagctggctagcatgggcatgtcccctatactaggtta; GST-RIP3, reverse: 5′agagccgcctcctcccgattttggaggatggtcgc; RIP3 (351–518 aa), forward: 5′tcgggaggaggcggctctaaactgaatctagaggagcctcc; RIP3 (1–518 aa), reverse: 5′tctagactcgagcggccgcttatttcccgctatgattataccaac. The GST-RIP3-C DNA was subcloned into pcDNA3.1(+) using Nhe I and Not I. The Gα13 gene in pCMV3 (Sino Biological) was amplified and subcloned into pcDNA3.1(+) using NheI and NotI with a His-tag at the C terminus of the Gα13 protein (forward primer: 5′gggagacccaagctggctagcatggcggacttcctgccgtcgc; reverse primer: 5′tctagactcgagcggccgcttaatgatgatgatgatgatgctgtagcataagctgctt). The constructs were verified by sequencing.
Expression and Purification of Proteins.
HEK 293T cells (American Type Culture Collection) were cultured in DMEM supplemented with 10% FBS and penicillin/streptomycin antibiotics at 37 °C in a 5% CO2 atmosphere. The HEK 293T cells were transfected by pcDNA3.1(+) containing target DNAs using Lipofectamine 2000 (Thermo Fisher Scientific). The cells were cultured for 48 h after transfection and then harvested. The cells were washed and resuspended in the lysis buffer (1× PBS pH 7.4 and 0.5% Nonidet P-40), containing 1× protease inhibitor mixture at a ratio of 1 mL of lysis buffer per 2 × 107 cells. The cells were lysed by sonication in five cycles (5 s on 30% power, 30 s off) on ice. The crude lysate was centrifuged at 20,000 × g for 30 min at 4 °C. The cleared lysate was then mixed with GST resin, followed by incubation for 2 h at room temperature. The resin was centrifuged, washed, and then resuspended in 500 μL of elution buffer (10 mM reduced glutathione and 50 mM Tris, pH 8.0), followed by another centrifugation at 2,000 × g for 5 min. The supernatant containing the purified protein was assayed by SDS/PAGE and Western blot analysis.
ELISA.
Proteins or BSA control (4 μg/mL) was immobilized onto microtiter plates at 37 °C for 1 h, and the wells were blocked with 3% BSA. Increasing concentrations of His-tagged Gα13 were incubated with immobilized RIP3 or BSA at 37 °C for 1 h, and the wells were washed three times with PBST (0.2% Tween-20). After incubation with mouse anti-His antibody (5 μg/mL) and HRP-conjugated goat anti-mouse antibody (1:8,000), standard washing substrate (tetramethylbenzidine; Thermo Fisher Scientific) was added to each well, and the plates were incubated at room temperature for 10 min. The reaction was stopped by addition of an equal volume of 2 M H2SO4. The plates were read at 450 nm in a Variskan Flash spectral scanning multimode reader (Thermo Fisher Scientific).
Transfection and Protein-Binding Assays.
The pcDNA3.1(+)-expressing His-Gα13 gene was cotransfected with the pcDNA3.1(+)-expressing GST or GST-RIP3 gene into HEK293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The cells were cultured for 48 h after transfection in DMEM supplemented with 10% FBS, then harvested and lysed with 1× lysis buffer (50 mM Tris⋅HCl pH 7.4, 0.5% Triton X-100, 150 mM NaCl, 10 mM MgCl2, 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF, and Protease Inhibitor Mixture). The lysate was centrifuged at 12,500 × g for 15 min at 4 °C. The clarified lysates were then mixed with glutathione-conjugated Sepharose beads and incubated at 4 °C overnight. The beads were washed with 1× lysis buffer three times, and the bead-bound proteins were analyzed by immunoblotting.
Platelet Spreading on Immobilized Fibrinogen.
Chamber slides with microtiter wells were coated with 10 μg/mL fibrinogen in 0.1 M NaHCO3 (pH 8.3) at 4 °C overnight. Washed platelets (2 × 107/mL) were allowed to adhere to and spread on fibrinogen-coated wells at 37 °C for 2 h with stimulation by thrombin. After washing, the cells were fixed, permeabilized, and stained with Alexa Fluor 488-conjugated phalloidin. Adherent platelets were viewed with an Olympus FluoView FV1000 confocal microscope. Images were acquired and the spreading area of single platelets was measured using ImageJ2x software, with pixel number as the unit of size. Ten randomly selected fields from at least three different tests were used for statistical analysis.
Clot Retraction.
Washed mouse platelets (4 × 108/mL) were resuspended in modified Tyrode’s buffer in unused aggregometer tubes and mixed with 150 μg/mL purified human fibrinogen. The clots were initiated by the addition of human α-thrombin to a final concentration of 1 U/mL, followed by incubation at 37 °C for 60 min. Clot retraction was monitored every 5 min and photographed. Clot size was quantified from photographs using Image J2x software.
Statistics.
All of the reported figures are derived from at least three independent experiments. Data are expressed as mean ± SEM. Statistical analysis was performed using Prism version 6.0 (GraphPad Software), and the data were compared using the unpaired Student t test (except for the data in Fig. 1, which were compared using the two-tailed Mann–Whitney U test). For all analyses, a P value <0.05 was considered to indicate statistical significance.
Acknowledgments
We thank Dr. Xiaodong Wang (National Institute of Biological Sciences) for his help with RIP3-deficient mice; Dr. Xiaoping Du (University of Illinois at Chicago) for critical discussion; and Dr. Chunyan He, Xiaojuan Zhao, and Guanglei Liu (Soochow University) for excellent technical assistance. This work was supported by grants from the Key Program of the National Natural Science Foundation of China (81130008, to K.D.), National Key Basic Research Program of China (2012CB526600, to K.D.), Priority Academic Program Development of Jiangsu Higher Education Institutions, Jiangsu Provincial Special Program of Medical Science (BL2012005), Jiangsu Province Key Medical Center (ZX201102), and Jiangsu Province Outstanding Medical Academic Leader Program (to K.D.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1610963114/-/DCSupplemental.
References
- 1.Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30(12):2341–2349. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Z, et al. Two waves of platelet secretion induced by thromboxane A2 receptor and a critical role for phosphoinositide 3-kinases. J Biol Chem. 2003;278(33):30725–30731. doi: 10.1074/jbc.M301838200. [DOI] [PubMed] [Google Scholar]
- 3.Kahn ML, et al. A dual thrombin receptor system for platelet activation. Nature. 1998;394(6694):690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- 4.Moers A, et al. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat Med. 2003;9(11):1418–1422. doi: 10.1038/nm943. [DOI] [PubMed] [Google Scholar]
- 5.Shen B, et al. A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature. 2013;503(7474):131–135. doi: 10.1038/nature12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gong H, et al. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin “outside-in” signaling. Science. 2010;327(5963):340–343. doi: 10.1126/science.1174779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr Opin Cell Biol. 2012;24(5):600–606. doi: 10.1016/j.ceb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meylan E, Tschopp J. The RIP kinases: Crucial integrators of cellular stress. Trends Biochem Sci. 2005;30(3):151–159. doi: 10.1016/j.tibs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Moriwaki K, Chan FK. RIP3: A molecular switch for necrosis and inflammation. Genes Dev. 2013;27(15):1640–1649. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. 2002;277(11):9505–9511. doi: 10.1074/jbc.M109488200. [DOI] [PubMed] [Google Scholar]
- 11.He S, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 12.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA. 2011;108(50):20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun L, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1-2):213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 14.Cho YS, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen W, et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem. 2013;288(23):16247–16261. doi: 10.1074/jbc.M112.435545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oberst A, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dillon CP, et al. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Reports. 2012;1(5):401–407. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X, et al. Investigation of receptor interacting protein (RIP3)-dependent protein phosphorylation by quantitative phosphoproteomics. Mol Cell Proteomics. 2012;11(12):1640–1651. doi: 10.1074/mcp.M112.019091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng L, Jin W, Wang X. RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc Natl Acad Sci USA. 2015;112(35):11007–11012. doi: 10.1073/pnas.1514730112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linkermann A, et al. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-α-induced shock. Mol Med. 2012;18:577–586. doi: 10.2119/molmed.2011.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woulfe D, et al. Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J Clin Invest. 2004;113(3):441–450. doi: 10.1172/JCI20267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, et al. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104(6):1703–1710. doi: 10.1182/blood-2003-10-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stojanovic A, et al. A phosphoinositide 3-kinase-AKT-nitric oxide-cGMP signaling pathway in stimulating platelet secretion and aggregation. J Biol Chem. 2006;281(24):16333–16339. doi: 10.1074/jbc.M512378200. [DOI] [PubMed] [Google Scholar]
- 24.Goggs R, et al. RhoG protein regulates platelet granule secretion and thrombus formation in mice. J Biol Chem. 2013;288(47):34217–34229. doi: 10.1074/jbc.M113.504100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pleines I, et al. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood. 2012;119(4):1054–1063. doi: 10.1182/blood-2011-08-372193. [DOI] [PubMed] [Google Scholar]
- 26.Jackson SP, Schoenwaelder SM. Procoagulant platelets: Are they necrotic? Blood. 2010;116(12):2011–2018. doi: 10.1182/blood-2010-01-261669. [DOI] [PubMed] [Google Scholar]
- 27.Usatyuk PV, et al. Role of c-Met/phosphatidylinositol 3-kinase (PI3k)/Akt signaling in hepatocyte growth factor (HGF)-mediated lamellipodia formation, reactive oxygen species (ROS) generation, and motility of lung endothelial cells. J Biol Chem. 2014;289(19):13476–13491. doi: 10.1074/jbc.M113.527556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997;389(6647):183–186. doi: 10.1038/38284. [DOI] [PubMed] [Google Scholar]
- 29.Gabbeta J, et al. Platelet signal transduction defect with Galpha subunit dysfunction and diminished Galphaq in a patient with abnormal platelet responses. Proc Natl Acad Sci USA. 1997;94(16):8750–8755. doi: 10.1073/pnas.94.16.8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naik MU, Stalker TJ, Brass LF, Naik UP. JAM-A protects from thrombosis by suppressing integrin αIIbβ3-dependent outside-in signaling in platelets. Blood. 2012;119(14):3352–3360. doi: 10.1182/blood-2011-12-397398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazharian A, et al. Protease-activating receptor-4 induces full platelet spreading on a fibrinogen matrix: involvement of ERK2 and p38 and Ca2+ mobilization. J Biol Chem. 2007;282(8):5478–5487. doi: 10.1074/jbc.M609881200. [DOI] [PubMed] [Google Scholar]
- 32.Kaiser WJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–31279. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moers A, Wettschureck N, Grüner S, Nieswandt B, Offermanns S. Unresponsiveness of platelets lacking both Galpha(q) and Galpha(13): Implications for collagen-induced platelet activation. J Biol Chem. 2004;279(44):45354–45359. doi: 10.1074/jbc.M408962200. [DOI] [PubMed] [Google Scholar]
- 34.Estevez B, et al. LIM kinase-1 selectively promotes glycoprotein Ib-IX-mediated TXA2 synthesis, platelet activation, and thrombosis. Blood. 2013;121(22):4586–4594. doi: 10.1182/blood-2012-12-470765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ni H, Papalia JM, Degen JL, Wagner DD. Control of thrombus embolization and fibronectin internalization by integrin alpha IIb beta 3 engagement of the fibrinogen gamma chain. Blood. 2003;102(10):3609–3614. doi: 10.1182/blood-2003-03-0850. [DOI] [PubMed] [Google Scholar]