

Abstract
The comparison of experimental and predicted kinetic isotope effects in the α-cleavage of alkoxy radicals is used here to judge the applicability of statistical rate theories. It is found that the governing rate theory and the statistical versus nonstatistical nature of the cleavage depend on the cleavage barrier and how much energy is imparted to the radical. The latter can then be controlled by changing the size of substituents in the system. With a large alkyl group substituent, the vibrational energy of the alkoxy radical is increased, but this energy is not statistically distributed, leading to a lower isotope effect than predicted by statistical theories. The observed isotope effect can be approximately rationalized using a semistatistical localized RRKM model.
In any reaction passing over an energy barrier, the products are initially imbued with excess energy. The partitioning of that energy between translational, rotational, and vibrational forms has long been a core interest of the field of gas-phase reaction dynamics, in part due to the ambition of selectively promoting or controlling reactions.1 Some general expectations for the energy partitioning are specified by the Polanyi rules, which relate the position of the transition state (TS) to vibrational versus translational energy in the reactant and products in the simplest atom-transfer reactions.2 Energy partitioning in larger molecules and in condensed phases is much less well understood.3 The literature provides no guidance as to how one might structurally control energy partitioning in an ordinary solution reaction or how this might be used to affect product selectivity.
We describe here a simple structural effect on the amount of vibrational energy that is partitioned to a reactive intermediate in an atom-transfer step and how that engenders nonstatistical dynamics4 and changes the selectivity of a reaction. The results show how ideas from collision dynamics can be used to influence complex organic reactions in solution.
The reaction of interest is the conversion of cycloalkyl hypochlorites (1a–c) to ω-chloroalkylketones (5a–c). This reaction involves a chain mechanism in which a chlorine atom is abstracted from 1 to afford the alkoxy radical 3.5 Radical 3 then undergoes a facile α-cleavage to afford the ring-opened radical 4. Chlorine atom transfer to 4 from 1 affords the product 5 and a new molecule of 3 to continue the chain. These reactions are clean and highly exothermic (37 kcal/mol for n = 2, 55 kcal/mol for n = 1 (CCSD(T)/cc-pvtz//M11L/6-311+G**)). The α-cleavage of alkoxy radicals is important in diverse areas of chemistry and has been assumed to be fully understood using statistical rate theories.6
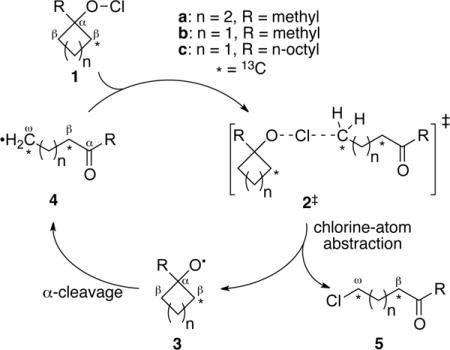
When a 13C is in the β-position of 1, the α-cleavage of 3 partitions the 13C between the β and ω positions of the ring-opened radical 4. The selectivity in this cleavage can be measured from the ratio of 13C in the two positions and expressed as an intramolecular kinetic isotope effect (KIE). This selectivity is readily determined by analysis of samples of 5 at natural abundance by NMR methodology.7 In all cases, the 13C content in the β position of 5 was in excess over that in the ω position, reflecting the qualitative expectation of a faster cleavage of a β-12C in 3. However, there were surprising variations in the magnitude of the excess.
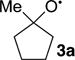
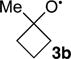
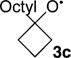
The results are summarized in Table 1. At 6.1%, the large KIE observed for α-cleavage in the five-membered ring 3a is comparable to that observed previously for the ring-opening of the cyclopropylcarbinyl radical, a high heavy-atom tunneling system. The KIE is then strikingly decreased for the methylcyclobutoxy radical 3b, at 4.4%. Most surprisingly, the KIE is further decreased to 2.9% for the octylcyclobutoxy radical 3c, less than half that observed for 3a.
Table 1.
Experimental and Predicted KIEs (42 °C)
| alkoxy radical | exptl KIEa | CVT/SCT KIE | α-cleavege barrierb (kcal/mol) | available vibrational energy | RRKM KIE |
|---|---|---|---|---|---|
|
1.061 (2) | 1.063 | 10.8 | 5.8 | – |
|
1.044 (1) | 1.060 | 3.0 | 4.8 | 1.043 |
|
1.029 (1) | 1.062 | 2.6 | 7.2 | 1.038 (1.033c) |
Based on the ratio of 13C in the β versus ω positions of 5. The 95% confidence limit on the last digit is shown in parentheses.
UM11L/6-311+G** potential energy barrier, in kcal/mol.
Prediction using a localized RRKM model in which the excess energy is limited to a molecular subset.
To interpret these results, we turned to computational and dynamic trajectory studies. Diverse DFT methods were explored in comparison with CCSD(T)/jun-cc-pvtz single-point energies for the 1a,b–5a,b energy surface. In this comparison, UM11L/6-311+G** calculations provided a highly accurate description of the α-cleavage (barriers within 0.1 kcal/mol, see the Supporting Information (SI)), but the chlorine-atom abstraction was more accurately modeled in UM11/6-31+G** calculations.
We first explored how the observations compared with predictions from transition state theory. Rate constants for the α-cleavage of β-13C-substituted 3a–c were calculated using canonical variational transition state theory (CVT) including small-curvature tunneling (SCT) using the GAUSSRATE/POLYRATE set of programs.8,9 The KIE predictions are subject to a series of complicating issues. For 3a, there are two competitive low-energy TSs. For 3b, the two Cα–Cβ bonds are not equivalent (differing by 0.04 Å), with bond-length isomers separated by a 0.4 kcal/mol barrier. This required allowance for the isotopically perturbed equilibrium between the isomers. For 3c, the octyl-group conformation desymmetrizes the ring opening, and it was assumed that conformational interconversion in the octyl group was slower than the very rapid ring opening. The tunneling contribution to the KIEs was in all cases substantial, at 1.0–1.2%. After allowing for each complication (see the SI for details), the predicted KIEs are shown in Table 1.
For 3a, the CVT/SCT-predicted KIE matches closely with the experimental value, within the error of the measurement. However, the experimental KIEs for 3b and 3c are far below the predicted values. The first-order interpretation of this difference is that it is associated with the differing barriers for α-cleavage in the systems. The cleavage barrier for 3a is relatively large, at 10.8 kcal/mol, and this leads to a lifetime that is sufficiently long for full energy equilibration to occur. As a result, the ring opening is governed by transition state theory and the CVT/SCT KIE prediction is accurate. With 3b and 3c, the α-cleavage barriers are much smaller. If the ring opening is faster than thermal equilibration, transition state theory is not applicable. This would cause the CVT/SCT predictions to be inaccurate.
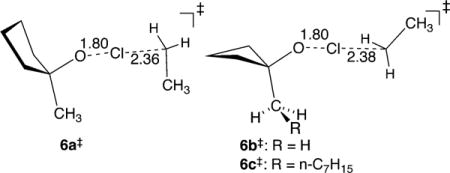
A more detailed interpretation of the results requires consideration of the amount of energy available to the intermediates 3a–c. The excess energy of ∼39 kcal/mol that is generated in the abstraction of a chlorine atom from 1 and 4 is partitioned into the two products, 3 and 5, and within each product that energy is partitioned into rotational, translational, and vibrational components (Figure 1). Of these, only the vibrational energy in 3 is available to promote α-cleavage. To assess this pivotal portion of the energy, we applied a modified version of the classical single-trajectory approximation of Hase.10 Series of trajectories were started from TSs 6a–c‡ for chlorine-atom abstraction from 1a–c by an ethyl radical, providing no zero-point energy for the real normal modes and a Boltzmann-random energy in the transition vector. The trajectories were then integrated forward in time, and the components of the energy were evaluated as the products 3 and chloroethane separated. Average energy partitionings were then calculated for each series.
Figure 1.
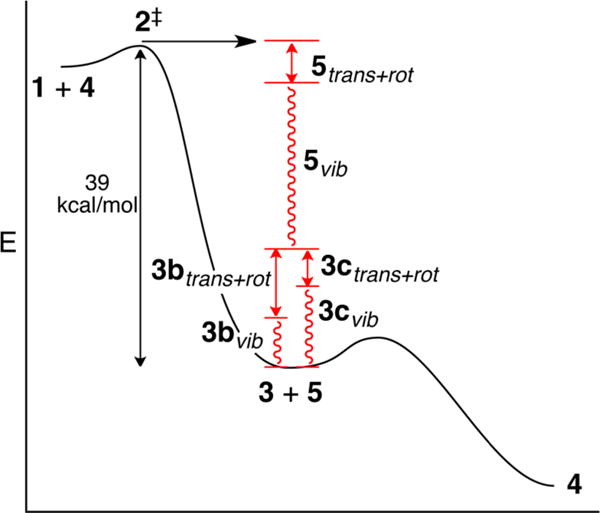
Energy partitioning in the chlorine-atom abstraction. Only the vibrational energy in 3 can promote the second step.
The exothermic atom abstractions 6a–c‡ have early TSs, so the Polanyi rules would predict that the largest proportion of the excess energy ends up as vibrational energy in 5. This prediction is correct; the chloroethane receives two-thirds of the energy, 26 kcal/mol, with 21 kcal/mol ending up as vibrational energy. The incipient alkoxy radical 3 receives 13 kcal/mol of excess energy. For 3a, 5.8 kcal/mol is put into vibrational energy, but this is not enough to overcome the α-cleavage barrier so the intermediate radical must await ordinary thermal activation. For 3b and 3c, however, their initial excess vibrational energy exceeds the cleavage barrier. This by itself would not guarantee that the α-cleavage would occur before thermal equilibration, but it signals the possibility.
For the 13 kcal/mol of excess energy in 3a–c, the partitioning depends strikingly on the structure. In particular, the presence of the octyl chain in 3c leads to a much higher proportion of the energy partitioned into vibrational energy (Table 1).
To explain this novel observation, we consider a limiting pure impulsive11 model for 3 in which the impulse from the repulsion between the oxygen and chlorine atoms after TS 2‡ acts only on these atoms, imparting a size-independent energy of EO to the oxygen atom, with all other atoms in the alkoxy fragment at rest. The initial momentum of the oxygen atom pO would be defined by eq 1, where mO is the oxygen-atom mass. Ignoring rotations for now, the EO would sometime later be partitioned into translational energy Etrans and vibrational energy Evib. Assuming that the molecular momentum pmol is conserved over short times in solution, so that pmol = pO, the Etrans would be defined by eq 2, where malkyl is the mass of the alkyl group in 3. In going from 3b to 3c, malkyl increases from 69 amu to 167, and Etrans would fall by more than a factor of 2. This model shows clearly why the translational energy should decrease as the alkyl group size increases from 3b to 3c, leaving more energy in Evib (eq 3). A parallel but more complex analysis can be made for rotational energy (see the SI). Overall, the effect in Table 1 is dramatic and has experimental consequences.
| (1) |
| (2) |
| (3) |
The effect of the differing excess energies in 3b and 3c was analyzed in two ways, statistically and with dynamic trajectories. If the excess energy in 3b and 3c is distributed statistically and if it is temporarily assumed that no energy is lost from 3 to the solvent on the time scale of the α-cleavage, then RRKM theory would apply. RRKM KIEs were predicted for 3b and 3c based on an energy distribution in each that is the sum of the canonical distribution and the excess vibrational energies derived above, including approximate SCT tunneling corrections of 1.002–1.004 (see the SI). As shown in Table 1, the RRKM KIE matches well with that observed for the α-cleavage of 3b. The RRKM-predicted KIE for 3c is however far too high versus experiment. Why?
Some insight into this question comes from trajectory studies. Quasiclassical direct-dynamics trajectories12 were initiated from the area of TSs 6b‡ and 6c‡. Each normal mode in 6b‡ and 6c‡ was given its zero-point energy (ZPE) plus a randomized excitation energy based on a Boltzmann distribution at 42 °C, with a random phase and sign for its initial velocity. The trajectories were then propagated forward and backward in time until either α-cleavage occurred to afford 4 or until 1 and ethyl radical were reformed. For both sets of trajectories, the ring-opening α-cleavage ensues within a few hundred femtoseconds after 3b/3c has separated from the chloroethane. However, the 3c α-cleavage occurs much more quickly; the average trajectory time starting from 6b‡ was 377 fs, while for 6c‡ the time is reduced to 259 fs. After formation of 3b and 3c, approximately 80 fs after 6b‡ and 6c‡, their decay was approximately exponential (see the SI) with half-lives of 200 and 125 fs, respectively. For comparison, the RRKM half-lives would be 340 and 300 fs, respectively. Both radicals are decaying faster than expected from their energies, but with 3c the α-cleavage is more accelerated.
Our hypothesis is that the energy in 3c is not statistically distributed and that little energy has been distributed to the octyl chain at the time of α-cleavage. The effect of the nonstatistical distribution of the energy is that the cyclobutyl ring moiety is much “hotter” than would be expected from the available vibrational energy. In the single-trajectory study starting from 6c‡, about 1 kcal/mol of the 7.2 kcal/mol of vibrational energy is passed down the octyl chain rapidly, within 100 fs, by ballistic energy transfer.13 Over the course of the next 300 fs, about 1 kcal/mol of additional energy makes its way into the chain. At equilibrium ∼4.8 kcal/mol of the vibrational energy, over half of the excess vibrational energy, would be in the alkyl chain, but 3c never lasts long enough for either the chain or the solvent to take up much energy. Instead, the nonstatistically localized excess energy promotes the rapid α-cleavage of 3c before equilibration can proceed.
We have previously applied a “localized RRKM” model,14 adapted from Rabinovitch,15 that assumes that the excess energy is localized within a “molecular subset ” of the molecule. This process allows an approximate statistical prediction of the rate or selectivity when only a portion of a larger molecule is vibrationally excited. In the current case, our molecule subset replaces the n-heptyl chain of 3c with a hydrogen atom (making it 3b). The model is then applied simply in an RRKM calculation by replacing the frequencies of 3c and its cleavage TS by those of 3b and its cleavage TS. When this is done, the predicted RRKM KIE is decreased to 1.033. While this is still higher than the experimental value, the model prediction is close enough to support the general idea that the low KIE with 3c is the result of a combination of greater excess energy in 3c relative to 3b and a nonstatistical distribution of that energy.
Overall, the governance of statistical rate theories in the α-cleavage of alkoxy radicals depends on the barrier for the cleavage, the amount of vibrational energy available from their formation, and the size of the system. With 3a, the barrier is larger than vibrational energy engendered by its formation, and TST governs the α-cleavage. With 3b, the available vibrational energy is greater than the barrier, but the system is small and RRKM theory provides a reasonable prediction of the selectivity. By increasing the size of the alkyl chain, the vibrational energy in 3c is increased and the α-cleavage occurs faster than equilibration of the vibrational energy. The α-cleavage selectivity becomes nonstatistical, but it can be approximately rationalized using a localized statistical model.
The results here illustrate one new rule with respect to energy partitioning in reactions and how control of that partitioning can lead to nonstatistical effects in experimental observations. We expect that other rules await discovery. On a more general level, our results show how the behavior of a reactive intermediate can depend on how it is formed, and we are pursuing reactions that will make use of this history-dependence to affect selectivity.
Supplementary Material
Acknowledgments
We thank the NIH (Grant GM-45617) for financial support. H.K. thanks the Japan Society for the Promotion of Science for a Postdoctoral Fellowship for Research Abroad.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b09052.
Complete descriptions of experimental procedures, calculations, and structures (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.(a) Zare RN. Science. 1998;279:1875–1879. doi: 10.1126/science.279.5358.1875. [DOI] [PubMed] [Google Scholar]; (b) Crim FF. Acc Chem Res. 1999;32:877–884. [Google Scholar]
- 2.(a) Polanyi JC, Wong WH. J Chem Phys. 1969;51:1439–1450. [Google Scholar]; (b) Polanyi JC. Acc Chem Res. 1972;5:161–168. [Google Scholar]
- 3.(a) Crim FF. Faraday Discuss. 2012;157:9–26. doi: 10.1039/c2fd20123b. [DOI] [PubMed] [Google Scholar]; (b) Ebrahimi M, Guo SY, McNab IR, Polanyi JC. J Phys Chem Lett. 2010;1:2600–2605. [Google Scholar]; (c) Liu J, Song K, Hase WL, Anderson SL. J Am Chem Soc. 2004;126:8602–8603. doi: 10.1021/ja048635b. [DOI] [PubMed] [Google Scholar]; (d) Kroes GJ, Gross A, Baerends EJ, Scheffler M, McCormack DA. Acc Chem Res. 2002;35:193–200. doi: 10.1021/ar010104u. [DOI] [PubMed] [Google Scholar]; (e) Halstead D, Holloway S. J Chem Phys. 1990;93:2859–2870. [Google Scholar]; (f) Voth GA, Hochstrasser RM. J Phys Chem. 1996;100:13034–13049. [Google Scholar]; (g) Campbell VL, Chen N, Guo H, Jackson B, Utz AL. J Phys Chem A. 2015;119:12434–12441. doi: 10.1021/acs.jpca.5b07873. [DOI] [PubMed] [Google Scholar]; (h) Xu L, Doubleday CE, Houk KM. J Am Chem Soc. 2010;132:3029–3037. doi: 10.1021/ja909372f. [DOI] [PubMed] [Google Scholar]; (i) Guo H, Jiang B. Acc Chem Res. 2014;47:3679–3685. doi: 10.1021/ar500350f. [DOI] [PubMed] [Google Scholar]
- 4.(a) Carpenter BK. Acc Chem Res. 1992;25:520–528. [Google Scholar]; (b) Debbert SL, Carpenter BK, Hrovat DA, Borden WT. J Am Chem Soc. 2002;124:7896–7897. doi: 10.1021/ja026232a. [DOI] [PubMed] [Google Scholar]; (c) Carpenter BK, Harvey JN, Orr-Ewing AJ. J Am Chem Soc. 2016;138:4695–4705. doi: 10.1021/jacs.6b01761. [DOI] [PubMed] [Google Scholar]; (d) Doubleday C, Jr, Bolton K, Hase WL. J Am Chem Soc. 1997;119:5251–5252. [Google Scholar]; (e) Doubleday C, Suhrada CP, Houk KN. J Am Chem Soc. 2006;128:90–94. doi: 10.1021/ja050722w. [DOI] [PubMed] [Google Scholar]; (f) Biswas B, Collins SC, Singleton DA. J Am Chem Soc. 2014;136:3740–3743. doi: 10.1021/ja4128289. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Chen Z, Nieves-Quinones Y, Waas JR, Singleton DA. J Am Chem Soc. 2014;136:13122–13125. doi: 10.1021/ja506497b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Oyola Y, Singleton D. J Am Chem Soc. 2009;131:3130–3131. doi: 10.1021/ja807666d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Thomas JR, Waas JR, Harmata M, Singleton DA. J Am Chem Soc. 2008;130:14544–14555. doi: 10.1021/ja802577v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Ussing BR, Hang C, Singleton DA. J Am Chem Soc. 2006;128:7594–7607. doi: 10.1021/ja0606024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Singleton DA, Hang C, Szymanski MJ, Greenwald EE. J Am Chem Soc. 2003;125:1176–1177. doi: 10.1021/ja027221k. [DOI] [PubMed] [Google Scholar]; (l) Yang Z, Yu P, Houk KN. J Am Chem Soc. 2016;138:4237–4242. doi: 10.1021/jacs.6b01028. [DOI] [PubMed] [Google Scholar]
- 5.Walling C, Padwa A. J Am Chem Soc. 1963;85:1593–1597. [Google Scholar]
- 6.(a) Orlando JJ, Tyndall GS, Wallington TJ. Chem Rev. 2003;103:4657–4690. doi: 10.1021/cr020527p. [DOI] [PubMed] [Google Scholar]; (b) Hayes CJ, Merle JK, Hadad CM. Adv Phys Org Chem. 2009;43:79–134. [Google Scholar]; (c) Inoue S, Kumagai T, Tamezawa H, Aota H, Matsumoto A, Yokoyama K, Matoba Y, Shibano M. Polym J. 2010;42:716–721. [Google Scholar]; (d) Dean RT, Fu S, Stocker R, Davies MJ. Biochem J. 1997;324:1–18. doi: 10.1042/bj3240001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hartung J, Gottwald T, Spehar K. Synthesis. 2002:1469–1498. [Google Scholar]
- 7.(a) Singleton DA, Szymanski MJ. J Am Chem Soc. 1999;121:9455–9456. [Google Scholar]; (b) Singleton DA, Schulmeier BE. J Am Chem Soc. 1999;121:9313–9317. [Google Scholar]; (c) Gonzalez-James OM, Zhang X, Datta A, Hrovat DA, Borden WT, Singleton DA. J Am Chem Soc. 2010;132:12548–12549. doi: 10.1021/ja1055593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng J, Zhang S, Corchado JC, Chuang YY, Coitino EL, Ellingson BA, Zheng J, Truhlar DG. GAUSSRATE, version 2009-A. University of Minnesota; Minneapolis, MN: 2010. [Google Scholar]
- 9.Zheng J, Zhang S, Lynch BJ, Corchado JC, Chuang YY, Fast PL, Hu W–P, Liu Y-P, Lynch GC, Nguyen KA, Jackels F, FernandezRamos A, Ellingson BA, Melissas VS, Villa J, Rossi I, Coitino EL, Pu J, Albu TV, Steckler R, Garrett BC, Isaacson AD, Truhlar DG. POLYRATE, version 2010. University of Minnesota; Minneapolis, MN: 2010. [Google Scholar]
- 10.Sun L, Park K, Song K, Setser DW, Hase WL. J Chem Phys. 2006;124:064313. doi: 10.1063/1.2166236. [DOI] [PubMed] [Google Scholar]
- 11.Busch GE, Wilson KR. J Chem Phys. 1972;56:3626–3638. [Google Scholar]; For an alternative approach based on the degrees of freedom, see:; Nogueira JJ, Hase WL, Martinez-Nunez E. J Phys Chem C. 2014;118:2609–2621. [Google Scholar]
- 12.Hase WL, Song KH, Gordon MS. Comput Sci Eng. 2003;5:36–44. [Google Scholar]
- 13.Schwarzer D, Kutne P, Schröder C, Troe J. J Chem Phys. 2004;121:1754–1764. doi: 10.1063/1.1765092. [DOI] [PubMed] [Google Scholar]
- 14.Quijano LMM, Singleton DA. J Am Chem Soc. 2011;133:13824–13827. doi: 10.1021/ja2043497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meagher JF, Chao KJ, Barker JR, Rabinovitch BS. J Phys Chem. 1974;78:2535–2543. [Google Scholar]; See also:; Rice OK. Z Phys Chem B. 1930;7:226–233. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.