

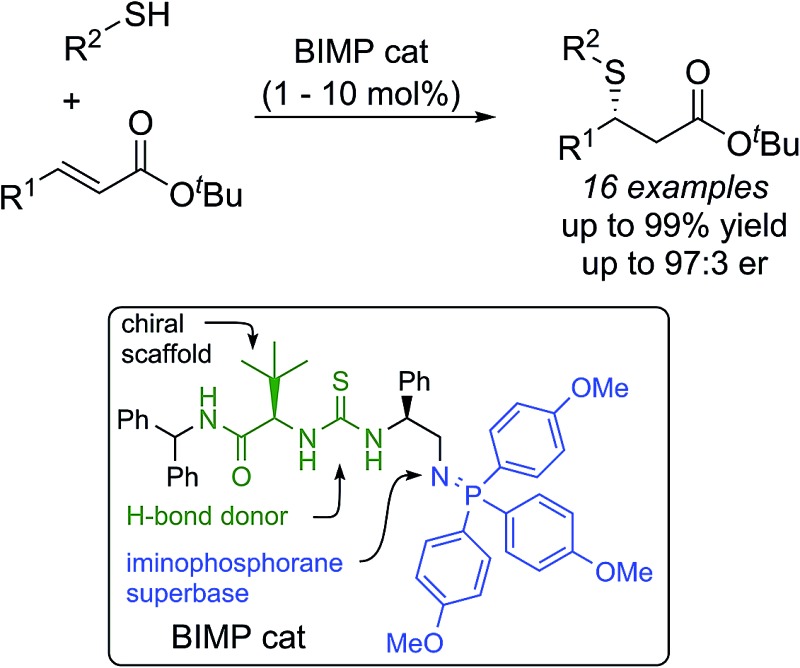
 The highly enantioselective sulfa-Michael addition of alkyl thiols to unactivated β-substituted-α,β-unsaturated esters catalyzed by a bifunctional iminophosphorane (BIMP) organocatalyst is described.
The highly enantioselective sulfa-Michael addition of alkyl thiols to unactivated β-substituted-α,β-unsaturated esters catalyzed by a bifunctional iminophosphorane (BIMP) organocatalyst is described.
Abstract
The highly enantioselective sulfa-Michael addition of alkyl thiols to unactivated β-substituted-α,β-unsaturated esters catalyzed by a bifunctional iminophosphorane (BIMP) organocatalyst is described. The low acidity of the alkyl thiol pro-nucleophiles is overcome by the high Brønsted basicity of the catalyst and the chiral scaffold/thiourea hydrogen-bond donor moiety provides the required enantiofacial discrimination in the addition step. The reaction is broad in scope with respect to the alkyl thiol and β-substituent of the α,β-unsaturated ester, affords sulfa-Michael adducts in excellent yields (up to >99%) and enantioselectivity (up to 97 : 3 er) and can operate down to 1 mol% catalyst loading.
Unactivated β-substituted-α,β-unsaturated esters, such as methyl crotonate, methyl cinnamate and their homologues, are a class of low reactivity electrophiles that offer a wealth of untapped potential in the field of enantioselective organocatalysis.1 To date, these esters have remained a persistent challenge as Michael acceptors in asymmetric catalysis using both metal-rich and metal-free catalyst systems, largely due to their low inherent electrophilicity2 and low propensity for catalyst activation and enantioface discrimination.3,4 They are commercial and cheap, or are readily prepared by a variety of standard methods and are stable. In contrast to commonly used (reactive) Michael acceptors such as nitroolefins, they lie at the bottom of the Mayr electrophile reactivity (E) scale,5,6 and unlike enal and enone Michael acceptors they cannot be activated through iminium ion formation with chiral amine catalysts.7 Related literature examples employ activated carboxylic derivatives8 such as N-enoyl imides, N-enoyl oxazolidinones, perfluorinated alkyl esters, thioamides, N-enoyl pyrroles and, most recently, aryl esters.9 Alternatively, activating substituents at the α- or β-positions can also be used to gain reactivity and/or stereoselectivity. To illustrate the case in point, to date there has not been a single report of a highly enantioselective addition of a pro-nucleophilic reagent [a carbon-centered (C–H) or heteroatom-centered (X–H) acid] to unactivated alkyl cinnamate or crotonate esters under organocatalytic conditions.10 Effectively, these cheap chemical feedstocks are out of reach of existing chiral organocatalysts and accordingly are a very attractive ‘simple’ target class of electrophiles for new enantioselective organocatalytic reaction development (Fig. 1).
Fig. 1. Bifunctional Brønsted base/H-bond donor organocatalytic SMA to α,β-unsaturated ester derivatives.

A proven strategy to overcome low substrate electrophilicity in base-catalyzed polar addition reactions is to increase the concentration of the nucleophilic conjugate base in the pot – and therefore the rate of the nucleophilic addition reaction – by enhancing the Brønsted basicity of the catalyst relative to tertiary amine catalysts.11–13 To this end, we disclosed that bifunctional iminophosphorane (BIMP) catalysts, containing a novel organosuperbase were highly efficacious in the first general enantioselective organocatalytic ketimine nitro-Mannich reaction.12b,d Likewise, very recently, high catalyst performance (in terms of reactivity and enantioselectivity) with a second generation BIMP catalyst was also witnessed in the first organocatalytic conjugate addition of alkyl thiols to unactivated α-substituted acrylate esters (such as methyl methacrylate).12e In both of these transformations an organosuperbase was demonstrated to be essential for reactivity.
We speculated that the reluctance of unactivated β-substituted-α,β-unsaturated esters to undergo organocatalytic Michael addition reactions could be overcome using our BIMP catalyst family. To exemplify this we chose the sulfa-Michael addition (SMA) of alkyl thiols as this is a reaction of central importance for the asymmetric construction of chiral sulfides possessing a stereogenic centre at the β-carbon and no organocatalytic enantioselective version has previously been reported.14,15 We reasoned that the high Brønsted basicity of our BIMP catalysts could activate the high pK a alkyl thiol pro-nucleophile (pK a(DMSO) = 17 for n-BuSH)16,17 and the modular design of the catalyst family, through its variable backbone scaffold, hydrogen-bond donor group and iminophosphorane superbase would expedite optimal catalyst identification. Herein, and as part of our research program towards the development of novel asymmetric reactions with challenging electrophile/pro-nucleophile combinations, we wish to report our investigations leading to the highly enantioselective SMA reaction of alkyl thiols to unactivated β-substituted-α,β-unsaturated esters.
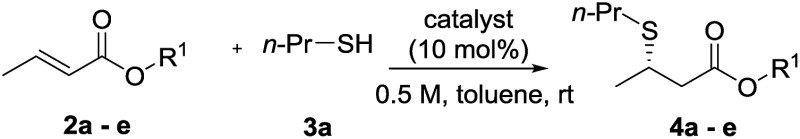
We chose commercially available methyl crotonate (2a) and 1-propanethiol (3a) as our model system and investigated reactivity using first generation BIMP catalyst 1a (Table 1, entry 1). In toluene, at room temperature using 10 mol% catalyst we were delighted to observe an exceptional reactivity profile; β-mercaptoester product 4a was afforded in near quantitative yield after only 2 hours with low but significant enantiocontrol (55 : 45 er).18 With good reactivity established we next investigated the performance of a small library of second generation BIMP catalysts featuring variations around the amide–thiourea motif that we recently reported12e (Table 1, entries 2–6). The modular design of our BIMP catalysts allowed rapid library generation and our attention focussed on the amide–thiourea moiety as the H-bond donor group and the tris-(4-methoxyphenylphosphine) derived iminophosphorane as the Brønsted basic group (Fig. 2).
Table 1. Catalyst screening studies and reaction optimization a .
| ||||||
| Entry | Cat. | R1 | Product | Time (h) | Yield b (%) | er c |
| 1 | 1a | Me | 4a | 2 | 94 | 55 : 45 |
| 2 | 1b | Me | 4a | 2 | 98 | 55 : 45 |
| 3 | 1c | Me | 4a | 2 | 94 | 52 : 48 |
| 4 | 1d | Me | 4a | 2 | 93 | 59 : 41 |
| 5 | 1e | Me | 4a | 2 | >99 | 75 : 25 |
| 6 | 1f | Me | 4a | 2 | 97 | 62 : 38 |
| 7 d | 1g | Me | 4a | 3 | >99 | 81 : 19 |
| 8 | 1g | Et | 4b | 3 | 95 | 84 : 16 |
| 9 | 1g | i-Pr | 4c | 3 | >99 | 85 : 15 |
| 10 | 1g | Bn | 4d | 3 | >99 | 81 : 19 |
| 11 d | 1g | t-Bu | 4e | 8 | 94 | 92 : 8 |
| 12 d , e | 1g | t-Bu | 4e | 8 | 95 | 94 : 6 |
| 13 f | 1g | t-Bu | 4e | 24 | 94 | 96 : 4 |
| 14 g | 1g | t-Bu | 4e | 72 | 94 | 97 : 3 |
aReactions were carried out with 0.20 mmol of 2 and 0.60 mmol of 3a.
bIsolated yield.
cDetermined by HPLC analysis on a chiral stationary phase.
dReaction performed on 0.10 mmol scale of 2a.
eReaction performed at 0 °C.
fReaction performed at 0 °C in Et2O.
gReaction performed at –15 °C in Et2O.
Fig. 2. Bifunctional iminophosphorane (BIMP) organocatalysts used in the optimization of the SMA reaction. PMP = p-methoxyphenyl.

Catalysts 1b–d possessing a thiourea constructed from two (S)-configured tert-leucine derived residues, the tris-(4-methoxyphenylphosphine)-derived iminophosphorane and a variable terminal amide group gave poor enantioselectivity in all cases (Table 1, entries 2, 3, and 4). When catalyst 1e – the diastereomer of 1d – was trialled however, a significant boost to the enantioselectivity was witnessed (Table 1, entry 5, 75 : 25 er).19
A comparison with an analogous catalyst possessing a phenylglycine and a tert-leucine residue (1g) resulted in a slight improvement to the enantioselectivity (Table 1, entry 7, 81 : 19 er). At this stage, the effect of varying the ester group of the crotonate on the enantioselectivity in the SMA was investigated. A range of simple, commercial or readily synthesized alkyl crotonate esters were trialled and a correlation between the size of the ester group and the enantioselectivity was observed – pleasingly tert-butyl crotonate (2e) afforded the product 4e in 92 : 8 er albeit in a slightly increased reaction time of 8 h (Table 1, entry 11). A reoptimization of the reaction conditions to 0.5 M in Et2O at 0 °C (Table 1, entries 12 & 13 and ESI†) resulted in a significant boost to the enantioselectivity (96 : 4 er) and cooling the reaction temperature further to –15 °C afforded β-mercaptoester 4e in 94% yield and 97 : 3 er (Table 1, entry 14).
With optimized reaction conditions established, the scope of the transformation with respect to the thiol pro-nucleophile and the α,β-unsaturated ester was investigated (Fig. 3). Minimal variation to the enantioselectivity was observed across a good range of linear (propyl to decyl) or branched (cyclic and acyclic) alkyl mercaptans. The reaction with 4-methoxybenzyl mercaptan was also well-tolerated and afforded the β-mercaptoester 4m in 99% yield and 95 : 5 er at –15 °C.
Fig. 3. Scope of the SMA of alkyl thiols to β-substituted-α,β-unsaturated esters. Reactions were carried out with 0.20 mmol 2 and 0.60 mmol 3. Yields are isolated yields and enantiomeric ratios were determined by HPLC analysis or GC analysis on a chiral stationary phase. aThe reaction was performed at –15 °C. bThe reaction was quenched after 96 h. cAbsolute configuration of 4n determined by chemical correlation (see ESI†).

Following investigation into the scope of the reaction with respect to the alkyl thiol, variation to the β-substituent of the α,β-unsaturated ester was subsequently examined using 1-propanethiol or 4-methoxybenzyl mercaptan as the sulfur-centred pro-nucleophile. We were pleased to observe that the excellent reactivity and selectivities were maintained when tert-butyl cinnamate, bearing a phenyl group at the β-position, was used as the electrophile to afford the desired β-mercaptoester 4n in 98% yield and 88 : 12 er.
Similarly, excellent yields of the β-mercaptoesters 4o–r were obtained from the corresponding primary alkyl β-substituted-α,β-unsaturated esters with very good levels of enantiocontrol. β-Mercaptoesters 4s and 4t containing a terminal N-Boc protected amine and TBS protected hydroxyl group respectively were also synthesized in good to excellent yields and excellent enantiomeric ratios.
Although the scope of the reaction was performed with 10 mol% catalyst loading, we were keen to demonstrate lower loadings were viable. Accordingly, and after reoptimization of the reaction conditions, to 5 M in Et2O at 0 °C, we were pleased to find β-mercaptoester 4e was afforded in near quantitative yield and 95 : 6 er on 7 mmol scale of tert-butyl crotonate (2e) using 1 mol% catalyst 1g (Scheme 1).
Scheme 1. Preparative scale synthesis of 4e.

To demonstrate synthetic utility of the β-mercaptoester products a selection of standard chemical transformations were carried out (Scheme 2). Thus β-mercaptoester 4e (95 : 5 er) was transesterified to the methyl ester 4a in a two step process; initial acidic cleavage of the tert-butyl ester and subsequent methyl ester formation under acidic conditions afforded 4a in 78% yield without compromising stereochemical integrity. Oxidation of 4e afforded sulfone 5a without any observable racemization in near quantitative yield. Finally, β-mercaptoester 4m was reduced to the alcohol in excellent yield, without appreciable loss of enantiopurity.20
Scheme 2. Derivatization. (a) TFA, Et2O, 0 °C to rt, then SOCl2, MeOH, 0 °C to rt, 78% yield over two steps, 94 : 6 er. (b) m-CPBA, CH2Cl2, 0 °C, 2 h, 96% yield, 94 : 6 er. (c) DIBAL-H, THF, –60 °C, 2 h, 93% yield, 93 : 7 er.

In summary, we have developed the first organocatalytic enantioselective SMA of alkyl thiols to unactivated β-substituted-α,β-unsaturated esters. Impressive reactivity and excellent levels of enantioselectivities were achieved across a range of linear, branched, cyclic alkyl and benzylic thiols, in SMA reactions to various β-substituted-α,β-unsaturated esters using a novel bifunctional iminophosphorane catalyst. This work demonstrates that the high reactivity of the BIMP catalysts enables low reactivity electrophiles such as β-substituted-α,β-unsaturated esters to undergo highly enantioselective conjugate addition reactions for the first time and thus represents a significant advance in the field. Work to uncover further capabilities of the BIMP catalyst family is ongoing in our laboratories and the results will be disclosed in due course.
Acknowledgments
We thank the EPSRC (Studentship and Doctoral Training Grant [EP/M50659X/1] to A. J. M. F.), AstraZeneca (Studentship to A. J. M. F.), the SCI (Postgraduate Scholarship to A. J. M. F.) and the China Scholarship Council-University of Oxford Scholarship (Studentship to J. Y.) for funding.
Footnotes
References
- For reviews on organocatalysis, see: ; (a) Dalko P. I., Moisan L. Angew. Chem., Int. Ed. 2004;43:5138. doi: 10.1002/anie.200400650. [DOI] [PubMed] [Google Scholar]; (b) Berkessel A. and Gröger H., Asymmetric Organocatalysis, Wiley VCH, Weinheim, 2005. [Google Scholar]; (c) List B., Yang J. W. Science. 2006;313:1584. doi: 10.1126/science.1131945. [DOI] [PubMed] [Google Scholar]; (d) Dondoni A., Massi A. Angew. Chem., Int. Ed. 2008;47:4638. doi: 10.1002/anie.200704684. [DOI] [PubMed] [Google Scholar]; (e) Bertelsen S., Jørgensen K. A. Chem. Soc. Rev. 2009;38:2178. doi: 10.1039/b903816g. [DOI] [PubMed] [Google Scholar]; (f) Palomo C., Oiarbide M., López R. Chem. Soc. Rev. 2009;38:632. doi: 10.1039/b708453f. [DOI] [PubMed] [Google Scholar]
- (a) Matsunaga S., Kinoshita T., Okada S., Harada S., Shibasaki M. J. Am. Chem. Soc. 2004;126:7559. doi: 10.1021/ja0485917. [DOI] [PubMed] [Google Scholar]; (b) López F., Harutyunyan S. R., Meetsma A., Minnaard A. J., Feringa B. L. Angew. Chem., Int. Ed. 2005;44:2752. doi: 10.1002/anie.200500317. [DOI] [PubMed] [Google Scholar]; (c) Pubill-Ulldemolins C., Bonet A., Bo C., Gulyás H., Fernández E. Chem.–Eur. J. 2012;18:1121. doi: 10.1002/chem.201102209. [DOI] [PubMed] [Google Scholar]
- Tomioka K. and Nagaoka Y., Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfatlz and H. Yamamoto, Springer, New York, 1999, vol. 3. [Google Scholar]
- For examples of enantioselective organocatalytic cycloaddition reactions using crotonate and cinnamate esters, see: ; (a) Mathieu B., de Fays L., Ghosez L. Tetrahedron Lett. 2000;41:9561. [Google Scholar]; (b) Ryu D. H., Lee T. W., Corey E. J. J. Am. Chem. Soc. 2002;124:9992. doi: 10.1021/ja027468h. [DOI] [PubMed] [Google Scholar]; (c) Gatzenmeier T., van Gemmeren M., Xie Y., Höfler D., Leutzsch M., List B. Science. 2016;351:949. doi: 10.1126/science.aae0010. [DOI] [PubMed] [Google Scholar]
- The electrophile-specific reactivity parameters E of trans-β-nitrostyrenes have been determined to be –15 < E < –12: Zenz I., Mayr H., J. Org. Chem., 2011, 76 , 9370 . [DOI] [PubMed] [Google Scholar]
- Preliminary studies indicate the electrophile-specific reactivity parameters E of ethyl cinnamate and ethyl crotonate are in the range of −25 < E < −23: H. Mayr, personal communication.
- For a review on iminium catalysis, see: Erkkilä A., Majander I., Pihko P., Chem. Rev., 2007, 107 , 5416 . [DOI] [PubMed] [Google Scholar]
- For reviews on organocatalytic conjugate addition reactions, see: ; (a) Vicario J. L., Badía D., Carrillo L. and Reyes E., Organocatalytic Enantioselective Conjugate Addition Reactions, Royal Society of Chemistry, Cambridge, 2010. [Google Scholar]; (b) Tsogoeva S. B. Eur. J. Org. Chem. 2007:1701. [Google Scholar]
- The organocatalytic enantioselective conjugate addition of benzyl mercaptan to activated aryl crotonate esters was reported with a N-heterocyclic carbene catalyst: Yuan P., Meng S., Chen J., Huang Y., Synlett, 2016, 27 , 1068 . [Google Scholar]
- To the best of our knowledge, only a single isolated example of a moderately enantioselective organocatalytic Michael addition to ethyl crotonate has been reported, see: Dong X., Fang X., Wang C.-J., Org. Lett., 2011, 13 , 4426 . [DOI] [PubMed] [Google Scholar]
- For reviews on Brønsted base H-bond donor bifunctional organocatalysts, see: ; (a) Takemoto Y. Org. Biomol. Chem. 2005;3:4299. doi: 10.1039/b511216h. [DOI] [PubMed] [Google Scholar]; (b) Connon S. J. Chem. Commun. 2008:2499. doi: 10.1039/b719249e. [DOI] [PubMed] [Google Scholar]; (c) Marcelli T., Hiemstra H. Synthesis. 2010:1229. [Google Scholar]
- For the use of chiral organosuperbase catalysts to enhance reactivity, see: ; (a) Bandar J. S., Lambert T. H. J. Am. Chem. Soc. 2012;134:5552. doi: 10.1021/ja3015764. [DOI] [PubMed] [Google Scholar]; (b) Núñez M. G., Farley A. J. M., Dixon D. J. J. Am. Chem. Soc. 2013;135:16348. doi: 10.1021/ja409121s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Takeda T., Terada M. J. Am. Chem. Soc. 2013;135:15306. doi: 10.1021/ja408296h. [DOI] [PubMed] [Google Scholar]; (d) Goldys A. M., Núñez M. G., Dixon D. J. Org. Lett. 2014;16:6294. doi: 10.1021/ol5029942. [DOI] [PubMed] [Google Scholar]; (e) Farley A. J. M., Sandford C., Dixon D. J. J. Am. Chem. Soc. 2015;137:15992. doi: 10.1021/jacs.5b10226. [DOI] [PubMed] [Google Scholar]; (f) Robertson G. P., Farley A. J. M., Dixon D. J. Synlett. 2016;27:21. [Google Scholar]; (g) Horwitz M. A., Zavesky B. P., Martinez-Alvarado J., Johnson J. S. Org. Lett. 2016;18:36. doi: 10.1021/acs.orglett.5b03127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the use of organosuperbases in synthesis, see: ; (a) Ishikawa T., Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts, Wiley, New York, 2009. [Google Scholar]; (b) Ishikawa T., Kumamoto T. Synthesis. 2006:737. [Google Scholar]; (c) Leow D., Tan C.-H. Chem.–Asian J. 2009;4:488. doi: 10.1002/asia.200800361. [DOI] [PubMed] [Google Scholar]; (d) Leow D., Tan C.-H. Synlett. 2010:1589. [Google Scholar]; (e) Ishikawa T. Chem. Pharm. Bull. 2010;58:1555. doi: 10.1248/cpb.58.1555. [DOI] [PubMed] [Google Scholar]; (f) Fu X., Tan C.-H. Chem. Commun. 2011;47:8210. doi: 10.1039/c0cc03691a. [DOI] [PubMed] [Google Scholar]; (g) Selig P. Synthesis. 2013;45:703. [Google Scholar]; (h) Krawczyk H., Dzięgielewski M., Deredas D., Albrecht A., Albrecht Ł. Chem.–Eur. J. 2015;21:10268. doi: 10.1002/chem.201500481. [DOI] [PubMed] [Google Scholar]; (i) Corey E. J., Grogan M. J. Org. Lett. 1999;1:157. doi: 10.1021/ol990623l. [DOI] [PubMed] [Google Scholar]; (j) Ishikawa T., Araki Y., Kumamoto T., Seki H., Fukuda K., Isobe T. Chem. Commun. 2001:245. [Google Scholar]; (k) Nugent B. M., Yoder R. A., Johnston J. N. J. Am. Chem. Soc. 2004;126:3418. doi: 10.1021/ja031906i. [DOI] [PubMed] [Google Scholar]; (l) Terada M., Ube H., Yaguchi Y. J. Am. Chem. Soc. 2006;128:1454. doi: 10.1021/ja057848d. [DOI] [PubMed] [Google Scholar]; (m) Uraguchi D., Sakaki S., Ooi T. J. Am. Chem. Soc. 2007;129:12392. doi: 10.1021/ja075152+. [DOI] [PubMed] [Google Scholar]; (n) Davis T. A., Wilt J. C., Johnston J. N. J. Am. Chem. Soc. 2010;132:2880. doi: 10.1021/ja908814h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Sohtome Y., Shin B., Horitsugi N., Takagi R., Noguchi K., Nagasawa K. Angew. Chem., Int. Ed. 2010;49:7299. doi: 10.1002/anie.201003172. [DOI] [PubMed] [Google Scholar]; (p) Davis T. A., Johnston J. N. Chem. Sci. 2011;2:1076. doi: 10.1039/C1SC00061F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Uraguchi D., Yoshioka K., Ueki Y., Ooi T. J. Am. Chem. Soc. 2012;134:19370. doi: 10.1021/ja310209g. [DOI] [PubMed] [Google Scholar]; (r) Corbett M. T., Uraguchi D., Ooi T., Johnson J. S. Angew. Chem., Int. Ed. 2012;51:4685. doi: 10.1002/anie.201200559. [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Misaki T., Jin N., Kawano K., Sugimura T. Chem. Lett. 2012;41:1675. [Google Scholar]; (t) Shubina T. E., Freund M., Schenker S., Clark T., Tsogoeva S. B. Beilstein J. Org. Chem. 2012;8:1485. doi: 10.3762/bjoc.8.168. [DOI] [PMC free article] [PubMed] [Google Scholar]; (u) Bandar J. S., Lambert T. H. J. Am. Chem. Soc. 2013;135:11799. doi: 10.1021/ja407277a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (v) Bandar J. S., Barthelme A., Mazori A. Y., Lambert T. H. Chem. Sci. 2015;6:1537. doi: 10.1039/c4sc02402h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (w) Uraguchi D., Yamada K., Ooi T. Angew. Chem., Int. Ed. 2015;54:9954. doi: 10.1002/anie.201503928. [DOI] [PubMed] [Google Scholar]; (x) Işik M., Unver M. Y., Tanyeli C. J. Org. Chem. 2015;80:828. doi: 10.1021/jo5022597. [DOI] [PubMed] [Google Scholar]; (y) Gao X., Han J., Wang L. Org. Lett. 2015;17:4596. doi: 10.1021/acs.orglett.5b02323. [DOI] [PubMed] [Google Scholar]; (z) Chen J., Meng S., Wang L., Tang H., Huang Y. Chem. Sci. 2015;6:4184. doi: 10.1039/c5sc00878f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on asymmetric sulfa-Michael additions, see: ; (a) Enders D., Lüttgen K., Narine A. A. Synthesis. 2007:959. [Google Scholar]; (b) Nishimura K., Ono M., Nagaoka Y., Tomioka K. J. J. Am. Chem. Soc. 1997;119:12974. [Google Scholar]; (c) Kanemasa S., Oderaotoshi Y., Wada E. J. Am. Chem. Soc. 1999;121:8675. [Google Scholar]; (d) Nishimura K., Ono M., Nagaoka Y., Tomioka K. Angew. Chem., Int. Ed. 2001;40:440. [PubMed] [Google Scholar]; (e) Hui Y., Jiang J., Wang W., Chen W., Cai Y., Lin L., Liu X., Feng X. Angew. Chem., Int. Ed. 2010;49:4290. doi: 10.1002/anie.201000105. [DOI] [PubMed] [Google Scholar]; (f) Bonollo S., Lanari D., Pizzo F., Vaccaro L. Org. Lett. 2011;13:2150. doi: 10.1021/ol200379r. [DOI] [PubMed] [Google Scholar]; (g) Kitanosono T., Sakai M., Ueno M., Kobayashi S. Org. Biomol. Chem. 2012;10:7134. doi: 10.1039/c2ob26264a. [DOI] [PubMed] [Google Scholar]; (h) Ogawa T., Kumagai N., Shibasaki M. Angew. Chem., Int. Ed. 2012;51:8551. doi: 10.1002/anie.201204365. [DOI] [PubMed] [Google Scholar]
- For a review on organocatalytic asymmetric SMA reactions, see: ; (a) Chauhan P., Mahajan S., Enders D. Chem. Rev. 2014;114:8807. doi: 10.1021/cr500235v. [DOI] [PubMed] [Google Scholar]; (b) Hiemstra H., Wynberg H. J. Am. Chem. Soc. 1981;103:417. [Google Scholar]; (c) Kumar A., Salunkhe R. V., Rane R. A., Dike S. Y. J. Chem. Soc., Chem. Commun. 1991:485. [Google Scholar]; (d) McDaid P., Chen Y., Deng L. Angew. Chem., Int. Ed. 2002;41:338. doi: 10.1002/1521-3773(20020118)41:2<338::aid-anie338>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]; (e) Li B.-J., Jiang L., Liu M., Chen Y.-C., Ding L.-S., Wu Y. Synlett. 2005:603. [Google Scholar]; (f) Leow D., Shishi L., Chittimalla S. K., Fu X., Tan C.-H. Angew. Chem., Int. Ed. 2008;47:5641. doi: 10.1002/anie.200801378. [DOI] [PubMed] [Google Scholar]; (g) Liu Y., Sun B., Wang B., Wakem M., Deng L. J. Am. Chem. Soc. 2009;131:418. doi: 10.1021/ja8085092. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Kimmel K. L., Robak M. T., Ellman J. A. J. Am. Chem. Soc. 2009;131:8754. doi: 10.1021/ja903351a. [DOI] [PubMed] [Google Scholar]; (i) Rana N. K., Selvakumar S., Singh V. K. J. Org. Chem. 2010;75:2089. doi: 10.1021/jo902634a. [DOI] [PubMed] [Google Scholar]; (j) Dai L., Wang S.-X., Chen F.-E. Adv. Synth. Catal. 2010;352:2137. [Google Scholar]; (k) Rana N. K., Singh V. K. Org. Lett. 2011;13:6520. doi: 10.1021/ol202808n. [DOI] [PubMed] [Google Scholar]; (l) Dai L., Yang H., Chen F. Eur. J. Org. Chem. 2011:5071. [Google Scholar]; (m) Palacio C., Connon S. Chem. Commun. 2012;48:2849. doi: 10.1039/c2cc17965b. [DOI] [PubMed] [Google Scholar]; (n) Uraguchi D., Kinoshita N., Nakashima D., Ooi T. Chem. Sci. 2012;3:3161. [Google Scholar]; (o) Dai L., Yang H., Niu J., Chen F. Synlett. 2012:314. [Google Scholar]; (p) Breman A. C., Smits J. M. M., de Gelder R., van Maarseveen J. H., Ingemann S., Hiemstra H. Synlett. 2012;23:2195. [Google Scholar]; (q) Fang X., Li J., Wang C.-J. Org. Lett. 2013;15:3448. doi: 10.1021/ol4015305. [DOI] [PubMed] [Google Scholar]; (r) Unhale R. A., Rana N. K., Singh V. K. Tetrahedron Lett. 2013;54:1911. [Google Scholar]; (s) Wang R., Liu J., Xu J. Adv. Synth. Catal. 2014;357:159. [Google Scholar]; (t) Phelan J. P., Patel E. J., Ellman J. A. Angew. Chem., Int. Ed. 2014;53:11329. doi: 10.1002/anie.201406971. [DOI] [PubMed] [Google Scholar]; (u) Fu N. K., Zhang L., Luo S. Z., Cheng J. P. Org. Lett. 2014;16:4626. doi: 10.1021/ol5022178. [DOI] [PubMed] [Google Scholar]
- Bordwell F. G., Hughes D. L. J. Org. Chem. 1982;47:3224. [Google Scholar]
- For comparison, the pK a of thiophenol in DMSO is 10.3. See ref. 16
- Bifunctional cinchonine derived bifunctional thiourea catalysts [890044-38-9] were found to be impotent in this transformation. After 7 days under analogous conditions no addition product 4a was observed by 1H NMR analysis of the crude reaction mixture
- The reaction with PhSH, 2a and catalyst 1e proceeded with 93% yield and 66 : 34 er
- The PMB thiol can be readily cleaved to afford the free mercaptan, see for example ref. 15g
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.