

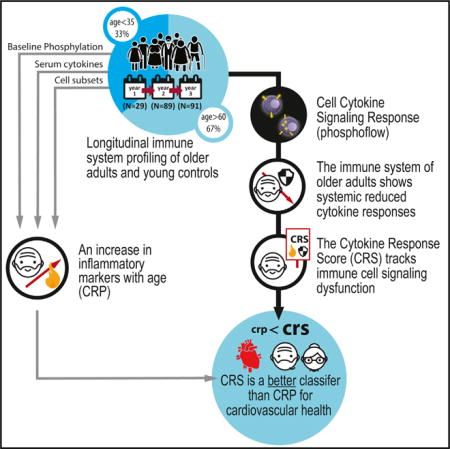
SUMMARY
Chronic inflammation, a decline in immune responsiveness, and reduced cardiovascular function are all associated with aging, but the relationships among these phenomena remain unclear. Here, we longitudinally profiled a total of 84 signaling conditions in 91 young and older adults and observed an age-related reduction in cytokine responsiveness within four immune cell lineages, most prominently T cells. The phenotype can be partially explained by elevated baseline levels of phosphorylated STAT (pSTAT) proteins and a different response capacity of naive versus memory T cell subsets to interleukin 6 (IL-6), interferon α (IFN-α), and, to a lesser extent, IL-21 and IFN-γ. Baseline pSTAT levels tracked with circulating levels of C-reactive protein (CRP), and we derived a cytokine response score that negatively correlates with measures of cardiovascular disease, specifically diastolic dysfunction and atherosclerotic burden, outperforming CRP. Thus, we identified an immunological link between inflammation, decreased cell responsiveness in the JAK-STAT pathway, and cardiovascular aging. Targeting chronic inflammation may ameliorate this deficiency in cellular responsiveness and improve cardiovascular function.
Graphical abstract
INTRODUCTION
Aging is associated with reduced immune responses, a phenomenon known as immunosenescence (Duraisingham et al., 2013). Multiple alterations in both the innate and adaptive arms of the immune system have been described, such as changes in immune cell-subset abundance and relative frequencies, decreased B and T cell proliferation, and reduced or altered function of multiple cell subsets (Furman et al., 2013; Weiskopf et al., 2009). While some of the mechanisms for this decline have been well studied, e.g., the decrease in naive T cells as a consequence of a reduction in thymic output (Palmer, 2013) and altered peripheral homeostasis (Qi et al., 2014), the changes observed in immune cell function are less well understood. Reductions in cellular responses with age, observed in multiple cells and pathways, have been noted (Furman et al., 2013; Furman et al., 2015; Longo et al., 2014; Longo et al., 2012), yet how these relate to other aspects of immune aging and whether they are associated with any other types of clinical pathology remain unknown.
Unlike acute inflammation, where high levels of inflammatory cells and cytokines are produced in response to a known cause such as infection or tissue injury, age-related chronic inflammation is characterized by a milder elevation of inflammatory components, a process known as “inflammaging” (Franceschi et al., 2000a), most prominently involving the pro-inflammatory proteins C-reactive protein (CRP) and interleukin-6 (IL-6) (Chovatiya and Medzhitov, 2014; Franceschi et al., 2000; Shaw et al., 2010; Wikby et al., 2006), whose increase has been associated with most age-related diseases, including cardiovascular disease (Frasca and Blomberg, 2016). A likely possibility for the link between low-grade chronic inflammation and age-related disease is cross-talk between an aging immune system and damaged tissue. In the case of cardiovascular disease, it is believed that innate sensing of danger signals from endothelial cells that have suffered from diverse perturbations (e.g., shear stress, hypoxia and nutrient deprivation following ischemia-reperfusion injury) induces an inflammatory state and activation of tissue repair processes (Epelman et al., 2015). In older adults suffering from chronic inflammation, this also seems to promote differentiation of adaptive immune cells with an accumulation of highly differentiated CD4+CD28− and CD8+CD28− T cells, considered to be hallmarks of immunosenescence and able to produce abundant quantities of inflammatory cytokines (Maly and Schirmer, 2015), which alone or in combination with other inflammatory markers (Morrisette-Thomas et al., 2014), have been suggested to predict both a risk of death and hospitalization (Adriaensen et al., 2015).
Despite our current understanding of such relationships, given the complexities and difficulties of studying the immune system in cardiac tissues in humans, we largely rely on the mechanisms observed in animal studies. Until recently, observational studies in humans have been conducted in a piecemeal fashion, providing us with little understanding about how the multiple age-associated immune components fit together in age-associated disease. While population and cross-sectional studies can be very important in discerning major trends, longitudinal studies are in many respects the ideal, because one is able to distinguish stable trends versus short-term fluctuations in a given subject.
Here, we broadly investigated immune cell signaling dysfunction and its association with inflammation and aging. We longitudinally sampled the peripheral blood of individuals of varying ages over the course of 3 years, sampled once a year as part of the Stanford-Ellison cohort. We profiled each sample for cell-subset specific JAK-STAT intra-cellular signaling responses to select pro- and anti-inflammatory cytokines. We focused on the STAT-signaling pathway because it is a sensitive measure for fluctuations in the levels of inflammatory mediators, such as those found in higher concentration in the circulation of aging individuals. We identified a systemic age-associated attenuation of cytokine responses, even at high concentrations, that is largely stable over 3 years. Complementary to this, we also measured 51 serum cytokines and broadly phenotyped immune cell subsets as part of our standard immune monitoring panel. Finally, we performed a comprehensive cardiovascular assessment of 40 of the older members of this cohort (Figure 1A). We show that this impaired cytokine response phenotype is driven by differences in naive and memory T cell subsets and elevated basal pSTAT levels and is clinically associated with markers of systemic, low-grade chronic inflammation, diastolic dysfunction, and increased atherosclerotic burden. We also derive a cytokine response score (CRS) that tightly associates immune health with cardiovascular function and may be useful clinically in the diagnosis and management of aging pathologies, especially as it can be monitored regularly with a blood draw. It also shows that lymphocytes, with their ability to circulate throughout the body, can serve as reporters of chronic inflammation, likely well in advance of clinical disease.
Figure 1. Phosphorylation of STATs in Response to Cytokine Stimulation Decreases with Age.
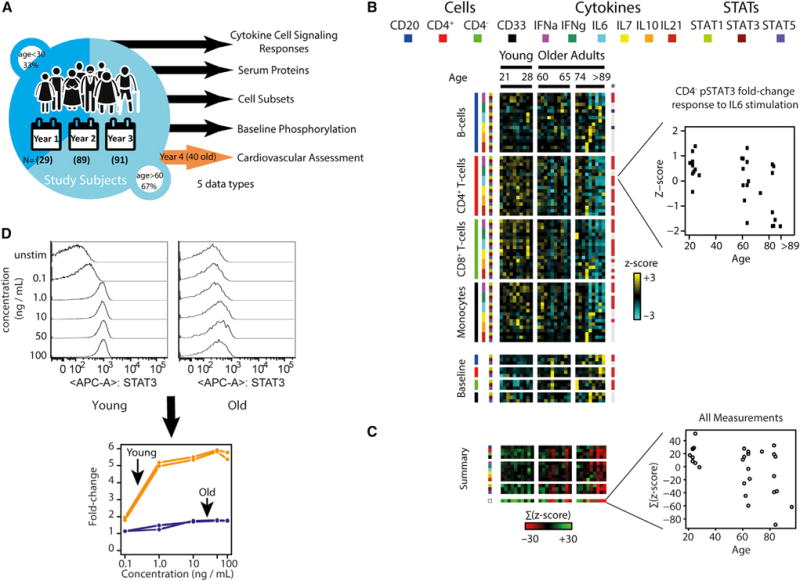
(A) Diagram of study design. Young and old adults were profiled repeatedly at yearly intervals for up to 3 years using multiplex immune assays. In the fourth year of the study, 40 older adult participants volunteered to undergo comprehensive cardiovascular monitoring.
(B) Heatmap of 72 cytokine stimulation assays measured across 29 individuals of different ages. Rows are assays color-coded by the condition (cell subset, cytokine, and pSTAT combination). Heatmap cells are colored according to the normalized fold change Z scores. Age-associated differences at a q ≤ 0.15 are marked on the right (red, decrease with age; purple, increase; gray, no significant difference). Inset of CD8 T cells stimulated by IL-6 and assayed for change in pSTAT3 abundance illustrates the reduced fold change response of some older adults.
(C) Individual age-associated cytokine response assays summarized by cell subset, stimuli or pSTAT protein. Reduction in responses is often systemic, with an individual exhibiting multiple dysfunctions. Inset shows a Z score summarization across individuals of all 72 assays. Older adults show reduced responses compared to young adults and high within-group variability.
(D) Reduced cellular response in old adults is independent of stimulus concentration. The entire 72-condition cytokine response assay was repeated at five different doses per stimuli for two young (orange) and old (blue) adults. Shown is an example histogram (top) and line plot visualization (bottom) of the fold change cytokine response as a function of cytokine stimuli concentration. A fold change of 1 signifies no change from baseline.
RESULTS
The Cytokine Signaling Response of Older Adults Is Systemically Reduced, Irrespective of Stimuli Dose
To identify age-associated differences in cellular immune responses, we performed cytokine stimulation assays on immune cell subsets isolated from peripheral blood samples of 29 self-identified healthy individuals aged 24–89 years (see Table S1 for demographics and Table S2 for types of assays run and the number of subjects per assay). For each individual, we performed 12 baseline phosphoprotein and 72 cytokine-response assays (four cell subsets, stimulated by six cytokines, and measuring the phosphorylation level of STAT1, STAT3, and STAT5; see Figure S1 for gating scheme) and detected an age-associated difference in 4 baseline and 39 responses at a false discovery rate of 5% (q ≤ 0.05) (Figure 1B; Table S3; STAR Methods) (Storey and Tibshirani, 2003). We observed age-associated differences for “canonical” (i.e., well established) cell-cytokine-pSTAT combinations, as well as other interactions that have been less studied and were generally of lower magnitude (Figure S2). In addition, the fold-change response was reduced between young (<40 years) and older adults (>60 years) across all assays identified as age-associated (with a mean reduction of 31%), differences that did not correlate with cytomegalovirus (CMV) seropositivity (Furman et al., 2015), diabetes status, or obesity (data not shown). Additionally, we observed a systemic (i.e., multiple combinations of cell subsets, stimuli and pSTAT were affected) reduction in cytokine response in the same individuals (Figure 1C).
Given the reports of increased inflammation with age, we examined whether older adults are functionally adapted to increased inflammation and require higher stimuli concentrations for an equivalent fold-change response to that observed in cells of young individuals. We repeated the stimulation assay for two young and two older adults at six different doses per each of the six stimuli. Older adults showed consistently decreased fold-changes as compared to the young, irrespective of dose level (Figures 1D and S3A), and all achieved saturation of fold-change pSTAT response at the highest stimulation dose (Figure S3B). Hence, the cell cytokine response difference between old and young adults is independent of the cytokine concentration. Taken together, we conclude that a systemic reduction in immune cell potential to respond to cytokine signals co-occurs with age, across multiple arms of the immune system, and independent of the stimuli concentration.
The Cytokine Signaling Response of Older Adults Is Reproducibly Reduced and Longitudinally Stable
To follow up this observation, we expanded the cohort in year 2 and for an additional year (91 individuals, 82 of whom returned the third year and 25 of whom also participated in the first year of the study; STAR Methods; Table S1). All the assays measured in year 1 were conducted for each individual in the second and third year with minor changes; new recruited participants donated blood and were assayed for pSTAT1, 3, and 5 responses in monocytes, B cells and CD4+ and CD8+ T cells stimulated by the same cytokine panel stimuli (with the exception of IL-7, for which we observed few age-dependent differences and thus replaced it with IL-2). Of the 60 cell-cytokine response assays run in each of the three study years, we applied a stringent cutoff to identify 17 cell-cytokine response assays as reproducibly age-associated and reduced with age (p value ≤ 0.05 on the Fisher’s combined probability test and a q-value < 5% in at least 2 years) (Figure 2A; Table S3). These included CD8+ T cell responses to IL-6, interferon α (IFN-α), IFN-γ, and IL-21 stimulations in pSTAT1, 3, and 5; CD4+ T cell responses to IL-6 and INF-α in pSTAT5; and B cell and monocyte responses to multiple stimuli in pSTAT3. In addition, we noted reproducible baseline levels of pSTAT1 elevation in B cells, CD4+ and CD8+ T cells, and, to a lesser extent, in pSTAT3. Notably, elevations in pSTAT5 baselines were not reproducibly detected, despite an age-associated pSTAT5 fold change reduction noted for multiple cell types and assays. Of note, in the majority of cases where no age-association was detected in the expanded cohort, this was primarily due either to a few older adults whose responses were similar to that of young adults or a few young individuals who exhibited a reduction in cytokine responses. This suggests that attenuation of cytokine responses, while age associated and predominant in older adults, is not exclusive to them and that not all cytokine response assays are temporally stable.
Figure 2. Population Stability of Age-Associated Differences in Cellular Response.
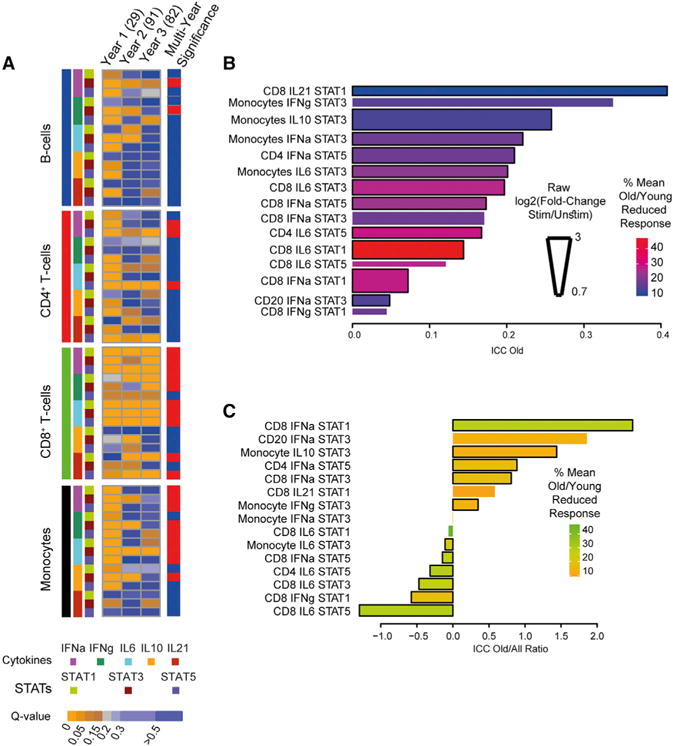
(A) Heatmap of 60 cytokine stimulations and 12 baseline measured over 3 years in partially overlapping individuals of different ages. Each row represents a single condition and is color-coded by the FDR (q-value) of the assay in a given cohort. A Fisher’s combined probability test, weighted for sample size each year, was performed on the multiple hypothesis adjusted p values of all assays in a year and is shown on the left. Red denotes a Fisher’s combined probability p value < 0.05.
(B) The intra-class correlation (ICC), a measure of within-subject stability, of age-associated cytokine response assays was found to be moderate to low for individual cytokine response assays when measured across older adults enrolled in the study for 3 years. Bar length denotes ICC magnitude, whereas bar width denotes measured (not normalized) fold change of response and color corresponds to the mean percent reduction in response of old compared to the young.
(C) The ratio of ICC values between old adults and all study participants is shown for all 15 ICC positive cytokine response assays. Bar color corresponds to the mean percent reduction in response of old versus young adults. A negative correlation exists between the cytokine response assay ICC score and the mean percent reduction in cytokine response (Spearman’s rho = −0.49, p = 0.058). Older adults tend toward increased stability (i.e., higher ICC), except in those assays in which they show a large reduction in response compared to the young.
The longitudinal nature of our experimental design, whereby most individuals returned from year to year over the course of 2–3 years, enabled us to assess the consistency of assays at the individual level across repeated visits. We next focused on older adults as we reasoned that their high between-individual variation compared to the young (Figures 1B and 1C, insets) could be leveraged toward understanding the biological and clinical contexts in which this systemic reduction in response occurs, with the narrower age span of older adults providing for finer resolution and the young individuals providing insights as controls. To do so, we computed the intra-class correlation (ICC) of each cytokine response assay across the 3 years of returning older adults (Figure 2B). The ICC provides a single summary measure of stability across multiple years of within-subject agreement in cytokine responses (between years) relative to the total observed variance (an ICC of 1 implies within-subject variation is non-existent; see STAR Methods). Of the 17 age-associated cytokine response assays, only 6 showed moderate consistency (ICC > 0.2) in ordering from year to year, with the remaining assays ICC values below that; for two (CD20+-IFNγ-pSTAT3 and CD8+-IL21-pSTAT5), the ICC score was below zero, implying rapid random change (i.e., year-to-year individual reordering), which may be due either to biological alterations in these older adults from one year to the next or to technical measurement noise. Of note, the conflict between stability and change was apparent in this analysis; the top most stable hit, CD8+-IL21-pSTAT1, was also the age-associated assay showing the smallest percent reduction in response between young and old adults. We also computed the ratio of ICC values between old adults and all study participants (Figure 2C). We noted that older adults tended toward increased stability (i.e., higher ICC), except in those assays in which they showed a large reduction in response compared to the young. Specifically, we detected a negative correlation between the cytokine response assay ICC score and the mean percent reduction in cytokine response of old versus young adults (Figure 2C; Spearman’s rho = −0.49, p = 0.058). This suggested that over time, an older adults’ cytokine response may continue degrading and hence that assays with low ICC values should be considered of relevance due to the real-world alterations of the human immune system, particularly with respect to the biology of aging, where fragility may lead to cytokine response instability. Taken together, we concluded that this set of reproducibly age-associated, pan-cell, pan-cytokine response assays strikes a balance between individual stability and biological decline and hence can be used to understand, at a systems level, the context and mechanism by which differences in cytokine response can arise.
Reduced Cytokine Responses Are Correlated with Increased Baseline Levels of pSTAT and Elevation in Memory Cell Populations
Given the systemic nature of this phenotype (Figure 1C), and the complexity of individual responses profiles (Figure 1B; individuals differed on the identity of assays in which their responses were most reduced), we reasoned that by summing across multiple assays we could buffer some of this biological and technical variation while capturing a system level complex phenotype. We defined a CRS as the sum of 15 reproducibly age-associated normalized cytokine responses whose ICC was positive (specifically, CD8: IFN-α pSTAT1, 3, and 5; CD8: IL-6 pSTAT1, 3, and 5, IFN-γ pSTAT1, IL-21 pSTAT1; CD4: IFN-α pSTAT5, IL-6 pSTAT5, CD20: IFN-α pSTAT1; and monocyte: IL-10 pSTAT3, IFN-γ pSTAT3, IFN-α pSTAT3, and IL-6 pSTAT3). Next, we identified correlates of the CRS in older adult by regressing the score against serum soluble proteins, cell subset, and baseline phosphoprotein levels, from samples collected from the same individuals at the same time point in each of the three assayed years (Table S4, significance estimated by permutation). Restricting our analysis to those features common to all three cohorts, we applied a stringent cutoff to identify features significantly associated with the CRS (p value ≤ 0.05 on the Fisher’s combined probability test and a q-value < 5% in at least 2 years). This procedure identified a set of ten features best correlated with the reduced response, which were predominantly cell-subset frequency alterations and baseline (unstimulated) pSTAT protein levels (Figure 3A). We identified the soluble inflammatory chemokine Rantes (CCL5), which is involved in the recruitment of leukocytes to the site of inflammation and in arterial tissue repair (von Hundelshausen et al., 2001), as having the strongest positive correlation with the CRS. Beyond this, the most prominent associations were the multiple elevations in baseline phosphoprotein levels, which negatively correlated with the CRS, and associations with cell subsets (B cell frequency and central memory and CD28− CD8+ T cell subset frequencies). This latter association with memory cell subsets suggested that the CRS may be correlated with the relative frequencies of cell subsets and that cell subsets may vary in their response to cytokine stimulations.
Figure 3. Reduced Responses to Cytokine Stimulation Are Primarily due to Elevation of Basal Levels of Phosphorylated STAT Proteins and Differences in Naive and Memory Cell Responses.
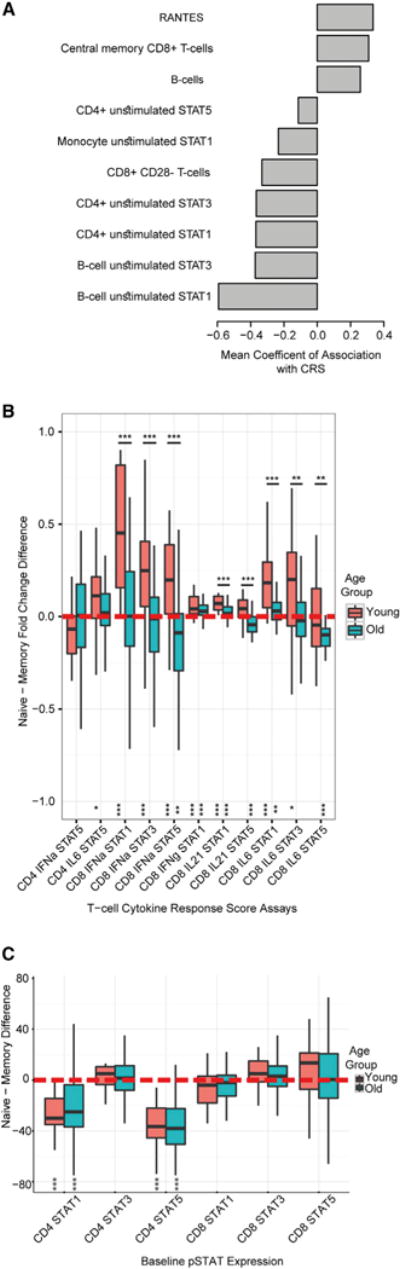
(A) The mean coefficient of association across three study years of immune measures found associated with the CRS (using a Fisher’s combined p value < 0.05 and a q-value < 5% in participants for least 2 of 3 years).
(B and C) Boxplots showing naive and memory CD4+ and CD8+ T cell cytokine responses of T cell member assays in the CRS (B) and baseline pSTAT levels (C) compared within an individual and between the young and old age groups. Zero (red dotted line) denotes no difference between naive and memory fold-change responses. Asterisks at the bottom denote significance of a paired within-individual naive memory comparison and those above boxplots denote between-group comparisons by Wilcoxon test (***p < 0.005, **p < 0.01, *p < 0.05).
To test this, and fitting with a naive-memory ratio shift being a hallmark of immunosenescence, we assayed cytokine responses of naive and memory CD4+ and CD8+ T cell populations in year 3 study subjects to understand naive-memory differences within an individual. As young and older adult immune system may be quite different from one another, we also analyzed the young as a control for the observations in the old (see STAR Methods). Comparing naive and memory cell subsets of stable cytokine-stimulation combinations that were included into the CRS, we observed that IL-6, IFN-α, and IL-21 stimulations in CD8+ cells in the young showed higher fold change STAT responses in naive cells than in the matched memory cell subsets of the same individual, as did CD4+ IL-6 pSTAT5 (Figure 3B; p < 0.05, Benjamini-Hochberg [BH] adjusted by Wilcoxon test; paired analysis for all assays, except for CD8+ IL-6 pSTAT5). However, naive cells of older adult individuals did not exhibit increased cytokine responses compared to memory cells in the same individual, except for IL-21 stimulations in CD8+ T cells and in CD8+ IL-6 pSTAT1 (p < 0.05, BH adjusted) and even exhibited a reversed signal (p < 0.05 by Wilcoxon test; paired analysis for cytokine responses of pSTAT5 in memory CD8+ T cells stimulated with IFN-α or IL-6 being higher than in naive cells). Interestingly, we noted differences in naive-memory responses between old and young were significant in all INF-α, IL-6, and IL-21 assays in CD8+ T cells (Figure 3Bl p < 0.01 by Mann-Whitney). To test whether differences in naive-memory response could be due to a difference in pSTAT baseline levels between the two cell types, we repeated the same analysis for unstimulated pSTAT level. In contrast to the fold change differences, which we primarily observed for CD8+ T cells, for baseline measures, we noted significant differences only in CD4+ T cells; specifically, higher pSTAT1 and pSTAT5 levels were found in CD4+ memory T cells. Taken together, this suggested that the observed response differences were not solely due to baseline naive-memory differences. Of note, although we noted differences in naive-memory response between young and old adults for almost all CRS stimulations, this was not the case for naive-memory baseline pSTAT differences, where young and old adults behaved similarly. In conjunction with this, as previously observed (Longo et al., 2014), we noted a reduction with age from young to older adults in 18 pSTAT signaling responses in CD4+ and CD8+ naive and memory cell stimulations (q < 0.05), primarily in IL-6 and IFN-α but also in IL-21 and IFN-γ (Table S5). Taken together, this suggests that the reduction in cytokine response with age is more dramatic in naive than in memory cell subsets and that naive-memory differences can only partially explain the observed between-individual differences in cell cytokine responses.
The second class of associated measures we identified with the CRS was an elevation in baseline phosphoprotein levels. Interestingly, across all 3 years, by far the strongest feature we found to be associated with the CRS was a negative correlation with baseline levels of pSTAT1 in B cells, even though the fold change reduction in B cell responses was smaller than most and only a single B cell cytokine-response assay met our stability and reproducibility criteria (Figure 2B). Baseline phosphoprotein levels may be expected to some extent to be negatively correlated with the fold change response to cytokines. However, careful analysis of our data suggested that this relation is observed much less often among older adults (r2 = 0.03 ± 0.01) than in the young (r2 = 0.2 ± 0.05) (Figure S4). Taken together, this suggests that variation in cytokine responses among older adults incorporates variation not only in baseline pSTAT levels but also in dysfunctional JAK-STAT signaling that is systemically shared across multiple lineages of the immune system.
Reduced Signaling Responses and Baseline Levels Correlate with Inflammation in Older Adults
An increase in inflammation with physiological age is well documented. Our CRS association analysis was restricted only to older adults, yet it identified inflammation-associated features such as the positive (i.e., improved cytokine response) association with Rantes and a negative association (i.e., poorer cytokine response) with an increased frequency of CD8+ CD28− T cells, a classic marker of immunosenescence and inflammaging (Weng et al., 2009). We thus wished to test whether immune cell signaling dysfunctions within our older adult cohort (CRS and baseline phosphoprotein levels) were associated with an established marker of clinical relevance to inflammation. To test this, we used measurements of highly sensitive CRP (hs-CRP), an established clinical marker of systemic inflammation, which we obtained as part of the assays conducted on year 2 subjects. Regression analysis revealed a significant association (p < 0.05) in older adults between CRP and the sum of pSTAT baseline levels across all immune cell types, but not with the CRS. In addition, we performed a supervised analysis in which we stratified older adults based on CRP level by standard levels of clinical risk (<1 mg/L versus >3 mg/L the for the hs-CRP assay) (Ridker, 2003; Ridker et al., 2000). This showed marked differences in sum of pSTAT baseline levels between groups with low and high inflammation, with individuals with high levels of CRP exhibiting elevated pSTAT baselines compared to those with low CRP values (Figure 4A; p = 0.01 by Wilcoxon test). For CRS, we observed a reciprocal trend (i.e., high CRP-low CRS and vice versa), yet with high variability, such that we detected no significant difference (Figure 4B; p = 0.49 by Wilcoxon test). Taken together, we conclude that immune cell signaling dysfunction partially captures known clinical marker of inflammation in older adults, which suggests that either pSTAT baseline levels or the CRS could be a useful indicator of unhealthy versus healthy aging.
Figure 4. Baseline Phosphoprotein Levels and Cytokine Response Are Correlated with Inflammation.
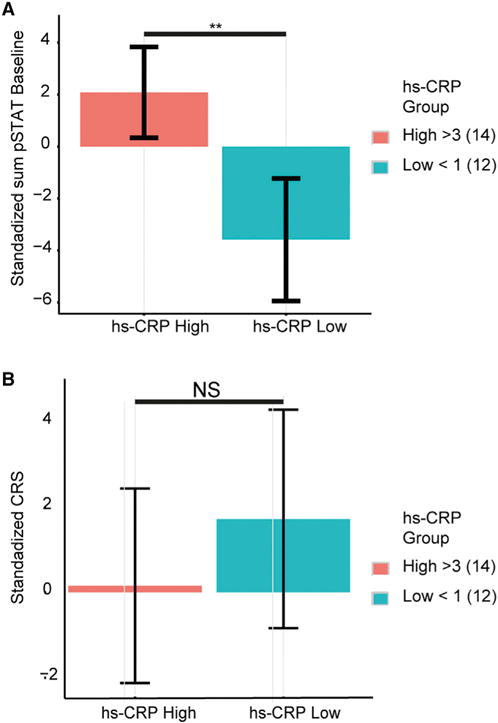
(A) Stratification of older adults based on standard levels of clinical risk for CRP (<1 mg/mL versus >3 mg/L for CRP) show significant differences in sum baseline cellular pSTAT levels (p = 0.01 by Wilcoxon test).
(B) A reciprocal, but not statistically significant, trend is observed for the CRS. Error bars depict SE.
Reduced Signaling Responses Are Associated with Atherosclerotic Burden and Diastolic Dysfunction
CRP and changes in immune subsets have been associated with cardiovascular aging (Howcroft et al., 2013; Okin and Medzhitov, 2012; Scrivo et al., 2011). Therefore, we sought to ask whether the cellular indicators of chronic inflammation described above, specifically diastolic dysfunction and atherosclerotic burden, two important parameters of cardiovascular aging, were also linked to cardiovascular disease. Approximately 12–16 months following their year 3 immune profile assessment, 40 older adults in our Stanford-Ellison cohort underwent cardiovascular phenotyping, including comprehensive clinical history, assessment of atherosclerotic plaque and arterial stiffness, and assessment of ventricular function including markers of ventricular relaxation (mitral annular velocity), abnormal filling pressures (E/e′ ratio surrogate), and left atrial (LA) remodeling (LA volume index) (Table S6; see STAR Methods for clinically established thresholds used in assessments). Subjects were graded by atherosclerotic burden into a clinical, sub-clinical, and minimal-inclusive burden and good cardiovascular health (n = 10, 7, and 5 and 18, respectively; see STAR Methods). For comparison purposes, when a cardiovascular parameter was known to be age related (e.g., aortic pulse wave velocity or mitral annular velocity), we age-adjusted both the immune signaling score and the assayed cardiovascular parameter.
We found a strong association between the CRS and the cardiovascular assessments of markers of ventricular/diastolic function, the strongest being with early diastolic mitral annular velocity (e′), a validated surrogate of ventricular relaxation for which we observed a nearly linear positive correlation with the CRS (Figure 5A; r = 0.62, p = 10−4). In addition, we observed a significantly reduced CRS in subjects with an increased ventricle filling pressure (Figure 5B; p = 0.04, measured by the E to e’ ratio, a validated surrogate of ventricle filling pressure) as well as in subjects with an enlarged LA volume index, a surrogate of atrial remodeling (Figure 5C; p = 0.02). We also observed a reduced CRS in subjects showing evidence of atherosclerotic burden (Figure 5D; p < 0.02 for age adjusted CRS). Interestingly those individuals who did not yet meet currently used criteria for sub-clinical cardiovascular disease but were no longer in good cardiovascular health also showed a poor CRS, suggesting its utility as a prognostic indicator of progressing cardiovascular disease. Moreover, the associations with cardiovascular functions were further supported by a negative correlation between the CRS and pulse wave velocity, a marker of central arterial stiffness, whereby individuals with an abnormally high level of stiffness had a trend towards reduced CRS scores (Figure 5E; p = 0.058 following age adjustment). Neither a single CRS member assay nor the baseline pSTAT levels performed as well as the CRS for stratifying these cardiovascular phenotypes (data not shown). Performing the same analysis without age-correcting the CRS showed qualitatively similar behavior (that is, a lower CRS score, i.e., a reduced fold-change response) in individuals meeting the abnormal criteria of each of these cardiovascular parameters assessed. In addition, to assess for confounding variables, we adjusted the CRS for CMV status, diabetes status, or BMI, all of which yielded qualitatively similar results following adjustment, suggesting neither could explain the association (Table S7). With respect to drugs, we detected no differences; however, individuals taking fish oil supplements (a source of omega 3 fatty acids with anti-inflammatory properties; Serhan et al., 2015) had a higher CRS (p = 0.04 by Wilcoxon test). Taken together, these results suggest a significant association between the CRS and both cardiac diastolic dysfunction and vascular atherosclerotic burden, two hallmarks of cardiovascular aging.
Figure 5. Reduced Signaling Responses Are Associated with Diastolic Dysfunction and Atherosclerotic Burden.
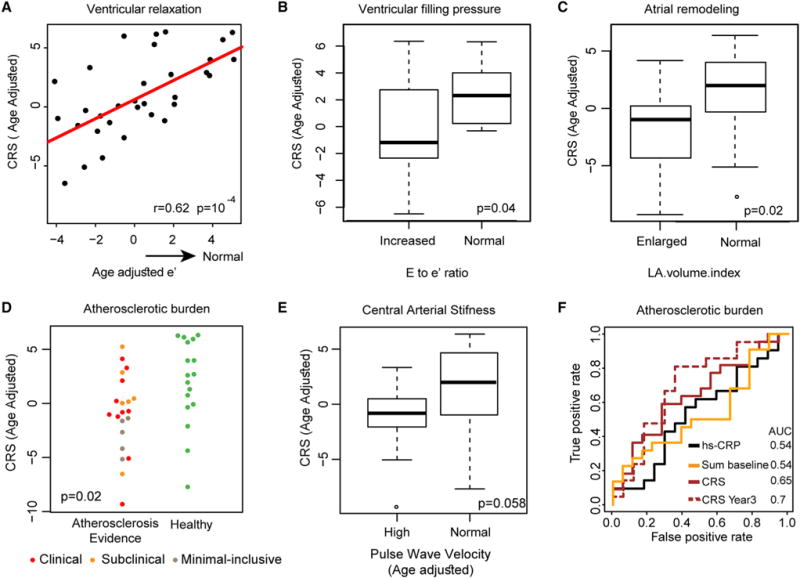
(A–C) The CRS is strongly associated with markers of diastolic function. (A) A positive linear correlation is observed between age adjusted e′, a sensitive measure of ventricular relaxation, and the age adjusted CRS (r = 0.62, p = 10−4). Age adjusted CRSs are significantly reduced in (B) individuals with increased E to e′ ratio (>9), a measure of increased ventricular filling pressure (p = 0.02) and in (C) individuals with a high left atrium volume index (>34), a measure of atrial remodeling (p = 0.02).
(D) Individuals with evidence of atherosclerotic burden (brown, orange, and red for minimal-inclusive, sub-clinical, and clinical subgroups) exhibit reduced CRS compared to those with healthy (green) cardiovascular aging (p = 0.02).
(E) Older adults exhibiting high central arterial stiffness as assessed by age-adjusted pulse wave velocity, a clinically validated measure (high > 10), show a reduced CRS (p = 0.058).
(F) ROC curve analysis comparing sensitivity and specificity of atherosclerosis burden classification by the CRS (as computed from year 2 or 3 data), the sum of pSTAT baseline data, and CRP in year 2. Both year 3 and year 2 CRS show improved classifier performance compared to that of CRP (AUC = 0.7, 0.65, 0.54, and 0.54, respectively for CRS year 3, CRS, sum of pSTAT baseline levels, and CRP).
To further test the sensitivity of immune signaling for classifying individuals for evidence of atherosclerotic burden versus health, we tested the performance of the CRS and baseline scores, compared with CRP based on year 2 values (CRP was only measured that year), almost 3 years prior to the cardiovascular assessment. Varying the classifier threshold, we computed the receiver operating characteristic (ROC) curves for each classifier, noting improved performance throughout the entire ROC range for the CRS scores compared to that of sum of pSTAT baseline and CRP (Figure 5F; area under the curve [AUC] = 0.65, 0.54, 0.54 for CRS, sum of pSTAT baseline and CRP). Performing this analysis when adjusting all tested measures for age yielded a similar ordering by performance but with lower AUCs. Along these lines, we found no associations between cardiovascular assessments and CRP levels, except for a weak correlation with e′ (p = 0.07), and the incorporation of CRP to any of the models for CRS and cardiovascular outcomes did not yield a better fit (p value for model comparison > 0.05 by likelihood ratio test). In addition, we tested classification performance based on year 3 CRS data, which were roughly 12 months more proximal to the measured cardiovascular parameters. This yielded an AUC of 0.7 (Figure 5F), suggesting that narrowing the time window between immune and cardiovascular monitoring could yield even better classification performance. We also observed better performance of CRS as a classifier compared the immune cell subset frequencies of CD8+ CD28− and CD4+ CD28− T cells, both classic immune measures that have previously been associated with immunosenescence and cardiovascular risk (Dumitriu et al., 2009; Goronzy and Weyand, 2013) (Figure S5), suggesting that functional cell cytokine response assays capture information of value beyond that obtained through typical phenotypic immune cell subset characterization or baseline phosphoprotein measures. Taken together, these results suggest that the CRS may be a better predictor of chronic inflammation and cardiovascular condition than other known immune-associated measures.
DISCUSSION
In this study, we used a systems immunology approach to profile the peripheral blood of generally healthy ambulatory donors of different ages across a number of technological platforms. Our aim was to identify robust and stable changes in cellular responses with age to better understand the biological context and clinical implications of reduced immune cell responsiveness with age. We focused on the lymphocytes and monocytes of the immune system, because they circulate throughout the body and likely reflect the overall inflammatory state of an individual. They are also easy to obtain in even a small blood sample and store well as peripheral blood mononuclear cells (PBMCs). We identify reduced signaling responses to cytokines with age, as we and others have previously observed (Furman et al., 2013, 2015; Longo et al., 2012, 2014), and show that this is a systemic and stable phenotype that is characteristic of many older adults, irrespective of the magnitude of stimulation. We summarize its most robust features in a CRS that aims to capture both the magnitude and system-wide effects of this phenotype in a simple scoring scheme while buffering for the biological and technical variation of any one single cell-cytokine-phosphoprotein stimulation.
We associate the dysfunction in immune cell signaling in older adults with an increase in CRP, a classic and widely used marker of inflammation. We further show that the CRS correlates with diastolic dysfunction and performs better as a classifier of atherosclerosis burden than CRP or other immune cell markers. This linkage to well-established metrics of cardiovascular status suggests the CRS may be a useful new proxy for healthy aging and is likely most relevant in complicated scenarios, such as those posed by diabetic subjects or patients with high levels of hypertension for whom standard cardiovascular assays are more difficult to perform. Beyond this, it would be important to determine the degree by which the reduced immune system responses precede early cardiovascular aging and, if so, to what extent. Interestingly, all the individuals who were classified as having early signs of sub-clinical atherosclerosis showed a strong reduction in their CRS, suggesting that this type of immune signaling dysfunction precedes clinically identifiable cardiovascular aging and hence could be an important early indicator of cardiovascular disease. Thus, while additional studies are needed to validate and properly employ this set of measurements in a clinical setting, the data presented here are encouraging as to its potential.
Although the JAK-STAT pathway has been shown to be robust against fluctuating environments (yielding a return to baseline levels at orders of magnitude differences in stimuli; Bachmann et al., 2011), our results suggest that low-grade chronic inflammation, as seen in aging, triggers an intrinsic pan-cell type defect in the JAK-STAT pathway, which fails to reduce baseline phosphoprotein levels following stimulation. It is possible that persistent in vivo exposure to CRP or other pro-inflammatory mediators known to increase with age, such as IL-6 (Michaud et al., 2013), may play a critical role in age-associated in vivo cell priming, where the effect of a first cytokine alters the response to a different one (Rawlings et al., 2004; Shuai and Liu, 2005; van Boxel-Dezaire et al., 2006). Although the kinetics of the priming effect are cell and cytokine dependent, its relation to the CRS, particularly in terms of IL-6 abundance, should be explored further. Irrespective, this suggests that the inherent flexibility of the JAK-STAT pathway is partially lost in a fraction of older adults, which is particularly important during infection and acute inflammation, since in both situations, cells are exposed to increases in cytokine bursts.
Our results lead to a model (Figure 6) of reduced cytokine responsiveness in chronic inflammatory states resulting from age-related changes in multiple physiological systems. In this scenario, circulating cells experience an inflammatory environment for long periods of time. Signaling pathways (e.g., JAK-STAT) lose functionality in the presence of repeated or long-duration inputs. Steady-state levels of the activated pSTATs rise, affecting the response potential. Considering their position between input and output suggests that elevated phosphoprotein levels at baseline form the link between well-known immunosenescence characteristics (i.e., low-grade chronic inflammation and unresponsive cells) and ultimately a degraded physiological outcome (e.g., poorer cardiovascular and immunological health), yet our results suggest that upstream defects in signaling are involved as well, as has been shown, for example, for CD4 naive T cell in older adults (Li et al., 2015). This model is consistent with a large body of data demonstrating that during aging, cells that have experienced DNA damage acquire a senescent phenotype and are able to produce high amounts of inflammatory mediators. This senescence-associated secretory phenotype (Tchkonia et al., 2013; Xu et al., 2015) may explain in part the age-related elevation in circulating cytokines and in baseline pSTAT levels and the lack of responses to in vitro cell stimulations we observe. Taken together, these results suggest that baseline phosphoprotein levels increase as a function of age, and either it or the CRS might constitute a sensitive readout for a pro-inflammatory state.
Figure 6. A Model Associating Elevated Baseline pSTAT Levels with Cellular Unresponsiveness and Chronic Pro-inflammation.
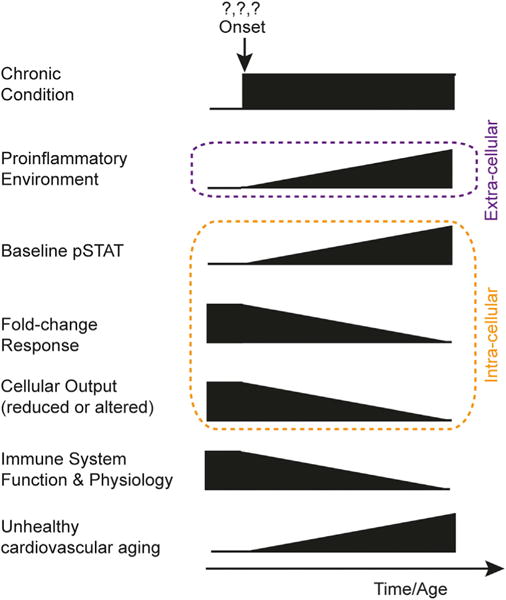
Proposed model for elevation of baseline pSTAT levels and its association with hallmark phenomenon of immunosenescence, an increased pro-inflammatory environment, unresponsive cells, and a clinical impact on immune response.
Although deciphering the exact conditions in which the JAK-STAT response failure occurs will require further investigation, evidence suggests that this process may be reversed, and targeting of pSTAT baseline levels may be one possibility. For example, Nadeau and colleagues have observed a reduction of pSTAT6 baseline levels in CD4+ memory T cells following oral corticosteroid treatment, a mainstay in severe asthma and asthma treatment (Gernez et al., 2007). A more drastic finding is that for both B and T cells, depletion of unresponsive memory cells from older mice drives the generation of naive cells, which appear to properly respond to stimuli (Haynes et al., 2005; Keren et al., 2011). These results would fit in the context of our proposed model, as the newly formed naive cells will experience the existing high pro-inflammatory environment, and therefore, the JAK-STAT response would be reset to baseline. An interesting point is that behavioral changes such as caloric restriction, known to be a potent anti-aging strategy that lowers chronic inflammation and prevents age-related chronic disease, also prevents age-related upregulation of SOCS proteins (Moyse et al., 2012), which may prevent baseline pSTAT elevation.
In summary, the comprehensive profiling described here on our longitudinal cohort empowers us to synthesize multiple observations within the context of aging of the effects of low-grade chronic inflammation on cellular responses and at least some significant clinical conditions. The model we present supports prior work on differing responses of JAK-STAT in acute versus chronic conditions (Stark and Darnell, 2012). Given that low-grade chronic inflammation is not restricted to aging and is also present in other physiological conditions, we speculate that this intrinsic pan-cell type feature of elevated baseline phosphorylation and the CRS could be generalized to a large number of chronic conditions, especially as altered cytokine/JAK-STAT system has been reported in HIV, Chagas disease (Albareda et al., 2015), arthritis (Sikora et al., 2012), asthma (Gernez et al., 2007), and possibly other cell-signaling pathways during aging (Sadighi Akha and Miller, 2005). Our findings presented here on the close relationship between the cellular immune state and cardiovascular decline suggest that it would be fruitful to explore the clinical relevance of cytokine responsiveness in other situations involving chronic inflammation.
STAR★METHODS
KEY RESOURCES TABLE
|
| ||
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|
| ||
| Antibodies | ||
|
| ||
| CD3 Pacific Blue | BD Biosciences | Cat. #558117; RRID: AB_397038 |
|
| ||
| CD4 PerCP-Cy5.5 | BD Biosciences | Cat. #341654; RRID:AB_400452 |
|
| ||
| CD20 PerCP-Cy5.5 | BD Biosciences | Cat. #558021; RRID:AB_396990 |
|
| ||
| CD33 PE-Cy7 | BD Biosciences | Cat. #333946; RRID:AB_399961 |
|
| ||
| pSTAT1 Alexa Fluor 488 | BD Biosciences | Cat. #612596; RRID:AB_399879 |
|
| ||
| pSTAT3 Alexa Fluor 647 | BD Biosciences | Cat. #557815; RRID: AB_647144 |
|
| ||
| pSTAT5 PE | BD Biosciences | Cat. #616567 |
|
| ||
| CD3 AmCyan | BD Biosciences | Cat. #339186; RRID:AB_647353 |
|
| ||
| CD4 Pacific Blue | BD Biosciences | Cat. #558116; RRID:AB_397037 |
|
| ||
| CD8 APC-H7 | BD Biosciences | Cat. #560179; RRID:AB_1645481 |
|
| ||
| CD28 APC | BD Biosciences | Cat. #559770; RRID:AB_398666 |
|
| ||
| CD27 PE | BD Biosciences | Cat. #555441; RRID:AB_395834 |
|
| ||
| CD45RA PE-Cy5 | BD Biosciences | Cat. #555490; RRID:AB_395881 |
|
| ||
| CD19 AlexaFluor700 | BD Biosciences | Cat. #557921; RRID:AB_396942 |
|
| ||
| CD56 PE | BD Biosciences | Cat. #563238 |
|
| ||
| CD33 PE-Cy7 | BD Biosciences | Cat. #333946; RRID:AB_399961 |
|
| ||
| TCRγδ APC | BD Biosciences | Cat. #555718; RRID:AB_398611 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| IFNα | PBL Interferon | Cat. #11105-1 |
|
| ||
| IFNγ | BD Biosciences | Cat. #554617 |
|
| ||
| IL-6 | BD Biosciences | Cat. #550071 |
|
| ||
| IL-7 | BD Biosciences | Cat. #554608 |
|
| ||
| IL-10 | BD Biosciences | Cat. #554611 |
|
| ||
| IL-21 | Life Technologies | Cat. #PHC0214 |
|
| ||
| IL-2 | BD Biosciences | Cat. #554603 |
|
| ||
| 16% paraformaldehyde | Alfa Aesar | Cat. #4368 |
|
| ||
| Methanol | Thermo Fisher | Cat. #A452SK-1 |
|
| ||
| Pacific Orange | Invitrogen | Cat. #P30253 |
|
| ||
| Alexa 750 | Invitrogen | Cat. #A20011 |
|
| ||
| C-reactive protein | Calbiotech | Cat#CR120C |
|
| ||
| CMV IgG | Calbiotech | Cat. #CM027G |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Human 42 plex Polystyrene kit | Millipore | H42; MPXHCYTO060KPMX42 |
|
| ||
| Human 51 plex Polystyrene kit | Affymetrix-Panomics | H50; PC009 |
|
| ||
| Human 51 plex Polystyrene kit | Affymetrix-Panomics | H51-1; PC009 |
|
| ||
| Deposited Data | ||
|
| ||
| Raw cell phenotyping, phosphoflow and cytokine data | Immport; RRID: SCR_012804 | SDY887,212,312 |
|
| ||
CONTACT FOR REAGENT AND RESOURCE SHARING
For reagent and resource requests please contact Professor Mark M. Davis, School of Medicine Stanford University and HHMI at mmdavis@stanford.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study Subjects
Peripheral blood samples were obtained at the Stanford Clinical and Translational Research Unit from adult participants (see Table S1 for demographics) coming in annually for three years as part of an influenza vaccine study. Total enrollment included 29 people the 1st year, expanded to 91 in the 2nd year (25 of which were returning subjects) and 82 in the 3rd year (all of which were returning subjects from Year 2). Informed consent was obtained from all of the subjects enrolled in this study, and the study protocol was approved by the Stanford University Administrative Panels on Human Subjects in Medical Research (IRB).
All volunteers were assessed as healthy at the time of initial enrollment based on an evaluation of their medical history and assessment of their vital signs. Exclusion criteria at time of enrollment were an active systemic or serious concurrent illness, a history of immunodeficiency, any known or suspected impairment of immunologic function, including clinically significant liver disease, diabetes mellitus treated with insulin, moderate to severe renal disease, blood pressure >150/95 mmHg at screening, chronic hepatitis B or C, recent or current use of immunosuppressive medication. Notably, a history of cardiovascular disease was not an exclusion criterion. In addition, none of the volunteers were recipients or donors of blood or blood products within the past 6 months and 6 weeks respectively nor showed any signs of febrile illness on day of baseline blood draw.
Sample Collection
In total, 120 ml whole blood (~40 mL/visit) whole blood was drawn per individual over three clinic visits and processed by standard procedures to PBMC and serum, if needed. Cells were frozen in DMSO with 10% FBS at −80°C overnight prior to transferring to liquid nitrogen.
All analyses described here were performed from samples drawn on the same visit (visit 1, baseline) with the exception of cell-subset phenotyping, which were analyzed from blood drawn 28 days after initial visit following verification that cell-subset frequencies do not change significantly from baseline at that time. CMV seropositivity was determined by standard ELISA using the Calbiotech CMV IgG ELISA kits (Calbiotech, Spring Valley, CA) as recommended by the manufacturer.
METHOD DETAILS
Cell Phenotyping and Cytokine Responses
For cell subset analysis, cells were thawed, washed twice with warm culture media and stained with the following antibody cocktail: CD3 AmCyan, CD4 PacificBlue, CD8 APCH7,CD28 APC, CD27 PE, CD45RA PE-Cy5,CD19Alexa Fluor700, CD56 PE, CD33PE-Cy7, TCRγδ APC, all reagents from BD Biosciences. Incubation with antibodies was performed for 30 min at 4°C. Cells were washed and resuspended in FACS buffer. Data were collected using DIVA software in an LRSII instrument (BD Biosciences). Data analysis was performed using FlowJo 8.8.6 by gating on live cells, then using double gating for singlet discrimination, followed by cell subset specific gating.
For cytokine response assays, thawed PBMC were rested for 1 hr in warm RPMI media with 10% FBS (culture media) before stimulation. Cells were distributed in 96-deep well blocks and stimulated for 15 min at 37°C with IFN-γ, IL6, IL7, IL10, IL21 at 50ng/ml or with 104 U/ml IFN-α (Following Year 1, IL7 stimulations were halted and IL2 added). After stimulation, cells were fixed with 1.5% PFA at room temperature for 10 min and washed with an excess of plain PBS. Cells were then span down at 2000 rpm for 5 min at 4°C and permeabilized with 100% cold methanol for 20 min on ice. Stimuli conditions were barcoded using a 3 × 3 matrix with Pacific Orange and Alexa Fluor 750 (Invitrogen Corp) at 0.03 and 0.04 ug/ml for low staining and 0.2 and 0.3 ug/ml for high staining, respectively (Krutzik and Nolan, 2006). Incubation with barcoding dyes was performed at 4°C for 30 min. After several washes with FACS buffer (PBS 2% FBS, 0.1% Na Azide) stimulated and barcoded cells were pooled into single tubes and stained for 30 min at 4°C with an antibody cocktail containing anti pSTAT1 Alexa Fluor 488, pSTAT3 Alexa Fluor 647, pSTAT5 PE (measuring pSTAT5A), CD3 Pacific Blue, CD4 PerCP-Cy5.5, CD20 PerCP-Cy5.5 and CD33 PE-Cy7 (all from BD Phosflow). After washing, cells were resuspended in FACS buffer and acquisition was performed on an LSRII instrument (BD Biosciences). Data were collected using DIVA software with analysis performed using FlowJo 8.8.6. by gating on live cells, then using double gating for singlet discrimination, followed by cell subset specific gating. Phosphorylation of STAT1, 3, and 5 proteins in B cells, CD4/CD8 T cells or monocytes was analyzed by debarcoding on stimuli-specific gating (Figure S1 for gating strategy). A drawback of this technology that limits the cell subset resolution is the availability of commercial antibodies that bind to epitopes resistant to methanol fixation. Therefore, we used antibodies to CD20, CD33, CD3 and CD4 to identify B cells, monocytes CD4 (CD3+ CD4+) and non-CD4 (CD3+ CD4−) T cells (Figure S1 for gating strategy). One caveat of this strategy is the contribution of γδ- and NK- T cells and double negative T cells to the CD8 signal, because all three subsets are CD3+ CD4−. Testing of pSTAT3 responses to IL-6 stimulations in young and old adults in an independent experiment showed double negative T cells responses mirrored closely those of CD8+ T cells, whereas gamma-delta and NKT cells constitute only a minority of lymphocytes. Gamma-delta and NKT cells constitute only a minority of lymphocytes while the frequency of CD8 T cells is often more than 30% of PBMC. We thus refer to the non-CD4 fraction as CD8 T cells. We used CD45RA to identify naive and memory T cells where assayed. Fold-change between stimulated and unstimulated conditions was calculated using the 90th percentile of the pSTAT1, 3, or 5 positive cells. Alternative use of the median fluorescent intensity yielded qualitatively similar results, namely it did not alter the observation of significant age dependent decline in phosphoprotein levels in response to cytokine stimulation.
For the dose-response assay, we measured three pairs of old and young individuals from year 1 that we paired on a single 96-deep well plate and stimulated for 15 min at 37°C. The protocol was identical to the one described above, with the exception of the cytokine stimulation doses: for IFN-γ, IL6, IL7, IL10, and IL21 we stimulated cells with 0.1, 1, 10, 50 and 100 ng/ml of cytokine whereas for IFN-α we stimulated with 0.12, 0.25, 0.5 1 and 2 × 104 U/ml. Baseline phosphorylated STAT levels were measured for each individual per plate in 5 replicates. Sample normalization was performed as described below, scaling between plates. To determine if a fold change difference exists for a given cell, cytokine, phosphoprotein assay between old and young we used a t test at the highest dose stimuli concentration and corrected for multiple hypotheses testing (Storey and Tibshirani, 2003). Of the three pairs of old-young samples we tested, one pair was missing a single, but different, measurement for each stimulus. Though the results were fully consistent with those observed in the two other old-young pairs (i.e., the young individual showed increased fold change for all doses for a large number of assays), we did not include results from this pair in any of the analyses reported here.
Serum Protein Levels
Serum samples were obtained by centrifugation of clotted blood and stored at −80°C before cytokine levels determination. Samples were measured by 42-plex kits (Millipore) in Year 1 and 51-plex kit (Panomics/Affymetrix) in Year 2 and 3. Kits were used according to manufacturer’s recommendations with modifications as described below: samples were mixed with antibody linked polystyrene beads on 96-well filter-bottom plates and incubated at room temperature for 2 hr followed by overnight incubation at 4°C. Room temperature incubation steps were performed on an orbital shaker at 500–600 rpm. Plates were vacuum filtered and washed twice with wash buffer, then incubated withbiotinylated detection antibody for 2 hr at room temperature. Samples were then filtered and washed twice as above and re-suspended in streptavidin-PE. After incubation for 40 min at room temperature, two additional vacuum washes were performed, and the samples re-suspended in Reading Buffer. Each sample was measured in duplicate. Plates were read using a Luminex Lab-Map200 instrument with a lower bound of 100 beads per sample per cytokine. The Luminex LabMap200 outputs the fluorescence intensity of each bead measured for a given cytokine in a sample. For each well, we considered the median fluorescence intensity (MFI) of all beads measured for that cytokine in a well as its abundance and averaged the MFI of the two replicates to obtain the abundance of a cytokine in a sample. Of note, detailed analyses of IL-6 serum sample data in this dataset and others we have profiled have shown that the Luminex platform is not well suited for the analysis of IL-6 from healthy human serum samples (Rosenberg-Hasson et al., 2014; Thompson et al., 2012). We have therefore decided to disregard the IL-6 serum cytokine measurements in our study.
High-sensitive C-reactive protein was measured from serum samples using a standard ELISA method (Calbiotech cat#CR120C) following the manufacturers’ recommendations.
Cardiovascular Phenotyping
Older adults in our study were contacted prospectively to participate in a comprehensive cardiovascular assessment at Stanford Cardiovascular Institute Biomarker and Phenotypic Core Laboratory. 40 of the study participants profiled agreed. Patients underwent assessment of both vascular and cardiac structure and function. We divided subjects into a clinical subgroup, defined as those having previously suffered from a myocardial infarction or had undergone a cardiovascular operative procedure; a sub-clinical subgroup which included those individuals with abnormal atherosclerotic plaque in the common or internal carotid artery or common femoral artery of greater than 30% diameter stenosis, and a healthy subgroup which included those individuals with no observed cardiovascular risk. Five individual were at the borderline of the established sub-clinical criteria but could not be classified as cardiovascularly healthy either.
Vascular studies included the measurement of both carotid intima-media thickness (cIMT) and central aortic PWV. We used a 9.0 MHz Philips linear array probe for carotid and femoral measurements. The cIMT was the average of the anterior, lateral, and posterior measurements and averaged for both the right and left carotid artery. Values above 0.7 were considered as high. Aortic PWV was calculated as the path length traveled and divided by transit time of the aortic pulse wave and reflects arterial stiffness (Reference Values for Arterial Stiffness’ Collaboration, 2010). Path length (D) was measured as the distance from the sternal notch to the femoral artery minus the echocardiographic distance from the sternal notch to proximal descending aorta. Doppler signals were recorded at 150 mm/s. The intersecting tangent method was used to measure the time from a reference ECG signal and the foot of the pulse wave. Heart rate had to be within 2 bpm to proceed for recording of the femoral signal. We considered raw PWV values above 10 as high. For cardiac imaging, we used the iE33 xMATRIX Echocardiography System manufactured by Philips (Andover, MA, USA). Echocardiographic studies were performed according to the American Society of Echocardiography (ASE) guidelines(Lang et al., 2005; Rudski et al., 2010) by our chief sonographer (Jospehine Puryear). LV dimensions and wall thickness were measured in the parasternal short axis view using M-mode tracings and averaged over 3 measurements for each. A value above 34 in the LA volume index was considered high. RWT was calculated as the diastolic (d) ratio of interventricular septal thickness and infero-lateral wall thickness divided by LV internal dimensions [RWT = (IVSd+LVPWd)/LVIDd]. LV mass (LVM) was estimated from M-mode linear LV diastolic measurements based on the ASE-recommended formula (LVM = 0.83 × (1.04[IVS+LVID+LVPW]3-LVID3) +0.6 where IVS = inter ventricular septum thickness and LVPW = LV posterior wall thickness). LV volumes were calculated with the Teichholz formula [Volume = (7/(2.4+LVID)) * LVID3, wherein LVID is the LV internal diameter] and mass to volume ratios were derived. These analyses were then analyzed by an expert cardiologist to yield a summary stratification assignment per individual on their cardiovascular health.
In the apical 4 chamber view LV ejection fraction (LVEF) was estimated with the biplane method of disc. Diastolic parameters were estimated using the ratio of LV early inflow velocity divided by early tissue Doppler velocity (E/e’)(Nagueh et al., 2009). We considered values above 9 as high. Operant end-diastolic elastance (Ed) was calculated as E/e’ ratio divided by SVI and represents a surrogate marker of stiffness (Redfield et al., 2005). The intra-class correlation coefficient was 0.92 for LV mass as measured by 2 independent observers in our laboratory.
For confounder effects we analyzed difference in CRS between older adults for every prescribed drug for which at least two patients self-reported being on. No significant differences were observed.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical analyses were performed using R. If not described here, statistical tests used and outcome are detailed in the Results section and in Figure legends.
Cytokine Response Normalization and Analysis
Across the three years, samples were analyzed at a single cytokine stimuli concentration, in assays performed over multiple days. Baseline phosphoprotein abundance in a given cell was measured in 6 replicates. Fold-change difference due to stimulation was computed as the ratio of the cell, cytokine stimulation, phosphoprotein measure to the raw, un-normalized, cell-phosphoprotein matching baseline that was measured on the same plate. For individual stability analysis (see below) we used ‘raw’ fold-change values, whereas for all assays, we day normalized by scaling each individual by the average fold-change for the assay measured on a given day. We tested each assay for day dependent differences. No significant differences between days were detected post-day normalization. Baseline measurements were similarly day normalized for all statistical analyses described.
In the dose-response assay, to determine if a fold-change difference exists for a given cell, cytokine, phosphoprotein assay between old and young we used a t test at the highest dose stimuli concentration and corrected for multiple hypotheses testing (Storey and Tibshirani, 2003). Of the three pairs of old-young samples we tested, one pair was missing a single, but different, measurement for each stimulus. Though the results were fully consistent with those observed in the two other old-young pairs (i.e., the young individual showed increased fold-change for all doses for a large number of assays), we did not include results from this pair in any of the analyses reported here.
Stable Age-Associated Response Identification
To identify age-associated cytokine responses, we used a linear regression model to identify measurements that showed a statistically significant change in expression with age. Our linear regression model accounted for both age and gender differences. Mathematically, our model takes the form:
Where Yij is the in individual i of measurement of j where j is either a baseline pSTAT value or fold change normalized phosphoflow measurement as described above. We applied the model to each data measurement. To compute p values of the beta coefficient of age and gender, we permuted the data measurement with respect to age and gender, 1,000 times, and then recomputed the regression. For each permutation we tested whether the absolute value of the permuted derived betas was greater or equal in size to the absolute value of the true beta coefficients. P-values for each beta were then calculated as the ratio of the number of times the betas from the permuted regressions exceeded the betas from the true data regression over the total number of trials. To correct for multiple hypothesis testing, we considered the regression p values for all measurements of a single data type simultaneously and calculated a q-value (Storey and Tibshirani, 2003) for each.
To identify reproducible age-associated cytokine responses, we computed a Fisher’s Combined Probability test for each assay across the three cohorts, on Benjamini-Hochberg adjusted p values of all assays in a given year. Assays were named reproducibly age-associated if their p value ≤ 0.05 on the Fisher’s combined probability test and a q-value below 5% in at least two years; the latter to avoid instances where a significant p value in the Fisher Combined Probability test was driven solely by a single year.
To assess individual stability of phenotypes by intra-class correlation we used the irr R package in a one-way model, based on ‘raw’ fold change values, and treating the different per-years rating as if coming from independent raters. The ICC is defined as the proportion of variance of an observation due to between-subject variability (as opposed to between years) in the true scores (responses) and was computed for two groups: once for older adults and once for all individuals as well as separate on young (below the age of 40) or older adults (> 60 yo). For each assay, we excluded results from individuals in the group who were not measured in all three years and mean centered the raw (i.e., not day normalized) data in a given year across the remaining individuals in the group.
Immune Signaling Dysfunction Analysis
We computed the CRS for each individual by summing the normalized values of the 15 reproducible age-associated cytokine responses whose ICC was positive, whereas for baseline, we summed all normalized baseline pSTAT values. Naive memory differences were assessed using a one way paired Wilcoxon test followed by a Benjamini-Hochberg p value adjustment.
DATA AND SOFTWARE AVAILABILITY
Data Resources
The accession number of the data reported in this paper is Immport: SDY887, SDY212 and SDY312 for Year 1, 2 and 3 of the study respectively.
Supplementary Material
Highlights.
A 3-year longitudinal study reveals systemic defects in STAT signaling in older adults
Defects are due to naive-memory cell differences and elevated basal pSTATs
A cytokine-response score (CRS) negatively correlates with cardiovascular disease
The CRS outperforms CRP in classifying individuals with an atherosclerotic burden
Acknowledgments
We thank A. Alpert, E. Starosvetsky, A. Morgan, N. Kotecha, P. Khatri, R. Kafri, S. Kim, R. Tibshirani, J. Goronzy, C. Weyand, and U. Rosenschien for illuminating discussions; J. Ptacek and G. Nolan for invaluable help with the cytokine response assays; research nurses S. Swope, C. Walsh, and S. Cathey, clinical research associates K. Spann, A. Gutierrez, and R. Fleischman, administrative associate T. Quan, and medical assistant/phlebotomist M. Ugur at the Stanford-LPCH Vaccine Program; A. Skrenchuk and B. Oskotsky for computational support; and the Hewlett Packard Foundation and Lucile Packard Children’s Hospital for computational resources. This work was supported in part by grants from the Ellison Medical Foundation, Howard Hughes Medical Institute, and National Institute of Allergy and Infectious Diseases (U19 AI057229 and U19 AI090019 to MMD and Bioinformatics Support Contract HHSN272201200028C to A.J.B.; BWF IRSA 1015009 and AHA 13EIA14420025 to J.C.W.), as well as Clinical and Translational Science Award 5UL1 RR025744 for the Stanford Center for Clinical and Translational Education and Research (Spectrum) from the National Center for Research Resources, National Institutes of Health. This trial is registered at http://www.clinicaltrials.gov as NCT01827462. S.S.S.-O. was supported in part by The Israeli Science Foundation (grant 1365/12). F.H. is supported by the Stanford Cardiovascular Institute. D.F. received support from the Stanford Center on Longevity. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.cels.2016.09.009.
AUTHOR CONTRIBUTIONS
Conceptualization, M.M.D., C.L.D., S.S.S.-O., and D.F.; Formal Analysis, S.S.S.-O., D.F., B.A.K., and Y.P.; Investigation, D.F., F.H., P.L., Y.-W.H., Y.R.-H., and F.A.G.G.; Resources, A.J.B., J.C.W., F.H., H.T.M., and C.L.D.; Writing – Original Draft, S.S.S.-O., D.F., B.A.K., A.J.B., and M.M.D.; Writing – Review & Editing, S.S.S.-O., D.F., and M.M.D.; Project Administration, S.M., and C.L.D.; Funding Acquisition, M.M.D., S.S.S.-O., F.H., A.J.B., and J.C.W.; Supervision, M.M.D., A.J.B., and Y.-h.C.
References
- Adriaensen W, Matheï C, Vaes B, van Pottelbergh G, Wallemacq P, Degryse JM. Interleukin-6 as a first-rated serum inflammatory marker to predict mortality and hospitalization in the oldest old: A regression and CART approach in the BELFRAIL study. Exp Gerontol. 2015;69:53–61. doi: 10.1016/j.exger.2015.06.005. [DOI] [PubMed] [Google Scholar]
- Albareda MC, Perez-Mazliah D, Natale MA, Castro-Eiro M, Alvarez MG, Viotti R, Bertocchi G, Lococo B, Tarleton RL, Laucella SA. Perturbed T cell IL-7 receptor signaling in chronic Chagas disease. J Immunol. 2015;194:3883–3889. doi: 10.4049/jimmunol.1402202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann J, Raue A, Schilling M, Böhm ME, Kreutz C, Kaschek D, Busch H, Gretz N, Lehmann WD, Timmer J, Klingmüller U. Division of labor by dual feedback regulators controls JAK2/STAT5 signaling over broad ligand range. Mol Syst Biol. 2011;7:516. doi: 10.1038/msb.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell. 2014;54:281–288. doi: 10.1016/j.molcel.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitriu IE, Araguás ET, Baboonian C, Kaski JC. CD4+ CD28 null T cells in coronary artery disease: when helpers become killers. Cardiovasc Res. 2009;81:11–19. doi: 10.1093/cvr/cvn248. [DOI] [PubMed] [Google Scholar]
- Duraisingham SS, Rouphael N, Cavanagh MM, Nakaya HI, Goronzy JJ, Pulendran B. Systems biology of vaccination in the elderly. Curr Top Microbiol Immunol. 2013;363:117–142. doi: 10.1007/82_2012_250. [DOI] [PubMed] [Google Scholar]
- Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015;15:117–129. doi: 10.1038/nri3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- Frasca D, Blomberg BB. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology. 2016;17:7–19. doi: 10.1007/s10522-015-9578-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman D, Jojic V, Kidd B, Shen-Orr S, Price J, Jarrell J, Tse T, Huang H, Lund P, Maecker HT, et al. Apoptosis and other immune biomarkers predict influenza vaccine responsiveness. Mol Syst Biol. 2013;9:659. doi: 10.1038/msb.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman D, Jojic V, Sharma S, Shen-Orr SS, Angel CJ, Onengut-Gumuscu S, Kidd BA, Maecker HT, Concannon P, Dekker CL, et al. Cytomegalovirus infection enhances the immune response to influenza. Sci Transl Med. 2015;7:281ra43. doi: 10.1126/scitranslmed.aaa2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gernez Y, Tirouvanziam R, Nguyen KD, Herzenberg LA, Krensky AM, Nadeau KC. Altered phosphorylated signal transducer and activator of transcription profile of CD4+CD161+ T cells in asthma: modulation by allergic status and oral corticosteroids. J Allergy Clin Immunol. 2007;120:1441–1448. doi: 10.1016/j.jaci.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goronzy JJ, Weyand CM. Understanding immunosenescence to improve responses to vaccines. Nat Immunol. 2013;14:428–436. doi: 10.1038/ni.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes L, Eaton SM, Burns EM, Randall TD, Swain SL. Newly generated CD4 T cells in aged animals do not exhibit age-related defects in response to antigen. J Exp Med. 2005;201:845–851. doi: 10.1084/jem.20041933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howcroft TK, Campisi J, Louis GB, Smith MT, Wise B, Wyss-Coray T, Augustine AD, McElhaney JE, Kohanski R, Sierra F. The role of inflammation in age-related disease. Aging (Albany, NY) 2013;5:84–93. doi: 10.18632/aging.100531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren Z, Naor S, Nussbaum S, Golan K, Itkin T, Sasaki Y, Schmidt-Supprian M, Lapidot T, Melamed D. B-cell depletion reactivates B lymphopoiesis in the BM and rejuvenates the B lineage in aging. Blood. 2011;117:3104–3112. doi: 10.1182/blood-2010-09-307983. [DOI] [PubMed] [Google Scholar]
- Krutzik PO, Nolan GP. Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat Methods. 2006;3:361–368. doi: 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
- Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, et al. Chamber Quantification Writing Group; American Society of Echocardiography’s Guidelines and Standards Committee; European Association of Echocardiography Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18:1440–1463. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Li G, Ju J, Weyand CM, Goronzy JJ. Age-associated failure to adjust type I IFN receptor signaling thresholds after T cell activation. J Immunol. 2015;195:865–874. doi: 10.4049/jimmunol.1402389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo DM, Louie B, Putta S, Evensen E, Ptacek J, Cordeiro J, Wang E, Pos Z, Hawtin RE, Marincola FM, Cesano A. Single-cell network profiling of peripheral blood mononuclear cells from healthy donors reveals age- and race-associated differences in immune signaling pathway activation. J Immunol. 2012;188:1717–1725. doi: 10.4049/jimmunol.1102514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo DM, Louie B, Ptacek J, Friedland G, Evensen E, Putta S, Atallah M, Spellmeyer D, Wang E, Pos Z, et al. High-dimensional analysis of the aging immune system: verification of age-associated differences in immune signaling responses in healthy donors. J Transl Med. 2014;12:178. doi: 10.1186/1479-5876-12-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maly K, Schirmer M. The story of CD4+ CD28- T cells revisited: solved or still ongoing? J Immunol Res. 2015;2015:348746. doi: 10.1155/2015/348746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud M, Balardy L, Moulis G, Gaudin C, Peyrot C, Vellas B, Cesari M, Nourhashemi F. Proinflammatory cytokines, aging, and age-related diseases. J Am Med Dir Assoc. 2013;14:877–882. doi: 10.1016/j.jamda.2013.05.009. [DOI] [PubMed] [Google Scholar]
- Morrisette-Thomas V, Cohen AA, Fülöp T, Riesco É, Legault V, Li Q, Milot E, Dusseault-Bélanger F, Ferrucci L. Inflamm-aging does not simply reflect increases in pro-inflammatory markers. Mech Ageing Dev. 2014;139:49–57. doi: 10.1016/j.mad.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyse E, Bédard K, Segura S, Mahaut S, Tardivel C, Ferland G, Lebrun B, Gaudreau P. Effects of aging and caloric restriction on brainstem satiety center signals in rats. Mech Ageing Dev. 2012;133:83–91. doi: 10.1016/j.mad.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Nagueh SF, Appleton CP, Gillebert TC, Marino PN, Oh JK, Smiseth OA, Waggoner AD, Flachskampf FA, Pellikka PA, Evangelista A. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr. 2009;22:107–133. doi: 10.1016/j.echo.2008.11.023. [DOI] [PubMed] [Google Scholar]
- Okin D, Medzhitov R. Evolution of inflammatory diseases. Curr Biol. 2012;22:R733–R740. doi: 10.1016/j.cub.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer DB. The effect of age on thymic function. Front Immunol. 2013;4:316. doi: 10.3389/fimmu.2013.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q, Zhang DW, Weyand CM, Goronzy JJ. Mechanisms shaping the naïve T cell repertoire in the elderly - thymic involution or peripheral homeostatic proliferation? Exp Gerontol. 2014;54:71–74. doi: 10.1016/j.exger.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- Redfield MM, Jacobsen SJ, Borlaug BA, Rodeheffer RJ, Kass DA. Age- and gender-related ventricular-vascular stiffening: a community-based study. Circulation. 2005;112:2254–2262. doi: 10.1161/CIRCULATIONAHA.105.541078. [DOI] [PubMed] [Google Scholar]
- Reference Values for Arterial Stiffness’ Collaboration. Determinants of pulse wave velocity in healthy people and in the presence of cardiovascular risk factors: ‘establishing normal and reference values’. Eur Heart J. 2010;31:2338–2350. doi: 10.1093/eurheartj/ehq165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM. Connecting the role of C-reactive protein and statins in cardiovascular disease. Clin Cardiol. 2003;26:III39–III44. doi: 10.1002/clc.4960261508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–1772. doi: 10.1161/01.cir.101.15.1767. [DOI] [PubMed] [Google Scholar]
- Rosenberg-Hasson Y, Hansmann L, Liedtke M, Herschmann I, Maecker HT. Effects of serum and plasma matrices on multiplex immunoassays. Immunol Res. 2014;58:224–233. doi: 10.1007/s12026-014-8491-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudski LG, Lai WW, Afilalo J, Hua L, Handschumacher MD, Chandrasekaran K, Solomon SD, Louie EK, Schiller NB. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr. 2010;23:685–713. doi: 10.1016/j.echo.2010.05.010. quiz 786–788. [DOI] [PubMed] [Google Scholar]
- Sadighi Akha AA, Miller RA. Signal transduction in the aging immune system. Curr Opin Immunol. 2005;17:486–491. doi: 10.1016/j.coi.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Scrivo R, Vasile M, Bartosiewicz I, Valesini G. Inflammation as “common soil” of the multifactorial diseases. Autoimmun Rev. 2011;10:369–374. doi: 10.1016/j.autrev.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Dalli J, Colas RA, Winkler JW, Chiang N. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim Biophys Acta. 2015;1851:397–413. doi: 10.1016/j.bbalip.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM. Aging of the innate immune system. Curr Opin Immunol. 2010;22:507–513. doi: 10.1016/j.coi.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- Sikora KA, Fall N, Thornton S, Grom AA. The limited role of interferon-g in systemic juvenile idiopathic arthritis cannot be explained by cellular hyporesponsiveness. Arthritis Rheum. 2012;64:3799–3808. doi: 10.1002/art.34604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark GR, Darnell JE., Jr The JAK-STAT pathway at twenty. Immunity. 2012;36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DK, Huffman KM, Kraus WE, Kraus VB. Critical appraisal of four IL-6 immunoassays. PLoS ONE. 2012;7:e30659. doi: 10.1371/journal.pone.0030659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, Weber C. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. 2001;103:1772–1777. doi: 10.1161/01.cir.103.13.1772. [DOI] [PubMed] [Google Scholar]
- Weiskopf D, Weinberger B, Grubeck-Loebenstein B. The aging of the immune system. Transpl Int. 2009;22:1041–1050. doi: 10.1111/j.1432-2277.2009.00927.x. [DOI] [PubMed] [Google Scholar]
- Weng NP, Akbar AN, Goronzy J. CD28(−) T cells: their role in the age-associated decline of immune function. Trends Immunol. 2009;30:306–312. doi: 10.1016/j.it.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikby A, Nilsson BO, Forsey R, Thompson J, Strindhall J, Löfgren S, Ernerudh J, Pawelec G, Ferguson F, Johansson B. The immune risk phenotype is associated with IL-6 in the terminal decline stage: findings from the Swedish NONA immune longitudinal study of very late life functioning. Mech Ageing Dev. 2006;127:695–704. doi: 10.1016/j.mad.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, White TA, Johnson KO, Stout MB, Mezera V, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci USA. 2015;112:E6301–E6310. doi: 10.1073/pnas.1515386112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.