

Abstract
Previous studies indicate that Kras is dispensable for fetal liver hematopoiesis, but its rolein adult hematopoiesis remains unclear. Here, we generated a Kras conditional knockout allele to address this question. Deletion of Kras in adult bone marrow is mediated by Vav-Cre or inducible Mx1-Cre. We find that loss of Kras leads to greatly reduced TPO signaling in hematopoietic stem cells (HSCs) and multipotent progenitors (MPPs), while SCF-evoked ERK1/2 activation is not affected. The compromised TPO signaling is associated with reduced long term- and intermediate-term HSC compartments and a bias towards myeloid differentiation in MPPs. Although GM-CSF-evoked ERK1/2 activation is only moderately decreased in Kras−/− myeloid progenitors, it is blunted in neutrophils and neutrophil survival is significantly reduced in vitro. At 9–12 months old, Kras conditional knockout mice develop profound hematopoietic defects, including splenomegaly, an expanded neutrophil compartment, and reduced B cell number. In a serial transplantation assay, the reconstitution potential of Kras−/− bone marrow cells is greatly compromised, which is attributable to defects in the self-renewal of Kras−/− HSCs and defects in differentiated hematopietic cells. Our results demonstrate that Kras is a major regulator of TPO and GM-CSF signaling in specific populations of hematopoietic cells and its function is required for adult hematopoiesis.
Keywords: Kras, Nras, adult hematopoiesis, hematopoietic stem cells, hematopoietic progenitor cells, stem cell self-renewal, cytokine signaling
Introduction
Mammalian hematopoiesis is a continuous production of blood cells for life. Definitive hematopoiesis, a process to give rise to hematopoietic stem cells (HSCs), occurs in two waves: the first wave happens in fetal liver during embryogenesis and the second wave occurs in adult bone marrow1. Fetal liver hematopoiesis and adult hematopoiesis are regulated by distinct cytokines. Stem cell factor (SCF) is important for both fetal liver and adult hematopoiesis, while thrombopoietin (TPO) is only essential to sustain adult hematopoiesis2–4. SCF signaling is mediated through its receptor, c-Kit, which is a receptor-like tyrosine kinase expressed in HSCs and all types of hematopoietic progenitors5. Upon ligand binding, c-Kit is activated and subsequently activates a wide array of downstream signaling-transducing pathways, including ERK1/2, AKT, and JAK/STAT. Interestingly, SCF stimulation appears to activate distinct pathways in different cell populations. For example, in erythroblasts, SCF induces bi-phase ERK1/2 activation (early and late stages) and sustained JAK2/STAT5 activation, while AKT phosphorylation is undetectable6, 7. In contrast, in bone marrow Lin− c-Kit+ cells, SCF robustly activates AKT, ERK1/2 is rapidly activated and its activation is sustained at a much lower level for hours, while STAT5 phosphorylation is not detectable (8, 9 and our unpublished results). Compared to the complex SCF signaling, TPO signaling transduced through its receptor, Mpl, is more consistent in different cell types. Mpl belongs to type 1 cytokine receptor family and depends on its associated JAK2 kinase to initiate signaling10. In both HSCs and platelets, TPO evokes activation of the ERK1/2, AKT, and JAK2/STAT3/5 pathways11, 12.
In addition to SCF and TPO, adult hematopoiesis is also regulated by cell lineage-specific cytokines. For example, GM-CSF regulates the survival, proliferation, differentiation and activation of macrophages and neutrophils13. Upon binding to its heteromeric receptor, which is composed of α (GM-CSFRα) and β subunit, the biological activities of GM-CSF are exerted through the JAK2/STAT5 and Ras/Raf/MEK/ERK pathways14. Although SCF, TPO, and GM-CSF are known to evoke the ERK1/2 pathway through Ras proteins, whether and how individual Ras isoforms distinctly involve in these signaling pathways during adult hematopoiesis remain largely elusive.
In mammals, there are 3 Ras genes (Hras, Nras, and Kras) encoding four homologous 21kD proteins: Hras, Nras, Kras4A and Kras4B15. The first 85 amino-terminal residues are identical and the middle 78 amino acids share an 85% – 90% identity among all Ras isoforms, while the last 25 amino acids at the carboxyl-terminus are highly variable16. Despite the high similarity in protein sequences and largely overlapping expression patterns, accumulating evidence suggest that the three Ras genes have distinct functions. Mice deficient for Hras, Nras, or Kras4a are viable and fertile17–19. Although Hras−/−; Nras−/− mice are viable, they are generated lower than the expected Mendelian ratio18. In contrast, Kras−/− mice die at mid-late gestation stages20, 21, indicating that only Kras4b is essential in mouse embryonic development. Replacing Kras with the Hras gene rescues embryonic lethality but not cardiovascular defects in Kras−/− mice, suggesting that Kras has a unique function in cardiovascular development22. Moreover, Nras−/−; Kras+/− mice die even earlier and display more severe phenotypes than Kras−/− mice, suggesting that Nras is partially redundant with Kras20.
Nras−/− and Kras−/− mice display distinct defects in hematopoiesis. Mice defective for Nras have lower numbers of CD8+ thymocytes, decreased thymocyte proliferation in vitro, and an increased sensitivity to influenza infection in the presence of low dose virus, indicating a role of Nras in adult T cell development and functions23. As for Kras, although E12.5 Kras−/− embryos have smaller and paler fetal livers than wild-type (WT) littermates, in vitro culture of fetal liver erythroid progenitors demonstrates that Kras deficiency leads to mildly delayed differentiation in early erythroid blasts, which is associated with down-regulated EPO-evoked Akt activation24, 25. Furthermore, Kras−/− fetal liver cells show reconstitution capability indistinguishable from control cells20. These data suggest that Kras plays a moderate role in fetal liver erythroid differentiation but is largely dispensable for fetal liver hematopoiesis. In contrast, chimeric mice generated from injection of Kras−/− embryonic stem cells (ESCs) into WT blastocysts show little contribution of Kras−/− cells to hematopoietic tissues even when these cells contribute to all the other tissues to a high degree20. However, whether Kras plays an important cell-autonomous role in adult hematopoiesis remains unclear.
Here, we generated a Kras conditional knockout allele to study Kras function in adult hematopoiesis. Kras is the major Ras isoform that is activated in whole bone marrow cells. However, loss of Kras does not trigger up-regulation or hyper-activation of Hras and Nras. Kras deficiency results in greatly reduced TPO signaling in HSCs and multipotent progenitors (MPPs), while SCF-evoked ERK1/2 activation in Kras−/− HSCs and MPPs remains indistinguishable from control cells. Frequencies of long term- and intermediate term-HSCs (LT- and IT-HSCs) in Kras−/− mice are significantly lower than those in control mice. In differentiated myeloid cells, GM-CSF-evoked ERK1/2 activation is blunted in Kras−/− mice but normal in Nras−/− mice. The survival of Kras−/− neutrophils is significantly reduced in vitro. At 9–12 months old, mice deficient for Kras expression in the hematopoietic compartment developed profound defects, including a dramatically expanded neutrophil compartment and significantly reduced number of B cells. To examine the self-renewal of Kras−/− and Nras−/− HSCs, we performed a serial competitive reconstitution assay. Kras−/− bone marrow cells show dramatically reduced reconstitution potential, while Nras−/− cells display a moderate reduction of reconstitution in the 2nd round of transplantation. Detailed analysis of donor-derived hematopoiesis in recipient mice suggests that defects in HSC self-renewal and differentiated hematopoietic cells contribute to the reduced reconstitution of Kras−/− cells. Our data suggest that Kras regulates the function of HSCs and differentiated myeloid cells through modulating cytokine signaling in adult hematopoiesis.
Materials and Methods
Mice
Kras conditional knockout allele was generated using λ-phage based recombineering technology26. Kras targeting vector was electroporated into V6.5 mouse embryonic stem cells (mESCs). Colonies resistant to G418 and gancyclovir were selected and screened using Southern blot analysis. Two out of 20 clones were correct. One of the correct clones was injected into C57BL/6 blastocysts to generate chimeric mice. Chimeric mice with high chimerism were further bred with C57BL/6 mice. Germ line transmission was confirmed by PCR using primers flanking the 5′ LoxP site as well as primers specific for Neo. The Neo selection cassette was subsequently removed by crossing the germ line-transmitted mice to Rosa26-FLPeR mice (Jackson Laboratories). The offsprings were backcrossed to C57BL/6 mice to separate Kras conditional knockout allele and Rosa26-FLPeR allele. The Kras conditional knockout mice were continuously backcrossed to C57BL/6. The following primers were used for genotyping: Kras 5′ LoxP forward (5′ –A GGGTAGGTGTTGGGATAGC- 3′), Kras 5′ LoxP reverse (5′ –GAGCCATTAGCTGCTACAAAACAG- 3′), Kras 3′ LoxP forward (5′ –GAGCAGC CCATGCTCTTAAC- 3′), Kras 3′ LoxP reverse (5′ -CCAATTAAAGGCCAACTGCT- 3′), neo forward (5′ ATTG AACAAGATGGATTGCAC 3′), and neo reverse (5′ TTC GTCCAGATCATCCTGATCGAC 3′).
All the mouse lines were maintained in pure C57BL/6 genetic background (>N10). Mice bearing conditional knockout Kras allele were crossed to Vav- or Mx1-Cre mice to generate Krasfl/fl; Vav-Cre or Krasfl/fl; Mx1-Cre mice, respectively. Vav-Cre mice were kindly provided by Dr. Chris Bradfield27. For all experiments on Krasfl/fl; Mx1-Cre mice, 8–10 weeks old control and compound mice were injected intraperitoneally with 7.5μg/g of polyinosinic-polycytidylic acid (pI-pC; GE Healthcare) every other day for 5 times. Treated mice were sacrificed 2 days after last pI-pC injection for analysis. The conditional oncogenic Nras allele (NrasLSL G12D) was described in28. The homozygous NrasLSL G12D/LSL G12D mice were used as Nras−/− mice. CD45.1+ congenic C57BL/6 recipient mice were purchased from NCI. All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and approved by an Animal Care and Use Committee at UW-Madison. The program is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
Serial BM competitive reconstitution assay
BM cells from donor mice (CD45.2+) were transplanted with competitor BM cells (CD45.1+) at 1:1 (1 million donor cells: 1 million competitor cells) or 3:1 (1.5 million donor cells: 0.5 million competitor cells) ration into individual lethally irradiated recipients. CD45.1+ recipient mice were irradiated with a Cesium source for 2 doses of 5 Gy (500 rads) each, delivered 3 hours apart.
For donor mice with Vav-Cre, PB samples were collected from recipient mice at 4, 8, and 16 weeks after transplantation and donor-derived cells were analyzed using a FACS Calibur (BD Biosciences). For donor mice with Mx1-Cre, recipient mice were injected with pI-pC 4 weeks after transplantation as described above. PB samples were collected from recipient mice at 4, 8, and 16 weeks after pI-pC injections and donor-derived cells were analyzed using a FACS Calibur (BD Biosciences).
Primary recipients were sacrificed 16 weeks after transplantation (Vav-Cre) or pI-pC injections (Mx1-Cre) to analyze donor-derived cells in BM and SP. Donor derived cells were enriched as CD45.2+ cells using an AutoMACS (Miltenyi Biotec). Recombination efficiency at the Kras locus was evaluated using primers Kras 5′ LoxP forward, Kras 3′ LoxP forward, and Kras 3′ LoxP reverse as described above. BM cells from 2 representative primary recipients per group (donor contribution in PB was close to the average of the group) were isolated and mixed. Two million of mixed cells were transplanted into individual lethally irradiated secondary recipients. Again, PB samples were collected from secondary recipients and analyzed at 4, 8, and 16 weeks after transplantation using a FACS Calibur (BD Biosciences). Secondary recipients were sacrificed 16 weeks after transplantation to analyze donor-derived cells in BM and SP.
Additional methods are described in Supplemental Methods
Results
Kras expression is efficiently deleted in adult hematopoietic cells using Vav-Cre or Mx1-Cre
To overcome the embryonic lethality of Kras germline knockout mice, we generated a Kras conditional knockout allele, in which exon 1 of Kras is flanked with a LoxP site using a recombineering technique (Fig. S1A)26. This allows spatially and temporally specific deletion of Kras upon expression of Cre recombinase. The correctly targeted mouse ESC clones were confirmed with Southern blot analysis using a 3′ external probe and PCR amplifying 5′ LoxP site (Fig. S1B). The correctly targeted mouse ESCs with normal karyotype were injected into V6.5 blastocysts and germline transmission of the engineered Kras allele was confirmed in multiple chimeric mice. Subsequently, these mice were crossed to Rosa26FLPeR/+ mice to remove PGK/EM7-neo selection cassette and the compound mice (Kras2lox, 2frt/+; Rosa26FLPeR/+) were further backcrossed to C57BL/6 mice to separate the two alleles (Fig. S1C). The generated Krasfl allele was continuously backcrossed to C57BL/6 background for more than 10 generations before conducting any experiments.
We used Vav-Cre and Mx1-Cre to delete Kras expression in adult hematopoietic system (Krasfl/fl; Vav-Cre and Krasfl/fl; Mx1-Cre mice, respectively). Vav-Cre drives Cre expression specifically in hematopoietic cells since E11.529, while polyinosinic-polycytidylic acid (pI-pC) injections trigger IFN signaling to induce expression of Mx1-Cre30. Both Vav-Cre and Mx1-Cre induced by 5 times of pI-pC injections deleted Kras exon1 in >95% of HSCs (Fig. S1D), which led to efficient removal of Kras expression in whole bone marrow cells (Fig. 1A). Consistent with a previous report20, deletion of Kras did not affect expression of Hras and Nras. More importantly, although Kras appeared to be the major Ras isoform activated in whole bone marrow cells, activation of Hras and Nras in Kras−/− cells was comparable to that in control cells (Fig. 1A). These results suggest that Kras deficiency does not trigger compensatory up-regulation or hyper-activation of Hras and Nras.
Figure 1. Krasfl/fl; Vav-Cre and pI-pC-treated Krasfl/fl; Mx1-Cre mice display abnormal hematopoiesis at 9–12 months old.
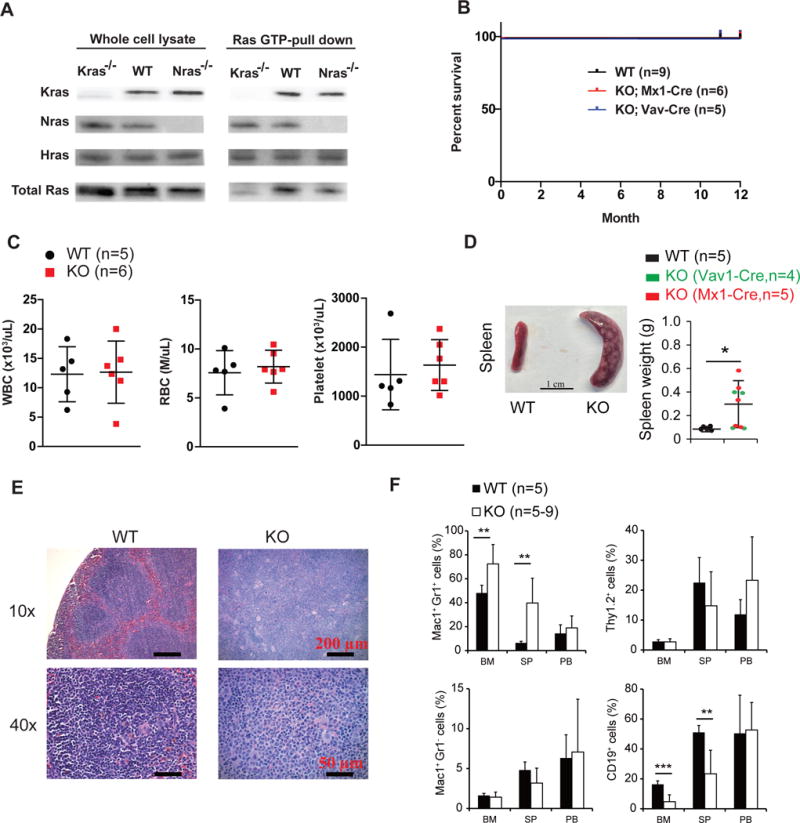
(A) Whole cell lysates were extracted from bone marrow cells of WT, Krasfl/fl; Vav-Cre, and NrasLSLG12D/LSL G12D (Nras−/−) mice at 8–10 weeks old. Ras-GTP was affinity purified from the whole cell lysates using a GST fusion with the Ras binding domain of Raf (Raf RBD) immobilized on agarose beads. Different Ras isoforms bound with GTP were analyzed using Western blot analysis. WT, wild-type. (B) Kaplan-Meier comparative survival analysis of different groups of animals Cumulative survival was plotted against days after birth. (C) CBC analysis was performed in 12-month-old mice. (D) Splenomegaly in a fraction of Krasfl/fl; Vav-Cre and pI-pC-treated Krasfl/fl; Mx1-Cre mice at 9–12 months old. (E) Representative histologic H&E sections of enlarged spleen in Krasfl/fl; Vav-Cre and pI-pC-treated Krasfl/fl; Mx1-Cre mice at 9–12 months old and of spleen in age-matched WT mice. (F) Flow cytometric analysis of neutrophils (Mac1+ Gr1+), monocytes (Mac1+ Gr1−), T cells (Thy1.2+), and B cells (CD19+) in bone marrow (BM), spleen (SP), and peripheral blood (PB). * P<0.05, ** P<0.01, *** P<0.001.
Kras deletion induces profound hematopoietic defects in 9–12 months old mice
Although both Krasfl/fl; Vav-Cre and pI-pC-treated Krasfl/fl; Mx1-Cre mice were born with expected Mendelian ratios and appeared overtly healthy for up to 12 months (Fig. 1B and 1C), 4/9 mice displayed significant splenomegaly with numerous expanded white blood cell foci at 9–12 months old (Fig. 1D). Histopathological evaluation of these enlarged spleens revealed that the tissue was heavily infiltrated with neutrophils (Fig. 1E). Unlike the neutrophils in mice with myeloproliferative neoplasms (MPNs) we previously characterized9, 31, the neutrophils in Krasfl/fl; Vav-Cre and pI-pC-treated Krasfl/fl; Mx1-Cre mice rarely infiltrated to other tissues (our unpublished observation) and displayed normal counts in peripheral blood (Fig. 1C and 1F). This might explain the overtly healthy condition of these mice. Nonetheless, all the Krasfl/fl; Vav-Cre and pI-pC-treated Krasfl/fl; Mx1-Cre mice exhibited profound hematopoietic defects at 9–12 months old regardless of their spleen size, including a greatly expanded neutrophil compartment and significantly reduced B cell compartment in bone marrow and spleen (Fig. 1F). These data suggest that Kras is required for normal adult hematopoiesis.
Kras deficiency significantly reduces LT- and IT-HSC compartments in adult mice
To determine how Kras deficiency affects adult hematopoiesis, we analyzed various hematopoietic tissues from 8–10 weeks old Krasfl/fl; Vav-Cre mice. The bone marrow cellularity and tissue weight (including spleen, thymus, and liver) in Krasfl/fl; Vav-Cre mice were indistinguishable from those in control mice (Fig. 2A). We first examined primitive hematopoietic compartment using LSK (Lin− Sca1+ c-Kit+) CD34 Flt3 CD49b (Fig. 2B). Long term-HSCs (LT-HSCs) are defined as Lin− Sca1+ c-Kit+ CD34− Flt3− CD49b−, intermediate term-HSCs (IT-HSCs) are Lin− Sca1+ c-Kit+ CD34− Flt3− CD49b+, short term-HSCs (ST-HSCs) are Lin− Sca1+ c-Kit+ CD34+Flt3− cells, and multipotent progenitors (MPPs) are Lin− Sca1+ c-Kit+ CD34+ Flt3+ 32–34. We found that in Krasfl/fl; Vav-Cre mice, LT- and IT-HSC compartments were significantly reduced, whereas ST-HSC, MPP, and LSK compartments were significantly expanded (Fig. 2B and 2C).
Figure 2. Kras deficiency significantly reduces LT- and IT-HSC compartments in adult mice.

Krasfl/fl; Vav-Cre (KO) and their littermate control mice (WT) were sacrificed 8–10 weeks after birth for analysis of different hematopoietic tissues. (A) Quantification of bone marrow cellularity and tissues weight (including spleen, thymus, and liver). (B, C) Representative gating strategy and quantification of long term (LT)-, intermediate term (IT)-, short term (ST)-HSCs (B), multipotent progenitors (MPPs), and LSK cells (Lin− Sca1+ c-Kit+) (C) in bone marrow. LT-HSCs are defined as LSK CD34− Flt3− CD49b−, IT-HSCs are defined as LSK CD34− Flt3− CD49b+, ST-HSCs are defined as LSK CD34+ Flt3−, and MPPs are defined as LSK CD34+ Flt3+. (D) Levels of phosphorylated ERK1/2 (pERK1/2) levels in response to TPO or SCF stimulation were measured using phospho-flow cytometry. Sorted CD150+ CD41− and CD150− CD41− cells were serum- and cytokine-starved for 30 minutes and stimulated with 0 or 20 ng/mL of TPO at 37°C for 10 minutes. HSCs (defined as [Lin CD48]−/low cKit+ cells from sorted CD150+ CD41− cells) and MPPs (defined as [Lin CD48]−/low cKit+ cells from sorted CD150− CD41− cells) were gated for data analysis. To quantify the activation of ERK1/2 in control cells and Kras deficient cells, median intensities of p-ERK1/2 at different TPO or SCF concentrations are compared to their respective control cells at 0 ng/ml, which is arbitrarily set at 1. Representative plots from one experiment are shown. The results are presented as means ± SD. * P<0.05, ** P<0.01, *** P<0.001.
Because hematopoietic stem and progenitor cells are tightly regulated by cytokines, we investigated whether loss of Kras disturbs cytokine-induced signaling. Indeed, TPO-evoked ERK1/2 activation was greatly reduced in HSCs deficient for Kras (Fig. 2D). Interestingly, despite the expanded MPP compartment in Krasfl/fl; Vav-Cre mice, these cells showed significantly lower TPO-evoked ERK1/2 activation than control MPPs (Fig. 2D). Concomitantly, TPO-evoked AKT and STAT5 activation was also significantly reduced in Kras deficient HSCs and MPPs (Fig. S2). In contrast, SCF-evoked ERK1/2 activation in Kras−/− HSCs and MPPs was comparable to that in control cells (Fig. 2D). Together, these results suggest that Kras regulates HSC compartment at least partially through modulating TPO signaling.
Loss of Kras leads to imbalanced myeloid progenitor and common lymphoid progenitor cells in adult mice
Since TPO-evoked ERK1/2 activation in Kras−/− HSCs and MPPs was significantly reduced compared to that in control cells, we investigated whether this signaling defect affected cell differentiation toward myeloid and lymphoid progenitors. We analyzed myeloid progenitor compartment (MP, Lin− Sca1− c-Kit+ IL7Rα−) and common lymphoid progenitors (CLPs, Lin− Sca1low c-Kitlow IL7Rα+) in control and Krasfl/fl; Vav-Cre mice. Our results demonstrated that MP compartment was significantly expanded in Krasfl/fl; Vav-Cre mice, owing to the expansion of granulocyte-macrophage progenitors (GMPs, Lin− Sca1− c-Kit+ IL7Rα− FcγRhi CD34+) (Fig. 3A). In contrast, CLP compartment in Krasfl/fl; Vav-Cre mice was significantly reduced compared to that in control mice. Consistent with increased GMPs, which are responsive to GM-CSF stimulation, the percentages of MPs (enriched in Lin−/low c-Kit+ cells) and myeloid precursors (enriched in Lin−/low c-Kit− cells) in response to GM-CSF were moderately but significantly increased in Krasfl/fl; Vav-Cre mice (Fig. 3B). However, the amplitude of ERK1/2 activation in these GM-CSF activated Krasfl/fl; Vav-Cre cells was moderately but significantly lower than that in control cells. These data suggest that loss of Kras leads to compromised TPO-evoked ERK1/2 activation in HSCs and MPPs, which might favor the differentiation toward MPs.
Figure 3. Loss of Kras leads to unbalanced myeloid progenitor and common lymphoid progenitor cells in adult mice.

Krasfl/fl; Vav-Cre (KO) and their littermate control mice (WT) were sacrificed 8–10 weeks after birth for flow cytometric analysis. (A) Quantification of myeloid progenitors (MPs), common myeloid progenitors (CMPs), granulocyte-macrophage progenitors (GMPs), megakaryocyte-erythroid progenitors (MEPs), and common lymphoid progenitors (CLPs) in bone marrow. (B) Whole bone marrow cells were serum- and cytokine-starved for 1 hour and stimulated with 0, 1, or 10 ng/mL of GM-CSF at 37°C for 10 minutes. Levels of phosphorylated ERK1/2 (pERK1/2) were measured using phospho-flow cytometry. Lin− cKit+ (enriched for myeloid progenitors) and Lin− cKit− (enriched for myeloid precursors) were gated for data analysis. Representative result from one mouse per group is shown. Percentage and median intensity of pERK1/2 in GM-CSF activated cells were further analyzed and quantified. The results are presented as means ± SD. * P<0.05, ** P<0.01.
Deletion of Kras blunts GM-CSF-evoked ERK1/2 activation in differentiated myeloid cells and decreases neutrophil survival
Consistent with the MP and CLP phenotypes, we found that in Krasfl/fl; Vav-Cre bone marrow and spleen, the frequency of differentiated myeloid cells was significantly increased, owing to the expanded neutrophils (Mac1+Gr1+) (Fig. 4A). Concomitantly, B-cell compartment was significantly reduced. Unlike Nras−/− mice23, in which T cell compartment is compromised, T-cell compartment in Krasfl/fl; Vav-Cre mice was comparable to that in control mice. GM-CSF-evoked ERK1/2 activation in Lin+ cells was blunted in Krasfl/fl; Vav-Cre mice (Fig. 4B). Further fractionation of Lin+ cells showed that ERK1/2 activation was not only dramatically decreased in myeloid cells from Krasfl/fl; Vav-Cre and pI-pC-treated Krasfl/fl; Mx1-Cre mice (Fig. 4B and S3), but also down-regulated in T cells (Fig. S4). In contrast, GM-CSF-evoked STAT5 activation in Lin+ cells from Krasfl/fl; Mx1-Cre mice was comparable to that in control cells (Fig. S3). To determine whether ERK1/2 signaling defect is specific for Kras−/− cells, we took advantage of the complete blockade of Nras expression in NrasLSL G12D/LSL G12D cells (Fig. S5A and S5B) and performed GM-CSF signaling study in these cells. Our results showed that GM-CSF-evoked ERK1/2 activation in NrasLSL G12D/LSL G12D myeloid cells was indistinguishable from that in control cells (Fig. S5C), suggesting that Kras is a major regulator of GM-CSF signaling in differentiated myeloid cells.
Figure 4. Deletion of Kras blunts GM-CSF-evoked ERK1/2 activation and reduces survival of differentiated myeloid cells.
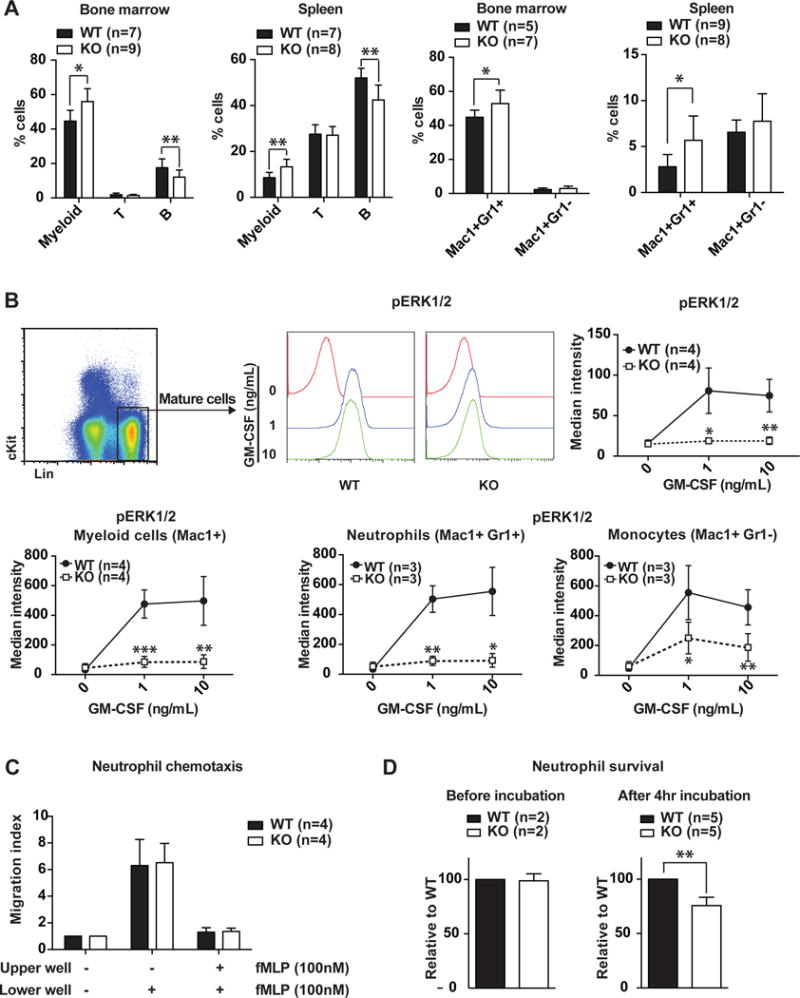
Krasfl/fl; Vav-Cre (KO) and their littermate control mice (WT) were sacrificed 8–10 weeks after birth for flow cytometric analysis. (A) Quantification of myeloid, T-, B-cells, neutrophils (Mac1+Gr1+) and monocytes (Mac1+Gr1−) in bone marrow and spleen. (B) Whole bone marrow cells were serum- and cytokine-starved for 1 hour and stimulated with 0, 1, or 10 ng/mL of GM-CSF at 37°C for 10 minutes. Levels of phosphorylated ERK1/2 (pERK1/2) were measured using phospho- flow cytometry. Differentiated cells (defined as Lin+), myeloid cells (defined as Mac1+), neutrophil cells (defined as Mac1+Gr1+), and monocytes (defined as Mac1+Gr1−) were gated for data analysis. (C, D) Neutrophils were enriched from bone marrow and analyzed for chemotaxsis capability using Trans-well migration assay (C) and for viability before and after 4 hr incubation using CellTiter Glo assay (D). The results are presented as means ± SD. * P<0.05, ** P<0.01, *** P<0.001.
Because ERK1/2 signaling regulates cell proliferation, survival, and migration, we investigated whether Kras deficiency affected any of these aspects in purified neutrophils. Our data showed that loss of Kras did not affect neutrophil chemotaxis in a trans-well migration assay but did significantly decrease neutrophil survival in vitro (Fig. 4C and 4D). Together, our results suggest that Kras mediates GM-CSF-evoked ERK1/2 activation to promote neutrophil survival.
Kras deficiency compromises HSC self-renewal
To determine the reconstitution potential of Kras−/− bone marrow cells, we performed a serial competitive reconstitution assay using control and Krasfl/fl; Vav-Cre cells together with competitor cells at 1:1 or 3:1 ratio (Fig. 5A). As expected, Kras−/−-derived cells were greatly under-represented in the peripheral blood of recipients, including both primary and secondary recipients and at both 1:1 and 3:1 ratios. The reduced reconstitution affected all lineages of cells, including myeloid, T, and B cells (Fig. 5B). However, unlike Mpl−/− cells, the reduced reconstitution of Kras−/− cells was still sustained at least for 2 rounds of transplantation. We repeated the same experimental procedure with Krasfl/fl; Mx1-Cre cells. Four weeks after transplantation, recipients were injected with pI-pC for 5 times to efficiently delete Kras expression (Fig. S6). The results were essentially same as those with Krasfl/fl; Vav-Cre cells. Consistent with non-transplanted Krasfl/fl; Vav-Cre mice, myeloid cell compartment was significantly expanded in Kras−/−-derived cells, while B cell compartment was significantly reduced (Fig. S7). In contrast to the greatly reduced reconstitution of Kras−/− cells, reconstitution capability of Nras−/− bone marrow cells was comparable to control cells in primary recipients but was moderately but significantly lower in secondary recipients (Fig. S3D). Our data suggest that Kras regulates HSC self-renewal and partially overlaps with Nras.
Figure 5. Kras−/− bone marrow cells show greatly reduced but sustainable reconstitution capability in serial competitive transplantation assay.
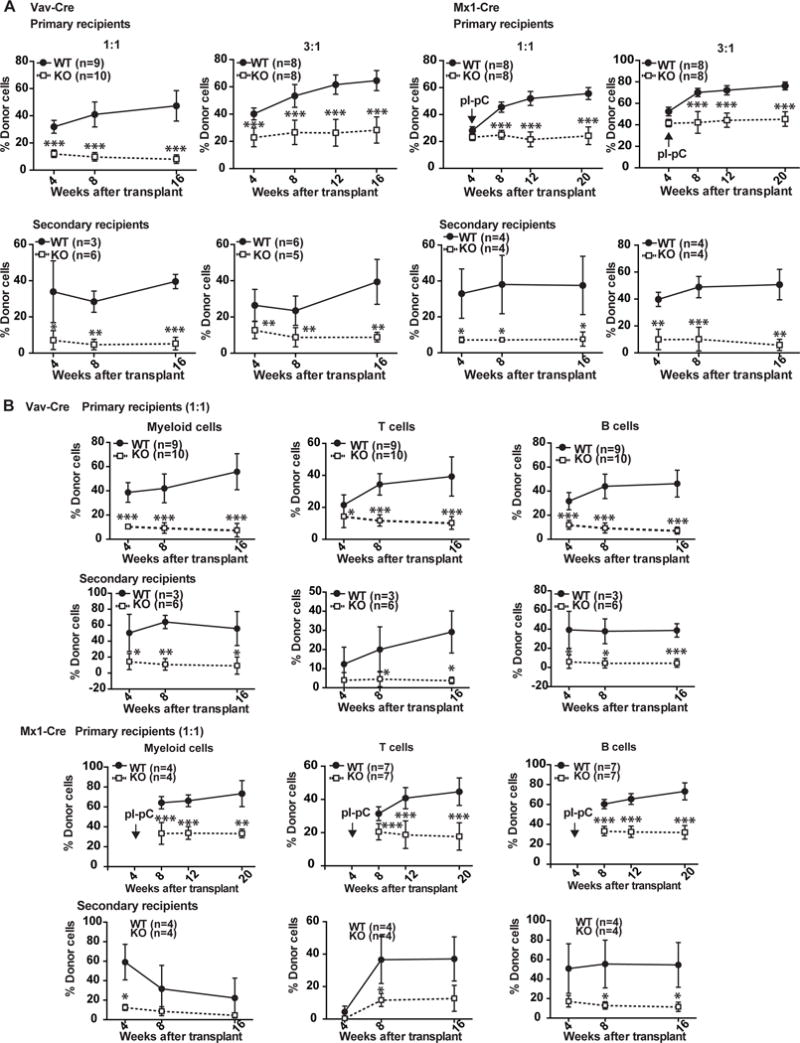
Serial bone marrow competitive reconstitution assay was performed as described in Materials and Methods. (A) Quantification of donor-derived cells in peripheral blood of primary and secondary recipients 4, 8, and 16 weeks after transplantation (Vav-Cre) or after pI-pC injections (Mx1-Cre). (B) Quantification of donor-derived myeloid, T, and B cells in peripheral blood of primary and secondary recipients 4, 8, and 16 weeks after transplantation (Vav-Cre) or after pI-pC injections (Mx1-Cre). The results are presented as means ± SD. * P<0.05, ** P<0.01, *** P<0.001.
We further analyzed Kras−/−-derived cells in primary and secondary recipients transplanted with Krasfl/fl; Vav-Cre cells 16 weeks after transplantation. Despite the dramatic reduction of Kras−/−-derived cells in peripheral blood (~17% of control cells), the representation of Kras−/−-derived cells in bone marrow was ~41% of that of control cells (Fig. 6A), suggesting an important role of Kras in promoting survival of differentiated blood cells in peripheral hematopoietic tissue. Based on LSK CD34 Flt3 CD49b markers, percentage of Kras−/−-derived LT-HSCs was ~50% of that of control LT-HSCs (Fig. 6B). Interestingly, this ratio maintained similarly through Kras−/−-derived LSK cells and was only moderately decreased to ~40% in Kras−/−-derived MPs (Fig. 6C). Consistent with our serial competitive reconstitution assay, Kras−/−-derived HSCs were further reduced in secondary recipients than those in primary recipients using LSK SLAM markers (Fig. 6B). Together, these results suggest that Kras plays an important role in regulating HSC self-renewal and survival of differentiated hematopoietic cells, while it is largely dispensable in intermediate progenitor cells.
Figure 6. Kras deficiency significantly reduces self-renewal capability of LT-HSCs.

Primary and secondary recipients transplanted with donor and competitor cells at 1:1 ration were sacrificed 16 weeks after transplantation for flow cytometric analysis of various hematopoietic tissues. (A) Quantification of donor-derived cells in bone marrow (BM), spleen (SP), and peripheral blood (PB). (B) Quantification of donor-derived HSCs, MPPs, and LSKs in BM. HSCs and MPPs are defined either based on LSK SLAM markers or based on LSK CD34 Flt3 markers. LT-, IT-, and ST-HSCs are defined as described in Figure 2 legend. (C) Quantification of donor-derived myeloid progenitors (MPs) and common lymphoid progenitors (CLPs) in BM. The results are presented as means ± SD. * P<0.05, ** P<0.01, *** P<0.001.
Discussion
In this study, we demonstrate that Kras plays an important role in adult hematopoiesis (Fig. 7). Loss of Kras leads to greatly compromised TPO signaling in HSCs and MPPs and GM-CSF-evoked ERK1/2 activation in differentiated myeloid cells. Consequently, Kras−/− HSCs show significantly reduced self-renewal potential, Kras−/− HSCs/MPPs demonstrate a differentiation bias toward myeloid lineage, and Kras−/− neutrophils display reduced survival in vitro. However, Kras is largely dispensable in intermediate progenitor cells (e.g. MPs). The signaling defects in Kras−/− cells leads to profound hematopoietic defects in 9–12 months old mice, including splenomegaly (partial penetrance), expanded neutrophil compartment, and reduced numbers of B cells. Our study reveals distinct Kras function in adult primitive hematopoietic compartment (including HSCs and MPPs) and differentiated cells (e.g. neutrophils and B cells).
Figure 7. Schematic illustration to summarize adult hematopoietic defects in Kras−/− mice.
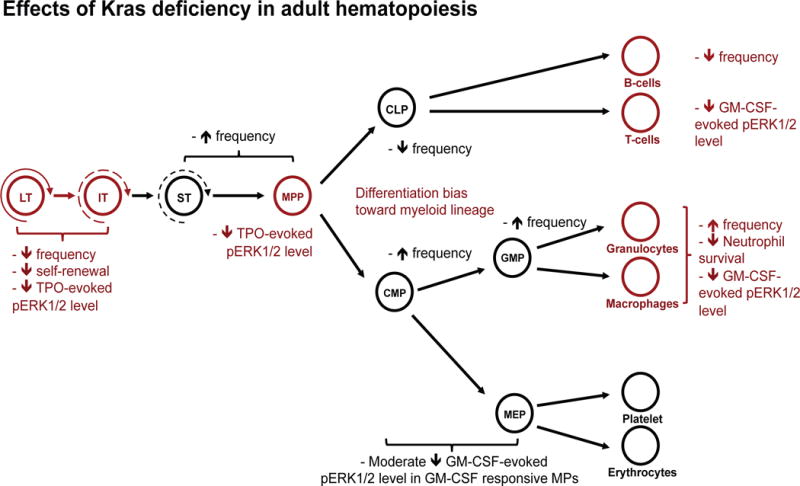
The proposed critical steps of adult hematopoiesis that require Kras function are highlighted in red font.
An earlier study suggests that Kras is dispensable for fetal liver hematopoiesis, but its role in adult hematopoiesis remains elusive20. In this study, we chose Vav-cre and Mx1-Cre to delete Kras expression in adult hematopoietic system. Vav-cre drives Cre expression exclusively in hematopoietic cells since E11.529. To avoid the potential carry-over effects of Kras deficiency in fetal liver hematopoiesis, we used the inducible Mx1-Cre to compliment studies with Vav-Cre. The results from both Cre systems are highly consistent with each other and also consistent with the earlier report20, suggesting that Kras plays an important function in adult hematopoiesis, distinct from that in fetal liver hematopoiesis.
Interestingly, we found that Kras is a major regulator for TPO signaling in HSCs and MPPs (Fig. 2D and S2). One possibility we favor is that lowered TPO-evoked ERK1/2 signaling in Kras−/− HSCs and MPPs results in reduced number of TPO-responsive cells, leading to compromised TPO signaling in general. In contrast, Kras is dispensable for SCF-evoked ERK1/2 activation in HSCs and MPPs (Fig. 2D). Surprisingly, Kras−/− LT- and IT-HSC compartments are significantly reduced, while MPP compartment is moderately expanded (Fig. 2). This could be due to the differential dependence of HSCs and MPPs on TPO signaling, which is supported by our previous TPO signaling study in these cells12. Although defective TPO signaling in Kras−/− MPPs could lead to lower number of TPO-responsive MPPs, the non-TPO responsive MPPs could still undergo expansion in response to the downstream hematopoietic defects (e.g. increased apoptosis in neutrophils), which is possibly mediated by unaffected SCF signaling. The impact of reduced TPO signaling on MPPs might be reflected on a lineage differentiation bias towards myeloid cells and in particular, GMPs (Fig. 3). The exact mechanism underlying the differential requirement of Kras in TPO versus SCF signaling is unclear. We suspect that this might be due to the elusive interaction between the ERK1/2 pathway and the JAK/STAT pathway that were suggested by previous studies9, 35, and the JAK/STAT pathway only directly involves in TPO receptor signaling but not in SCF receptor signaling in bone marrow stem/progenitor cells. Nonetheless, our observation is consistent with previous reports that Kras is dispensable for SCF-evoked ERK1/2 activation in fetal liver erythroid progenitors24 and oncogenic Kras does not cause hyper-activation of SCF-evoked ERK1/2 signaling in adult hematopoietic stem/progenitor cells8, 36. Clearly, TPO-evoked ERK1/2 activation is largely Kras-dependent, while SCF-evoked ERK1/2 activation is mainly Kras-independent. Because only SCF but not TPO signaling is required for fetal liver hematopoiesis2, 4, we believe that the essential role of Kras in TPO but not in SCF signaling explains its differential function in adult versus fetal liver hematopoiesis.
Kras−/− bone marrow cells show greatly reduced but sustained reconstitution in serially transplanted recipients (Fig. 5), which is attributed by defects in two major cell types. First, Kras deficiency leads to greatly reduced TPO-evoked ERK1/2 activation in HSCs. ERK1/2 signaling is recently linked to HSC self-renewal and differentiation. In Erk1−/−; Erk2−/− mice, HSC activity is completely abolished37. Moderate hyperactivation of ERK1/2 by endogenous NrasG12D/+ leads to increased HSC self-renewal with a myeloid differentiation bias38, while strong hyperactivation of ERK1/2 by NrasG12D/G12D results in decreased HSC self-renewal with a lymphoid differentiation bias39. Not surprisingly, in non-transplanted Krasfl/fl; Vav-Cre mice, frequency of LT-HSCs was significantly lower than that of control HSCs (Fig. 2B). In serially transplanted recipients, Kras−/−-derived HSCs were further reduced in secondary recipients than those in primary recipients, suggesting reduced HSC self-renewal potential (Fig. 6B). Noticeably, Kras deficiency did not completely abolish HSC self-renewal, likely owing to the partial functional redundancy with Nras; Nras deficiency resulted in moderately decreased HSC self-renewal (Fig. S5D).
Second, percentage of Kras−/−-derived cells in peripheral blood of recipients was significantly further reduced than that in bone marrow (Fig. 6A), suggesting that loss of Kras affects egress/migration and/or survival of differentiated hematopoietic cells. We show that Kras is the major Ras isoform activated in differentiated myeloid cells (Fig. 1E). Consistent with this result, GM-CSF-evoked ERK1/2 activation was blunted in Kras−/− myeloid cells (Fig. 4B and S3) but was normal in Nras−/− cells (Fig. S5C). The defective cytokine signaling did not affect Kras−/− neutrophil migration but decreased neutrophil survival (Fig. 4C and 4D). Although Krasfl/fl; Vav-Cre and pI-pC treated Krasfl/fl; Mx1-Cre mice are overtly healthy and maintain a normal CBC result up to 1 year, their hematopoietic compartments in bone marrow and spleen display profound defects (Fig. 1). The partially penetrant splenomegaly developed in these 9–12 months old mice suggest that they are highly prone to myeloid diseases. Indeed, we recently found that loss of WT Kras promotes oncogenic Kras-induced MPN in a cell autonomous manner40. Moreover, B cell compartment was significantly reduced in Krasfl/fl; Vav-Cre mice and in Kras−/−-derived cells in transplant recipients (Fig. 1F, 4A and S7). Consistent with our finding, recent work from Chen and colleagues showed that the ERK1/2 pathway is down-regulated in Kras−/− B cells, which is associated with B cell differentiation defects at both pre-B cell and mature B cell stages41. Together, these data indicate loss of Kras leads to various defects in differentiated hematopoietic cells, which could contribute to reduced reconstitution of Kras−/−-derived cells in recipients.
Although Kras−/− MPs showed moderate reduction in GM-CSF-evoked ERK1/2 activation, Kras is largely dispensable in these cells. This conclusion is supported by two observations. First, in Krasfl/fl; Vav-Cre mice, MP compartment was significantly expanded, which could compensate for the defective differentiated myeloid cells. Second, in recipients, the reconstitution of Kras−/−-derived MPs maintained at a similar level as Kras−/−-derived HSCs, suggesting no further significant reduction of MP genesis and survival. We believe that loss of Kras in MPs could be compensated by Nras.
Conclusion
In summary, our study demonstrates the important function of Kras in adult hematopoiesis. Loss of Kras leads to greatly reduced cytokine-evoked ERK1/2 activation in HSCs, MPPs, and differentiated myeloid cells. Reduced HSC self-renewal and myeloid cell survival contribute to the greatly reduced reconstitution of Kras−/−-derived bone marrow cells in recipient mice. Consequently, older mice deficient for Kras in hematopoietic compartment develop profound hematopoietic defects and are prone to myeloid diseases.
Supplementary Material
Significance Statement.
Adult hematopoiesis is regulated by multiple cytokines, including stem cell factor (SCF), thrombopoietin (TPO), and granulocyte macrophage colony-stimulating factor (GM-CSF). Although all of them are known to activate ERK1/2 through Ras proteins, whether and how individual Ras isoforms distinctly involve in these signaling pathways during adult hematopoiesis remain largely elusive. Here, using a novel Kras conditional knockout allele we generated, we demonstrate that Kras is dispensable for SCF-evoked ERK1/2 activation but is required for TPO signaling in adult HSCs. Consequently, Kras deficiency leads to reduced long-term and intermediate-term HSC compartments, compromised HSC self-renewal, and a differentiation bias toward myeloid lineages. In addition, Kras loss leads to blunted GM-CSF-evoked ERK1/2 signaling in neutrophils and reduced neutrophil survival. Our study reveals a unique function of Kras in adult hematopoiesis, mainly in HSCs and differentiated blood cells.
Acknowledgments
We are grateful to Dr. Chris Bradfield for providing the Vav-Cre mice. We would like to thank the University of Wisconsin Carbone Comprehensive Cancer Center (UWCCC) for use of its Shared Services to complete this research. This work was supported by R01 grants R01CA152108 and R01HL113066, and a Scholar Award from the Leukemia & Lymphoma Society to J.Z. This work was also supported in part by NIH/NCI P30 CA014520-UW-Comprehensive Cancer Center Support.
Footnotes
Conflict of interest: We declare that no conflict of interest exists.
Authorship Contributions
A. Damnernsawad and J. Zhang: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, and final approval of the manuscript; G. Kong, Z. Wen, A. Rajagopalan, and X. You: collection and/or assembly of data, data analysis and interpretation and final approval of the manuscript; Y. Liu, J. Wang, Y. Zhou, H. Luo, E. A. Ranheim, and Q. Chang: data analysis and interpretation and final approval of the manuscript.
References
- 1.Dzierzak E, Speck NA. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol. 2008;9:129–136. doi: 10.1038/ni1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qian H, Buza-Vidas N, Hyland CD, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell. 2007;1:671–684. doi: 10.1016/j.stem.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Thoren LA, Liuba K, Bryder D, et al. Kit regulates maintenance of quiescent hematopoietic stem cells. J Immunol. 2008;180:2045–2053. doi: 10.4049/jimmunol.180.4.2045. [DOI] [PubMed] [Google Scholar]
- 4.Ding L, Saunders TL, Enikolopov G, et al. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–462. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor ML, Metcalfe DD. Kit signal transduction. Hematol Oncol Clin North Am. 2000;14:517–535. doi: 10.1016/s0889-8588(05)70294-x. [DOI] [PubMed] [Google Scholar]
- 6.Weiler SR, Mou S, DeBerry CS, et al. JAK2 is associated with the c-kit proto-oncogene product and is phosphorylated in response to stem cell factor. Blood. 1996;87:3688–3693. [PubMed] [Google Scholar]
- 7.Varricchio L, Tirelli V, Masselli E, et al. The expression of the glucocorticoid receptor in human erythroblasts is uniquely regulated by KIT ligand: implications for stress erythropoiesis. Stem cells and development. 2012;21:2852–2865. doi: 10.1089/scd.2011.0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Meter ME, Diaz-Flores E, Archard JA, et al. K-RasG12D expression induces hyperproliferation and aberrant signaling in primary hematopoietic stem/progenitor cells. Blood. 2007;109:3945–3952. doi: 10.1182/blood-2006-09-047530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang JY, Liu YG, Li ZY, et al. Endogenous oncogenic Nras mutation leads to aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood. 2010;116:5991–6002. doi: 10.1182/blood-2010-04-281527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chou FS, Mulloy JC. The thrombopoietin/MPL pathway in hematopoiesis and leukemogenesis. Journal of cellular biochemistry. 2011;112:1491–1498. doi: 10.1002/jcb.23089. [DOI] [PubMed] [Google Scholar]
- 11.Tong W, Lodish HF. Lnk inhibits Tpo-mpl signaling and Tpo-mediated megakaryocytopoiesis. J Exp Med. 2004;200:569–580. doi: 10.1084/jem.20040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du J, Wang J, Kong G, et al. Signaling profiling at the single cell level identifies a distinct signaling signature in murine hematopoietic stem cells. Stem Cells. 2012;30:1447–1454. doi: 10.1002/stem.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hercus TR, Thomas D, Guthridge MA, et al. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood. 2009;114:1289–1298. doi: 10.1182/blood-2008-12-164004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi Y, Liu CH, Roberts AI, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell research. 2006;16:126–133. doi: 10.1038/sj.cr.7310017. [DOI] [PubMed] [Google Scholar]
- 15.Barbacid M. ras genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 16.Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012 doi: 10.1182/blood-2012-05-378596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umanoff H, Edelmann W, Pellicer A, et al. The murine N-ras gene is not essential for growth and development. Proc Natl Acad Sci U S A. 1995;92:1709–1713. doi: 10.1073/pnas.92.5.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esteban LM, Vicario-Abejon C, Fernandez-Salguero P, et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol Cell Biol. 2001;21:1444–1452. doi: 10.1128/MCB.21.5.1444-1452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plowman SJ, Williamson DJ, O’Sullivan MJ, et al. While K-ras is essential for mouse development, expression of the K-ras 4A splice variant is dispensable. Mol Cell Biol. 2003;23:9245–9250. doi: 10.1128/MCB.23.24.9245-9250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson L, Greenbaum D, Cichowski K, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koera K, Nakamura K, Nakao K, et al. K-ras is essential for the development of the mouse embryo. Oncogene. 1997;15:1151–1159. doi: 10.1038/sj.onc.1201284. [DOI] [PubMed] [Google Scholar]
- 22.Potenza N, Vecchione C, Notte A, et al. Replacement of K-Ras with H-Ras supports normal embryonic development despite inducing cardiovascular pathology in adult mice. EMBO Rep. 2005;6:432–437. doi: 10.1038/sj.embor.7400397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez de Castro I, Diaz R, Malumbres M, et al. Mice deficient for N-ras: impaired antiviral immune response and T-cell function. Cancer Res. 2003;63:1615–1622. [PubMed] [Google Scholar]
- 24.Zhang J, Lodish HF. Identification of K-ras as the major regulator for cytokine-dependent Akt activation in erythroid progenitors in vivo. Proc Natl Acad Sci U S A. 2005;102:14605–14610. doi: 10.1073/pnas.0507446102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Lodish HF. Endogenous K-ras signaling in erythroid differentiation. Cell Cycle. 2007;6:1970–1973. doi: 10.4161/cc.6.16.4577. [DOI] [PubMed] [Google Scholar]
- 26.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stadtfeld M, Graf T. Assessing the role of hematopoietic plasticity for endothelial and hepatocyte development by non-invasive lineage tracing. Development. 2005;132:203–213. doi: 10.1242/dev.01558. [DOI] [PubMed] [Google Scholar]
- 28.Haigis KM, Kendall KR, Wang Y, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600–608. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gan T, Jude CD, Zaffuto K, et al. Developmentally induced Mll1 loss reveals defects in postnatal haematopoiesis. Leukemia. 2010;24:1732–1741. doi: 10.1038/leu.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuhn R, Schwenk F, Aguet M, et al. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 31.Wang JY, Liu YG, Li ZY, et al. Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood. 2011;118:368–379. doi: 10.1182/blood-2010-12-326058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osawa M, Hanada K, Hamada H, et al. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273:242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- 33.Yang L, Bryder D, Adolfsson J, et al. Identification of Lin(−)Sca1(+)kit(+)CD34(+)Flt3- short-term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated transplant recipients. Blood. 2005;105:2717–2723. doi: 10.1182/blood-2004-06-2159. [DOI] [PubMed] [Google Scholar]
- 34.Benveniste P, Frelin C, Janmohamed S, et al. Intermediate-term hematopoietic stem cells with extended but time-limited reconstitution potential. Cell Stem Cell. 2010;6:48–58. doi: 10.1016/j.stem.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 35.Li Q, Bohin N, Wen T, et al. Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature. 2013;504:143–147. doi: 10.1038/nature12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du J, Liu Y, Meline B, et al. Loss of CD44 attenuates aberrant GM-CSF signaling in Kras G12D hematopoietic progenitor/precursor cells and prolongs the survival of diseased animals. Leukemia. 2013;27:754–757. doi: 10.1038/leu.2012.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Staser K, Park SJ, Rhodes SD, et al. Normal hematopoiesis and neurofibromin-deficient myeloproliferative disease require Erk. J Clin Invest. 2013;123:329–334. doi: 10.1172/JCI66167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J, Kong G, Liu Y, et al. Nras G12D/+ promotes leukemogenesis by aberrantly regulating haematopoietic stem cell functions. Blood. 2013;121:5203–5207. doi: 10.1182/blood-2012-12-475863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong G, Wunderlich M, Yang D, et al. Combined MEK and JAK inhibition abrogates murine myeloproliferative neoplasm. J Clin Invest. 2014;124:2762–2773. doi: 10.1172/JCI74182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong G, Chang Y-I, Damnernsawad A, et al. Loss of wild-type Kras promotes activation of all Ras isoforms in oncogenic Kras-induced leukemogenesis. Leukemia. 2016 doi: 10.1038/leu.2016.40. in print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Zheng Y, You X, et al. Kras is critical for B cell lymphopoiesis. Journal of Immunology. 2016 doi: 10.4049/jimmunol.1502112. in print. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.