

SUMMARY
Genetically modified T cells expressing chimeric antigen receptors (CARs) demonstrate robust responses against lineage restricted, non-essential targets in hematologic cancers. However, in solid tumors, the full potential of CAR T cell therapy is limited by the availability of cell surface antigens with sufficient cancer-specific expression. The majority of CAR targets have been normal self-antigens on dispensable hematopoietic tissues or overexpressed shared antigens. Here, we established that abnormal self-antigens can serve as targets for tumor rejection. We developed a CAR that recognized cancer-associated Tn glycoform of MUC1, a neoantigen expressed in a variety of cancers. Anti-Tn-MUC1 CAR T cells demonstrated target-specific cytotoxicity and successfully controlled tumor growth in xenograft models of T cell leukemia and pancreatic cancer. These findings demonstrate the therapeutic efficacy of CAR T cells directed against Tn-MUC1 and present aberrantly glycosylated antigens as a novel class of targets for tumor therapy with engineered T cells.
INTRODUCTION
Chimeric antigen receptor (CAR) therapies have emerged as a potent new tool for hematologic malignancies (Brentjens et al., 2013; Grupp et al., 2013; Kalos et al., 2011; Kochenderfer et al., 2010; Porter et al., 2011). The major scientific issue facing the field with engineered T cells is whether this technology can be applied to solid tumors (June et al., 2015). To date, CARs have mostly targeted shared antigens found on nonessential tissues, such as CD19 on B lymphocytes. However, epithelial malignancies mostly exist within essential tissues and most epithelial tumor-associated antigens are shared proteins also found less abundantly in normal tissues. Although these shared proteins are overexpressed in malignancy, the immune system is tolerant to them because of thymic deletion and other post-thymic mechanisms. In addition, CAR and T cell receptor (TCR) therapies developed against these shared proteins have been met with serious adverse events. For example, when a CAR targeting her2/neu was tested, the patient died from cardiopulmonary toxicity within days (Morgan et al., 2010). In contrast, vaccines to her2/neu have been given to patients with a high degree of safety (Emens et al., 2009), and millions of patients have been treated with passive transfer therapy with trastuzumab with a favorable safety profile. The explanation for the differential toxicity between adoptive cell therapy and vaccines and antibody therapy is likely that the CAR T cells are simply more potent.
One well-characterized cellular process involved in differential processing following malignant transformation is protein glycosylation. Glycosylation also has a role in regulating immune tolerance, as reviewed (Rabinovich and Croci, 2012). Protein glycosylation is initiated with the covalent linkage of glycans to asparagine residues (N-linked) or serine (Ser) or threonine (Thr) residues (O-linked). Here, we focus on O-linked glycosylation, which is initiated with the addition of N-Acetylgalactosamine (GalNAc) to Ser or Thr residues by approximately 20 human polypeptide GalNAc-transferases (GalNAc-Ts) (Bennett et al., 2012). In normal cells GalNAc residues attached to the protein backbone are further elongated by the T synthase to form the Core 1 structure (Gal-GalNAc-α-Ser/Thr) (Ju et al., 2002). The Core 1 synthase, C1Gal-T1, requires the chaperone protein, Cosmc, to prevent rapid degradation (Ju and Cummings, 2002).
Changes in cell surface glycosylation have been shown to increase tumorigenesis and metastasis (Ohtsubo and Marth, 2006; Ren et al., 2014; Tamura et al., 2014; Tarp and Clausen, 2008; Taylor-Papadimitriou et al., 1999). The most prevalent aberrant glycoforms found in cancer are the Tn (GalNAcα1-O-Ser/Thr) and sialyl-Tn (STn) (NeuAcα2-6-GalNAcα1-O-Ser/Thr) glycoforms (Springer, 1984). Several mechanisms might result in accumulation of Tn glycoforms, including loss of T synthase activity (Ju et al., 2002) due to mutations or epigenetic silencing of COSMC and ectopic expression of GalNAc-Ts (Gill et al., 2013; Ju et al., 2008; Radhakrishnan et al., 2014; Schietinger et al., 2006). Somatic mutation of COSMC can lead to loss of tolerance in the bone marrow lineage with resultant hemolytic anemia and IgA nephrophathy (Berger, 1999; Ju and Cummings, 2005). Hypoxic conditions often found in tumors might alter expression of glycosyltransferases (Kannagi et al., 2010), including sialyltransferases such as ST6GalNAcI to create sialyl-Tn antigens. Glycosylation changes also alter cell adhesion and motility, which increase the metastatic potential of the tumor cell (Gill et al., 2013; Radhakrishnan et al., 2014; Ren et al., 2014; Tamura et al., 2014). Tn and STn antigen expression is correlated with adverse outcome and decreased patient survival in breast cancer, gastric cancer, endometrial cancer, and oral squamous cell carcinoma, among other cancers (Cazet et al., 2010; Itzkowitz, 2003; Lin et al., 2014; Ohno et al., 2006; Victorzon et al., 1996). Aberrant expression of Tn and STn glycoforms have in particular been found on the cell membrane mucin MUC1, which is a large protein with tandem repeated sequences carrying O-glycans overexpressed in most adenocarcinomas (Finn et al., 2011; Graham et al., 1996; Tarp and Clausen, 2008; Taylor-Papadimitriou et al., 1999). In health, the Tn antigen is not expressed and humans have natural anti-Tn IgM antibodies. However, exposure of Tn in cancer cells might lead to loss of immunological tolerance to Tn-glycopeptide epitopes, induction of IgG antibodies (Ju et al., 2008; Schietinger et al., 2006; Wandall et al., 2010) and immunopathology (Berger, 1999; Ju and Cummings, 2005).
We previously demonstrated that it is safe to elicit IgG antibodies to the Tn-MUC1 epitope recognized by 5E5 using a glycopeptide vaccine (Sabbatini et al., 2007) and we documented the existence of spontaneous IgG antibodies to this in some cancer patients (Wandall et al., 2010). Here, we develop and characterize a novel CAR based on a monoclonal antibody (5E5) specific to a Tn-MUC1 glycopeptide epitope widely expressed by adenocarcinomas (Sørensen et al., 2006; Tarp et al., 2007). This CAR is able to eliminate Tn-MUC1-expressing tumors in mouse models of leukemia and pancreatic cancer. We propose that cancer-specific neoepitopes formed by aberrant glycosylation might serve as targets for CAR T cell therapy for a variety of adenocarcinomas and bone-marrow-derived cancers.
RESULTS
Jurkat T Cell Leukemia Contains Mutations in COSMC and Expresses Tn Antigen Epitopes
Previous work has shown that the Jurkat T cell leukemia line contains a one base nucleotide deletion that results in a frameshift and early truncation of the T synthase chaperone protein, COSMC, responsible for preventing rapid degradation of the T synthase (Ju and Cummings, 2002). We also identified an alanine (A) to valine (V) substitution (nucleotide C428T) in our Jurkat E6-1 line of unknown effect on gene function that was not previously reported (Figure S1A). Using an in vitro fluorescence assay to measure T synthase activity, we detected negligible enzymatic activity in Jurkat cell extracts, compared with robust activity in 293T cell extracts (Figure S1B). These results confirm the COSMC truncation in Jurkat leukemia cells is correlated with the activity of T synthase.
We performed flow cytometric analysis of the Jurkat cell line immunostained with two characterized Tn antigen-specific antibodies, 5E5 and BaGs3 (Figure S1C). 5E5 mAb was developed and characterized for its specificity to the Tn/STn glycopeptide epitope on MUC1 (Sørensen et al., 2006). BaGs3 is an anti-Tn mAb (Borgert et al., 2012). Both 5E5 and BaGs3 mAbs demonstrated staining on the surface of Jurkat cells, indicating broad aberrant O- glycosylation of proteins in these cells.
5E5 Exhibits High Affinity for Tn-MUC1
The specificity of antibodies used to construct chimeric antigen receptors is critical because any cross-reactivity to cell surface proteins on normal tissues can be toxic as a T cell therapy. Thus, we evaluated the specificity of the 5E5 mAb and CAR in vitro with a 60-mer non-glycosylated MUC1 peptide (MUC1-60-mer) and corresponding Tn-glycopeptide (MUC1-9Tn) as well as a control mucin, sheep-derived submaxillary mucin (OSM). The OSM mucin is heavily glycosylated with STn O-glycans and serves as a general STn hapten antigen recognized by most anti-STn antibodies.
Neuraminidase treated Asialo-OSM (aOSM) serves as a general Tn hapten. Using previously described antibodies to the T, Tn, and STn carbohydrate haptens 3C9, 5F4, and 3F1 mAbs, respectively (Madsen et al., 2013; Radhakrishnan et al., 2014), we confirmed by ELISA that none of the antigens contained Core 1 structure (Figure 1A, top left panel). MUC1-9Tn and aOSM both contain Tn antigen (Figure 1A, top right panel), and OSM contains STn antigen (Figure 1A, bottom left panel). The unglycosylated MUC1-60-mer peptide was not recognized by any of these antibodies. The 3F1 antibody demonstrated low-level binding to aOSM peptide at higher concentrations, suggesting that the peptide contained some residual sialyation. The 5E5 mAb displayed high affinity for the MUC1-9Tn peptide and no reactivity against the non-glycosylated MUC1-60-mer peptide (Figure 1A, bottom right panel), indicating that the antibody detects the Tn-MUC1 glycopeptide specific epitope without reactivity with the unglycosylated peptide backbone. 5E5 does weakly react with the Tn hapten at high density on aOSM (Sørensen et al., 2006), and due to the high affinity of the 5E5 mAb, we titrated the glycopeptides to 233fg/mL in order to identify a decrease in antibody binding (Figure 1A, bottom right panel). We observed low-level binding of 5E5 mAb to aOSM compared with the MUC1-9Tn, which is in striking contrast to the reactivity of the 5F4 mab to the Tn hapten.
Figure 1. 5E5 mAb and 5E5 CAR Specifically Recognize the Tn-MUC1, but Not the Peptide Alone.
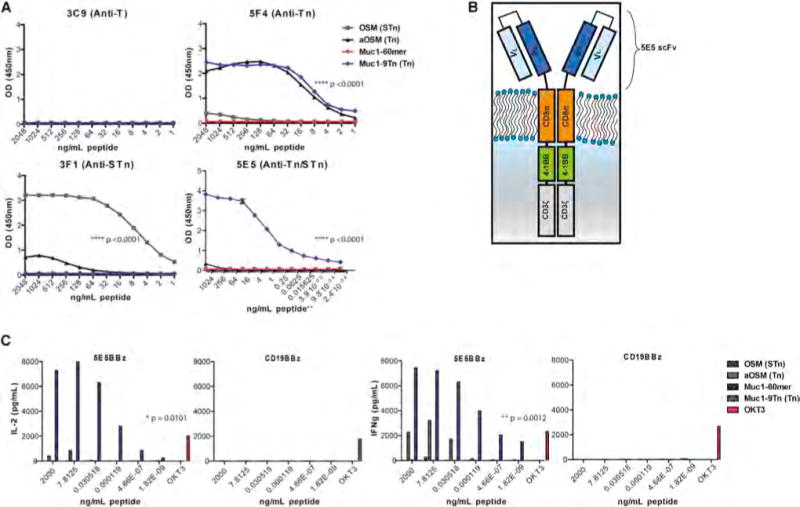
(A) ELISA-peptide assay of glycopeptides OSM, aOSM, Muc1-9Tn, and unconjugated Muc1-60-mer with antibodies 3C9, 5F4, 3F1, and 5E5. The peptides were coated in the plate wells at concentrations of 2,000 ng/mL in bicarbonate buffer and halved in dilutions until 1 ng/mL. The bottom right panel used starting concentration of 2,000 ng/mL and diluted 8-fold until 233 fg/mL (noted with **). Statistical significance is calculated using an unpaired t test comparing 5F4 ELISA for Muc1-9Tn and Muc1-60-mer, 3F1 ELISA for OSM and aOSM, and 5E5 ELISA for Muc1-9Tn and Muc1-60-mer. p < 0.0001.
(B) Graphical representation of the chimeric antigen receptor developed using the 5E5 scFv, CD8α hinge, and transmembrane domain, 4-1BB and CD3zeta endodomain.
(C) Indirect ELISA assays quantifying the cytokine production in supernatant from 5E5 CAR and CD19 CAR T cells cultured on peptide-bound plates for 24 hr. Peptides were plated starting at concentrations of 2,000 ng/mL and diluted 256-fold until 1.82 ag/mL. 10 μg/mL of OKT3 mAb was used as a control T cell stimulant. Statistical significance of 5E5 CAR T cells on Muc1-9Tn versus Muc1-60-mer peptide is p = 0.0101 for IL-2 secretion and p = 0.0012 for IFN-γ secretion.
We next characterized the binding of the 5E5 mab and scFv to normal human tissues and cancer. Immunostaining of normal human tissues with 5E5 mAb demonstrates no binding to most tissues, including tonsil, lymph node, prostate, adrenal, liver, heart, breast, spinal cord, cerebellum, cervix, testes, thyroid, endometrium, thymus, muscle, colon, esophagus, parathyroid, placenta, spleen, and adipose (Figure S2A). In contrast, the tissue microarray for stomach, lung, pancreas, and kidney did stain with 5E5 mAb. However, with confocal microscopy, the 5E5 staining pattern was found to be largely intracellular and colocalized with a marker of the cis-Golgi network, GM130, in contrast to normal MUC1 expression, stained with HMFG1 mAb, which localizes at the membrane and often colocalizes with EpCAM staining (Figure S2C). In contrast to normal human tissue, 5E5 mAb has intense binding to the plasma membranes of human breast cancer and colocalizes with EpCAM staining (Figure S2B). These results provide support for cancer-specific expression of Tn-MUC1 and further demonstrate that there is virtually non-existent expression of this epitope on the cellular surface of normal tissues.
We next synthesized the variable heavy and variable light chains of the 5E5 mAb, linked with a 3X(GGGGS) spacer and incorporated the single chain variable fragment (scFv) into a CAR backbone containing a CD8α hinge and transmembrane domain, a 4-1BB co-stimulatory domain and a CD3zeta activation domain (Figure 1B). The same structural backbone of the 5E5 CAR was used for the CD19 targeting CAR used for leukemia (Imai et al., 2004; Milone et al., 2009), which has demonstrated robust clinical activity in patients with leukemia (Maude et al., 2014). Normal human donor T cells were stimulated with anti-CD3 and anti-CD28 mAb-coated magnetic beads and transduced with a lentiviral vector encoding either CD19 CAR or 5E5 CAR (Figure S3A, S3B). CAR T cells were plated on OSM, aOSM, MUC1-60-mer, or MUC1-9Tn peptide-coated plates for 24 hr and the culture media was assessed for the cytokines interleukin-2 (IL-2) and interferon-gamma (IFN-γ) (Figure 1C). One μg/mL of anti-CD3ε mAb, OKT3, was used as a positive control for T cell stimulation. 5E5 CAR T cells secreted high quantities of IL-2 and IFN-γ in response to MUC1-9Tn but not the non-glysosylated Muc1-60-mer. IFN-γ secretion was also observed from 5E5 CAR T cells in response to aOSM peptide, which expresses Tn antigen on the sheep mucin, suggesting that while 5E5 CAR demonstrates a high affinity for Tn-MUC1, it might also demonstrate low level recognition for Tn antigen on other mucin-like glycoproteins, consistent with Figure 1A. In contrast, CD19 CAR T cells demonstrated no reactivity to any of the peptides but showed similar cytokine secretion in response to OKT3 stimulation, compared with 5E5 CAR T cells, which specifically produced cytokines in response to Tn-MUC1.
Similar to CD19 CAR T cells, 5E5 CAR T cells demonstrated no reactivity against normal human primary cardiac, aortic and pulmonary myocytes, renal epithelial cells, pulmonary endothelial cells, or osteoblasts in chromium release assays (Figure 2), while 5E5 CAR T cells and CD19 CAR T cells lysed positive controls Jurkat E6-1 and Jurkat 19COSMC cells, respectively. In contrast, a CAR that demonstrates normal tissue toxicity, MC813-70, demonstrated lysis of all evaluated normal primary cells.
Figure 2. Evaluation of 5E5 CAR T Cells Reactivity to a Panel of Human Primary Cells.
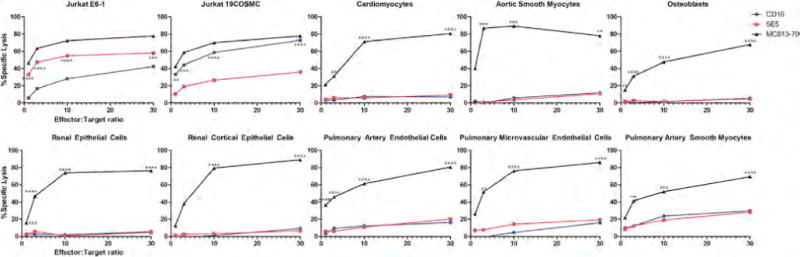
5E5 and CD19 CAR T cells were tested in a chromium release lysis assays at effector:target ratios of 1:1 to 30:1. MC813-70 CAR T cells known to exhibit normal tissue toxicity were used as a positive control. In addition the various CAR T cells were tested on wild-type Jurkat and Jurkat with reconstituted COSMC. Statistical comparisons are between 5E5 CAR and CD19 CAR in the positive controls Jurkat E6-1 and Jurkat 19COSMC. All other comparisons are made between MC813-70 and 5E5. ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001.
5E5 CAR T Cells Induce Anti-Tumor Responses
CD19-specific CD19 CAR T cells or Tn-MUC1-specific 5E5 CAR T cells were cultured overnight with K562 leukemia cell line, Jurkat leukemia cell line, or NNP4 primary ovarian cancer cells collected from a malignant pleural effusion, and analyzed for intracellular cytokine production and degranulation via flow cytometry (Figure 3A). While 5E5 CAR T cells exhibited no cytokine production over background controls in the absence of target, they did respond to K562 cells, Jurkat cells and NNP4 cells. CD19 CAR T cells did not react to any of the tumor cell lines, indicating low levels of alloreactivity under these conditions.
Figure 3. Antitumor Efficacy of 5E5 CAR T Cells In Vitro and In Vivo.
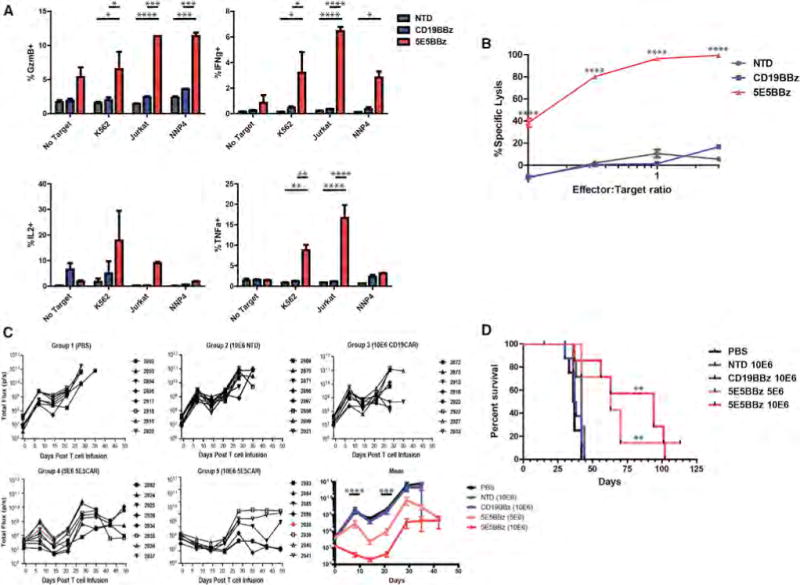
(A) Analysis of the cytokine-producing T cells from an intracellular cytokine assay of T cells, CAR-transduced or non-transduced (NTD), cultured with K562, K562-meso, Jurkat, and NNP4 cells. NNP4 is a sample of primary epithelial ovarian cancer cells obtained from a pleural tap. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001. Data are plotted as mean ± SEM.
(B) In vitro cytotoxicity assay of T cells, 5E5 CAR, CD19 CAR, or NTD, cultured with Jurkat cell line at the indicated effector-to-target ratios. **** = p < 0.0001
(C) Serial bioluminescence imaging of NSG mice injected intravenously with 5 × 106 CBG+ Jurkat cells and infused with PBS or the indicated T cells, 1 × 107 or 5 × 106 cells as indicated, 8 days after tumor engraftment. Two mice, one in group 4 and one in group 5, died due to conditions not related to treatment and are indicated as red lines. The bottom right panel displays the mean bioluminescence by treatment group. n = 8 mice per group. See also Figure S4A. Statistical significance calculated between CD19 CAR and 5E5 CAR (5 × 106 and 1 × 107). There is no significance observed between 5 × 106 and 1 × 107 dose of 5E5 CAR. *** = p < 0.001, **** = p < 0.0001.
(D) Kaplan-Meyer survival curve of mice bearing Jurkat tumor by treatment group. Median survival of PBS group = 36.5, NTD group = 42, CD19 CAR group = 37.5, 5 × 106 5E5 CAR group = 63 and 1 × 107 5E5 CAR group = 94 days post T cell infusion. Comparison of survival of CD19 CAR group to 5 × 106 5E5 CAR group is p = 0.0016 and for 1 × 107 5E5 CAR group is p = 0.0014. No statistical significance between 5E5 CAR doses.
Jurkat cells were then transduced with lentiviral vector encoding GFP and Click Beetle Green (CBG) luciferase, connected by a T2A signal peptide (GFP-T2A-CBG), for bioluminescence assays. Normal donor T cells, transduced with 5E5 CAR, CD19 CAR, or not transduced, were cultured with the luminescent Jurkat cells overnight at effector-to-target (E:T) ratios of 0.1:1 to 3:1 and the remaining luminescence was measured. 5E5 CAR T cells exhibited potent cytotoxicity against Jurkat cells at all E:T ratios, in contrast with non-transduced controls (Figure 3B).
A xenograft model of T cell leukemia was established in immune-compromised NOD-SCID-Gamma (NSG) mice by intravenously injecting 5 × 106 Jurkat GFP-T2A-CBG cells. When the mean bioluminescence of tumors reached ~5 × 107 photons/second 8 days after tumor injection, mice were injected intravenously with non-transduced normal human donor T cells, CD19 CAR T cells, or 5E5 CAR T cells, where all cell populations were 50% CD4 and 50% CD8, and, in transduced groups (CD19 and 5E5), CD4+ T cells were 60% CAR+ and CD8+ T cells were 25% CAR+. Treated mice were imaged weekly for tumor bioluminescence until all control mice had reached pre-approved IACUC morbidity endpoints (Figure S4A). Median survival of mice treated with non-transduced and CD19 CAR T cells was 42 and 37.5 days, respectively, with all control-treated mice dead within 36 days post T cell infusion. In contrast, mice treated with 5E5 CAR T cells had a dose dependent increase in median survival of 63 and 94 days. 5E5 CAR T cell treated mice demonstrated a reduction in tumor growth over all other groups throughout the experiment (Figure 3C). Mice treated with 5E5 CAR T cells at either dose also demonstrated significantly increased survival over all control mice (Figure 3D) (p = 0.0014). As far as we are aware, these are the first results showing control of Jurkat leukemia cells by adoptive transfer, although others have shown in vitro antitumor effects (Katsuhara et al., 2015).
Rescue of COSMC in Jurkat Cells Abolishes Recognition by 5E5 CAR T Cells
To restore COSMC function, Jurkat GFP-T2A-CBG cells were transduced with a lentiviral vector encoding COSMC and CD19 lacking the intracellular domain (CD19t), as a selection marker, separated by a P2A signal peptide (CD19t-P2A-COSMC), and sorted for CD19 expression. Following restoration of COSMC, the isogenic Jurkat cells no longer stained positive with 5E5 mAb, suggesting that the MUC1-Tn antigen is no longer exposed (Figure 4A and Figure S5A). In addition, a reduction in αGalNAc (Tn) residues on the surface was observed, as measured by Vicia Villosa lectin (VVA) binding (Figure S5B). The T synthase assay showed enzyme activity in COSMC-restored Jurkat cell extracts was similar to the high T synthase activity observed in 293T cell extracts (Figure 4B), demonstrating that COSMC expression also restored T synthase activity to the Jurkat CD19t-P2A-COSMC cells. Taken together, these results demonstrate that reconstitution of COSMC expression in Jurkat cells restores T synthase activity and eliminates the aberrantly glycosylated Tn-MUC1 antigen that 5E5 mAb recognizes.
Figure 4. Restoration of COSMC Expression Eliminates 5E5 mAb Staining, Increases T Synthase Activity, and Prevents 5E5 CAR T Cell-Induced Cytotoxicity.
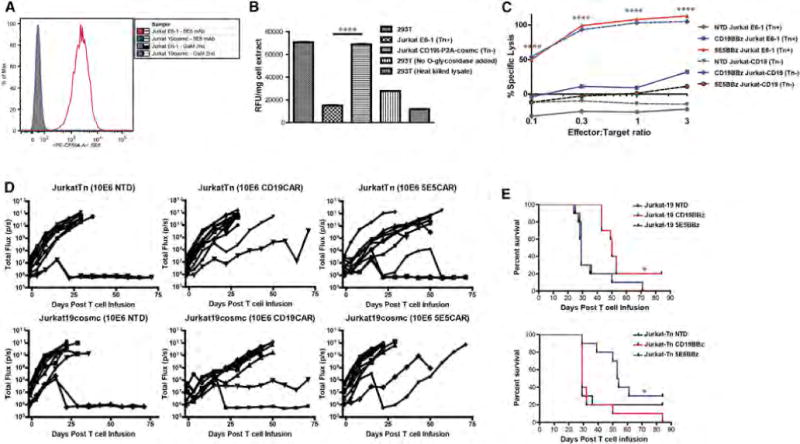
(A) Histogram analysis of GFP+ Jurkat cells and GFP+ Jurkat CD19t-P2A-COSMC cells stained with Goat-anti-mouse-PE alone or 5E5 mAb + GAM-PE. See also Figure S5A.
(B) T synthase activity of cell extracts from 293T, Jurkat cells, and CD19t-P2A-cosmc Jurkat cells using an in vitro fluorescent assay. p < 0.0001. Data are plotted as mean ± SEM.
(C) In vitro cytotoxicity assay of T cells, NTD, CD19BBz, or 5E5BBz, cultured with either Jurkat cells or Jurkat CD19t-P2A-COSMC cells at the indicated effector-to-target ratios. p < 0.0001.
(D) Serial bioluminescence imaging of NSG mice injected intravenously with 5 × 106 CBG+ Jurkat cells or Jurkat CD19t-P2A-COSMC cells and infused with 1 × 107 of the indicated T cells 8 days after tumor engraftment. Each symbol represents one animal. n = 8 animals per group. See also Figure S4B.
(E) Kaplan-Meyer survival curve of mice bearing Jurkat and CD19t-P2A-cosmc+ Jurkat tumors by treatment group. Median survival for Jurkat-19COSMC NTD group = 29, Jurkat-19COSMC CD19 CAR group = 50, and Jurkat-19COSMC 5E5 CAR group = 29 days post T cell infusion. Significance of overall survival for Jurkat-19COSMC experiment p = 0.0117. Median survival of Jurkat-Tn NTD group = 29, Jurkat-Tn CD19 CAR group = 29, and Jurkat-Tn 5E5 CAR group = 53.5 days post T cell infusion. Significance of overall survival for Jurkat-Tn experiment p = 0.0199.
An in vitro cytotoxicity assay of the isogenic Jurkat cells cultured with non-transduced, CD19 CAR, or 5E5 CAR-transduced T cells demonstrated that 5E5 CAR T cells do not exhibit cytotoxicity against Jurkat cells with restored T synthase activity (Figure 4C). However, CD19 CAR T cells were able to lyse the Jurkat CD19t-P2A-COSMC cells, demonstrating that the COSMC restoration does not alter the resistance or susceptibility of Jurkat cells to cytolysis by CAR T cells. Complementary to these single target experiments, 5E5 CAR T cells were able to distinguish Tn-MUC1 positive Jurkat cells (E6-1) from Tn-MUC1 negative Jurkat cells (CD19t-P2A-COSMC) within the same well (Figure S5C), demonstrating that the antigen is not transmitted to other cells by shedding or other means and suggesting that CAR T cells directed against MUC1 might have low bystander toxicity when differential O-glycosylation is targeted.
Xenograft models using Jurkat and Jurkat CD19t-P2A-COSMC cells also demonstrated that 5E5 CAR cytotoxicity is specific to defective O-linked glycosylation caused by COSMC-deficiency. Mice receiving wild-type Jurkat (Tn-MUC1 positive) cells and treated with 5E5 CAR T cells demonstrated increased survival over control treated mice, and likewise, mice receiving Jurkat CD19t-P2A-COSMC (Tn-MUC1 negative) cells treated with CD19 CAR T cells also showed anti-tumor recognition and increased survival over control treated groups (Figures 4D and 4E, Figure S4B). Immunohistochemical analysis of bone marrow, spleen, pancreas, liver, and kidney from mice bearing no tumor, Jurkat E6-1 tumor, or Jurkat CD19t-P2A-COSMC tumor revealed that human T cells home to the bone marrow and spleen, regardless of CAR or tumor, and 5E5 CAR T cells are not found within murine normal tissue (Figure S4C). Importantly, 5E5 CAR T cells had no effect on mice with COSMC-restored Jurkat tumors and similarly for CD19 CAR T cells within mice bearing Jurkat E6-1 tumors. These unresponsive treatment groups showed an abundance of GFP+ Jurkat cells within the bone marrow, highlighting the inability of the T cells to eradicate the tumor in these cases.
5E5 CAR T Cells Recognize Multiple Different Cancers
5E5 CAR T cells, along with CD19 CAR T cells and non-transduced T cells were cultured with a panel of human leukemia, pancreatic, and breast cancer cell lines (Figure 5A). 5E5 CAR T cells secreted IFN-γ when cultured with two of the leukemia lines tested, the pancreatic cancer cell lines Capan-2 and Hs766T, and the breast cancer cell lines MDA-MB-453 and MCF7. However, not all tumor cells tested stimulated 5E5 CAR T cells, perhaps due to checkpoint expression or lack of antigen expression. Panc1, for instance, does not express MUC1. The O-glycophenotype of the cancer cell lines was interrogated with immunostaining and quantitative PCR (Figures 5B and 5C). Staining by 5E5 mAb, 3F1 mAb, and VVA lectin, as well as relative expression of ST6GalNAc1, necessary for formation of STn antigen, correlated with stimulation of 5E5 CAR T cells. The lack of response by the CD19 CAR T cells indicates that the allogeneic effect was low under these conditions.
Figure 5. 5E5 CAR T Cells Are Reactive against Multiple Tumor Histotypes, which Can Be Predicted from O-Glycophenotyping and O-Glycotransferase Expression.
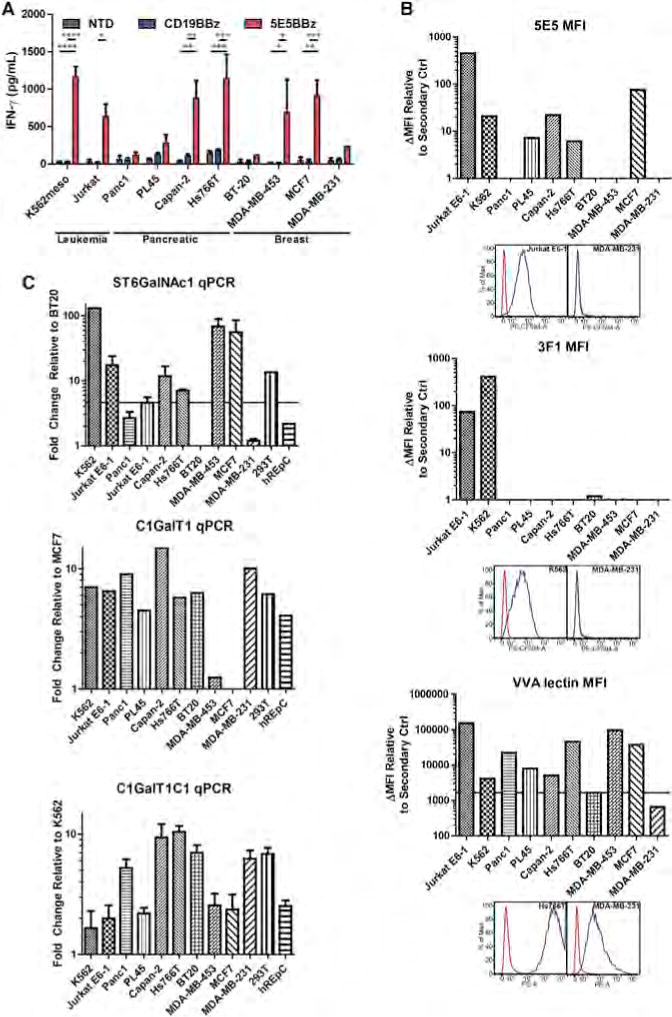
(A) Indirect ELISA quantifying T cell (NTD or transduced with the indicated CAR) secretion of IFN-γ during 24 hr culture with the indicated tumor cell lines. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001. Data are plotted as mean ± SEM.
(B) Cancer cell lines from A were immunostained with 5E5 mAb, 3F1 mAb, and VVA lectin and flow cytometric analysis was performed. Representative histograms of positive and negative cell lines are presented below each MFI plot. The ΔMFI was calculated by adjusting for MFI of secondary antibody stained cells and plotted accordingly. Cell lines stained brightly for 5E5, 3F1, or VVA lectin are likely to generate an immunostimulatory reaction from 5E5 CAR T cells. A caveat to this finding is the high VVA expression of Panc1 does not correlate with Figure 5A, yet Panc1 is known to not expression MUC1.
(C) Relative quantitative PCR was performed on cDNA prepared from the cell lines evaluated in (A). Expression of ST6GalNAc1 mRNA correlated with cytokine secretion and reactivity of 5E5 CAR T cells to these cell lines. Expression of C1GalT1 (T synthase) was also severely reduced in breast cancer cell lines MDA-MB-453 and MCF7. T synthase is necessary to convert O-linked Tn antigens into Core 1 glycans. Also, low expression of C1GalT1C1 (Cosmc) mRNA is observed in 5E5 CAR-stimulating leukemia and breast cancer cell lines. Data are plotted as mean ± SEM.
Using the pancreatic cancer cell line Hs766T, we established a disseminated tumor xenograft model by injecting mice intraperitoneally with 1 × 105 luciferase-expressing tumor cells. Tumor engrafted in many areas of the peritoneal cavity, including in the mouse pancreas. Three weeks post tumor engraftment, when the mean tumor bioluminescence reached approximately ~5 × 107 photons/second, mice were treated intravenously with PBS or non-transduced T cells, CD19 CAR or 5E5 CAR T cells. The injected T cells were 50% CD4+ and 50% CD8+ and CD4+ T cells were 60% CAR+ and CD8 T cells were 25% CAR+. The 5E5 CAR T cells had a potent antitumor effect as mice treated with 5E5 CAR T cells survived or 113 days post T cell infusion, at the termination of the experiment (Figure 6A, 6B, 6C). The 5E5 CAR treated mice had 100% survival, compared with 40% and 33% survival of mice treated with non-transduced and CD19 CAR T cells, respectively (p < 0.02). Immunohistochemical analysis demonstrated that 5E5 CAR T cells accumulate abundantly within the Hs766T tumor, while only small quantities of CD19 CAR T cells are found within the tumor (Figure 6D).
Figure 6. 5E5 CAR T Cell-Treated Mice Exhibited Superior Tumor Rejection and Prolonged Survival against Disseminated Pancreatic Cancer.
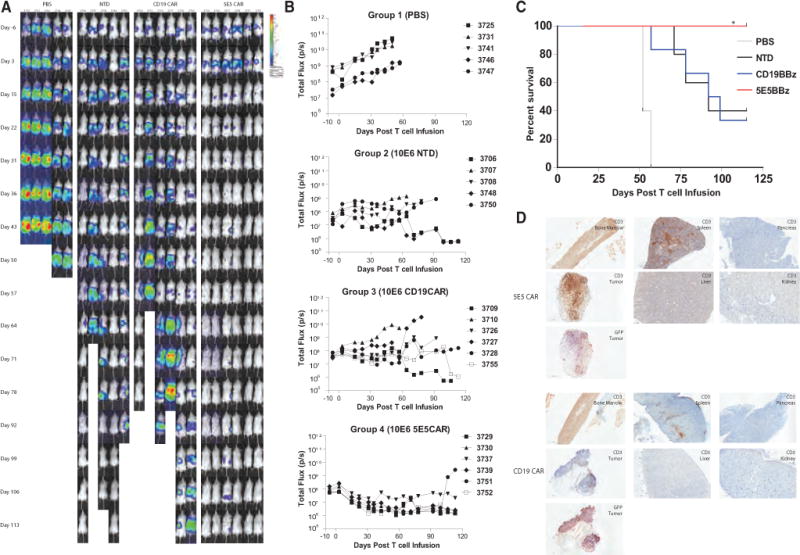
(A) Mice were injected with 1 × 105 CBG+ Hs766T cells and imaged 6 days prior to T cell infusion and serially after treatment until 113 days post T cell infusion.
(B) Serial bioluminescence of NSG mice injected intraperitoneally with 1 × 105 CBG+ Hs766T metastatic pancreatic cancer cell line and infused with PBS or 106 of the indicated T cells when the mean tumor bioluminescence was in the 107–108 p/s range.
(C) Kaplan-Meyer survival curve of mice bearing the Hs766T tumor by treatment group (N = 6 mice per CD19 CAR and 5E5 CAR groups, N = 5 mice per PBS and NTD groups). Median survival of CD19 CAR group is 95.5 days and undefined for 5E5 CAR group. Significance of survival between CD19 CAR and 5E5 CAR group is p = 0.0183.
(D) Immunohistochemistry of bone marrow, spleen, pancreas, liver, kidney, and tumor from mice bearing Hs766T tumor subcutaneously and treated with CD19 CAR or 5E5 CAR. Anti-human CD3 staining revealed human T cells in the bone marrow and spleen of all mice. Tumors from mice treated with 5E5 CAR T cells contained significantly more T cells than mice treated with CD19 CAR T cells.
DISCUSSION
Our studies demonstrate that a CAR targeting a common tumor aberrant O-glycosylation site, the Tn and STn antigen on MUC1, is able to specifically recognize multiple types of tumors in vitro, and demonstrates anti-tumor efficacy in murine models of cancers as diverse as leukemia and pancreatic cancer. A variety of experimental approaches, including isogenic Jurkat cells, indicate that the CAR is specific for the Tn-MUC1 epitope. Loss of COSMC expression is common among malignancies, as demonstrated in The Cancer Genome Atlas, with epigenetic silencing of COSMC found more commonly than genomic mutations (Mi et al., 2012; Radhakrishnan et al., 2014).
Perhaps the greatest challenge to the field of engineered T cell therapy has been the identification of a “universal” CAR. We found that the 5E5 CAR T cells recognized and exhibited cytotoxicity against the Jurkat T cell leukemia cell line, the K562 myelogenous leukemia cell line, pancreatic cancer cell lines, and breast cancer cell lines. The 5E5 mAb has also been reported to stain multiple myeloma and ovarian cancer (Andrulis et al., 2014; Cloosen et al., 2006; Van Elssen et al., 2010), suggesting that the 5E5 CAR could have widespread use for a variety of cancers.
A previous study showed that the 237 CAR, which recognizes a murine tumor-specific glycopeptide epitope (Schietinger et al., 2006), mediated T cell lysis of Jurkat cells (Stone et al., 2012). In that case, recognition was proposed to be due to expression of the Tn antigen linked to a protein distinct from the original 237-target antigen, a glycosylated peptide from the murine OTS8 protein. Importantly, normal primary T cells are COSMC-proficient and therefore do not express Tn-MUC1 recognized by the 5E5 mAb or CAR.
A previous CAR T cell trial targeting carboxy anhydrase IX, an enzyme overexpressed in kidney cancer uncovered low-level expression of the target in normal biliary tree, resulting in serious liver toxicity (Lamers et al., 2013). We were unable to detect expression of Tn-MUC1 epitope on primary cells from healthy donors. While there was staining of some organs by the 5E5 mab in tissue microarrays, confocal microscopy indicates that the staining is intracellular and colocalizes with the Golgi complex. Whether small amounts of surface expression of the 5E5 epitope occurs in non-cancerous tissues is unknown and can only be determined in a clinical trial.
The occurrence of an autoimmune syndrome upon somatic loss of COSMC in hematopoietic stem cells further suggests that the Tn epitope is not expressed in healthy adults, and is reassuring because the syndrome is rare (Berger, 1999). This combined with the observations that Tn is often overexpressed in tumors suggest that targeting of Tn with CAR T cells has promise for cancer therapy (Tarp and Clausen, 2008). The potential of CAR T cells engineered to target aberrantly glycosylated cancer-specific proteins might provide a safer alternative than targeting shared, overexpressed tumor antigens. In summary, our results provide rationale for a clinical trial to target solid tumors that express Tn-MUC1 with CAR T cells.
EXPERIMENTAL PROCEDURES
Vector Design
The cDNA sequence of the variable light and heavy chains (scFv) of the 5E5 mAb (Sørensen et al., 2006) were separated by a 3xGGGGS linker and custom synthesized by GeneArt (Life Technologies). The scFv was cloned into a CAR-encoding lentivirus backbone, containing the CD8a leader sequence, a portion of the CD8α extracellular domain and transmembrane domain, and 4-1BB and CD3zeta endodomains (5E5-41BB-CD3z) in the pTRPE vector: a custom synthesized third generation lentiviral production vector using the EF1α promoter based on the similar pELNS vector as previously described (Carpenito et al., 2009). The genomic DNA of Cosmc was amplified from primary human T cells with a forward primer containing the P2A sequence, and then cloned into pTRPE. cDNA of CD19, truncated after the transmembrane domain as previously described (Milone et al., 2009), was amplified and cloned into the pTRPE-P2A-cosmc vector to create pTRPE-CD19t-P2A-COSMC.
T Cell Transduction and Expansion
Lentiviral supernatant was generated from 293T cells transfected with pTRPE-5E5 scFv-41BB-CD3z, pELPS-anti-CD19-41BB-CD3z, or pELNS-SS1 scFv-41BB-CD3z plasmids, plus gag/pol, env, and Vsvg as previously described (Parry et al., 2003) and collected at 24 and 48 hr. Normal donor T cells were positively selected from leukapheresis products using anti-CD4 and anti-CD8 microbeads (Miltenyi), expanded in vitro with anti-CD3/CD28 magnetic beads (Life Technologies), cultured in RPMI-1640 supplemented with 10% fetal calf serum (FCS), HEPES, penicillin and streptomycin (R10) and 100 IU/mL IL-2 for 10–17 days. T cells were transduced with lentiviral supernatant 16 hr after bead stimulation.
Cells
The Jurkat E6-1 and K562 cell lines were obtained from ATCC and maintained in R10. The Capan-2, Panc-1, PL45, Hs766T, BT-20, MCF-7, MDA-MB-231, and MDA-MB-453 cell lines were also obtained from ATCC and maintained in DMEM supplemented with 10% FCS, penicillin and streptomycin. NNP4 primary ovarian cancer cells were obtained for a pleural tap of an ovarian cancer patient at the University of Chicago Hospitals. K562meso was generated through transduction of K562 cell line with lentiviral supernatant containing mesothelin cDNA expressed by the EF1α promoter. Luciferase-expressing cell lines were generated through transduction of the parental cell line with lentiviral supernatant containing Click Beetle Green luciferase-T2A-GFP and sorted for GFP expression on the BD Influx (BD Biosciences). Cell lines expressing COSMC were generated by transducing the parental cell lines with lentiviral supernatant containing CD19t-P2A-cosmc and sorted on the BD Influx for expression of CD19.
Flow Cytometry
Anti-human antibodies were purchased from BioLegend, eBioscience, or Becton Dickinson. The 5E5 mAb was developed as previously described (Sørensen et al., 2006; Tarp et al., 2007). BaGs3 was a gift from Dr. Richard Cummings at Emory University. For staining, cells were washed with phosphate-buffered saline (PBS) containing 2% FCS, and stained on ice. In all analyses, singlets were gated on using FSC-H versus FSC-A and SSC-H versus SSC-A, followed by gating based on forward versus side scatter characteristics. Surface expression of CD19, SS1, and 5E5 CAR was detected by staining with a biotin-conjugated goat anti-mouse F(ab)2 antibody (Jackson ImmunoResearch) and PE-conjugated streptavidin. Flow cytometry was performed on a 4-laser LSRII or LSRFortessa (Becton Dickinson).
Cytotoxicity Assay
Cytotoxicity assays were performed using luciferase-expressing cell lines. T cells were incubated with cancer cell lines at indicated effector-target ratios for 16 hr. Cells were then washed once in PBS, lysed in luciferase cell culture lysis reagent (Promega), and subsequently mixed with luciferase assay reagent (Promega). Luminescence of the lysates was analyzed using a plate spectrophotometer. Specific lysis of each sample was calculated using the luminescence of target cells alone, and Triton-X lysed target cells, corresponding to 0% lysis and 100% lysis respectively.
Cytotoxicity of CAR T cells cultured with normal human primary cells was evaluated in a 4 hr 51Cr release assay. One million target cells were labeled with 51Cr (50 mCi) for 1 hr at 37°C. After labeling, 1 × 104 labeled target cells were plated in each well of a 96-well plate and effector cells were added in a volume of 100 μl at indicated E:T ratios. Cells were co- incubated for 4 hr at 37°C and 30 μl of supernatant was collected and transferred onto the filter of a LumaPlate. Radioactivity on dried LumaPlate was measured using a β-emission reading liquid scintillation counter and the percentage specific lysis was determined as mentioned above.
Intracellular Cytokine Staining
Human T cells, CAR transduced or non-transduced, were co-cultured with target cells (tumors, cell lines, or human primary cells) in a 1:1 ratio, at 2 × 106 cells/mL in 96-well round bottom tissue-culture plates at 37 degrees, 5% CO2 for 16 hr, in RPMI-1640 plus 10% FBS, in the presence of CD107a mAb and golgi-inhibitors monensin and brefeldinA. Cells were washed, stained with live/dead viability stain, followed by surface staining for CD3 and CD8, then fixed, permeabilized, and intracellularly stained for IFN-γ, TNF-α, IL-2, and GzmB. Cells were analyzed by 9-color flow cytometry (Beckton Dickinson LSRFortessa), and gated on live, single-cell lymphocytes, and CD3-positive lymphocytes.
Immunofluorescence Microscopy
Formalin fixed normal human kidney, normal human pancreas, and human breast cancer were obtained from the Tumor Tissue and Biospecimen Bank at the Hospital of the University of Pennsylvania. The tissue was paraffin-embedded, sectioned, and immunostained with antibodies to Tn-MUC1 5E5 mAb, anti-MUC1 HMFG1 mAb (Abcam), rabbit anti-human EpCAM (Abcam), and rabbit anti-human cis-Golgi GM130 (Abcam). The sections were stained with secondary anti-mouse and anti-rabbit Ig and imaged on a Leica SP8 confocal microscope.
Cytokine Secretion
Human T cell effectors were incubated with target cells at a 2:1 ratio in R10 for 16 hr. Supernatant was analyzed for IFN-γ using the Human IFNγ DuoSet ELISA (R&D Systems). Effector cells were also cultured on glycopeptide-coated plates for 24 hr and the supernatant was analyzed for IFN-γ or IL-2 using the Human IL-2 DuoSet ELISA (R&D Systems).
Peptide ELISA
Synthetic glycopeptides were prepared as previously described (Tarp et al., 2007). Lyophilized peptides were resuspended in PBS and coated on 96-well plates overnight at 4°C at the indicated concentrations. Plates were blocked with 10 mg/mL BSA in PBS-Tween for 1 hr at 37°C, incubated with primary antibody for 1 hr, followed by HRP-conjugated anti-mouse IgG secondary antibody for 30 min, then developed with TMB substrate solution (Pierce) for 20 min, and the reaction was stopped with 2N H2SO4. The absorbance was read at 450 nm using a plate spectrophotometer.
Xenograft Models
NSG mice were provided by the University of Pennsylvania Stem Cell & Xenograft Core. Jurkat and Jurkat CD19-P2A-COSMC xenograft models were established by injecting 5 million luciferase-expressing cells in 100 μl of PBS in the lateral tail vein. Hs766T xenograft model was established by injection of 100,000 luciferase-expressing cells in 100 μl PBS intraperitoneally. When the mean total flux of the tumor was between 107 and 108 photos/sec, 10 million 5E5 CAR, CD19 CAR, or NTD T cells in 100 μl of PBS, were injected once in the lateral tail vein. Bioluminescence was measured through serial weekly imaging on a Xenogen IVIS-200 Spectrum camera. Mice were euthanized when moribund or upon the development of hind-limb paralysis or accumulated abdominal ascites. All experiments were performed via protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
T Synthase Assay
Cell lines were lysed in 1X RIPA (Cell Signaling) supplemented with phenylmethanesulfonylfluoride (PMSF) and the protein concentration was quantified by the DC Protein Assay (BioRad). The T synthase activity of the cell lines was assessed according to the protocol by Ju et al. (2011). Briefly, 30 μg of protein extract was incubated with GalNAc-4MU, UDP-galactose, MnCl2, and O-glycosidase for one hour at 37°C. The reaction was stopped with the addition of 1 M glycine-NaOH pH 10 and the subsequent fluorescence of each sample was measured at excitation of 355 nm and emission of 460 nm on the plate spectrophotometer.
Supplementary Material
In Brief.
Posey and colleagues developed a CAR T cell therapy to break immune tolerance to solid tumors by targeting an aberrantly glycosylated, cancer-specific glycoprotein in multiple cancer histotypes and demonstrated efficacy and safety in tumors as diverse as leukemia and pancreatic cancer.
Highlights.
Cancer cells of many tissues express an abnormal glycoform of MUC1, Tn-MUC1
Normal human tissue does not express detectable Tn-MUC1 on the cellular surface
CAR T cells are engineered to target Tn-MUC1 lyse tumor cells in vitro and in vivo
Abnormal glycoform epitopes are valid clinical targets for CAR T cells
Acknowledgments
We are grateful for advice from Michael Milone and Robert Vonderheide, the Perelman School of Medicine Stem Cell & Xenograft Core, the Human Immunology Core, the Cancer Histology Core, and for staff in the animal facility for assistance with experiments. Supported in part by a grant from Novartis and National Institutes of Health (5R01CA120409, C.H.J.; 5R01CA037156, H.S. and H.C.; and DP2 CA174502, L.A.J.), The Danish Research Councils (DFF – 4004-00397B, C.S.), and The Danish National Research Foundation (DNRF107, H.C.). A.D.P. and C.H.J. are members of the Parker Institute for Cancer Immunotherapy, which supported the University of Pennsylvania Cancer Immunotherapy Program. The University of Copenhagen has patented the 5E5 antibody and antigen epitope. The University of Chicago has filed a patent on the 5E5 CAR and an invention disclosure has been filed on these studies. The University of Pennsylvania has entered into a strategic alliance with Novartis for the development of chimeric antigen receptors.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, A.D.P., H.S., H.C., and C.H.J.; Methodology, A.D.P.; Investigation, R.D.S., A.C.B., U.M., C.S., M.D.F., A.P.C., T.J.C., D.S., J.S., T.D.M., B.E., K.S., and J.D.S; Writing – Original Draft, A.D.P.; Writing – Review & Editing, A.D.P., R.D.S., A.C.B., B.E., D.M.K., R.Y., B.K., H.S., H.C., L.A.J., and C.H.J.; Funding Acquisition, L.A.J., H.S., H.C., C.S., and C.H.J.; Resources, C.S., U.M., H.C., K.S., and H.S.; Supervision, L.A.J. and C.H.J.
This arrangement is managed in accordance with the University of Pennsylvania’s Conflict of Interest Policy. The authors are in compliance with this policy.
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2016.05.014.
References
- Andrulis M, Ellert E, Mandel U, Clausen H, Lehners N, Raab MS, Goldschmidt H, Schwartz-Albiez R. Expression of Mucin-1 in multiple myeloma and its precursors: correlation with glycosylation and sub-cellular localization. Histopathology. 2014;64:799–806. doi: 10.1111/his.12330. [DOI] [PubMed] [Google Scholar]
- Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, Tabak LA. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology. 2012;22:736–756. doi: 10.1093/glycob/cwr182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger EG. Tn-syndrome. Biochim Biophys Acta. 1999;1455:255–268. doi: 10.1016/s0925-4439(99)00069-1. [DOI] [PubMed] [Google Scholar]
- Borgert A, Heimburg-Molinaro J, Song X, Lasanajak Y, Ju T, Liu M, Thompson P, Ragupathi G, Barany G, Smith DF, et al. Deciphering structural elements of mucin glycoprotein recognition. ACS Chem Biol. 2012;7:1031–1039. doi: 10.1021/cb300076s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazet A, Julien S, Bobowski M, Burchell J, Delannoy P. Tumour-associated carbohydrate antigens in breast cancer. Breast Cancer Res. 2010;12:204. doi: 10.1186/bcr2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloosen S, Gratama J, van Leeuwen EB, Senden-Gijsbers BL, Oving EB, von Mensdorff-Pouilly S, Tarp MA, Mandel U, Clausen H, Germeraad WT, Bos GM. Cancer specific Mucin-1 glycoforms are expressed on multiple myeloma. Br J Haematol. 2006;135:513–516. doi: 10.1111/j.1365-2141.2006.06331.x. [DOI] [PubMed] [Google Scholar]
- Emens LA, Asquith JM, Leatherman JM, Kobrin BJ, Petrik S, Laiko M, Levi J, Daphtary MM, Biedrzycki B, Wolff AC, et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: a chemotherapy dose-ranging factorial study of safety and immune activation. J Clin Oncol. 2009;27:5911–5918. doi: 10.1200/JCO.2009.23.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn OJ, Gantt KR, Lepisto AJ, Pejawar-Gaddy S, Xue J, Beatty PL. Importance of MUC1 and spontaneous mouse tumor models for understanding the immunobiology of human adenocarcinomas. Immunol Res. 2011;50:261–268. doi: 10.1007/s12026-011-8214-1. [DOI] [PubMed] [Google Scholar]
- Gill DJ, Tham KM, Chia J, Wang SC, Steentoft C, Clausen H, Bard-Chapeau EA, Bard FA. Initiation of GalNAc-type O-glycosylation in the endoplasmic reticulum promotes cancer cell invasiveness. Proc Natl Acad Sci USA. 2013;110:E3152–E3161. doi: 10.1073/pnas.1305269110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RA, Burchell JM, Taylor-Papadimitriou J. The polymorphic epithelial mucin: potential as an immunogen for a cancer vaccine. Cancer Immunol Immunother. 1996;42:71–80. doi: 10.1007/s002620050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- Itzkowitz S. Colon carcinogenesis in inflammatory bowel disease: applying molecular genetics to clinical practice. J Clin Gastroenterol. 2003;36:S70–S74. doi: 10.1097/00004836-200305001-00012. discussion S94-76. [DOI] [PubMed] [Google Scholar]
- Ju T, Cummings RD. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc Natl Acad Sci USA. 2002;99:16613–16618. doi: 10.1073/pnas.262438199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju T, Cummings RD. Protein glycosylation: chaperone mutation in Tn syndrome. Nature. 2005;437:1252. doi: 10.1038/4371252a. [DOI] [PubMed] [Google Scholar]
- Ju T, Cummings RD, Canfield WM. Purification, characterization, and subunit structure of rat core 1 Beta1,3-galactosyltransferase. J Biol Chem. 2002;277:169–177. doi: 10.1074/jbc.M109056200. [DOI] [PubMed] [Google Scholar]
- Ju T, Lanneau GS, Gautam T, Wang Y, Xia B, Stowell SR, Willard MT, Wang W, Xia JY, Zuna RE, et al. Human tumor antigens Tn and sialyl Tn arise from mutations in Cosmc. Cancer Res. 2008;68:1636–1646. doi: 10.1158/0008-5472.CAN-07-2345. [DOI] [PubMed] [Google Scholar]
- Ju T, Xia B, Aryal RP, Wang W, Wang Y, Ding X, Mi R, He M, Cummings RD. A novel fluorescent assay for T-synthase activity. Glycobiology. 2011;21:352–362. doi: 10.1093/glycob/cwq168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: a race to the finish line. Sci Transl Med. 2015;7:280ps7. doi: 10.1126/scitranslmed.aaa3643. [DOI] [PubMed] [Google Scholar]
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells expressing chimeric receptors establish memory and potent antitumor effects in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannagi R, Sakuma K, Miyazaki K, Lim KT, Yusa A, Yin J, Izawa M. Altered expression of glycan genes in cancers induced by epigenetic silencing and tumor hypoxia: clues in the ongoing search for new tumor markers. Cancer Sci. 2010;101:586–593. doi: 10.1111/j.1349-7006.2009.01455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuhara A, Fujiki F, Aoyama N, Tanii S, Morimoto S, Oka Y, Tsuboi A, Nakajima H, Kondo K, Tatsumi N, et al. Transduction of a novel HLA-DRB1*04:05-restricted, WT1-specific TCR gene into human CD4+ T cells confers killing activity against human leukemia cells. Anticancer Res. 2015;35:1251–1261. [PubMed] [Google Scholar]
- Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, den Bakker M, Oosterwijk E, Debets R, Gratama JW. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Ho JY, Hsieh MT, Chiang HL, Chuang JM, Whang-Peng J, Chang YC, Tseng YH, Chen SF, Nieh S, Hwang J. Reciprocal relationship of Tn/NF-κB and sTn as an indicator of the prognosis of oral squamous cell carcinoma. Histopathology. 2014;64:713–721. doi: 10.1111/his.12309. [DOI] [PubMed] [Google Scholar]
- Madsen CB, Lavrsen K, Steentoft C, Vester-Christensen MB, Clausen H, Wandall HH, Pedersen AE. Glycan elongation beyond the mucin associated Tn antigen protects tumor cells from immune-mediated killing. PLoS ONE. 2013;8:e72413. doi: 10.1371/journal.pone.0072413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi R, Song L, Wang Y, Ding X, Zeng J, Lehoux S, Aryal RP, Wang J, Crew VK, van Die I, et al. Epigenetic silencing of the chaperone Cosmc in human leukocytes expressing tn antigen. J Biol Chem. 2012;287:41523–41533. doi: 10.1074/jbc.M112.371989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Molecular therapy: the journal of the American Society of Gene Therapy. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S, Ohno Y, Nakada H, Suzuki N, Soma G, Inoue M. Expression of Tn and sialyl-Tn antigens in endometrial cancer: its relationship with tumor-produced cyclooxygenase-2, tumor-infiltrated lymphocytes and patient prognosis. Anticancer Res. 2006;26(6A):4047–4053. [PubMed] [Google Scholar]
- Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol. 2003;171:166–174. doi: 10.4049/jimmunol.171.1.166. [DOI] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich GA, Croci DO. Regulatory circuits mediated by lectin-glycan interactions in autoimmunity and cancer. Immunity. 2012;36:322–335. doi: 10.1016/j.immuni.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan P, Dabelsteen S, Madsen FB, Francavilla C, Kopp KL, Steentoft C, Vakhrushev SY, Olsen JV, Hansen L, Bennett EP, et al. Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc Natl Acad Sci USA. 2014;111:E4066–E4075. doi: 10.1073/pnas.1406619111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren D, Jia L, Li Y, Gong Y, Liu C, Zhang X, Wang N, Zhao Y. ST6GalNAcII mediates the invasive properties of breast carcinoma through PI3K/Akt/NF-κB signaling pathway. IUBMB Life. 2014;66:300–308. doi: 10.1002/iub.1268. [DOI] [PubMed] [Google Scholar]
- Sabbatini PJ, Ragupathi G, Hood C, Aghajanian CA, Juretzka M, Iasonos A, Hensley ML, Spassova MK, Ouerfelli O, Spriggs DR, et al. Pilot study of a heptavalent vaccine-keyhole limpet hemocyanin conjugate plus QS21 in patients with epithelial ovarian, fallopian tube, or peritoneal cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:4170–4177. doi: 10.1158/1078-0432.CCR-06-2949. [DOI] [PubMed] [Google Scholar]
- Schietinger A, Philip M, Yoshida BA, Azadi P, Liu H, Meredith SC, Schreiber H. A mutant chaperone converts a wild-type protein into a tumor-specific antigen. Science. 2006;314:304–308. doi: 10.1126/science.1129200. [DOI] [PubMed] [Google Scholar]
- Sørensen AL, Reis CA, Tarp MA, Mandel U, Ramachandran K, Sankaranarayanan V, Schwientek T, Graham R, Taylor-Papadimitriou J, Hollingsworth MA, et al. Chemoenzymatically synthesized multimeric Tn/STn MUC1 glycopeptides elicit cancer-specific anti-MUC1 antibody responses and override tolerance. Glycobiology. 2006;16:96–107. doi: 10.1093/glycob/cwj044. [DOI] [PubMed] [Google Scholar]
- Springer GF. T and Tn, general carcinoma autoantigens. Science. 1984;224:1198–1206. doi: 10.1126/science.6729450. [DOI] [PubMed] [Google Scholar]
- Stone JD, Aggen DH, Schietinger A, Schreiber H, Kranz DM. A sensitivity scale for targeting T cells with chimeric antigen receptors (CARs) and bispecific T-cell Engagers (BiTEs) OncoImmunology. 2012;1:863–873. doi: 10.4161/onci.20592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura F, Sato Y, Hirakawa M, Yoshida M, Ono M, Osuga T, Okagawa Y, Uemura N, Arihara Y, Murase K, et al. RNAi-mediated gene silencing of ST6GalNAc I suppresses the metastatic potential in gastric cancer cells. Gastric cancer: official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association. 2014 doi: 10.1007/s10120-014-0454-z. [DOI] [PubMed] [Google Scholar]
- Tarp MA, Clausen H. Mucin-type O-glycosylation and its potential use in drug and vaccine development. Biochim Biophys Acta. 2008;1780:546–563. doi: 10.1016/j.bbagen.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Tarp MA, Sørensen AL, Mandel U, Paulsen H, Burchell J, Taylor-Papadimitriou J, Clausen H. Identification of a novel cancer-specific immunodominant glycopeptide epitope in the MUC1 tandem repeat. Glycobiology. 2007;17:197–209. doi: 10.1093/glycob/cwl061. [DOI] [PubMed] [Google Scholar]
- Taylor-Papadimitriou J, Burchell J, Miles DW, Dalziel M. MUC1 and cancer. Biochim Biophys Acta. 1999;1455:301–313. doi: 10.1016/s0925-4439(99)00055-1. [DOI] [PubMed] [Google Scholar]
- Van Elssen CH, Frings PW, Bot FJ, Van de Vijver KK, Huls MB, Meek B, Hupperets P, Germeraad WT, Bos GM. Expression of aberrantly glycosylated Mucin-1 in ovarian cancer. Histopathology. 2010;57:597–606. doi: 10.1111/j.1365-2559.2010.03667.x. [DOI] [PubMed] [Google Scholar]
- Victorzon M, Nordling S, Nilsson O, Roberts PJ, Haglund C. Sialyl Tn antigen is an independent predictor of outcome in patients with gastric cancer. International journal of cancer. Journal international du cancer. 1996;65:295–300. doi: 10.1002/(SICI)1097-0215(19960126)65:3<295::AID-IJC3>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Wandall HH, Blixt O, Tarp MA, Pedersen JW, Bennett EP, Mandel U, Ragupathi G, Livingston PO, Hollingsworth MA, Taylor-Papadimitriou J, et al. Cancer biomarkers defined by autoantibody signatures to aberrant O-glycopeptide epitopes. Cancer Res. 2010;70:1306–1313. doi: 10.1158/0008-5472.CAN-09-2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.