

ABSTRACT
Reactive oxygen species (ROS) production and oxidative stress (OS) in adipose tissue are associated with obesity and insulin resistance (IR). The nature of this relationship i.e., cause and effect or consequence has not been clearly determined. We provide evidence that elevated mitochondrial ROS generated by adipocytes from mice with diet-induced obesity (DIO) represents an adaptive mechanism that precipitates fatty acid oxidation, mitochondrial biogenesis, and mitochondrial uncoupling in an effort to defend against weight gain. Consistent with that, mice with adipocyte-specific deletion of manganese superoxide dismutase (MnSOD) exhibit increased adipocyte superoxide generation and are protected from weight gain and insulin resistance which otherwise develops in wild-type (WT) mice that consume an obesogenic diet. The defense mechanism displayed by MnSOD-deficiency in fat cells appears to be mediated by a dual effect of ROS on inefficient substrate oxidation through uncoupling of oxidative phosphorylation and enhanced mitochondrial biogenesis. The aim of this commentary is to summarize and contextualize additional evidence supporting the importance of mitochondrial ROS in the regulation of mitochondrial biogenesis and the modulation of uncoupling protein 1 (UCP1) expression and activation in both white and brown adipocytes.
KEYWORDS: mitochondria, brown adipose tissue, MnSOD, oxidative modification, proton leak, superoxide, ROS, Uncoupling protein 1 (UCP1)
Mitochondria are the major sites of substrate oxidation in mammalian cells and a major source of ROS.1-3 Mitochondria can generate superoxide and hydrogen peroxide (H2O2) from at least 11 different oxidoreductases associated with substrate catabolism and the electron transport chain (ETC).4 ROS generation by mitochondria is a tightly controlled process i.e., low levels for cellular signaling are allowed whereas high levels that can damage the cellular milieu are mitigated by endogenous antioxidant enzymes. Importantly, there is substantial uncertainly regarding the different mechanisms controlling increased mitochondrial ROS generation in vivo, as well as the type of ROS species that are relevant for signaling in different aspects of physiology. Superoxide is the short-lived proximal ROS generated by mitochondrial oxidoreductases, and is rapidly converted to H2O2 by manganese superoxide dismutase (MnSOD) in the mitochondrial matrix.5 Superoxide that is generated and released in the mitochondrial intermembrane space by complex III and the enzyme p66Shc is converted to H2O2 by the Cu,ZnSOD enzyme.6-8 Matrix H2O2 is degraded by a glutathione-dependent system catalyzed by glutathione peroxidases and the peroredoxin-thioredoxin system,9 whereas cytosolic degradation of H2O2 is mainly controlled by catalase, cytosolic peroxidases, and peroxiredoxins.10 Distinct from other antioxidant enzymes, MnSOD is important due to its localization in the mitochondrial matrix, and to its high affinity for superoxide in that compartment.5 The physiologic relevance of MnSOD was demonstrated by the robust phenotype of mice lacking the in vitro Sod2 gene (encoding MnSOD). In this regard, Sod2-deficient mice die within 1–18 d from dilated cardiomyopathy and neurodegenerative abnormalities, highlighting the importance of this enzyme in the maintenance of organ function.11
Obesity is a growing epidemic driving a rise in the incidence of type 2 diabetes, cardiovascular diseases and cancer.12 Obesity results from an energy imbalance wherein storage of fat in white adipose tissue (WAT) exceeds energy expenditure. Recent studies have associated systemic OS with obesity-related complications13 as enhanced adipose OS is linked to inflammation, adipokine dysregulation and insulin resistance.14-16 While these studies demonstrated a correlation exists between increased adipose OS and the incidence of obesity and insulin resistance, a cause and effect has not been demonstrated in vivo. Furthermore, the use of knockout or transgenic animals targeting antioxidant enzymes at the whole body level makes it difficult to precisely define the contribution to weight gain from adipose tissue OS per se. In addition, ROS, in the form of H2O2, stimulate adipogenesis in vitro,17 and could explain why fat accumulation is increased in mouse models with whole body deletion of certain antioxidant enzymes.15,16,18,19 Thus, defining the precise role of adipocyte ROS in the regulation of its metabolism and function is crucial for developing novel therapeutic strategies to combat obesity.
AdSod2 KO increases energy expenditure and protects against DIO
To define the specific contribution of mitochondria-generated ROS in adipocytes to the pathogenesis of obesity and insulin resistance, we generated the first mouse model wherein MnSOD could be specifically deleted in adipocytes i.e., AdSod2 KO mice. We originally hypothesized that AdSod2 KO mice would develop obesity due to superoxide-mediated damage to mitochondrial components, leading to reduced substrate oxidation and lipid accumulation in adipocytes. Instead, increased superoxide generation by adipocytes from AdSod2KO vs. WT mice stimulated fatty acid (FA) oxidation and increased mitochondrial biogenesis in adipocytes, and these effects were most prominent in mice that consumed high-fat diet (HFD) vs. standard chow.20 Specifically, enhanced FA oxidation in white adipose tissue (WAT) and brown adipose tissue (BAT) of AdSod2 KO mice increased metabolic rate and energy expenditure and protected the mice from weight gain and insulin resistance that otherwise developed in WT mice that consumed HFD. Elevated energy expenditure observed in AdSod2 KO mice occurred despite BAT atrophy, and is not secondary to shivering-induced thermogenesis. Moreover, energy expenditure was still elevated even when adrenergic input to BAT and shivering-induced thermogenesis were blocked by housing the AdSod2 KO mice at thermoneutrality. Therefore, AdSod2 KO was sufficient to drive increased adipose energy expenditure in the absence of systemic adrenergic cues. Most importantly, when adipose progenitors were isolated from these mice and induced to differentiate in vitro, a marked increase in mitochondrial proton leak was observed in cells isolated from AdSod2 KO vs. WT mice, supporting a cell autonomous effect. This evidence for increased mitochondrial uncoupling in adipocytes from the mutant mice was associated with a robust induction of uncoupling protein 1 (UCP1) expression in WAT and BAT. Collectively, these data clearly demonstrate that enhanced mitochondrial ROS triggers an adaptive mechanism that protects against diet-induced fat accrual through the stimulation of FA oxidation, enhanced mitochondrial proliferation, and the induction of mitochondrial uncoupling (Fig. 1).
Figure 1.
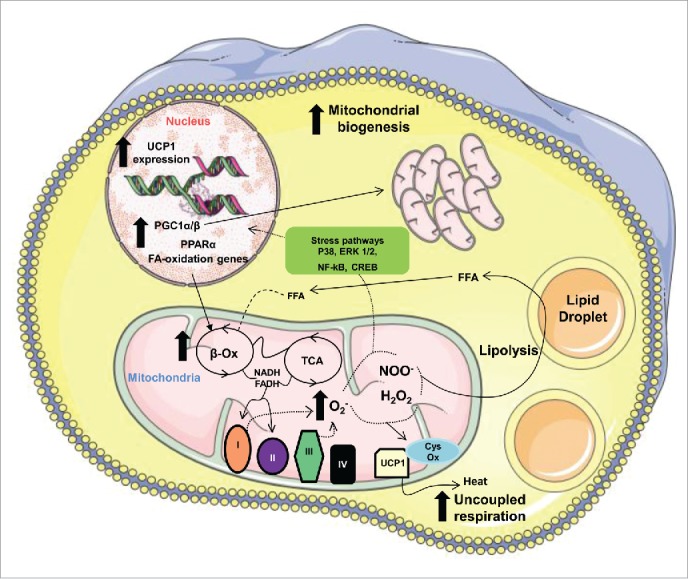
Schematic diagram of the effects of MnSOD deletion in adipocytes. The free fatty acids (FFA) released after lipolysis are oxidized in the mitochondria through an inefficient process involving active UCP1. The inefficient FA oxidation is supported by ROS-mediated transcriptional regulation of mitochondrial biogenesis and UCP1 expression.
Adipocyte OS induced by SOD2 KO signals to drive mitochondrial biogenesis in WAT
Studies in mice have shown that during the development of DIO, there is an early increase in ROS levels in WAT followed by a later induction of mitochondrial biogenesis.21 Mitochondrial biogenesis is orchestrated by a transcriptional cascade involving peroxisome proliferator-activated receptor γ coactivators 1α (PGC-1α), PGC-1β, PGC-1-related coactivator (PRC), estrogen-related receptor α (ERRα), nuclear respiratory factors 1 (NRF1) and 2 (NRF2) and mitochondrial transcription factor A (Tfam).22 Mitochondrial biogenesis was enhanced in WAT of the AdSod2 KO mice and was associated with increased transcription of PGC-1α, PGC-1β and nuclear and mitochondrial-encoded genes, implying a causal role for ROS in the induction of mitochondrial biogenesis. Lack of both PGC-1α and PGC-1β impairs differentiation-induced mitochondria biogenesis and mitochondrial respiration in brown preadipocytes.23 Mice lacking PGC-1α in fat cells revealed a role for this co-activator in the orchestration of the transcription of mitochondrial and FA oxidation genes and the regulation of glucose homeostasis and insulin sensitivity in response to high fat feeding.24 Indeed, during acute or chronic cold acclimation in rodents, an elevation in ROS generation and a shift toward a more oxidized environment was observed in BAT and was associated with mitochondrial proliferation.25-28 Furthermore, agents known to deplete glutathione (GSH) such as buthionine sulfoximine (BSO) increased the expression of PGC-1α both in WAT and in BAT.29 In addition, ROS in the form of H2O2 was shown to directly induce the expression of PGC-1α and PGC-1β in brown-fat fibroblasts through a mechanism involving the Cre-binding protein (CREB).30 Whether the induction of mitochondrial biogenesis observed in WAT of the AdSod2 KO mice is CREB-mediated is unknown, but studies in our laboratory are actively investigating this possibility.
The induction of mitochondrial biogenesis in WAT of AdSod2 KO mice was only observed when the animals were maintained on a high fat diet (HFD). Moreover, while AdSod2 KO appeared to increase basal superoxide levels, HFD was sufficient to drive superoxide higher but to similar extents in both WT and KO mice.20 However, it is unknown if the levels of other ROS species such as H2O2 or peroxynitrite, previously shown to drive mitochondrial proliferation.30,31 are different in AdSod2 KO vs. WT mice on HFD, and this is a topic for future study. Furthermore, the lack of difference in superoxide levels in WAT between KO and WT mice fed HFD could be explained by the higher mitochondrial uncoupling capacity and ROS mitigation in the KO mice. Further studies using mitochondrial-targeted antioxidant, and mitochondrial uncoupling inhibitors, and redox proteomic methods32 are required to define the role of ROS in the promotion of mitochondrial biogenesis in AdSod2 KO mice.
Another important finding in the AdSod2 KO mouse model is the induction of UCP1 expression both in WAT and BAT, and the increase in mitochondrial uncoupling in WAT in mice that consumed HFD. Interestingly, despite elevated UCP1 expression in BAT, AdSod2 KO mice were cold-intolerant, probably due to the significant atrophy and loss of lipid stores in this depot. The mechanisms underlying BAT atrophy in AdSod2 KO mice are not known, but similar finding was reported in the aP2-Ucp transgenic mice.33,34 Despite the atrophy of BAT, UCP1 content was preserved in aP2-Ucp transgenic mice and increased in AdSod2 KO mice, suggesting that atrophy is caused by a reduction in the total number of cells or a depletion in the lipid stores. In the AdSod2 KO model, the reduction in cellularity in BAT could be caused by reduced proliferation or increased cell death as a result of excessive UCP1-mediated mitochondrial uncoupling and a depletion of ATP necessary for these processes. UCP1 induction in WAT of AdSod2 KO mice occurred independently of the diet and does not correlate with the induction of mitochondrial biogenesis, suggesting ROS as the underlying trigger. Indeed, H2O2 treatment induced UCP1 expression in brown-fat fibroblasts even when PGC-1α was ablated, thus separating UCP1 induction from mitochondrial biogenesis.30 Moreover, the peroxisome proliferator-activated receptor γ (PPAR γ) agonist rosiglitazone was shown to induce UCP1 expression along with mitochondrial biogenesis in WAT, but the abundance of UCP1 mRNA was greater than any mitochondrial transcripts, indicating specific activation of this gene.35 Consistent with this idea, recent studies demonstrated that ROS are required for both the induction of UCP1 expression in BAT and WAT, as well as increased UCP1-dependent respiration. In this regard, overexpression of the antioxidant molecule Sestrin2 resulted in a reduction of UCP1 expression in BAT, an effect similar to the use of the compounds that modulate cellular redox status such as butylated hydroxyanisole (BHA) and N-Acetylcysteine (NAC) in mice.36 In this study, it was shown that cold-induced ROS-mediated UCP1 expression in BAT was mediated through p38 mitogen-activated protein kinase (p38MAPK) pathway. Moreover, mice deficient in nuclear factor-erythroid 2-related factor 2 (NRF2), which controls expression of antioxidant genes, display a 2-fold induction of Ucp1 mRNA in WAT and in isolated fibroblasts vs. WT mice, and this effect is mitigated when mutant cells are treated with antioxidants.18 Surprisingly, MnSOD deficiency in skin tissue, which enhanced superoxide levels, also induced UCP1 expression through a PPARα-dependent mechanism.37 On balance, when our data are taken together with what currently is known in the literature, it appears that a common ROS-dependent mechanism for the induction of UCP1 expression exists in WAT and BAT. Future investigations from our laboratory will define the signaling pathways involved in the induction of UCP1 transcription in both WAT and BAT of the AdSod2 KO mice.
AdSOD2 deletion increases uncoupled respiration in cells and in vivo
In addition to the induction of UCP1 transcription in WAT and BAT of AdSod2 KO mice, we showed that this resulted in increased energy expenditure at both the whole body and adipocyte-autonomous level. Specifically, HF-fed but not chow-fed AdSod2 KO mice exhibited elevation in energy expenditure in vivo, enhanced mitochondrial uncoupling, and increased leak respiration in isolated preadipocytes in vitro. In interpreting these findings it is important to note that increased UCP1 gene and protein expression alone are not sufficient to explain increased energy expenditure in the AdSod2KO model. Although UCP1 facilitates uncoupled respiration, in the cellular milieu, it is maintained in a purine-nucleotide bound state,38 which in the absence of FA renders it inactive.39 So, current evidence suggests that in the native mitochondrial and adipocyte environment, UCP1 does not drive uncoupled respiration basally, instead requiring activation (for example by adrenergic stimulus or increased local FA). Therefore, diet-induced initiation of mitochondrial uncoupling in Sod2 KO adipocytes presumably involves modulation in the activation status of UCP1 and UCP1-dependent respiration. This is further supported by the observation that, in AdSod2 KO adipocytes, leak respiration specifically is enhanced while chemically uncoupled maximal respiration is unaffected. So, enhancement of respiration is attributable to a specific effect on the ATP synthase uncoupled component.
How OS stimulates UCP1-dependent respiration in AdSod2 KO mice is an outstanding question. More generally, the role of ROS and related redox-active signaling modalities in regulation of UCP1 activity has been the subject of longstanding studies. The importance of superoxide.38,40-42 and other redox-active species (e.g. 4-HNE43,44) in modulating UCP1 function is the subject of debate. Relatedly, a role for thiol redox status (which depends on local ROS modulation) has been suggested in BAT mitochondrial UCP1 uncoupling.26 Studies on the role of OS and UCP1 activation have relied largely on the use of isolated mitochondria and reconstituted in vitro systems. Therefore, the extent to which physiologically relevant ROS production, redox tone, and local UCP1 environment are faithfully recapitulated is unclear. An advantage of the AdSOD2 KO model is that OS-dependent effects on leak respiration and UCP1-dependent thermogenesis are monitored under native conditions (both in adipocytes and in vivo).
Indeed, recent observations have suggested that redox regulation of UCP1-dependent respiration is particularly relevant when studied in adipocytes and in vivo where both redox homeostasis and free fatty-acid concentrations are subject to strict endogenous regulation; parameters that are necessarily divergent from in vitro conditions. Specifically, a role was recently demonstrated for mitochondrial ROS induction and protein thiol redox tone in supporting UCP1-dependent thermogenesis upon adrenergic stimulus in vivo.28 In this study, the role of mitochondrial ROS in supporting increased energy expenditure was shown to genetically require UCP1 in vivo and in brown adipocytes, and involve direct modification by ROS of a functional cysteine residue (Cys253) on UCP1. Interestingly, mutagenesis of this redox-sensitive site on UCP1 renders it less sensitive to acute activation by adrenergic stimulus in adipocytes, suggesting that this ROS-modified site acts as a rheostat to tune UCP1 activity. More generally, this study demonstrated that numerous BAT mitochondrial metabolic enzymes are similarly modified by ROS in vivo during thermogenesis and could be similarly functionally regulated by these modifications. It would therefore be of great interest to examine ROS-mediated post-translational modifications of UCP1 and other metabolic targets in genetic models of increased OS such as the AdSod2 KO model, to determine whether these modifications play a role in driving increased energy expenditure. More generally, the AdSod2KO model now provides an elegant system with which to examine the molecular mechanisms of ROS-mediated activation of thermogenic gene programs and active thermogenic respiration on a cellular and organismal level.
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases grants 1-R01-DK-098646–01A1 to SB and by the Human Frontiers Science Foundation to ETC.
References
- [1].Loschen G, Flohe L, Chance B. Respiratory chain linked H(2)O(2) production in pigeon heart mitochondria. FEBS Lett 1971; 18:261-4; PMID:11946135; http://dx.doi.org/ 10.1016/0014-5793(71)80459-3 [DOI] [PubMed] [Google Scholar]
- [2].Loschen G, Azzi A, Richter C, Flohe L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett 1974; 42:68-72; PMID:4859511; http://dx.doi.org/ 10.1016/0014-5793(74)80281-4 [DOI] [PubMed] [Google Scholar]
- [3].Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J 1972; 128:617-30; PMID:4404507; http://dx.doi.org/ 10.1042/bj1280617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 2016; 100:14-31; PMID:27085844. [DOI] [PubMed] [Google Scholar]
- [5].McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 1969; 244:6049-55; PMID:5389100 [PubMed] [Google Scholar]
- [6].St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 2002; 277:44784-90; PMID:12237311; http://dx.doi.org/ 10.1074/jbc.M207217200 [DOI] [PubMed] [Google Scholar]
- [7].Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, C Contursi, Pelliccia G, Luzi L, Minucci S, Marcaccio M, et al.. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005; 122:221-33; PMID:16051147; http://dx.doi.org/ 10.1016/j.cell.2005.05.011 [DOI] [PubMed] [Google Scholar]
- [8].Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem 2001; 276:38388-93; PMID:11507097; http://dx.doi.org/ 10.1074/jbc.M105395200 [DOI] [PubMed] [Google Scholar]
- [9].Collins Y, Chouchani ET, James AM, Menger KE, Cocheme HM, Murphy MP. Mitochondrial redox signalling at a glance. J Cell Sci 2012; 125:801-6; PMID:22448036; http://dx.doi.org/ 10.1242/jcs.098475 [DOI] [PubMed] [Google Scholar]
- [10].Andriantsitohaina R, Duluc L, Garcia-Rodriguez JC, Gil-del Valle L, Guevara-Garcia M, Simard G, Soleti R, Su DF, Velásquez-Pérez L, Wilson JX, et al.. Systems biology of antioxidants. Clin Sci (Lond) 2012; 123:173-92; PMID:22494160; http://dx.doi.org/ 10.1042/CS20110643 [DOI] [PubMed] [Google Scholar]
- [11].Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, et al.. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 1995; 11:376-81; PMID:7493016; http://dx.doi.org/ 10.1038/ng1295-376 [DOI] [PubMed] [Google Scholar]
- [12].Smyth S, Heron A. Diabetes and obesity: the twin epidemics. Nat Med 2006; 12:75-80; PMID:16397575; http://dx.doi.org/ 10.1038/nm0106-75 [DOI] [PubMed] [Google Scholar]
- [13].Keaney JF Jr., Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, Massaro JM, Sutherland P, Vita JA, Benjamin EJ, et al.. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol 2003; 23:434-9; PMID:12615693; http://dx.doi.org/ 10.1161/01.ATV.0000058402.34138.11 [DOI] [PubMed] [Google Scholar]
- [14].Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004; 114:1752-61; PMID:15599400; http://dx.doi.org/ 10.1172/JCI21625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chang YC, Yu YH, Shew JY, Lee WJ, Hwang JJ, Chen YH, Chen YR, Wei PC, Chuang LM, Lee WH. Deficiency of NPGPx, an oxidative stress sensor, leads to obesity in mice and human. EMBO Mol Med 2013; 5:1165-79; PMID:23828861; http://dx.doi.org/ 10.1002/emmm.201302679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Huh JY, Kim Y, Jeong J, Park J, Kim I, Huh KH, Kim YS, Woo HA, Rhee SG, Lee KJ, et al.. Peroxiredoxin 3 is a key molecule regulating adipocyte oxidative stress, mitochondrial biogenesis, and adipokine expression. Antioxid Redox Signal 2012; 16:229-43; PMID:21902452; http://dx.doi.org/ 10.1089/ars.2010.3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tormos KV, Anso E, Hamanaka RB, Eisenbart J, Joseph J, Kalyanaraman B, Chandel NS. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab 2011; 14:537-44; PMID:21982713; http://dx.doi.org/ 10.1016/j.cmet.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schneider K, Valdez J, Nguyen J, Vawter M, Galke B, Kurtz TW, Chan JY. Increased energy expenditure, Ucp1 expression, and resistance to diet-induced obesity in mice lacking nuclear factor-erythroid-2-related transcription factor-2 (Nrf2). J Biol Chem 2016; 291:7754-66; PMID:26841864; http://dx.doi.org/ 10.1074/jbc.M115.673756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li Y, Mouche S, Sajic T, Veyrat-Durebex C, Supale R, Pierroz D, Ferrari S, Negro F, Hasler U, Feraille E, et al.. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int J Obes (Lond) 2012; 36:1503-13; PMID:22430302; http://dx.doi.org/ 10.1038/ijo.2011.279 [DOI] [PubMed] [Google Scholar]
- [20].Han YH, Buffolo M, Pires KM, Pei S, Scherer PE, Boudina S. Adipocyte-specific deletion of manganese superoxide dismutase protects from diet-induced obesity through increased mitochondrial uncoupling and biogenesis. Diabetes 2016; 65:2639-51; PMID:27284109; http://dx.doi.org/ 10.2337/db16-0283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang PW, Kuo HM, Huang HT, Chang AY, Weng SW, Tai MH, Chuang JH, Chen IY, Huang SC, Lin TK, et al.. Biphasic response of mitochondrial biogenesis to oxidative stress in visceral fat of diet-induced obesity mice. Antioxid Redox Signal 2014; 20:2572-88; PMID:24111683; http://dx.doi.org/ 10.1089/ars.2013.5334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 2008; 88:611-38; PMID:18391175; http://dx.doi.org/ 10.1152/physrev.00025.2007 [DOI] [PubMed] [Google Scholar]
- [23].Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab 2006; 3:333-41; PMID:16679291; http://dx.doi.org/ 10.1016/j.cmet.2006.04.002 [DOI] [PubMed] [Google Scholar]
- [24].Kleiner S, Mepani RJ, Laznik D, Ye L, Jurczak MJ, Jornayvaz FR, Estall JL, Chatterjee Bhowmick D, Shulman GI, Spiegelman BM. Development of insulin resistance in mice lacking PGC-1alpha in adipose tissues. Proc Natl Acad Sci U S A 2012; 109:9635-40; PMID:22645355; http://dx.doi.org/ 10.1073/pnas.1207287109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Barja de Quiroga G, Lopez-Torres M, Perez-Campo R, Abelenda M, Paz Nava M, Puerta ML. Effect of cold acclimation on GSH, antioxidant enzymes and lipid peroxidation in brown adipose tissue. Biochem J 1991; 277(Pt 1):289-92; PMID:1854342; http://dx.doi.org/ 10.1042/bj2770289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mailloux RJ, Adjeitey CN, Xuan JY, Harper ME. Crucial yet divergent roles of mitochondrial redox state in skeletal muscle vs. brown adipose tissue energetics. FASEB J 2012; 26:363-75; PMID:21940996; http://dx.doi.org/ 10.1096/fj.11-189639 [DOI] [PubMed] [Google Scholar]
- [27].Sekhar BS, Kurup CK, Ramasarma T. Generation of hydrogen peroxide by brown adipose tissue mitochondria. J Bioenerg Biomembr 1987; 19:397-407; PMID:3624219; http://dx.doi.org/ 10.1007/BF00768542 [DOI] [PubMed] [Google Scholar]
- [28].Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J, Pierce KA, Laznik-Bogoslavski D, Vetrivelan R, Pierce KA, et al.. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016; 532:112-6; PMID:27027295; http://dx.doi.org/ 10.1038/nature17399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lettieri Barbato D, Tatulli G, Maria Cannata S, Bernardini S, Aquilano K, Ciriolo MR. Glutathione decrement drives thermogenic program in adipose cells. Sci Rep 2015; 5:13091; PMID:26260892; http://dx.doi.org/ 10.1038/srep13091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, et al.. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006; 127:397-408; PMID:17055439; http://dx.doi.org/ 10.1016/j.cell.2006.09.024 [DOI] [PubMed] [Google Scholar]
- [31].Marine A, Krager KJ, Aykin-Burns N, Macmillan-Crow LA. Peroxynitrite induced mitochondrial biogenesis following MnSOD knockdown in normal rat kidney (NRK) cells. Redox Biol 2014; 2:348-57; PMID:24563852; http://dx.doi.org/ 10.1016/j.redox.2014.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chouchani ET, James AM, Fearnley IM, Lilley KS, Murphy MP. Proteomic approaches to the characterization of protein thiol modification. Curr Opin Chem Biol 2011; 15:120-8; PMID:21130020; http://dx.doi.org/ 10.1016/j.cbpa.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Stefl B, Janovska A, Hodny Z, Rossmeisl M, Horakova M, Syrovy I, Bémová J, Bendlová B, Kopecký J. Brown fat is essential for cold-induced thermogenesis but not for obesity resistance in aP2-Ucp mice. Am J Physiol 1998; 274:E527-33; PMID:9530137 [DOI] [PubMed] [Google Scholar]
- [34].Kopecky J, Rossmeisl M, Hodny Z, Syrovy I, Horakova M, Kolarova P. Reduction of dietary obesity in aP2-Ucp transgenic mice: mechanism and adipose tissue morphology. Am J Physiol 1996; 270:E776-86; PMID:8967465 [DOI] [PubMed] [Google Scholar]
- [35].Wilson-Fritch L, Nicoloro S, Chouinard M, Lazar MA, Chui PC, Leszyk J, Straubhaar J, Czech MP, Corvera S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest 2004; 114:1281-9; PMID:15520860; http://dx.doi.org/ 10.1172/JCI21752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ro SH, Nam M, Jang I, Park HW, Park H, Semple IA, Kim M, Kim JS, Park H, Einat P, et al.. Sestrin2 inhibits uncoupling protein 1 expression through suppressing reactive oxygen species. Proc Natl Acad Sci U S A 2014; 111:7849-54; PMID:24825887; http://dx.doi.org/ 10.1073/pnas.1401787111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xu Y, Miriyala S, Fang F, Bakthavatchalu V, Noel T, Schell DM, Wang C, St Clair WH, St Clair DK. Manganese superoxide dismutase deficiency triggers mitochondrial uncoupling and the Warburg effect. Oncogene 2015; 34:4229-37; PMID:25362851; http://dx.doi.org/ 10.1038/onc.2014.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nicholls DG. The physiological regulation of uncoupling proteins. Biochimica et biophysica acta 2006; 1757:459-66; PMID:16725104; http://dx.doi.org/ 10.1016/j.bbabio.2006.02.005 [DOI] [PubMed] [Google Scholar]
- [39].Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 2012; 151:400-13; PMID:23063128; http://dx.doi.org/ 10.1016/j.cell.2012.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, et al.. Superoxide activates mitochondrial uncoupling proteins. Nature 2002; 415:96-9; PMID:11780125; http://dx.doi.org/ 10.1038/415096a [DOI] [PubMed] [Google Scholar]
- [41].Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology 2011; 26:192-205; PMID:21670165; http://dx.doi.org/ 10.1152/physiol.00046.2010 [DOI] [PubMed] [Google Scholar]
- [42].Silva JP, Shabalina IG, Dufour E, Petrovic N, Backlund EC, Hultenby K, Wibom R, Nedergaard J, Cannon B, Larsson NG. SOD2 overexpression: enhanced mitochondrial tolerance but absence of effect on UCP activity. EMBO J 2005; 24:4061-70; PMID:16281056; http://dx.doi.org/ 10.1038/sj.emboj.7600866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Malingriaux EA, Rupprecht A, Gille L, Jovanovic O, Jezek P, Jaburek M, Pohl EE. Fatty acids are key in 4-hydroxy-2-nonenal-mediated activation of uncoupling proteins 1 and 2. PloS one 2013; 8:e77786; PMID:24204965; http://dx.doi.org/ 10.1371/journal.pone.0077786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shabalina IG, Petrovic N, Kramarova TV, Hoeks J, Cannon B, Nedergaard J. UCP1 and defense against oxidative stress. 4-Hydroxy-2-nonenal effects on brown fat mitochondria are uncoupling protein 1-independent. J Biol Chem 2006; 281:13882-93; PMID:16543238; http://dx.doi.org/ 10.1074/jbc.M601387200 [DOI] [PubMed] [Google Scholar]