

In this review, Liu and Hebrok outline our current understanding of the β-cell maturation process, emphasize recent literature on novel heterogeneity markers, and offer perspectives on reconciling the findings from these two areas.
Keywords: β-cell, maturation, β-cell heterogeneity, pancreatic islets, stem cell differentiation, aging
Abstract
Following differentiation during fetal development, β cells further adapt to their postnatal role through functional maturation. While adult islets are thought to contain functionally mature β cells, recent analyses of transgenic rodent and human pancreata reveal a number of novel heterogeneity markers in mammalian β cells. The marked heterogeneity long after maturation raises the prospect that diverse populations harbor distinct roles aside from glucose-stimulated insulin secretion. In this review, we outline our current understanding of the β-cell maturation process, emphasize recent literature on novel heterogeneity markers, and offer perspectives on reconciling the findings from these two areas.
β Cells within the pancreatic islets of Langerhans are the only cells within the body that secrete the hormone insulin to control blood glucose levels. To maintain glucose homeostasis, β cells should secrete insulin in response to high levels of glucose to initiate clearance but suppress insulin secretion at low glucose levels to allow for basal bodily functions. Mammalian β cells gain the ability to differentially respond to low and high glucose levels after birth in a process known as functional maturation (Box 1). While the physiological phenomenon of β-cell maturation has been documented for decades in mammalian species (Lavine et al. 1971; King et al. 1986; Bliss and Sharp 1992), we are only beginning to understand the molecular mechanisms associated with the process (Fig. 1).
Box 1
Functional maturation—The process by which differentiated β cells acquire glucose-responsive insulin secretion.
Maturation driver—A biomolecule that is sufficient to induce functional maturation in immature β cells.
Maturation marker—A biomolecule whose expression is correlated to induction of, but is not sufficient to induce, functional maturation in immature β cells.
Disallowed genes—Genes that are repressed in β cells but expressed, often highly, in a variety of other tissues or in undifferentiated immature β cells.
Molecular heterogeneity—Differential expression of specific proteins or patterns of proteins between β-cell subpopulations.
Cellular heterogeneity—Mixed composition of mature β-cell subpopulations with molecular heterogeneity.
Figure 1.

Molecular and metabolic changes associated with functional maturation of rodent β cells. Both immature and mature β cells are lineage-specified but differ in their insulin secretion response to glucose. (A) Mature β cells contain more insulin granules and secrete more insulin when stimulated with high levels of glucose. (B) One of the major differences between immature and mature β cells is the switch in expression from high-affinity hexokinase (HK) to low-affinity glucokinase (GCK). Glucose metabolism is thus only activated in mature cells in the presence of elevated levels of glucose. (C) Glucose-responsive respiration is also enhanced through the up-regulation of metabolic components involved in the tricarboxylic acid (TCA) cycle, oxidative phosphorylation (OxPhos), and electron transport chain (ETC). (D) Changes in the expression pattern of microRNAs (miRs) during β-cell maturation directly regulate metabolic gene targets as well as disallowed genes (Box 1). (E) In addition to expressing canonical β-cell transcription factors that are present in immature cells (Pdx1, Nkx6.1, and Isl1), mature β cells express additional maturation genes such as MafA, urocortin3 (Ucn3), and estrogen-related receptor γ (Errγ). β-Cell-disallowed genes Hk1, monocarboxylate carrier 1 (Mct1), lactate dehydrogenase A (Ldha), repressor element 1 silencing transcription factor (Rest), and platelet-derived growth factor receptor α (Pdgfrα) are also down-regulated during maturation.
Although glucose-responsive insulin secretion appears to be the ultimate function of β cells, we are concurrently discovering new subpopulations within adult β cells based on novel molecular properties. These subpopulations express varying degrees of genes associated with β-cell functional maturation and may also retain nonsecretory roles important for overall islet function. A better understanding of how and why these different β-cell subpopulations arise in mature adult islets may further elucidate the effects of these β-cell subtypes on islet health.
Toward reconciling the coexistence of β-cell maturation and novel β-cell heterogeneity markers demonstrated in recent literature, we first present an overview of molecular mechanisms involved in β-cell maturation. We then take a closer look at three studies published within the past year that emphasize novel subpopulations of β cells in adult islets. Finally, we close with a summary of how defined compositions of β cells with different sensitivities to glucose affect the overall function of mouse islets as a case study approach to quantitatively interrogate the effect of β-cell subtypes on islet function.
Mechanisms underlying β-cell maturation
Immature β cells in newborn mammals are lineage-specified, as they share many molecular features with mature adult β cells. Both types of β cells express the canonical β-cell transcription factors Pdx1, Nkx6.1, Nkx2.2, Isl1, and Pax6 (Conrad et al. 2014), although increases in some of these genes have been observed in mature cells (Yoshihara et al. 2016). Both β-cell types produce insulin but not other hormones. Immature β cells, however, have a decreased glucose threshold for stimulated insulin secretion and thus secrete insulin even at low levels of glucose (Blum et al. 2012).
The basic molecular circuitry that couples glucose to insulin secretion is similar in immature and mature β cells (Fig. 1). Glucose initiates the insulin secretion circuitry when taken into cells by glucose transporters (McCulloch et al. 2011). Glycolysis converts glucose to pyruvate, which in turn activates the tricarboxylic acid (TCA) cycle and oxidative phosphorylation in the mitochondria to generate ATP. Increased ATP production in mitochondria triggers closure of ATP-sensitive potassium (KATP) channels and membrane depolarization. Subsequent calcium (Ca2+) influx through voltage-gated Ca2+ channels leads to exocytosis of insulin granules from the β cell. Immature and mature β cells release similar amounts of insulin when depolarized independent of glucose sensing and metabolism, although mature cells contain larger numbers of insulin granules (Fig. 1A; Blum et al. 2012). However, specific molecular changes during β-cell maturation alter glucose sensitivity at various points in the transition between the immature and mature states. These changes are described in the following sections.
The mechanisms involved in maturation are currently being explored in both mouse and human cell models. In rodents, birth to postnatal weaning is generally accepted as the transition period during which β cells functionally mature (Lavine et al. 1971; Bliss and Sharp 1992; Jacovetti et al. 2015; Stolovich-Rain et al. 2015). Much of the information that we have about β-cell maturation has thus been from studying islets from neonatal mice and rats as they are weaned from a milk to chow diet during the second to fourth weeks of life. Similar data from humans are more difficult to collect. Recently, however, directed differentiation protocols for human pluripotent stem cells (hSCs) have achieved monohormonal β-like cells that have transcriptional profiles and limited glucose responsiveness somewhat similar to immature β cells (Hrvatin et al. 2014; Pagliuca et al. 2014; Rezania et al. 2014; Russ et al. 2015). Genetic and pharmacological manipulation of these human-derived model systems can complement studies in rodents. Understanding the mechanisms behind β-cell maturation will be important as we continue to investigate therapeutic opportunities for addressing β-cell dysfunction in type 1 and type 2 diabetes (T1D and T2D). Studying maturation in the hSC models especially has clear implications for both basic and islet replacement translational research (Johnson 2016).
Maturation-associated metabolic changes
One of the known ways in which mature β cells differ from immature β cells is in their metabolic machinery. The first step and kinetic bottleneck of glycolysis is performed by hexokinases. Four mammalian hexokinases exist, but mature β cells express only hexokinase IV, also known as glucokinase (GCK). Compared with the other hexokinases, GCK has a low affinity for glucose and thus catalyzes the phosphorylation of glucose at higher concentrations of glucose than the other hexokinases (Moukil et al. 2000). In this way, GCK serves as a high-glucose sensor (Liang et al. 1991; Piston et al. 1999). The expected lower glycolytic activity of mature β cells in basal glucose conditions is consistent with the observation that mature islets have lower levels of oxygen consumption, a readout of downstream oxidative phosphorylation, than immature islets at basal levels of glucose (Stolovich-Rain et al. 2015). Thus, the switch from high-affinity hexokinases to GCK explains in part the higher threshold of glucose required for insulin secretion in mature β cells (Fig. 1B).
In addition to the GCK enzyme necessary for appropriate glucose sensing, the expression of many other downstream components that couple the metabolism of glucose to the insulin exocytotic machinery also increases during β-cell maturation (Fig. 1C; Rorsman et al. 1989; Welsh et al. 1989; Hellerstrom and Swenne 1991; Jermendy et al. 2011). Genes for metabolic enzymes involved in glycolysis, TCA cycle, oxidative phosphorylation, and electron transport chain and for components directly involved in insulin granule exocytosis show enhanced expression during rodent β-cell maturation (Jacovetti et al. 2015; Stolovich-Rain et al. 2015; Yoshihara et al. 2016). A number of these genes have been interrogated in genetic mouse models to demonstrate their importance in general β-cell function (Remedi and Nichols 2009). Similar widespread changes also occur during human β-cell maturation, as genome-wide transcriptional analysis comparing insulin-positive cells from fetal (immature) and adult human β cells revealed enrichment of gene ontology terms associated with glucose metabolism and insulin processing in mature cells (Hrvatin et al. 2014). In sum, while the switch from hexokinase to GCK regulates glucose sensing in β cells, expression changes in a plethora of other molecular players are also necessary for β-cell maturation.
Active investigation is under way to understand how the multitude of metabolic, exocytotic, and other cell physiological changes is exacted at the molecular level during the weaning window in rodents. One recently explored mechanism that may contribute to the global changes associated with β-cell maturation lies in post-transcriptional gene regulation by microRNAs (miRNAs or miRs). Islet miRNA profiling showed distinct expression patterns before and after weaning in rat pups (Fig. 1D; Jacovetti et al. 2015). Knockdown of specific miRs in the miR-17/92, miR-181, and miR-106b/25 families (which decrease in expression upon β-cell maturation) enhanced glucose-stimulated insulin secretion (GSIS) in immature rat islets, demonstrating their inhibitory role on the functional maturation process. These miRNAs could directly repress expression of mitochondrial metabolism genes (including phosphofructokinase, glycerol-3-phosphate dehydrogenase, and malate dehydrogenase), were predicted to bind additional regulators of metabolism and insulin secretion, and have additional nonmetabolic target genes (Jacovetti et al. 2015). However, expression of mimics of miR-17/92, miR-181, and miR-106b/25 family members in mature adult islets had little consequence on insulin secretion, suggesting that complete functional maturation also involves other mechanisms (Jacovetti et al. 2015).
Drivers and markers of β-cell maturation
While many proteins have been found to be important for specification of the β-cell lineage (Conrad et al. 2014; Nair and Hebrok 2015), only a handful have been specifically linked to the process of β-cell maturation (Fig. 1E). We distinguish below between active maturation drivers (Box 1), which are sufficient to induce functional maturation in immature β cells, and passive maturation markers (Box 1), which are correlated to induction of, but are not sufficient to induce, functional maturation in immature β cells.
MAFA is a basic leucine zipper transcription factor that is up-regulated in the presence of glucose and can activate the insulin gene (Kataoka et al. 2002; Olbrot et al. 2002; Matsuoka et al. 2004). MAFA protein and mRNA expression gradually increase, and MAFA localization shifts from cytoplasmic to nuclear over the first few weeks of life in rat pups (Artner et al. 2010; Matsuoka et al. 2010; Aguayo-Mazzucato et al. 2011). These changes in MAFA occur over the time frame during which GSIS becomes apparent. Adenoviral-mediated overexpression of Mafa in immature postnatal day 2 (P2) rat islets increased nuclear localization of MAFA and induced mature GSIS behavior despite insulin expression and content levels well below those found in adult islets (Aguayo-Mazzucato et al. 2011). A positive effect of Mafa expression on GSIS has also been demonstrated using insulin-expressing cells and inducible MAFA; expression of wild-type Mafa increased, while expression of dominant-negative Mafa decreased GSIS in INS-1 cells (Wang et al. 2007). Importantly, when Pdx1, another β-cell transcription factor known to regulate insulin, was overexpressed, P2 islets did not show improved GSIS but instead increased insulin secretion at both basal and stimulatory glucose levels. Mafa thus appears to be a driver of maturation that specifically improves the glucose sensitivity of insulin secretion machinery in a number of models for immature β cells, likely because it transcriptionally regulates a number of genes related to insulin secretion (Matsuoka et al. 2004; Zhang et al. 2005; Wang et al. 2007). Based on its importance in mouse models, MAFA expression has recently become a focus in efforts to derive β cells from human stem cells (Rezania et al. 2014). However, it is possible that human β-cell maturation does not depend specifically on MAFA, as adult human β cells also express MAFB (Dai et al. 2012; Arda et al. 2016).
As MAFA has been found to be an important transcription factor for maturation in rodents, the physiological regulators for Mafa expression are also maturation drivers. Along these lines, thyroid hormone signaling has been linked to Mafa expression. Treatment of isolated immature rat islets, fetal human islets, and hSC-derived β-like cells with tri-iodothyronine (T3) leads to increased Mafa expression and improved GSIS (Aguayo-Mazzucato et al. 2013, 2015). The specific in vivo role of T3 signaling in β-cell maturation is supported by the fact that expression and processing of T3 in postnatal rat islets track with the β-cell maturation process and that expression of T3 receptors in rat islets is highly up-regulated during the second and third weeks of life (Aguayo-Mazzucato et al. 2013).
Another recently described driver of β-cell functional maturation is the nuclear receptor estrogen-related receptor γ (ERRγ). Similar to Mafa, Errγ expression in islets increases between the neonatal and adult stages (Yoshihara et al. 2016). β-Cell-specific knockout of Errγ in mice results in morphologically normal islets that nonetheless have significantly lower glucose-responsive insulin secretion compared with wild-type islets. Intriguingly, overexpression of Errγ in functionally immature β-like cells derived from human induced pluripotent stem cells leads to glucose responsiveness and improved mitochondrial morphology (Yoshihara et al. 2016). Errγ overexpression also led to a metabolic gene expression profile more similar to that of human adult islets, although the stem cell-derived β-like cells still expressed lower mRNA levels of insulin processing and KATP channel components (Yoshihara et al. 2016).
NEUROD is another transcription factor that has been associated with β-cell maturation in genetic mouse models. Mice with conditional knockout of NeuroD in insulin-expressing cells have normal-sized islets with wild-type mRNA levels of β-cell genes Pdx1, Nkx6.1, and MafA (Gu et al. 2010). However, NeuroD knockout islets exhibit immature insulin secretion; they secrete higher basal and lower glucose-stimulated levels of insulin (Gu et al. 2010). The immature secretion phenotype is mirrored by enhanced expression of glycolytic genes and glucose metabolism, with higher oxygen consumption at basal levels and lower oxygen consumption at high levels of glucose. While islet size is unperturbed, conditional NeuroD knockout also affected the cellular architecture of islets (non-β endocrine cells were found within the core instead of predominantly at the islet periphery) and decreased Ins1 but not Ins2 expression, which suggests additional defects in islet development apart from glucose sensing. Although NeuroD overexpression studies were not done to test whether it can directly induce maturation, these findings do suggest that NeuroD is involved in molecular processes that are necessary for β cells to achieve mature glucose-sensitive function.
In contrast to the maturation driver functions of MAFA and ERRγ, Urocortin3 (UCN3), a secreted protein in the corticotropin-releasing factor hormone family (Kuperman and Chen 2008), serves as a marker of β-cell maturation (van der Meulen and Huising 2014). Ucn3 is highly expressed in adult mouse and human β cells (Blum et al. 2012; van der Meulen et al. 2012). Ucn3 expression increases over the maturation period in mouse β cells as well as over the in vivo maturation of hSC-derived insulin-expressing cells after kidney graft transplantation (Blum et al. 2012; van der Meulen et al. 2012). Ucn3 expression is specific to β cells in adult mouse islets but marks both β and α cells in adult primate islets (Blum et al. 2012; van der Meulen et al. 2012). UCN3 injections enhance plasma levels of insulin in rats, but treatment with UCN3 does not promote maturation of stem cell-derived insulin-expressing cells (Li et al. 2003; Blum et al. 2012). While Ucn3 expression in β cells is intriguing, the peptide may act elsewhere in the islet, as its cognate receptor was recently found specifically on δ cells (van der Meulen et al. 2015). Interestingly, UCN3 secretion from the mouse MIN6 β-cell line is induced with glucose (Li et al. 2007). Glucose-induced UCN3 secretion is blocked when cells are treated with diazoxide, an activator of KATP channels, indicating that UCN3 secretion relies on KATP channel activity that is downstream from glucose metabolism. Thus, UCN3 activity is an interesting indicator of functional glucose metabolism and sensing in mature β cells, although it does not likely act directly on β cells to induce maturation.
Down-regulation of β-cell-specific disallowed genes
Mature β cells are defined by not only the presence of maturity genes but also the repression of specific genes termed disallowed genes (Fig. 1E; Box 1; Rutter et al. 2015; Lemaire et al. 2016). By analyzing the mRNA expression arrays from various mouse tissues, a number of laboratories have identified genes that are specifically down-regulated in adult islet cells (Pullen et al. 2010; Thorrez et al. 2011). Of these disallowed genes, the genes for high-affinity hexokinase 1 (HK1) and lactate dehydrogenase A (LDHA) are highly expressed in other tissues as well as immature β cells but become repressed in the postnatal islet coincident with β-cell maturation (Thorrez et al. 2011; Dhawan et al. 2015; Yoshihara et al. 2016). The repression of Hk and Ldha genes appears to involve epigenetic regulation during β-cell development and maturation (van Arensbergen et al. 2010; Dhawan et al. 2015). Additional disallowed genes for monocarboxylate carrier (MCT1), repressor element 1 silencing transcription factor (REST), and platelet-derived growth factor receptor α (PDGFRα) polypeptide are also targets of miR-29b-3p, a miR that increases in expression during in vivo rat β-cell maturation (Jacovetti et al. 2015). This finding that miRNAs regulate disallowed genes is in line with another report of improper regulation of disallowed genes in islets from mice deficient for Dicer, a ribonuclease involved in the processing of miRNAs (Martinez-Sanchez et al. 2015). Hence, different mechanisms down-regulate this subset of disallowed genes during β-cell maturation.
The repression of Hk1, Mct1, and Ldha is consistent with the metabolic regulation associated with β-cell maturation. Given the high affinity of HK1 for glucose, repression of its expression is in line with the lower glucose sensitivity of mature cells. MCT1 is a transporter for pyruvate, which feeds into the TCA cycle after conversion to acetyl-coenzyme A, and lactate, which can be converted into pyruvate. Since lactate and pyruvate can be produced by other tissues, such as muscle during exercise, repression of Mct1 in mature β cells prevents such metabolic interference that could otherwise promote insulin secretion in the absence of glucose. Ldha repression further prevents conversion of exogenous lactate into pyruvate, which could similarly dysregulate insulin secretion.
Repression of Rest in islets has a more indirect impact on β-cell function. Insulin secretion requires fusion of insulin granules to the plasma membrane. Proteins including Ca2+-sensing synaptotagmins and SNARE complexes orchestrate the membrane fusion. Primary β cells overexpressing Rest showed decreased expression of a number of these fusion proteins as well as impaired insulin secretion (Martin et al. 2008). Rest overexpression in β cells also leads to glucose intolerance in mice (Martin et al. 2008). These data suggest that the exocytotic machinery necessary for proper insulin secretion can be expressed only in the absence of REST. Rest repression may also play a role in postnatal β-cell survival, as Rest-overexpressing mice have decreased β-cell mass due to apoptosis (Martin et al. 2012).
PDGFRα is a tyrosine kinase receptor involved in cell division in mouse β cells (Chen et al. 2011). Pdgfrα expression in islets decreases with age and is down-regulated in adult versus juvenile islets for both mice and humans (Chen et al. 2011). Up-regulation of miR-29b-3p, which can repress Pdgfrα expression, accompanies additional miRNA changes that suppress proliferation during weaning. These observations are in line with decreased β-cell proliferative capacity during the third week of life (Jacovetti et al. 2015).
Pressing questions related to β-cell maturation
β-Cell functional maturation is proving to be a straightforward physiological change with complex molecular underpinnings that demand further investigation. While a number of molecular drivers described here appear sufficient for a glucose-stimulated insulin response when exogenously introduced into immature β cells, a precise and detailed time line of when these drivers appear in the natural maturation process in relation to one another could better inform on how drivers and markers are related. While some factors, such as MAFA, are known to be responsive to T3 and glucose, the changes in other molecules, such as miRNAs, are associated only with shifts in nutrient source. Identification of specific maturation-inhibiting or maturation-inducing factors in milk or chow, respectively, as well as their cellular sensors could aid in the characterization of the molecular sequence of events. The specific timing and intervals at which different molecules are required for maturation will also be important to determine. For instance, the inability of miR-17/92, miR-181, and miR-106b/25 family member mimics to inhibit insulin secretion function in adult islets when knockdown of the same miRNAs could improve function in immature islets suggests that these miRNAs either are only unidirectionally effective at a specific stage in the transition from immature to mature cells or become redundant when other maturation landmarks are in place (Jacovetti et al. 2015). Defining which genes or combination of genes are imperative for functional maturity is therefore an ongoing effort.
Carefully outlining the requirement of drivers and markers for β-cell maturation will directly affect efforts at generating hSC-derived β cells. Most information regarding β-cell maturation has been derived from studies in rodents. Neither the exact timing nor the mechanistic underpinnings of in vivo human β-cell maturation have been explored in detail. While previously published reports using hSC-derived insulin-producing cells have looked at a few of the maturation genes described here, hSC-derived cells could be more thoroughly characterized to align with primary mature cells (Johnson 2016; Lemaire et al. 2016). Although we focused here on the initial functional maturation period soon after birth, β cells continue to develop and improve in secretory function months after weaning in rodents (Bliss and Sharp 1992; Avrahami et al. 2015; Stolovich-Rain et al. 2015; Helman et al. 2016). Similarly, a recent study on human β cells in islets from juvenile (>0.5 but <9 yr of age) and adult (>28 yr of age) donors revealed decreases in genes related to cell cycle and increases in genes involved in hormone regulation in adult versus juvenile β cells (Arda et al. 2016). A separate gene expression study on single human endocrine cells found that, while the gene expression signature of adult β cells excludes α-cell genes, juvenile β cells can misexpress adult α-cell genes (Wang et al. 2016b). These studies in human β cells indicate that, similar to rodent β cells, β-cell development is a lifelong process (Helman et al. 2016). To generate maximally functional β cells from hSC sources, we may also need to consider additional phases of mature β-cell aging that exist beyond the initial functional maturation period.
Many of the currently defined maturation genes were identified in multicellular islets or total β-cell populations. Population-based analysis could mask any variations in gene expression of minor subpopulations of cells. Below, we look more closely at molecular subtypes that have been described within the β-cell population in adult islets.
β-Cell heterogeneity—recent findings and implications for islet function
After β-cell maturation, pancreatic islets are capable of sensing glucose and responding with insulin secretion to maintain homeostatic levels of glucose. However, when examined at the single-cell level, individual β cells can be incredibly molecularly diverse. For instance, expression of β-cell proteins (such as insulin and Pdx1), glucose metabolism components (such as GCK and GLUT2), and cell–cell adhesion molecules (such as E-cadherin and NCAM) can vary between individual β-cells in rodent and human islets (Heimberg et al. 1993; Roscioni et al. 2016; Wang et al. 2016b). These proteins positively correlate with β-cell secretory function, likely due to the direct role of these proteins in β-cell–cell adhesion and metabolism. However, over the past year, a number of groups have identified additional markers of heterogeneity within the β-cell population with less obvious ties to insulin secretion pathways. Below, we highlight three recent studies that have used novel markers of β-cell heterogeneity to identify β-cell subtypes associated with islet health or function (Fig. 2).
Figure 2.
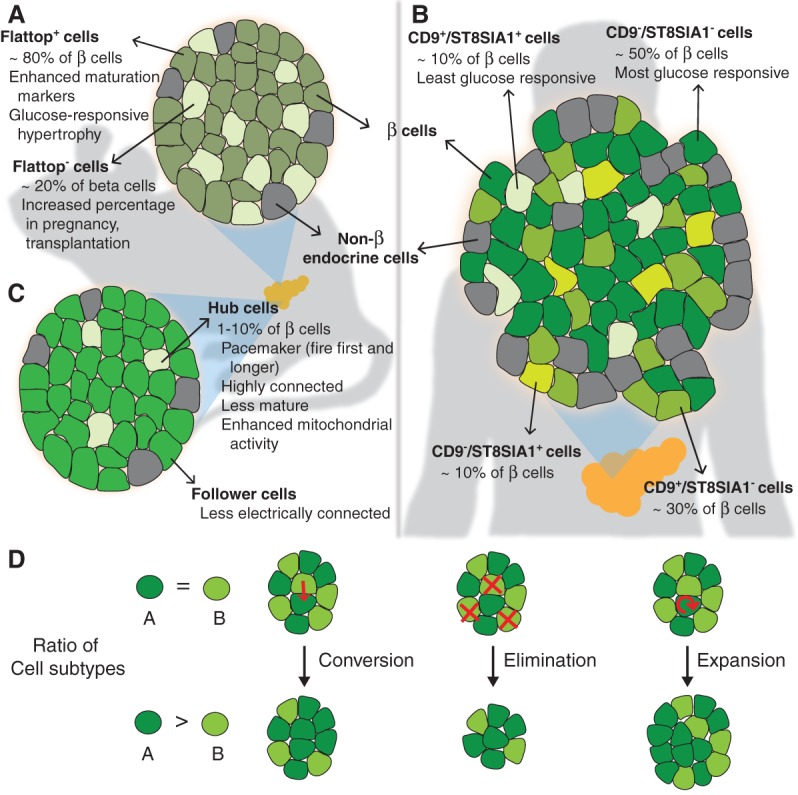
Markers of β-cell heterogeneity in adult islets. Three recent studies identified novel markers of β-cell heterogeneity and new β-cell subtypes (shades of green) in adult islets. (A) Flattop, a Wnt/planar cell polarity (PCP) pathway effector, marks the major subpopulation of mouse β cells with a mature transcriptional profile. The ratio of Flattop-positive (Flattop+) to Flattop-negative (Flattop−) cells can change in response to different physiological contexts. (B) Differential expression of CD9 and ST8SIA1 demarcate four molecularly and functionally distinct subsets of human β cells. (C) Hub cells are highly connected pacemaker cells that instigate electrical signaling in follower cells in mouse islets. Hub cells express lower levels of some β-cell proteins and have enhanced metabolic response to glucose. (D) Processes such as conversion, selective elimination, and selective expansion can change the relative ratios of β-cell subtypes. Whether these processes occur for the different subtypes described during islet development, maturation, or disease progression remains to be determined.
One recently highlighted marker of islet cell heterogeneity is Flattop (FLTP) (Fig. 2A; Bader et al. 2016; Roscioni et al. 2016). FLTP is an effector of the Wnt/planar cell polarity (PCP) pathway that is necessary for collective epithelial organization and architecture (Gegg et al. 2014). Using an islet-specific Fltp reporter mouse model, Bader et al. (2016) identified clear molecular and physiological differences between FLTP-positive (FLTP+) and FLTP-negative (FLTP−) β-cell populations. FLTP+ cells increase in number through islet development and constitute the majority (∼80%) of β cells in adult animals. The investigators found that compared with FLTP− cells, FLTP+ cells express higher mRNA levels of maturation genes, such as Ucn3 and MafA, as well as mRNAs and mitochondrial proteins associated with oxidative phosphorylation and insulin secretion (Bader et al. 2016). FLTP knockout mice displayed very mild islet function phenotypes (Bader et al. 2016), suggesting that FLTP is a passive marker of β-cell maturation similar to UCN3. Interestingly, both FLTP− and FLTP+ lineage populations can expand but in response to different stimuli. Ki67 immunostaining analysis showed that FLTP− cells are more proliferative in homeostatic conditions (∼8% Ki67+ compared with ∼2% Ki67+ in FLTP+ population at P11) and during pregnancy (∼4% Ki67+ compared with <2% Ki67+ in FLTP+ population). The percentage of FLTP− cells also initially increases after islet transplantation to the iris. As the investigators observed a decrease in islet size during the early stages after transplantation, the relative increase in FLTP− cells may have resulted from specific loss of FLTP+ cells. In contrast, FLTP+ cells expand during high-fat diet-induced islet hypertrophy, in line with their relatively heightened metabolic capacity. While the extent of FLTP heterogeneity was not explored in the human context, the investigators did find a positive correlation between islet mRNA expression of the human FLTP homolog and the glucose tolerance of human patients; healthy donors expressed higher levels than patients with prediabetes or T2D.
The second study made use of a combination of antibodies against different cell surface antigens. Dorrell et al. (2011) isolated the β-cell population using a previously described panel of antibodies that allows separation of different cell types in human islets. By additionally staining the purified β-cell population with antibodies against CD9 and ST8SIA1, the investigators found four antigenically distinct populations of β cells in healthy human islets (Fig. 2B; Dorrell et al. 2016). RNA sequencing (RNA-seq) analysis of the different β-cell subsets indicated that all four expressed canonical β-cell genes, such as INS, PDX1, MAFA, and NKX6.1. The β-cell subsets are molecularly distinct populations, however, as the investigators found >100 other genes that were differentially expressed between CD9-positive and CD9-negative or ST8SIA1-positive and ST8SIA1-negative populations. In particular, gene ontology terms related to protein secretion were specifically associated with the transcriptional profile of ST8SIA1− cells. The subsets also differ in their functional capacity. Insulin secretion analysis on the purified and reaggregated β-cell subtypes revealed differential GSIS; ST8SIA1−/CD9− β cells had the lowest basal secretion and the highest stimulation index. Interestingly, islets from patients with T2D showed aberrant distribution of the β-cell subtypes compared with those from healthy patients. This last finding suggests an association between the pattern of β-cell subtype distribution and islet health, although whether these changes occur before or after the onset of T2D remains to be determined.
The third study investigated heterogeneity in electrical-coupling capacity among β cells. Combining elegant live-cell imaging, correlation analysis, and light-controlled molecular systems, Johnston et al. (2016) identified and characterized a minor population (1%–10%) of highly connected and electrochemically distinct β cells, termed hub cells, within intact mouse islets (Fig. 2C). Compared with nonhub cells (also termed “follower” cells), hub cells exhibit accelerated and extended Ca2+ signaling on exposure to stimulatory levels of glucose and also have many electrically coupled cellular connections. Although glucose-stimulated human islets displayed a Ca2+ signaling activation network consistent with the existence of a population of more connected β cells, hub cells were not specifically identified or studied in human islets. By illuminating single cells in mouse islets expressing a light-gated chloride channel, the investigators could selectively inhibit Ca2+ signaling in individual cells. Whereas inactivating individual follower cells had no significant effect on synchronized Ca2+ signaling dynamics, inactivating hub cells disrupted the synchronization of Ca2+ signaling across the islet. Similar results were observed for zinc coreleased with insulin; hub cell inactivation dramatically decreased zinc secretion across the entire islet. Hub cell activity was dependent on gap junctions and could be blocked upon islet treatment with proinflammatory cytokines or lipotoxicity. Hub cells thus serve as a type of pacemaker with a central function of synchronizing glucose-stimulated Ca2+ signaling and insulin granule secretion of networks of follower cells across an islet. The identification and characterization of hub cells thus reveals a novel mechanism for the concerted insulin secretion from multiple β cells within rodent islets.
Closer inspection of the metabolic and protein expression profiles of hub and follower cells revealed a unique signature of β-cell-associated proteins in hub cells. While morphologically normal, hub cell mitochondria became more active and hyperpolarized on glucose stimulation than those in follower cells. Hub cells were insulin monohormonal but produced less insulin and contained fewer insulin granules compared with other β cells. Hub cells expressed lower levels of PDX1 and significantly lower levels of NKX6.1 than follower cells, suggesting a slightly undifferentiated phenotype. However, hub cells were likely not progenitor cells, since they were also negative for endocrine progenitor factor Neurogenin3. In sum, hub cells appear to be β cells with more metabolic capacity but diminished expression of insulin and some β-cell transcription factors.
Despite the differences in approaches and the mode of cellular heterogeneity (Box 1) discovered, the heterogeneity studies above raise many similar additional questions.
When and why do the distinct subpopulations of β cells arise in the islet?
While the transgenic reporter mouse allowed for monitoring of both FLTP subtypes from embryonic to adult stages (Bader et al. 2016), we await further analysis to understand when hub cells and the different ST8SIA1/CD9 β-cell subtypes arise. It is possible that FLTP− and hub cells represent a more immature cell type, as they express lower levels of β-cell and maturation genes and proteins. However, it is equally possible that these cells underwent the maturation process and then lost some of their maturation signature through a dedifferentiation process, similar to what has been observed in stressed β cells (Talchai et al. 2012; Puri et al. 2015). More thorough investigation of the temporal pattern of heterogeneity marker expression through β-cell development, β-cell maturation, and diabetes onset will allow us to discriminate between these possibilities.
How similar or overlapping are these different subpopulations?
Because each of the studies described above focused on distinct heterogeneity markers, we can only speculate on how one β-cell subtype aligns with subtypes found in a different study. The observation of lower expression levels of β-cell maturation genes in both the FLTP− and hub cells begs the question of whether these populations overlap. However, it is unlikely that these populations are one and the same due to the differences in percentages observed (FLTP− ∼20%; hub cells <10%), although hub cells could be a FLTP− subset. Interestingly, another recent study found enhanced PAX4 expression in ∼30% of β cells (Mellado-Gil et al. 2016). This PAX4-expressing population also replicated during pregnancy and may overlap with the FLTP− β-cell population. Conversely, it is possible that the minor populations identified here (i.e., hubs, FLTP−, and ST8SIA1+ and/or CD9+ β cells) are exclusive from one another. As more molecular indicators of β-cell heterogeneity are discovered, cross-comparison of indicator expression between studies may lead to further insights into specialized β-cell subsets.
In the near future, it is possible that even these general subsets of β cells will be further distinguished into distinct molecular populations. This prospect is especially likely as new technologies and methodologies allowing for single-cell analysis of pancreas cells become more widespread (Muraro et al. 2016; Segerstolpe et al. 2016; Wang et al. 2016a). Of note, the wealth of data from single-cell methods and analyses has led the larger scientific community to carefully consider which components of a cell's molecular signature are indicative of a permanent identity and which components are expressed at different levels across individual cells because these components correlate with dynamic permissive states (Wagner et al. 2016). Studies of cellular heterogeneity are uncovering similar challenges at two biological scales. At the molecular level, we strive to determine which markers of β-cell heterogeneity signify a truly different type of β cell versus markers that correlate with β-cell states that are dynamic and fluid. In addition, at the cellular level, we hope to identify β-cell subtypes that have a true consequence on specific islet behaviors (e.g., insulin secretion, maintenance of mass, response to insult, etc.) (discussed further below). Deconvoluting β-cell heterogeneity at both of these levels will be required to understand how β-cell subtypes relate to each other.
Can cells of one subtype convert to another subtype? What do changes in subtype ratios indicate?
Another question regarding β-cell heterogeneity is whether a cell of one β-cell subtype can convert to a different β-cell subtype. Conversion of FLTP− to FLTP+ cells under different physiological stressors could be tracked using the reporter mouse model, but lineage tracing of the hub cells and ST8SIA1/CD9 β-cell subtypes was not possible. Hub cells were demonstrated to be stable over the course of a few hours (Johnston et al. 2016). We will be interested to learn whether hub cell fate in vivo is stable over longer periods (for days, weeks, or longer) and whether the number of hub cells in an islet is fixed and/or predetermined. Plasticity in hub cell fate could be especially pertinent to rescuing islet functionality in diabetes should it be possible to generate new hub cells or convert followers to hubs. Understanding whether the relative subtype ratios change as a result of physiological stimuli may have additional translational impacts. For instance, determining whether changes in ST8SIA1/CD9 β-cell subtypes coincide or precede initial glucose intolerance in prediabetic patients or mouse models could lead to development of new diagnostic tools. In addition, understanding the mechanism by which ratiometric changes occur could reveal temporal- or context-dependent roles for the different β-cell subtypes. For example, conversion, selective expansion, and selective elimination are three possible ways in which subtype ratios in an islet can shift (Fig. 2D). The subpopulation that is targeted for these processes in different contexts will likely need to be determined on a case-by-case basis.
How do these markers of heterogeneity and subpopulations affect β-cell or islet function?
Within this small survey of studies, in all cases, the major cell subtype (i.e., FLTP+, ST8SIA1−/CD9−, and follower cells) comprised functionally mature cells as judged by β-cell and maturation gene or protein expression. The predominance of these glucose-responsive insulin-secreting cells in healthy islets underscores hormone secretion as the principal function of the islet. However, what remains to be explored is whether the different minor β-cell subpopulations (especially FLTP− and ST8SIA1+ and/or CD9+ β cells) serve specialized roles in either insulin secretion or nonsecretory functions. The rare hub cells are presumably less insulin secretory than follower cells (as they express lower levels of insulin) but clearly play a critical role in synchronizing activation and insulin secretion of other β cells. Similarly, the FLTP− subpopulation, which has a less mature gene expression profile than FLTP+ cells, contains more proliferative (Ki67+) cells and may be important in maintaining functional islet mass. While percentage shifts in the individual ST8SIA1/CD9 human β-cell subtypes were not consistent, in most cases, the combined percentage of the three less insulin secretory subtypes (ST8SIA1+ or CD9+) were higher in T2D islets compared with healthy donor islets (Dorrell et al. 2016). The specific roles for these three subpopulations in healthy islets remain to be determined but will be important for understanding how their increased presence impacts overall islet function. Additional roles for heterogeneous β-cell subpopulations that have been suggested in the literature include secreting insulin in response to nonglucose stimuli, preventing insulin secretion from neighboring cells until the high-glucose threshold is reached, and allowing for timely secretion shutoff across the islet after glucose stimulation (Giordano et al. 1991; Speier et al. 2007). Associating FLTP−, ST8SIA1+, or CD9+ and yet to be discovered β-cell subtypes with these and other roles will be important in our efforts to understand the cellular requirements for a healthy islet and whether changes in the cellular composition of an islet leads to loss of tissue function and diabetes.
Studying effects of heterogeneity on islet function: case study of KATP channel mutants
In light of the continual discovery of new heterogeneity markers and β-cell subtypes, it will be important to develop model systems in which to directly test how heterogeneity markers impact β-cell and islet function. In some cases, a reductionist approach, such as the one used to purify CD9/ST8SIA1 subtypes for secretion studies, may be useful in understanding how each population behaves. Such an approach depends on validated molecular heterogeneity (Box 1) markers for subtypes of interest, tractable tools for live-cell separation, and sufficient starting material to produce enough purified cells for downstream analysis. Unfortunately, purification of β-cell subtypes removes them from the native islet microenvironment. Cell isolation not only disrupts the cell–cell and cell–matrix interactions of β cells in the islet but also eliminates signals from non-β cells (such as other endocrine, endothelial, and neuronal cells) that may be important for the function of the islet as a whole. An alternative approach would be to use genetic methods to design model islets with defined numbers of different β-cell subtypes. This approach would depend on choosing an appropriate β-cell promoter and genetic recombination strategy for heterogeneity marker expression. The major advantage of a genetic approach is that the islet microenvironment is maintained, while the percentages and ratios of β-cell subtypes are manipulated.
It is instructive to consider a model example of how such a genetic mosaic approach was used to study the effect of β-cell heterogeneity in glucose sensitivity and Ca2+ activation on overall islet activation (Fig. 3). β-Cell heterogeneity in glucose sensing and insulin secretion has long been appreciated in studies of individual islet cells (Salomon and Meda 1986; Stefan et al. 1987; Kiekens et al. 1992; Van Schravendijk et al. 1992; Bennett et al. 1996; Roscioni et al. 2016). As described above, insulin secretion is initiated by increases in intracellular Ca2+ through voltage-dependent Ca2+ channels. These Ca2+ channels are in turn activated when KATP channels close, preventing potassium efflux and allowing for membrane depolarization. Islet β cells therefore need functional KATP channels to appropriately respond to increased ATP production resulting from glucose metabolism (Fig. 3A). To empirically test the extent to which cellular heterogeneity in glucose-dependent ATP production and Ca2+ signaling capacity impacts islet function, a number of groups have made use of mosaic islets from transgenic mice expressing mutant KATP channels in β cells.
Figure 3.

Effects of cellular heterogeneity in expression of KATP channel mutants on islet Ca2+ activation. (A) Dissociated β cells expressing wild-type KATP channel subunit Kir6.2 (Kir6.2WT) should be inactive in Ca2+ signaling at low glucose but active at high glucose (green curve). (B) In contrast, β cells expressing Kir6.2[ΔN30,K185Q] are inexcitable; they are resistant to depolarization and Ca2+ activation from increases in ATP resulting from increased metabolism in high glucose (red line). (C) β Cells expressing mutant Kir6.2[AAA] can be depolarized independently of glucose metabolism and are constitutively active (yellow line) even under low-glucose conditions compared with wild-type cells (green dashed curve). (D) Despite the glucose-independent behaviors of individual β cells expressing mutant Kir6.2, multicellular islets containing 70% β cells expressing constitutively active Kir6.2[AAA] (green and yellow curve) or up to 20% β cells expressing inactive Kir6.2[ΔN30,K185Q] (red and green curve) respond to low and high glucose similar to islets containing 100% Kir6.2WT β cells (green curve). Mosaic islets maintain glucose responsiveness largely due to cell–cell gap junction coupling. The figure was adapted and created by us from data from Rocheleau et al. (2006), Benninger et al. (2011), and Hraha et al. (2014).
A recent study made use of a mouse model with Cre-inducible expression of an overactive ATP-insensitive KATP channel component, Kir6.2[ΔN30,K185Q], that renders cells resistant to depolarization and thus inexcitable (Fig. 3B; Remedi et al. 2009; Benninger et al. 2011; Hraha et al. 2014). By crossing Kir6.2 conditional mutant alleles into a Pdx-CreER strain, the investigators could induce different percentages of inexcitable β cells in islets by altering the number of tamoxifen treatments (Hraha et al. 2014). Analysis of single-cell Ca2+ signaling activation on glucose stimulation in transgenic islets revealed that overall islet activation (read as percentage of Ca2+-active cells) was robust and similar to wild type in islets with <20% inexcitable cells (Fig. 3D). However, activation across the islet quickly deteriorated with >20% inexcitable cells. Islets with 1%–20% inexcitable cells performed similarly to wild-type islets in in vivo glucose regulation, in vitro Ca2+ signaling activation, and GSIS. Despite seemingly normal global function, Ca2+ imaging of individual β cells in the 1%–20% inexcitable islets revealed less sustained Ca2+ activation than cells in wild-type islets. Taken together, these findings suggest that, although even small numbers of electrically defective β cells can affect Ca2+ signaling dynamics at the individual cell level, robust glucose-stimulated depolarization across an islet requires only ∼80% electrically functional β cells. We point out that this study may overestimate the number of inexcitable cells that are permissive for overall islet function in light of the discovery of hub cells. Hub cells were only recently described, so the extent of mutant channel expression in hub and follower cells was not determined. However, considering the pacemaker properties of hub cells, one would surmise that mutant channel expression in hub cells would drastically affect global islet activation. Based on their lower expression of Pdx1, hub cells may not have been as efficiently targeted for mutant channel expression as follower cells with the Pdx1-CreER driver.
The previous study demonstrated the effect of heterogeneity in Ca2+ activation competency on islet activation in high-glucose conditions. Use of a different mutant KATP transgenic mouse has also provided insight into how Ca2+ signaling heterogeneity affects the suppression of insulin secretion in islets at basal glucose conditions. In this study, transgenic mice expressing a dominant-negative Kir6.2[AAA] KATP channel component (constitutively closed, allowing for easier or constitutive Ca2+ channel activation) and a GFP reporter in 70% of β cells were studied (Fig. 3C; Koster et al. 2002; Rocheleau et al. 2006). When transgenic islets expressing the inactive KATP channel were dissociated to single cells, individual GFP+ cells expressing the mutant channel exhibited sporadic Ca2+ signaling in both low and high concentrations of glucose, as expected from decoupling of glucose metabolism from channel gating (Rocheleau et al. 2006). However, intact transgenic islets with 70% of β cells expressing the Kir6.2[AAA] KATP channel had near-normal insulin secretion and Ca2+ signaling in response to low and high glucose (Fig. 3D). While the normal response to stimulatory glucose is not surprising because the cells expressing the mutant channel are easier to activate, the finding that the secretion of an islet with mostly hyperactive cells could be suppressed at basal glucose suggests that the 30% wild-type glucose-responsive β cells could silence the aberrant electrical capacity in neighboring mutant cells. The role of cell–cell coupling in the nonresponsiveness of transgenic islets at basal levels of glucose was directly probed by treating intact islets with a gap junction inhibitor in low-glucose conditions. The uncoupled transgenic islets in low glucose displayed aberrant and localized Ca2+ signaling, as would be expected with loss of cell–cell regulation. This work suggests that 30% electrically competent β cells can effectively silence dysregulated neighboring cells and underscores the power of gap–junction coupling in whole-islet suppression at basal conditions.
These mouse islet studies using mutant KATP channels have been insightful in defining the impressive levels of functional heterogeneity (up to 20% Kir6.2[ΔN30,K185Q] or >70% Kir6.2[AAA]) that can be tolerated for overall islet function (Fig. 3). Cell–cell coupling in intact islets could maintain near-normal insulin secretion behaviors despite the drastic perturbations to the membrane potential of the cells expressing mutant KATP channels. We consider it likely that many of the molecular heterogeneities now being discovered will be found (independently or even in concert) to be less consequential for glucose sensing and subsequent insulin secretion than disrupting the function of KATP channels. It is thus possible that even high percentages of novel β-cell subpopulations will have minimal effects on normal insulin secretion in a healthy islet. Different subpopulations may also have roles in other contexts. For instance, some heterogeneity markers may indicate subpopulations more resistant to stress that can maintain or replenish islet mass on tissue injury. Conversely, other heterogeneity markers may indicate β cells with subpar properties. Such defective cells could persist innocuously in healthy islets but contribute to islet dysfunction and disease when subpopulation ratios change in response to additional tissue insults. Mimicking physiological challenges and monitoring additional islet parameters may be required to fully elucidate the role of novel β-cell subtypes.
For now, it is unclear whether the results regarding β-cell heterogeneity and islet activation found in mice will translate to humans. Human islets are more variable in size and cellular architecture and generally have fewer (∼50% compared with ∼90% in mice) and more disperse β cells than rodent islets (Brissova et al. 2005; Cabrera et al. 2006; Bosco et al. 2010; Levetan and Pierce 2013; Bonner-Weir et al. 2015). In line with a more diffuse β-cell–cell intercommunication network than mouse islets, human islets display more localized Ca2+ activation synchronization (Johnston et al. 2016). With a less cohesive β-cell network, it is possible that changes in the cellular composition of β cells will have more drastic effects in human islets compared with mice. As directed differentiation protocols for generating β cells from hSC sources continue to improve, we may soon begin to use genetic manipulations similar to those in mouse models to address the effects of cell heterogeneity on communal Ca2+ signaling or insulin secretion behaviors in a human context.
Conclusion
Global research efforts continue to refine our understanding of the maturation process and the defining features of a mature β cell. Findings over the past few years have clarified the molecular changes in various signaling pathways within different compartments in the cell that accompany the acquisition of glucose-responsive insulin secretion. While we have begun to characterize the mechanistic relationship between certain players, such as disallowed genes and mitochondrial metabolism, work remains to be done to explain how other maturation drivers and markers relate to the concerted establishment of glucose sensing, metabolism, and insulin exocytotic machineries during the maturation transition.
In parallel, new modes of β-cell heterogeneity are continually being discovered. Understanding the role of different β-cell subpopulations on islet function or health should remain a priority as we delve into an era of single-cell-resolution analyses. The β-cell subpopulations reviewed here express varied and sometimes low levels of β-cell maturation genes, lending credence to the idea that functional islets may contain specialized β-cell subtypes in addition to a majority of functionally mature secretory cells. We anticipate that future studies on the specific roles of β-cell subpopulations in islets will refine our idea of what constitutes a healthy post-maturation islet.
Acknowledgments
We thank Dr. Gopika Nair, Dr. Audrey Parent, and Dr. Sapna Puri for critically reading the manuscript. We thank the National Institutes of Health for supporting the post-doctoral fellowship of J.S.E.L. (T32 CA177555) and the work in the laboratory of M.H. (R01 DK105831 and R01 DK108666).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.294389.116.
References
- Aguayo-Mazzucato C, Koh A, El Khattabi I, Li WC, Toschi E, Jermendy A, Juhl K, Mao K, Weir GC, Sharma A, et al. 2011. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat β cells. Diabetologia 54: 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguayo-Mazzucato C, Zavacki AM, Marinelarena A, Hollister-Lock J, El Khattabi I, Marsili A, Weir GC, Sharma A, Larsen PR, Bonner-Weir S. 2013. Thyroid hormone promotes postnatal rat pancreatic β-cell development and glucose-responsive insulin secretion through MAFA. Diabetes 62: 1569–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguayo-Mazzucato C, DiIenno A, Hollister-Lock J, Cahill C, Sharma A, Weir G, Colton C, Bonner-Weir S. 2015. MAFA and T3 drive maturation of both fetal human islets and insulin-producing cells differentiated from hESC. J Clin Endocrinol Metab 100: 3651–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arda HE, Li L, Tsai J, Torre EA, Rosli Y, Peiris H, Spitale RC, Dai C, Gu X, Qu K, et al. 2016. Age-dependent pancreatic gene regulation reveals mechanisms governing human β cell function. Cell Metab 23: 909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, Magnuson MA, Stein R. 2010. MafA and MafB regulate genes critical to β-cells in a unique temporal manner. Diabetes 59: 2530–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrahami D, Li C, Zhang J, Schug J, Avrahami R, Rao S, Stadler MB, Burger L, Schubeler D, Glaser B, et al. 2015. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved β cell function. Cell Metab 22: 619–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader E, Migliorini A, Gegg M, Moruzzi N, Gerdes J, Roscioni SS, Bakhti M, Brandl E, Irmler M, Beckers J, et al. 2016. Identification of proliferative and mature β-cells in the islets of Langerhans. Nature 535: 430–434. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Jetton TL, Ying G, Magnuson MA, Piston DW. 1996. Quantitative subcellular imaging of glucose metabolism within intact pancreatic islets. J Biol Chem 271: 3647–3651. [DOI] [PubMed] [Google Scholar]
- Benninger RK, Remedi MS, Head WS, Ustione A, Piston DW, Nichols CG. 2011. Defects in β cell Ca2+ signalling, glucose metabolism and insulin secretion in a murine model of K(ATP) channel-induced neonatal diabetes mellitus. Diabetologia 54: 1087–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss CR, Sharp GW. 1992. Glucose-induced insulin release in islets of young rats: time-dependent potentiation and effects of 2-bromostearate. Am J Physiol 263: E890–E896. [DOI] [PubMed] [Google Scholar]
- Blum B, Hrvatin SS, Schuetz C, Bonal C, Rezania A, Melton DA. 2012. Functional β-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol 30: 261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner-Weir S, Sullivan BA, Weir GC. 2015. Human islet morphology revisited: human and rodent islets are not so different after all. J Histochem Cytochem 63: 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco D, Armanet M, Morel P, Niclauss N, Sgroi A, Muller YD, Giovannoni L, Parnaud G, Berney T. 2010. Unique arrangement of α- and β-cells in human islets of Langerhans. Diabetes 59: 1202–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM, Powers AC. 2005. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem 53: 1087–1097. [DOI] [PubMed] [Google Scholar]
- Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, Caicedo A. 2006. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci 103: 2334–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, Schorle H, Sage J, Kim SK. 2011. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature 478: 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad E, Stein R, Hunter CS. 2014. Revealing transcription factors during human pancreatic β cell development. Trends Endocrinol Metab 25: 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Brissova M, Hang Y, Thompson C, Poffenberger G, Shostak A, Chen Z, Stein R, Powers AC. 2012. Islet-enriched gene expression and glucose-induced insulin secretion in human and mouse islets. Diabetologia 55: 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, Bhushan A. 2015. DNA methylation directs functional maturation of pancreatic β cells. J Clin Invest 125: 2851–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell C, Schug J, Lin CF, Canaday PS, Fox AJ, Smirnova O, Bonnah R, Streeter PR, Stoeckert CJ Jr, Kaestner KH, et al. 2011. Transcriptomes of the major human pancreatic cell types. Diabetologia 54: 2832–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell C, Schug J, Canaday PS, Russ HA, Tarlow BD, Grompe MT, Horton T, Hebrok M, Streeter PR, Kaestner KH, et al. 2016. Human islets contain four distinct subtypes of β cells. Nat Commun 7: 11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg M, Bottcher A, Burtscher I, Hasenoeder S, Van Campenhout C, Aichler M, Walch A, Grant SG, Lickert H. 2014. Flattop regulates basal body docking and positioning in mono- and multiciliated cells. Elife 3: e03842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano E, Bosco D, Cirulli V, Meda P. 1991. Repeated glucose stimulation reveals distinct and lasting secretion patterns of individual rat pancreatic B cells. J Clin Invest 87: 2178–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Stein GH, Pan N, Goebbels S, Hornberg H, Nave KA, Herrera P, White P, Kaestner KH, Sussel L, et al. 2010. Pancreatic β cells require NeuroD to achieve and maintain functional maturity. Cell Metab 11: 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimberg H, De Vos A, Vandercammen A, Van Schaftingen E, Pipeleers D, Schuit F. 1993. Heterogeneity in glucose sensitivity among pancreatic β-cells is correlated to differences in glucose phosphorylation rather than glucose transport. EMBO J 12: 2873–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellerstrom C, Swenne I. 1991. Functional maturation and proliferation of fetal pancreatic β-cells. Diabetes 40: 89–93. [DOI] [PubMed] [Google Scholar]
- Helman A, Avrahami D, Klochendler A, Glaser B, Kaestner KH, Ben-Porath I, Dor Y. 2016. Effects of ageing and senescence on pancreatic β-cell function. Diabetes Obes Metab 18: 58–62. [DOI] [PubMed] [Google Scholar]
- Hraha TH, Westacott MJ, Pozzoli M, Notary AM, McClatchey PM, Benninger RK. 2014. Phase transitions in the multi-cellular regulatory behavior of pancreatic islet excitability. PLoS Comput Biol 10: e1003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrvatin S, O'Donnell CW, Deng F, Millman JR, Pagliuca FW, DiIorio P, Rezania A, Gifford DK, Melton DA. 2014. Differentiated human stem cells resemble fetal, not adult, β cells. Proc Natl Acad Sci 111: 3038–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacovetti C, Matkovich SJ, Rodriguez-Trejo A, Guay C, Regazzi R. 2015. Postnatal β-cell maturation is associated with islet-specific microRNA changes induced by nutrient shifts at weaning. Nat Commun 6: 8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermendy A, Toschi E, Aye T, Koh A, Aguayo-Mazzucato C, Sharma A, Weir GC, Sgroi D, Bonner-Weir S. 2011. Rat neonatal β cells lack the specialised metabolic phenotype of mature β cells. Diabetologia 54: 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD. 2016. The quest to make fully functional human pancreatic β cells from embryonic stem cells: climbing a mountain in the clouds. Diabetologia 59: 2047–2057. [DOI] [PubMed] [Google Scholar]
- Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, Piemonti L, Marchetti P, Bugliani M, Bosco D, et al. 2016. β Cell hubs dictate pancreatic islet responses to glucose. Cell Metab 24: 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M, Handa H. 2002. MafA is a glucose-regulated and pancreatic β-cell-specific transcriptional activator for the insulin gene. J Biol Chem 277: 49903–49910. [DOI] [PubMed] [Google Scholar]
- Kiekens R, In ’t Veld P, Mahler T, Schuit F, Van De Winkel M, Pipeleers D. 1992. Differences in glucose recognition by individual rat pancreatic B cells are associated with intercellular differences in glucose-induced biosynthetic activity. J Clin Invest 89: 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RA, Smith RM, Dahlenburg GW. 1986. Long term postnatal development of insulin secretion in early premature infants. Early Hum Dev 13: 285–294. [DOI] [PubMed] [Google Scholar]
- Koster JC, Remedi MS, Flagg TP, Johnson JD, Markova KP, Marshall BA, Nichols CG. 2002. Hyperinsulinism induced by targeted suppression of β cell KATP channels. Proc Natl Acad Sci 99: 16992–16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperman Y, Chen A. 2008. Urocortins: emerging metabolic and energy homeostasis perspectives. Trends Endocrinol Metab 19: 122–129. [DOI] [PubMed] [Google Scholar]
- Lavine RL, Chick WL, Like AA, Makdisi TW. 1971. Glucose tolerance and insulin secretion in neonatal and adult mice. Diabetes 20: 134–139. [DOI] [PubMed] [Google Scholar]
- Lemaire K, Thorrez L, Schuit F. 2016. Disallowed and allowed gene expression: two faces of mature isletβ cells. Annu Rev Nutr 36: 45–71. [DOI] [PubMed] [Google Scholar]
- Levetan CS, Pierce SM. 2013. Distinctions between the islets of mice and men: implications for new therapies for type 1 and 2 diabetes. Endocr Pract 19: 301–312. [DOI] [PubMed] [Google Scholar]
- Li C, Chen P, Vaughan J, Blount A, Chen A, Jamieson PM, Rivier J, Smith MS, Vale W. 2003. Urocortin III is expressed in pancreatic β-cells and stimulates insulin and glucagon secretion. Endocrinology 144: 3216–3224. [DOI] [PubMed] [Google Scholar]
- Li C, Chen P, Vaughan J, Lee KF, Vale W. 2007. Urocortin 3 regulates glucose-stimulated insulin secretion and energy homeostasis. Proc Natl Acad Sci 104: 4206–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Jetton TL, Zimmerman EC, Najafi H, Matschinsky FM, Magnuson MA. 1991. Effects of alternate RNA splicing on glucokinase isoform activities in the pancreatic islet, liver, and pituitary. J Biol Chem 266: 6999–7007. [PubMed] [Google Scholar]
- Martin D, Allagnat F, Chaffard G, Caille D, Fukuda M, Regazzi R, Abderrahmani A, Waeber G, Meda P, Maechler P, et al. 2008. Functional significance of repressor element 1 silencing transcription factor (REST) target genes in pancreatic β cells. Diabetologia 51: 1429–1439. [DOI] [PubMed] [Google Scholar]
- Martin D, Allagnat F, Gesina E, Caille D, Gjinovci A, Waeber G, Meda P, Haefliger JA. 2012. Specific silencing of the REST target genes in insulin-secreting cells uncovers their participation in β cell survival. PLoS One 7: e45844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Sanchez A, Nguyen-Tu MS, Rutter GA. 2015. DICER inactivation identifies pancreatic β-cell ‘disallowed’ genes targeted by microRNAs. Mol Endocrinol 29: 1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka TA, Artner I, Henderson E, Means A, Sander M, Stein R. 2004. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc Natl Acad Sci 101: 2930–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka TA, Kaneto H, Miyatsuka T, Yamamoto T, Yamamoto K, Kato K, Shimomura I, Stein R, Matsuhisa M. 2010. Regulation of MafA expression in pancreatic β-cells in db/db mice with diabetes. Diabetes 59: 1709–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch LJ, van de Bunt M, Braun M, Frayn KN, Clark A, Gloyn AL. 2011. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic β cells: implications for understanding genetic association signals at this locus. Mol Genet Metab 104: 648–653. [DOI] [PubMed] [Google Scholar]
- Mellado-Gil JM, Jimenez-Moreno CM, Martin-Montalvo A, Alvarez-Mercado AI, Fuente-Martin E, Cobo-Vuilleumier N, Lorenzo PI, Bru-Tari E, Herrera-Gomez Ide G, Lopez-Noriega L, et al. 2016. PAX4 preserves endoplasmic reticulum integrity preventing β cell degeneration in a mouse model of type 1 diabetes mellitus. Diabetologia 59: 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moukil MA, Veiga-da-Cunha M, Van Schaftingen E. 2000. Study of the regulatory properties of glucokinase by site-directed mutagenesis: conversion of glucokinase to an enzyme with high affinity for glucose. Diabetes 49: 195–201. [DOI] [PubMed] [Google Scholar]
- Muraro MJ, Dharmadhikari G, Grun D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJ, et al. 2016. A single-cell transcriptome atlas of the human pancreas. Cell Syst 3: 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair G, Hebrok M. 2015. Islet formation in mice and men: lessons for the generation of functional insulin-producing β-cells from human pluripotent stem cells. Curr Opin Genet Dev 32: 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olbrot M, Rud J, Moss LG, Sharma A. 2002. Identification of β-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci 99: 6737–6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. 2014. Generation of functional human pancreatic β cells in vitro. Cell 159: 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piston DW, Knobel SM, Postic C, Shelton KD, Magnuson MA. 1999. Adenovirus-mediated knockout of a conditional glucokinase gene in isolated pancreatic islets reveals an essential role for proximal metabolic coupling events in glucose-stimulated insulin secretion. J Biol Chem 274: 1000–1004. [DOI] [PubMed] [Google Scholar]
- Pullen TJ, Khan AM, Barton G, Butcher SA, Sun G, Rutter GA. 2010. Identification of genes selectively disallowed in the pancreatic islet. Islets 2: 89–95. [DOI] [PubMed] [Google Scholar]
- Puri S, Folias AE, Hebrok M. 2015. Plasticity and dedifferentiation within the pancreas: development, homeostasis, and disease. Cell Stem Cell 16: 18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remedi MS, Nichols CG. 2009. Hyperinsulinism and diabetes: genetic dissection of β cell metabolism-excitation coupling in mice. Cell Metab 10: 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remedi MS, Kurata HT, Scott A, Wunderlich FT, Rother E, Kleinridders A, Tong A, Bruning JC, Koster JC, Nichols CG. 2009. Secondary consequences of β cell inexcitability: identification and prevention in a murine model of K(ATP)-induced neonatal diabetes mellitus. Cell Metab 9: 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O'Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. 2014. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 32: 1121–1133. [DOI] [PubMed] [Google Scholar]
- Rocheleau JV, Remedi MS, Granada B, Head WS, Koster JC, Nichols CG, Piston DW. 2006. Critical role of gap junction coupled KATP channel activity for regulated insulin secretion. PLoS Biol 4: e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, Arkhammar P, Bokvist K, Hellerstrom C, Nilsson T, Welsh M, Welsh N, Berggren PO. 1989. Failure of glucose to elicit a normal secretory response in fetal pancreatic β cells results from glucose insensitivity of the ATP-regulated K+ channels. Proc Natl Acad Sci 86: 4505–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscioni SS, Migliorini A, Gegg M, Lickert H. 2016. Impact of islet architecture on β-cell heterogeneity, plasticity and function. Nat Rev Endocrinol 12: 695–709. [DOI] [PubMed] [Google Scholar]
- Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, Guo T, Puri S, Haataja L, Cirulli V, et al. 2015. Controlled induction of human pancreatic progenitors produces functional β-like cells in vitro. EMBO J 34: 1759–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter GA, Pullen TJ, Hodson DJ, Martinez-Sanchez A. 2015. Pancreatic β-cell identity, glucose sensing and the control of insulin secretion. Biochem J 466: 203–218. [DOI] [PubMed] [Google Scholar]
- Salomon D, Meda P. 1986. Heterogeneity and contact-dependent regulation of hormone secretion by individual B cells. Exp Cell Res 162: 507–520. [DOI] [PubMed] [Google Scholar]
- Segerstolpe A, Palasantza A, Eliasson P, Andersson EM, Andreasson AC, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, et al. 2016. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 24: 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speier S, Gjinovci A, Charollais A, Meda P, Rupnik M. 2007. Cx36-mediated coupling reduces β-cell heterogeneity, confines the stimulating glucose concentration range, and affects insulin release kinetics. Diabetes 56: 1078–1086. [DOI] [PubMed] [Google Scholar]
- Stefan Y, Meda P, Neufeld M, Orci L. 1987. Stimulation of insulin secretion reveals heterogeneity of pancreatic B cells in vivo. J Clin Invest 80: 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolovich-Rain M, Enk J, Vikesa J, Nielsen FC, Saada A, Glaser B, Dor Y. 2015. Weaning triggers a maturation step of pancreatic β cells. Dev Cell 32: 535–545. [DOI] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, Accili D. 2012. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150: 1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorrez L, Laudadio I, Van Deun K, Quintens R, Hendrickx N, Granvik M, Lemaire K, Schraenen A, Van Lommel L, Lehnert S, et al. 2011. Tissue-specific disallowance of housekeeping genes: the other face of cell differentiation. Genome Res 21: 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Arensbergen J, Garcia-Hurtado J, Moran I, Maestro MA, Xu X, Van de Casteele M, Skoudy AL, Palassini M, Heimberg H, Ferrer J. 2010. Derepression of Polycomb targets during pancreatic organogenesis allows insulin-producing β-cells to adopt a neural gene activity program. Genome Res 20: 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Huising MO. 2014. Maturation of stem cell-derived β-cells guided by the expression of urocortin 3. Rev Diabet Stud 11: 115–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Xie R, Kelly OG, Vale WW, Sander M, Huising MO. 2012. Urocortin 3 marks mature human primary and embryonic stem cell-derived pancreatic α and β cells. PLoS One 7: e52181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Donaldson CJ, Caceres E, Hunter AE, Cowing-Zitron C, Pound LD, Adams MW, Zembrzycki A, Grove KL, Huising MO. 2015. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat Med 21: 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Schravendijk CF, Kiekens R, Pipeleers DG. 1992. Pancreatic β cell heterogeneity in glucose-induced insulin secretion. J Biol Chem 267: 21344–21348. [PubMed] [Google Scholar]
- Wagner A, Regev A, Yosef N. 2016. Revealing the vectors of cellular identity with single-cell genomics. Nat Biotechnol 34: 1145–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Brun T, Kataoka K, Sharma AJ, Wollheim CB. 2007. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 50: 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Golson ML, Schug J, Traum D, Liu C, Vivek K, Dorrell C, Naji A, Powers AC, Chang KM, et al. 2016a. Single-cell mass cytometry analysis of the human endocrine pancreas. Cell Metab 24: 616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Schug J, Won KJ, Liu C, Naji A, Avrahami D, Golson ML, Kaestner KH. 2016b. Single-cell transcriptomics of the human endocrine pancreas. Diabetes 65: 3028–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh N, Svensson C, Welsh M. 1989. Content of adenine nucleotide translocator mRNA in insulin-producing cells of different functional states. Diabetes 38: 1377–1380. [DOI] [PubMed] [Google Scholar]
- Yoshihara E, Wei Z, Lin CS, Fang S, Ahmadian M, Kida Y, Tseng T, Dai Y, Yu RT, Liddle C, et al. 2016. ERRγ is required for the metabolic maturation of therapeutically functional glucose-responsive β cells. Cell Metab 23: 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K, et al. 2005. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol 25: 4969–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]