

Guo et al. show that liver tumor-initiating cells (TICs) actively recruit M2 macrophages from as early as the single-cell stage. Elimination of TIC-associated macrophages abolishes tumorigenesis in a manner dependent on the immune system.
Keywords: tumor-initiating cell, macrophage, liver cancer, immunosurveillance, YAP, Hippo pathway
Abstract
Tumor infiltrated type II (M2) macrophages promote tumorigenesis by suppressing immune clearance, promoting proliferation, and stimulating angiogenesis. Interestingly, macrophages were also found to enrich in small foci of altered hepatocytes containing liver tumor-initiating cells (TICs). However, whether and how TICs specifically recruit macrophages and the function of these macrophages in tumor initiation remain unknown due to technical difficulties. In this study, by generating genetically defined liver TICs, we demonstrate that TICs actively recruit M2 macrophages from as early as the single-cell stage. Elimination of TIC-associated macrophages (TICAMs) abolishes tumorigenesis in a manner dependent on the immune system. Mechanistically, activation of the Hippo pathway effector Yes-associated protein (YAP) underlies macrophage recruitment by TICs. These results demonstrate for the first time that macrophages play a decisive role in the survival of single TICs in vivo and provide a proof of principle for TIC elimination by targeting YAP or M2 macrophages.
Tumor cells display extensive and dynamic cross-talk with inflammatory cells in the tumor microenvironment (Mantovani et al. 2008; Grivennikov et al. 2010). Macrophages are a major type of immune cell that participate in this tumor microenvironment interplay. For instance, in tissues with chronic inflammation caused by pathogen infection, smoking, or obesity, macrophages are capable of creating an environment that promotes cellular acquisition of tumorigenic mutational hits (Grivennikov et al. 2010). Macrophages involved in these tumor-initiating systematic inflammations are of the immune-activated (M1) type and are not specifically associated with tumor cells. Interestingly, established tumors also actively recruit macrophages (tumor-associated macrophages [TAMs]) and educate them to be protumorigenic (Noy and Pollard 2014). TAMs adopt a trophic immunosuppressive phenotype that is functionally reminiscent of the alternatively activated type II (M2) macrophages (Wynn et al. 2013). TAMs promote tumor progression by facilitating angiogenesis, promoting proliferation and invasion, and suppressing immunosurveillance. It was generally believed that the recruitment of macrophages into established tumors was mediated by factors such as CSF1, VEFGA, and CCL2 secreted from the altered and often hypoxic tumor microenvironment (Grivennikov et al. 2010). However, macrophages were also found to enrich in carcinogen diethylnitrosamine (DEN)-induced small foci of altered hepatocytes (FAHs) containing tumor-initiating cells (TICs) in which a typical hypoxic tumor microenvironment was absent (He et al. 2013). This observation suggests that, during tumorigenesis, macrophages could be recruited at a much earlier stage. However, due to technical difficulties involved in the tracing and manipulation of individual TICs in vivo, the mechanism of recruitment and the function of these TIC-associated macrophages (TICAMs) have not been determined experimentally.
Interestingly, DEN-induced FAHs not only express liver progenitor markers but also exhibit activated Yes-associated protein (YAP) (He et al. 2013). YAP is a transcription coactivator and a major effector of the Hippo pathway, which plays a fundamental role in limiting organ size (Pan 2010; Halder and Johnson 2011; Yu et al. 2015). The Hippo pathway kinases Lats1/2 phosphorylate YAP on five serine residues, which results in its cytoplasmic retention and degradation (Dong et al. 2007; Zhao et al. 2007, 2010; Hao et al. 2008; Oka et al. 2008). YAP promotes cell proliferation and inhibits apoptosis by regulating gene expression, largely through TEAD family transcription factors (Zhao et al. 2008; Galli et al. 2015; Zanconato et al. 2015). Interestingly, YAP also induces dedifferentiation of mature hepatocytes into liver progenitor cells (oval cells) (Yimlamai et al. 2014). Furthermore, the deregulation of the Hippo pathway has been observed in many human cancers, and the potent tumorigenic activity of YAP has been demonstrated in genetically engineered mouse models, particularly in the liver (Dong et al. 2007; Zhou et al. 2009; Lee et al. 2010; Lu et al. 2010; Song et al. 2010; Yimlamai et al. 2015; Zhang et al. 2015). These facts prompted us to investigate whether YAP plays a role in the interplay between TICs and macrophages and determine the role of TICAMs in tumorigenesis.
Results
YAP activation recruits macrophages to single TICs
Recent reports have demonstrated hydrodynamic tail vein injection (HDI) (Liu et al. 1999) of plasmids as a convenient approach for genetic modification in the mouse liver (Chen and Calvisi 2014; Xue et al. 2014). To investigate whether YAP activation in hepatocytes causes macrophage recruitment, we constructed a piggyBac (PB) transposon element (Ding et al. 2005) expressing human YAP (Fig. 1A). Coinjection of the transposon with a PB transposase plasmid via HDI led to transient expression in ∼20% (Supplemental Fig. S1A,B) and stable integration in <1% of hepatocytes (data not shown). The expression of the YAP-5SA active mutant (Zhao et al. 2007) (serine-to-alanine mutation at all five Hippo pathway phosphorylation sites; used in the following study unless specified) induced liver tumors in a course of 3 mo and induced large tumors within 4 mo in all of the >30 mice examined (Fig. 1B; Supplemental Fig. S2), while the expression of wild-type YAP, the TEAD-binding-deficient YAP-5SA-S94A mutant (Zhao et al. 2008), or control red fluorescent protein (RFP) did not induce tumors. Immunohistochemistry (IHC) with an antibody recognizing human but not mouse YAP confirmed that the tumors originated from human YAP-expressing hepatocytes (Fig. 1C). These active YAP-induced tumors are highly proliferative, as indicated by proliferation marker Ki67. These tumors are also poorly differentiated, as indicated by the expression of hepatic progenitor cell (oval cell) and bile duct marker keratin 19 (K19) and the loss of hepatocyte differentiation marker HNF4α (Fig. 1C). By analyzing liver slices at post-injection day 10, we found massive recruitment of CD45+ leukocytes to YAP+ cells (Fig. 1D). More strikingly, the recruitment of macrophages began as early as post-injection day 2 at the singe-cell stage (Fig. 1E). A previous report has demonstrated that YAP expression dedifferentiates hepatocytes into progenitor cells so that they could self-renew, differentiate, and repopulate damaged livers (Yimlamai et al. 2014). Indeed, YAP activation quickly induced K19 in hepatocytes (Fig. 1F), although full conversion to oval cell morphology required more time (day 20). We also noticed that many YAP+ cells exhibited aberrant morphology from post-injection day 5 to day 15, often with enlarged nuclei and spread cytoplasm with vacuoles (Fig. 1F). Such morphology is reminiscent of cellular senescence or failure of cytokinesis in proliferating cells. However, the aberrant morphology was largely resolved before day 20, and the remaining clones quickly expanded, indicating that YAP expression overrides or circumvents senescence. While oncogene-induced senescence recruits lymphocytes through the senescence-associated secretory phenotype (SASP) (Kang et al. 2011), YAP-induced macrophage recruitment begins before the appearance of the aberrant morphology and persists after its resolution (Fig. 1F), suggesting a more direct mechanism. Further analysis of dissociated liver cells by flow cytometry confirmed that expression of active YAP, but not the inactive mutant, caused a threefold increase of CD45+ leukocytes (Fig. 1G), consisting mainly of CD11b+/F4/80+ macrophages, whereas Gr-1+/Ly6c+ myeloid-derived suppressor cells (MDSCs), NK cells, and T cells were rare (Fig. 1G). This is in contrast to immune cells recruited by oncogene RAS-induced SASP, which consisted largely of neutrophils and lymphocytes and, to a much smaller extent, monocytes/macrophages (Kang et al. 2011). These data indicate that YAP activation is sufficient to recruit TICAMs to liver TICs.
Figure 1.

YAP activation strongly recruits macrophages to TICs. (A) Plasmids expressing the PB transposase and human YAP from a transposon element were delivered by HDI. (B) Active YAP induces liver tumorigenesis. Representative livers at 4 mo after injection. n = 6 mice. (C) Pathological analysis of YAP-induced liver tumors. Haematoxylin and eosin (HE) and IHC staining of liver tumors induced by the HDI of active YAP. Results are representative of more than five independent experiments. (D) YAP expression recruits leukocytes. Post-injection day 10 liver sections of YAP-5SA-injected mice were stained for CD45 and human YAP. Results are representative of more than five independent experiments. (E,F) Time course of macrophage recruitment and hepatocyte dedifferentiation induced by YAP. Serial liver sections of YAP-5SA-injected mice were stained for human YAP, F4/80, and K19. Results are representative of three independent experiments. (G) Flow cytometry analysis of immune cells recruited by YAP expression. Dissociated hepatic cells from YAP-5SA-injected mice at post-injection day 10 were stained, and CD45+ leukocytes were further analyzed for macrophage, MDSC, and lymphocyte markers. Results are representative of three independent experiments.
To determine whether the activation of endogenous Yap could also generate TICs and recruit TICAMs, we injected plasmids expressing the Cas9 enzyme and single-guide RNAs (sgRNAs) targeting Lats1/2 (Fig. 2A). In contrast to the traditional liver-specific Lats1/2 knockout mouse model (Lee et al. 2016; Yi et al. 2016), in which liver failure is caused by deficient hepatocyte maturation, our approach benefited from the mosaic nature of the model, and we observed tumorigenesis within 8 mo (Fig. 2B). The tumors resembled those induced by active YAP in the expression of K19 and were infiltrated by macrophages (Fig. 2C). To confirm the cellular origin of the tumors, we cloned the fragments of Lats1 and Lats2 predicted to be targeted by the sgRNAs from total tumor DNA and sequenced 20 clones for both Lats1 and Lats2. We found that 45% of clones carried insertions/deletions (indels) for Lats1 and that 50% of clones carried indels for Lats2 (Fig. 2D,E). However, no indels were found for either Lats1 or Lats2 in control-injected livers. Furthermore, Yap phosphorylation was markedly inhibited in tumors, indicating functional inactivation of Lats1/2 (Fig. 2F). Thus, successful Lats1/2 knockout by a Cas9-mediated strategy led to tumorigenesis. Clonal activation of endogenous Yap at an earlier stage was indicated by its translocation from the cytoplasm to the nucleus in RFP-positive clones, which was accompanied by the recruitment of CD45+ cells (Fig. 2G). However, limited by the efficiency of Cas9-mediated gene editing, not all RFP-positive cells exhibited enhanced nuclear Yap (data not shown). This suggests that, as a surrogate for sgRNA expression, RFP is not a reliable indicator of Lats1/2 knockout. In addition, despite extensive trials, we did not find an antibody suitable for IHC staining of mouse Lats1/2. We thus prepared an anti-active YAP antibody recognizing S127 unphosphorylated YAP. The specificity of this antibody in Western blotting and IHC staining was fully validated (Supplemental Fig. S3A,B). Using this new reagent, we found that there were, on average, 4.4 ± 1.6 (mean ± SD) clones positive for active Yap in every 3.7 mm2 view of liver sections (Supplemental Fig. S3C). These clones were also K19+ and were surrounded by CD45+/F4/80+ macrophages (Fig. 2H). Lats1/2 are activated by upstream kinases Mst1/2, and Mst1/2 knockout also leads to Yap activation and liver enlargement followed by tumorigenesis (Zhou et al. 2009; Lu et al. 2010; Song et al. 2010). We therefore examined whether hepatocyte-specific Mst1/2 knockout recruits macrophages before tumorigenesis (Fig. 2I). Indeed, ablation of Mst1/2 using traditional genetic engineering approaches also led to strong infiltration of macrophages (Fig. 2J). These data indicate that deregulation of the endogenous Hippo pathway converts hepatocytes to TICs and leads to TICAM recruitment.
Figure 2.

Inactivation of the endogenous Hippo pathway recruits TICAMs. (A) Design of Cas9-mediated Lats1/2 knockout through HDI. (B) Lats1/2 knockout induces liver tumorigenesis. Representative livers 8 mo after injection, n = 5 mice. (C) HE and IHC staining of nontumor tissue and liver tumors induced by Lats1/2 knockout. Results are representative of three independent experiments. (D,E) The fragments of Lats1 (D) and Lats2 (E) genes targeted by sgRNAs were PCR-amplified from total tumor genomic DNA and sequenced. Representative sequences are shown. Red arrow lines denote predicted Cas9 cutting sites. (F) Tumors induced by Lats1/2 knockout exhibit a low YAP phosphorylation level as determined by Western blotting. Results are representative of two independent experiments. (G) Enhanced nuclear Yap level and CD45+ leukocyte recruitment to clones induced by Lats1/2 knockout. Immunofluorescence staining of post-injection day 30 serial liver sections for Yap and CD45. The arrow indicates an RFP-positive cell with nuclear Yap, and the arrowhead indicates an RFP-negative cell with cytoplasmic Yap. Results are representative of three independent experiments. (H) IHC staining of Lats1/2 knockout clones in post-injection day 30 serial liver sections. Note the oval cell-like morphology of cells with active Yap. Results are representative of three independent experiments. (I) Liver-specific Mst1/2 double knockout increases liver size. Livers at 2 mo of age were imaged. Images are representative of five mice. (J) Enhanced macrophage infiltration in Mst1/2 double-knockout livers. Sections of livers the same as those in I were stained for macrophage marker F4/80. Results are representative of three independent experiments.
YAP recruits TICAMs through induction of Ccl2 and Csf1 expression
We next determined how YAP expression recruits TICAMs. We found that the TEAD-binding-deficient 5SA-S94A mutant was defective for TICAM induction (Fig. 3A) and that the coexpression of VGLL4, a YAP-competitive TEAD-binding protein (Koontz et al. 2013; Jiao et al. 2014; Zhang et al. 2014), blocked YAP-induced TICAM recruitment (Fig. 3B). These data demonstrate that YAP–TEAD transcriptional activity mediates TICAM recruitment and exclude the role of a direct immunoreaction to human YAP protein in this process. Cytokine arrays indicated an induction of Ccl2 and Csf1 by active YAP (Fig. 3C), which was confirmed at the mRNA level (Fig. 3D). In these experiments, TICAMs were eliminated by the ablation of the mouse immune system through total body irradiation (Fig. 3A), thus excluding the possibility of TICAM-derived cytokines. Chromatin immunoprecipitation (ChIP) indicated that TEAD1 and YAP, but not the S94A mutant, bound directly to the promoter of Ccl2 (Fig. 3E,F). Indeed, YAP and TEAD1 also strongly activated a Ccl2 reporter gene (Fig. 3G). Therefore, Ccl2 is a direct YAP–TEAD target gene in TICs. The association of YAP and TEAD with the Csf1 promoter was not significant (Fig. 3E,F), and thus Csf1 expression might be induced through alternative mechanisms. Furthermore, inactivation of the endogenous Hippo pathway by Mst1/2 knockout in the liver also leads to robust Ccl2 induction and a milder but significant induction of Csf1 expression (Fig. 3H). These data indicate that Ccl2 and Csf1 are YAP target genes.
Figure 3.

YAP activation recruits TICAMs through induction of Ccl2 and Csf1 expression. (A) Elimination of TICAM recruitment by blocking TEAD binding to YAP or irradiation of mice. Human YAP IHC staining of the indicated liver sections at post-injection day 5. Note the presence or absence of macrophages around YAP-positive cells. Results are representative of three independent experiments. (B) VGLL4 inhibits TICAM recruitment by YAP expression. YAP-5SA and HA-VGLL4 were coinjected. Post-injection day 5 liver sections were IHC-stained for human YAP and HA-VGLL4. The arrowhead indicates a clone expressing both YAP and VGLL4. The arrow indicates a clone expressing YAP only. Images are representative of images from three mice. (C) YAP-induced secretion of Ccl2 and Csf1 as determined by cytokine arrays of whole-liver lysates at post-injection day 5 in a single experiment. (D) YAP stimulates Ccl2 and Csf1 mRNA levels, as determined by real-time RT–PCR in post-injection day 5 livers. Three technical replicates from two mice. The experiment was reproduced three times in the laboratory. (E) YAP binds to the Ccl2 promoter. Liver lysates from 5SA-injected or 5SA-S94A-injected mice the day after injection were subjected to ChIP using control IgG or anti-human YAP. Three PCR technical replicates from a single experiment representative of three independent experiments. (F) TEAD1 binds to the Ccl2 promoter. Liver lysates from YAP-5SA-injected mice the day after injection were subjected to ChIP using control IgG or anti-TEAD1. Three PCR technical replicates from a single experiment representative of three independent experiments. (G) YAP and TEAD1 activate the Ccl2 promoter in the luciferase reporter assay. Three biological replicates from a single experiment representative of two independent experiments. (H) Enhanced Ccl2 and Csf1 mRNA levels in Mst1/2 double-knockout livers. Ccl2 and Csf1 expression in livers of 2-mo-old wild-type and Mst1/2 double-knockout mice were determined by RT–PCR. Three technical replicates from two mice in a single experiment. (I) YAP recruits immune cells expressing Ccr2 and TAM markers. The mRNA levels of the indicated genes were determined in immune cells enriched by Percoll fractionation. Three technical replicates from a single experiment representative of two independent experiments. (J) The treatment scheme for the Ccr2 depleter mice. (K) Human YAP and F4/80 IHC staining of YAP-injected Ccr2 depleter mouse liver sections at post-injection day 4. Results are representative of three independent experiments. Values represent mean + SD.
Ccl2 attracts cells expressing its receptor, Ccr2, from the circulation. Consistently, TICAM-enriched nonparenchymal cells isolated from YAP-injected livers expressed high levels of Ccr2 (Fig. 3I). In addition, these cells also exhibited elevated expression of markers expressed in TAMs (Fig. 3I; Franklin et al. 2014; Roszer 2015). To evaluate the role of Ccl2–Ccr2 in TICAM recruitment, we used the Ccr2 depleter mice (Hohl et al. 2009), which lost Ccr2+ macrophages after the injection of diphtheria toxin (DT) (Fig. 3J; Supplemental Fig. S4A). We observed that DT injection eliminated YAP-induced TICAMs in Ccr2 depleter mice but not nontransgenic control mice (Fig. 3K; Supplemental Fig. S4B). These data indicate that YAP-induced Ccl2 expression plays a critical role in TICAM recruitment.
TICAMs are required for tumorigenesis
We further investigated how YAP-induced TICAMs would affect tumorigenesis. However, depletion of all Ccr2+ cells in YAP-injected mice caused early lethality around post-injection day 10 and prevented further analysis of tumorigenesis. Examination of the livers from these mice revealed the massive expansion of YAP+ cells (Supplemental Fig. S4C), and thus the mice likely died from liver failure due to YAP-induced hepatocyte dedifferentiation. This would suggest that TICAMs play a tumor-suppressive role by eliminating YAP+ TICs. However, we noticed that normal liver macrophages (Kupffer cells) were also eliminated in the Ccr2 depleter mice at the dose of DT that we used (Supplemental Fig. S4B). It has been reported that depletion of Kupffer cells dramatically increases liver tumorigenesis from xenograft cancer cells (Heuff et al. 1993; Oosterling et al. 2005). Thus, the functional role of TICAMs needs to be dissected from that of Kupffer cells using a more specific approach.
We thus combined Ccl2/Csf1 shRNAs with YAP in a single transposon element to achieve simultaneous YAP expression and Ccl2/Csf1 knockdown after hydrodynamic delivery (Fig. 4A,B), which indeed blocked YAP-induced TICAM recruitment (Fig. 4C). Two different combinations of Ccl2 and Csf1 shRNAs also largely repressed TICAM recruitment, albeit less completely, thus supporting the specificity of our shRNAs (Fig. 4D). Since the shRNAs were expressed in the same cells as YAP, these data further confirm that TICAMs are recruited by Ccl2/Csf1 secreted by YAP+ TICs. Strikingly, the elimination of TICAMs blocked tumorigenesis from YAP-induced TICs (Fig. 4E,F). To determine whether macroscopically invisible nodules exist in livers expressing YAP and Ccl2/Csf1 shRNAs together, we stained liver sections for human YAP. To our surprise, we did not find any YAP+ nodules when Ccl2 and Csf1 shRNAs were expressed (Fig. 4G). Thus, unlike liver residential Kupffer cells, TICAMs are required for YAP-induced tumorigenesis.
Figure 4.
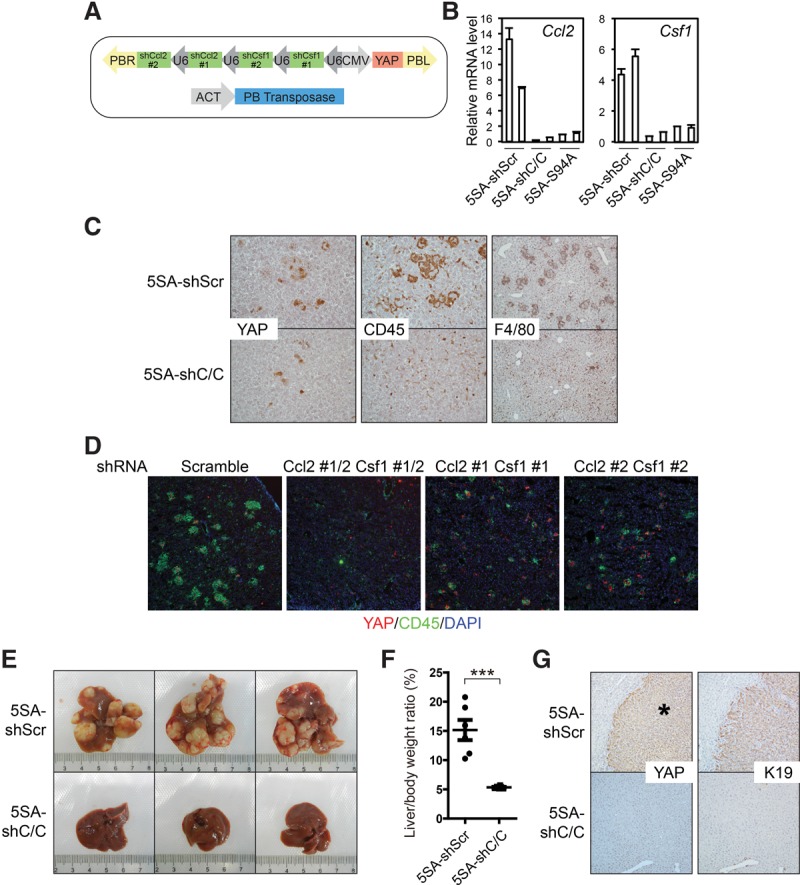
YAP-induced TICAMs are essential for tumorigenesis. (A) Design of the plasmids for Ccl2 and Csf1 knockdown together with YAP expression. (B) shRNA expression normalizes YAP-induced Ccl2 and Csf1 mRNA levels, as determined at post-injection day 3. Three technical replicates from two mice. The experiment was duplicated in the laboratory. (C) Knockdown of Ccl2 and Csf1 eliminates YAP-induced TICAMs. IHC staining of post-injection day 4 liver sections of mice injected with YAP-5SA-shScramble or YAP-5SA-shCcl2/Csf1. Images are representative of more than five mice. (D) Two different combinations of Ccl2 and Csf1 shRNAs eliminate TICAM recruitment by YAP expression. Post-injection day 3 liver sections were stained for CD45 and human YAP. Images are representative of two mice. (E) Ccl2/Csf1 knockdown blocks YAP-induced tumorigenesis. Representative liver photographs 4 mo after injection. n = 6 mice. The experiments were reproduced three times in the laboratory. (F) Quantification of the liver/body weight ratio of the mice in E. n = 6. P-values were calculated by Student's t-test. (***) P < 0.001. (G) The absence of human YAP-positive nodules in YAP-5SA-shCcl2/Csf1-injected livers at 4 mo after injection. Liver sections of mice injected with YAP-5SA-shScramble or YAP-5SA-shCcl2/Csf1 were IHC-stained for human YAP and K19. The asterisk denotes a tumor. Images are representative of three mice.
TICAMs protect TICs from immune clearance
The complete elimination of YAP-expressing nodules by Ccl2/Csf1 knockdown suggests that YAP+ TICs were eliminated at an early stage. To determine the effects of TICAM eradication on TICs, we traced Ccl2/Csf1 knockdown YAP+ clones up to 10 d after injection. The number of clones expressing YAP together with scramble shRNAs dropped gradually mainly due to the cessation of transient YAP expression from unintegrated plasmids. However, clones with Ccl2/Csf1 knockdown exhibit a marked decrease in clone number (Fig. 5A,B). Rare leftover YAP+ clones were K19− non-TIC single cells, likely due to unfaithful YAP gene integration during transposition and abortive hepatocyte dedifferentiation (Fig. 5C). Therefore, YAP+ TICs were eliminated without TICAMs. Notably, Ccl2/Csf1 shRNAs did not affect the number of control RFP-expressing clones (Fig. 5D), thus excluding the role of nonspecific toxicity of shRNA expression in the clearance of YAP+ clones.
Figure 5.

YAP-induced TICAMs protect TICs from immune clearance. (A) Ccl2/Csf1 knockdown leads to the clearance of YAP-positive cells. Liver sections at post-injection day 2 to day 10 were stained for human YAP. Results are representative of three independent experiments. (B) Quantification of YAP-positive clones in the same liver sections as those in A. Six views on sections from two mice for each genotype per time point were quantified. The experiments were reproduced three times. P-values were calculated by Student's t-test. (**) P < 0.01; (***) P < 0.001. (C) IHC staining of the remaining clones in serial liver sections of YAP-5SA-shCcl2/Csf1-injected mice at post-injection day 10. Results are representative of three independent experiments. (D) The expression of Ccl2 and Csf1 shRNAs does not cause cell clearance due to toxicity. Transposon elements carrying RFP alone or together with scrambled or Ccl2/Csf1 shRNAs were injected. The liver sections from post-injection day 2 to day 15 were quantified for RFP-positive clones. n = 2 mice in a single experiment. P-values > 0.05, as calculated by Student's t-test. (E) Experiments similar to those in B except that Rag1−/− mice were used. P-values > 0.05, as calculated by Student's t-test. The experiments were duplicated in the laboratory. (F) Rag1 knockout rescues TICAM-negative YAP-expressing clones. The liver sections of injected Rag1−/− mice from post-injection day 2 to day 10 were stained for human YAP. Results are representative of two independent experiments. (G) TICAMs are not required for the proliferation of YAP-expressing cells. Liver sections of injected Rag1−/− mice at post-injection day 10 were stained for human YAP and Ki67. Ki67+ YAP-expressing cells are denoted by arrowheads, a Ki67+ macrophage is denoted by an arrow, and a Ki67+ YAP− hepatocyte is denoted by a double arrow. Results are representative of three independent experiments. (H) The experiments were similar to those in B, except that p53 knockout mice were used. P-values > 0.05, as calculated by Student's t-test. The experiments were reproduced three times. (I) Knockout of p53 rescues YAP-induced tumorigenesis after the elimination of TICAMs. Representative liver photographs at 2 mo after injection. n = 3 mice. (J) Ccl2/Csf1 knockdown does not block YAP-induced hepatocyte dedifferentiation. The liver sections of injected p53−/− mice at post-injection day 10 were IHC-stained for the differentiation markers HNF4α and K19. Results are representative of three independent experiments. Values represent mean + SD.
We further asked how YAP+ TICs were eliminated without TICAMs. We observed that the elimination of TICAM-free YAP+ TICs was blocked in Rag1−/− mice lacking mature B and T lymphocytes, which play a major role in anti-cancer immunosurveillance (Fig. 5E,F; Miller and Sadelain 2015). In addition, by quantification, we found that while TICAM-free YAP+ clones have statistically fewer YAP+ cells, they retain the ability to grow (Fig. 5F; Supplemental Fig. S5A). Ki67 staining indicated that when Ccl2 and Csf1 were intact, YAP+ cells could proliferate, while surrounding TICAMs were also highly proliferative (Fig. 5G). In addition, some YAP− hepatocytes adjacent to YAP+ clones were also proliferating, suggesting that there is a non-cell-autonomous effect of YAP expression to promote cell proliferation. However, loss of TICAMs in Rag1−/− mice did not block YAP+ TICs from proliferating (Fig. 5G). These results indicate that while secreted factors from TICs or TICAMs may have an additional role in promoting cell proliferation non-cell-autonomously, TICAMs are not required for TICs to proliferate or evade senescence. p53 plays an important role in the T-cell-mediated death of transformed cells (Thiery et al. 2015). Hence, we found that p53 knockout also blocked the elimination of YAP+ TICs (Fig. 5H; Supplemental Fig. S5B) and rescued tumorigenesis in the absence of TICAMs (Fig. 5I). In addition, loss of TICAMs did not inhibit the dedifferentiation of YAP+ clones in p53−/− mice (Fig. 5J), suggesting that TICAMs are not required for the conversion of hepatocytes into TICs. These findings demonstrate that YAP-induced TICAMs prevent the immunosurveillance-dependent and p53-dependent clearance of TICs.
YAP activation and macrophage recruitment during liver tumor initiation
So far, genetic alterations of Hippo pathway components in human liver cancer were found to be rare (Zhao et al. 2007). However, YAP activation in the same context is much more prevalent (Zhao et al. 2007). Therefore, we investigated whether the deregulation of pathways commonly mutated in human hepatocellular carcinoma (HCC), such as the MAPK and PI3K–AKT–mTOR pathways (Totoki et al. 2014; Schulze et al. 2015), leads to YAP activation. Expression of the active KRAS-G12V mutant led to tumorigenesis within 6 mo in wild-type ICR mice (Fig. 6A), although, in a previous report, a similar NRAS-G12V mutant failed to induce tumorigenesis in the C.B-17 mice (Kang et al. 2011). KRAS-induced tumors were positive for HA tag staining, indicating that they originated from KRAS-expressing TICs (Fig. 6B). However, Yap was not activated in KRAS-induced tumors, as indicated by immunoblotting with antibodies against total Yap protein and active YAP (Fig. 6C). We further examined the effect of constitutively active AKT and EGFR, which induced profound tumorigenesis within 5 mo in combination but not individually (Fig. 6D). The origin of these tumors from AKT/EGFR-expressing hepatocytes was also confirmed by HA tag staining (Fig. 6E). Interestingly, AKT/EGFR-induced tumors exhibited consistently elevated YAP protein and active YAP levels when compared with adjacent normal tissues (Fig. 6F). We also noticed that inhibition of Mob phosphorylation, a marker of Mst1/2 kinase activity (Zhou et al. 2009), did not always accompany Yap activation, suggesting alternative mechanisms for Yap activation (Fig. 6F). We thus examined whether Yap activation happened at the tumor initiation stage in response to AKT/EGFR expression. Interestingly, while some HA tag+ cells remained negative for active Yap, a fraction of them exhibited Yap activation from the single-cell stage (Fig. 6G), suggesting that these cells might be TICs for later tumorigenesis. More importantly, these Yap-activated cells were surrounded by F4/80+ macrophages (Fig. 6G). These data suggest that pathologically relevant oncogenes such as AKT/EGFR induce Yap activation, which recruits TICAMs for TIC survival and tumorigenesis. Hepatocytes expressing other oncogenes such as RAS may evade immunosurveillance through alternative mechanisms.
Figure 6.

YAP activation and macrophage recruitment during mouse and human liver tumor initiation. (A) Liver tumorigenesis induced by HDI of HA-KRAS-G12V. A representative liver at 170 d after injection is shown. n = 3 mice. (B) HE and anti-HA tag IHC staining of sections of tumors induced by KRAS. Images are representative of three mice. (C) KRAS-induced tumors do not exhibit YAP activation. Normal liver and tumor tissues from two mice were analyzed by Western blotting. (D) Liver tumorigenesis induced by HDI of active AKT and EGFR. Representative livers at 140 d after injection with the indicated plasmids are shown. n = 4 mice. (E) HE and anti-HA tag IHC staining of sections of tumors induced by active AKT and EGFR. Images are representative of three mice. (F) YAP activation in tumors induced by active AKT and EGFR. Normal liver and three tumor tissues from each of the four active AKT- and EGFR-injected mice were analyzed by Western blotting. (G) AKT/EGFR-induced endogenous Yap activation correlates with macrophage recruitment. Representative clones that were negative (left panel) or positive (right panel) for active Yap on liver sections at post-injection day 5. Images are representative of two independent experiments. (H) HE and IHC staining of human liver high-grade dysplastic nodules (HGDNs). Four cases with YAP activation and macrophage enrichment are shown. HGDNs are indicated by asterisks. (I) Quantification of active YAP and CD68 staining intensity in H. “H” indicates HGDNs, and “N” indicates adjacent normal tissue. (J) A scatter plot matrix is shown between pairs of CCL2 and YAP or CTGF mRNA expression data in 371 HCC tumors. Pearson correlation R-value and P-value are indicated.
We further investigated whether YAP was activated during human HCC initiation. While accurate identification of single-cell stage TICs in human livers remains impractical, we could pinpoint larger precancerous lesions such as high-grade dysplastic nodules (HGDNs), which, in 60%–80% of cases, progress into HCC within 5 yr (Borzio et al. 2003). By analyzing eight cases of HGDNs, we found that YAP was activated in four cases in which the enrichment of CD68+ macrophages was also evident (Fig. 6H,I). In two more cases, the YAP activation was marginal, and, in the other two cases, YAP was similarly activated in adjacent tissue. The enrichment of macrophages was not significant in the latter four cases (data not shown). Thus, in a limited number of HGDN cases, YAP activation correlates with macrophage recruitment in precancerous human liver lesions and thus possibly plays a role in human HCC initiation.
Because YAP-induced CCL2 expression plays a key role in the recruitment of macrophages during liver tumor initiation, we also asked whether YAP activity correlates with CCL2 expression in human HCC. By analyzing a cohort of 371 HCC cases from The Cancer Genome Atlas (TCGA) database, we found that the mRNA level of CCL2 does not significantly correlate with the mRNA level of YAP (Fig. 6J). Interestingly, the CCL2 mRNA level correlates with the mRNA level of the classical YAP target gene CTGF (Fig. 6J). The discrepancy is possibly due to nontranscriptional mechanisms for YAP deregulation in cancer. Therefore, CCL2 expression in HCC likely correlates with YAP activity.
Discussion
The role of TAMs in liver cancer development has been examined in various mouse models, including DEN-driven HCC or orthotopic HCC xenograft models (Wan et al. 2015). In these models, the depletion of TAMs represses tumorigenesis and metastasis, indicating an important role of macrophages in liver tumorigenesis. However, these studies are not specific enough to provide insights into the role of macrophages that associate with TICs at the tumor initiation stage. The recent development of the HDI method allows genetic engineering and tracing of single hepatocytes in vivo. Using this approach, the study of the RAS oncogene reveals senescence as a critical mechanism of tumor suppression (Narita and Lowe 2005; Kang et al. 2011). However, senescence also impedes generation of TICs via expression of oncogenes, including RAS, thus preventing study of the interplay between TICs and the microenvironment. Nevertheless, the senescence barrier is inevitably breached during human tumorigenesis so that TICs emerge and tumors grow. Importantly, we found that activation of YAP efficiently surpasses senescence and generates proliferative TICs with progenitor cell properties. This breakthrough made it possible for us to demonstrate directly for the first time that TICs actively recruit TICAMs to evade immune clearance starting from the single-cell stage and that the transcriptional activation of CCL2/CSF1 by YAP underlies the recruitment of TICAMs (Fig. 7). Interestingly, elimination of all Ccr2+ cells leads to fast expansion of YAP-expressing cells, indicating that other Ccr2+ cells, such as normal Kupffer cells, play an important role in tumor suppression. This highlights the importance of specifically targeting TICAMs for the inhibition of tumorigenesis. It is interesting to note that the context-dependent role of the CCL2–CCR2 axis in tumorigenesis was also reported recently in the clearance of senescent hepatocytes and the promotion of tumor cell survival and growth (Eggert et al. 2016). This further supports that nonselective inhibition of CCL2–CCR2 may cause adverse effects and that specific targeting of TICAMs might be a viable option.
Figure 7.
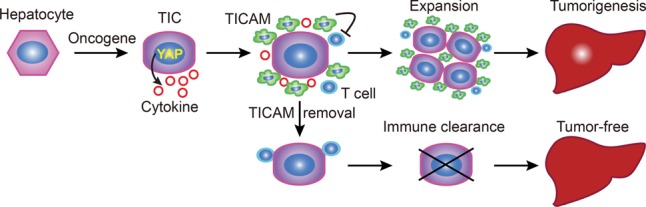
A model for the role of YAP and TICAMs in the development of liver TICs into tumors.
The Hippo pathway has well-known cell-autonomous functions in development and tumorigenesis (Yu et al. 2015). In Drosophila, the Hippo pathway inhibits cell proliferation and promotes apoptosis through inhibition of two Yorkie (YAP homolog) target genes: cyclin E and death-associated inhibitor of apoptosis 1 (Diap1) (Pan 2010). Additionally, the Hippo pathway represses self-renewal and expansion of tissue-specific progenitor cells (Yu et al. 2015). These cell-autonomous mechanisms are believed to play a key role in tumorigenesis induced by deregulation of the Hippo pathway. Nevertheless, recent research demonstrated that, in prostate cancer, YAP stimulates chemokine CXCL5 expression, thus recruiting MDSCs to promote tumor progression (Wang et al. 2016). However, the functional roles of YAP during tumor initiation have not been determined. In this study, we unveiled that the YAP–TEAD transcriptional complex directly or indirectly activates transcription of chemokine CCL2 and growth factor CSF1, respectively, and thus promotes TIC survival and tumorigenesis. We propose that both YAP-induced cell-autonomous dedifferentiation of hepatocytes and non-cell-autonomous recruitment of TICAMs are critical for tumorigenesis. Additionally, we found that YAP activation is induced by certain pathologically relevant oncogenes at the tumor initiation stage and that YAP activation was found in precancerous human liver lesions. Thus, deregulation of YAP due to mutation of non-Hippo pathway oncogenes and tumor suppressors likely plays a broader role in liver tumor initiation.
This study relied on hepatocyte genetic engineering by HDI of a PB transposon plasmid toolkit, a versatile method in genetic modifications. First, it achieves both gain and loss of functions. For example, we demonstrated that expression of active YAP or knockout of its upstream inhibitors, Lats1/2, leads to similar phenotypes. Second, it enables convenient combinational genetic modifications, which, in traditional genetically engineered mouse models, requires tedious crosses of mouse lines. For instance, we could coexpress two oncogenes or express an oncogene while knocking down its downstream effectors, thus enabling functional dissection of signaling cascades. The usefulness of Cas9-mediated knockout in combinational genetic modification is still limited by unsatisfactory gene-editing efficiency. The HDI approach has an additional advantage in that TICs generated with this method are surrounded by genetically normal cells, which is important when investigating tumor cell interplay with the microenvironment, as it more precisely mimics the mosaic nature of human tumorigenesis. We anticipate this toolkit to be broadly useful for investigating liver tumorigenesis and liver TICs in vivo.
Treatments for liver cancer after diagnosis are astonishingly limited (Gravitz 2014). Thus, it is important to prevent the disease by, for example, vaccination against the hepatitis B virus and changing dietary habits. However, due to various reasons, a large population is still under the shadow of high-risk factors for liver cancer. Hence, eliminating TICs before their development into tumors presents an attractive preventative measure. Our discovery of the non-cell-autonomous function of YAP in TICAM recruitment and the decisive role of TICAMs on TIC survival and liver tumorigenesis suggest targeting YAP or TICAMs as a new approach for immunoprevention of liver cancer and potentially other cancers.
Materials and methods
Mice and HDI
All animal study protocols were approved by the Zhejiang University Animal Care and Use Committee. Four-week-old male ICR mice were purchased from the Shanghai SLAC Laboratory Animal Company. Rag1−/− mice were purchased from the Mode Animal Research Centre of Nanjing University. Ccr2 depleter mice were kindly provided by Dr. Eric G. Pamer. p53−/− mice were from Dr. Yong Cang. Mst1/2F/F mice obtained from Dr. Yingzi Yang and Dr. Zengqiang Yuan were crossed with Albumin-Cre mice obtained from Dr. Yong Cang for liver-specific Mst1/2 knockout. For HDI, plasmid DNA suspended in sterile Ringer's solution in a volume equal to 10% of the body weight was injected in 5–7 sec via the tail vein of 4-wk-old male ICR mice or the indicated mice. No statistical method was used to predetermine sample size. No randomization or blinding was used. The amount of injected DNA was 50 μg of total transposon plasmids together with 10 µg of PB transposase plasmids. When specified, mice were X-ray-irradiated by using 9 Gy for 4.5 min on a RadSource 2000 irradiator 12 h before HDI.
IHC and immunofluorescence staining
Mouse livers were fixed in neutral buffered formalin for 24 h at room temperature and then embedded and processed according to standard protocols. The sections were deparaffinized through graded ethanol solutions. After an antigen retrieval procedure of 30 min using target retrieval solution (DAKO), the sections were stained with specific antibodies using the avidin–biotin complex system (Vector Laboratories). 3,3′-diaminobenzidine (DAB) was used as the substrate. Cell nuclei were counterstained with hematoxylin. To quantify active YAP and CD68 stainings, we cropped areas of HGDNs from images and quantified the staining intensity by the IHC profiler plug-in of ImageJ. The nuclear-stained image mode and cytoplasmic stained image mode were used for the quantification of active YAP and CD68, respectively. The staining intensities in normal tissue regions were quantified in a similar way. For immunofluorescence staining, frozen sections were fixed with 4% paraformaldehyde and then incubated with primary and Alexa fluor 488- and Alexa fluor 594-conjugated secondary antibodies.
Flow cytometry
Mouse livers were perfused using the two-step collagenase technique. The cell suspension was passed through a 70-µm cell strainer (Corning). The single-cell suspension was centrifuged at 300g for 5 min at 4°C. The cell pellet was then resuspended in FACS buffer (1% BSA in PBS) and incubated with the indicated antibodies for 30 min on ice. The signal was detected by using a Beckman FC 500 MCL and was analyzed using CXP software.
Luciferase assay
For the Ccl2 and Csf1 promoter reporter assays, 293T cells from laboratory stock were transfected with the reporter CMV-β-gal and the indicated plasmids. Thirty-six hours after transfection, the cells were lysed, and the luciferase activity was assayed using a luciferase assay system (Promega) per the manufacturer's instructions. All of the luciferase activities were normalized to β-galactosidase activity.
Immunoblotting and cytokine array
Immunoblotting was performed using standard protocols. Briefly, the cells were lysed with 1% SDS lysis buffer, and the protein concentration was determined using a BCA assay kit (Pierce). Protein samples were resolved on SDS-PAGE and then transferred to PVDF membranes. The membranes were blocked with 5% BSA, incubated sequentially with primary and secondary antibodies, and then washed. Protein expression was determined by using ECL detection reagent (Pierce).
YAP-induced cytokines in TICs were determined using the Mouse Cytokine Array Panel A from R&D Systems according to the manufacturer's instructions. Briefly, mice were irradiated to ablate the immune system 12 h before HDI. At post-injection day 5, the livers were dissected and homogenized. The homogenates were mixed with a cocktail of biotinylated detection antibodies. The sample/antibody mixture was then incubated with the array membrane. After a wash to remove unbound material, streptavidin-HRP and chemiluminescent detection reagents were added sequentially.
Human specimens and their analysis
All HGDN samples used in this study were obtained during liver transplantations or liver resections performed at the Eastern Hepatobiliary Surgery Hospital (Shanghai, China) from 2010 to 2014. These samples were obtained with informed consent according to the Eastern Hepatobiliary Surgery Hospital Research Ethics Committee. Each HGDN specimen was diagnosed consistently by two senior pathologists according to previously reported criteria for HGDN (Borzio et al. 2003). mRNA sequencing data of tumors from patients with HCC (n = 371) were downloaded from TCGA database. Pearson correlation was performed between gene CCL2 and gene CTGF or YAP using their log2 values in GraphPad Prism 5.0.
Supplementary Material
Acknowledgments
We thank Dr. Yong Cang for critical reading of the manuscript, Dr. Eric G. Pamer for Ccr2 depleter mice, Dr. Yong Cang for p53 knockout mice and Albumin-Cre mice, Dr. Yingzi Yang and Zengqiang Yuan for Mst1/2F/F mice, Dr. Tian Xu and Dr. Xiaohui Wu for the PB system, and Dr. Tong Ji for assistance on IHC. Research in the laboratory of B.Z. is supported by grants from the National Natural Science Foundation of China Excellent Yong Scholars Project (31422036), the State Key Development Program for Basic Research of China (2013CB945303), the National Natural Science Foundation of China General Projects (31471316), the International Collaboration Project (31661130150), the Fundamental Research Funds for Central Universities of China (2015XZZX004-17), the Qianjiang Scholar Plan of Hangzhou, the Thousand Young Talents Plan of China, and the Newton Advanced Fellowship from the Academy of Medical Sciences UK. B.Z. and X.G. designed the study. X.G., Y.Z., H.Y., S.S., X.D., X.J., and L.L. performed experiments and analyzed data. Y.Y., X.B., and L.C. performed histological evaluations. F.J. and J.J performed TCGA database HCC gene expression analysis. X.-G.G., X.-H.F., T.L., and H.W. provided reagents and conceptual advice. B.Z. and X.G. wrote the manuscript with comments from all authors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.294348.116.
References
- Borzio M, Fargion S, Borzio F, Fracanzani AL, Croce AM, Stroffolini T, Oldani S, Cotichini R, Roncalli M. 2003. Impact of large regenerative, low grade and high grade dysplastic nodules in hepatocellular carcinoma development. J Hepatol 39: 208–214. [DOI] [PubMed] [Google Scholar]
- Chen X, Calvisi DF. 2014. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol 184: 912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. 2005. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 122: 473–483. [DOI] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. 2007. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130: 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, Medina-Echeverz J, Longerich T, Forgues M, Reisinger F, et al. 2016. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 30: 533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. 2014. The cellular and molecular origin of tumor-associated macrophages. Science 344: 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli GG, Carrara M, Yuan WC, Valdes-Quezada C, Gurung B, Pepe-Mooney B, Zhang T, Geeven G, Gray NS, de Laat W, et al. 2015. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell 60: 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravitz L. 2014. Liver cancer. Nature 516: S1. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. 2010. Immunity, inflammation, and cancer. Cell 140: 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G, Johnson RL. 2011. Hippo signaling: growth control and beyond. Development 138: 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Chun A, Cheung K, Rashidi B, Yang X. 2008. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem 283: 5496–5509. [DOI] [PubMed] [Google Scholar]
- He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, Shalapour S, Seki E, Yost SE, Jepsen K, et al. 2013. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 155: 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuff G, Oldenburg HS, Boutkan H, Visser JJ, Beelen RH, Van Rooijen N, Dijkstra CD, Meyer S. 1993. Enhanced tumour growth in the rat liver after selective elimination of Kupffer cells. Cancer Immunol Immunother 37: 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M, Pamer EG. 2009. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe 6: 470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, He F, Wang Y, Zhang Z, Wang W, et al. 2014. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 25: 166–180. [DOI] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. 2011. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479: 547–551. [DOI] [PubMed] [Google Scholar]
- Koontz LM, Liu-Chittenden Y, Yin F, Zheng Y, Yu J, Huang B, Chen Q, Wu S, Pan D. 2013. The hippo effector yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev Cell 25: 388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KP, Lee JH, Kim TS, Kim TH, Park HD, Byun JS, Kim MC, Jeong WI, Calvisi DF, Kim JM, et al. 2010. The Hippo–Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci 107: 8248–8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Park JO, Kim TS, Kim SK, Kim TH, Kim MC, Park GS, Kim JH, Kuninaka S, Olson EN, et al. 2016. LATS–YAP/TAZ controls lineage specification by regulating TGFβ signaling and Hnf4α expression during liver development. Nature communications 7: 11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Song Y, Liu D. 1999. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther 6: 1258–1266. [DOI] [PubMed] [Google Scholar]
- Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, Halder G, Finegold MJ, Lee JS, et al. 2010. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci 107: 1437–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. 2008. Cancer-related inflammation. Nature 454: 436–444. [DOI] [PubMed] [Google Scholar]
- Miller JF, Sadelain M. 2015. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell 27: 439–449. [DOI] [PubMed] [Google Scholar]
- Narita M, Lowe SW. 2005. Senescence comes of age. Nat Med 11: 920–922. [DOI] [PubMed] [Google Scholar]
- Noy R, Pollard JW. 2014. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41: 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Mazack V, Sudol M. 2008. Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J Biol Chem 283: 27534–27546. [DOI] [PubMed] [Google Scholar]
- Oosterling SJ, van der Bij GJ, Meijer GA, Tuk CW, van Garderen E, van Rooijen N, Meijer S, van der Sijp JR, Beelen RH, van Egmond M. 2005. Macrophages direct tumour histology and clinical outcome in a colon cancer model. J Pathol 207: 147–155. [DOI] [PubMed] [Google Scholar]
- Pan D. 2010. The hippo signaling pathway in development and cancer. Dev Cell 19: 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszer T. 2015. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm 2015: 816460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. 2015. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 47: 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, Park O, Chang J, Simpson RM, et al. 2010. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci 107: 1431–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery J, Safta TB, Ziani L, Chouaib S. 2015. Mechanisms of cytotoxic lymphocyte-mediated apoptosis and relationship with the tumor suppressor p53. Crit Rev Immunol 35: 433–449. [DOI] [PubMed] [Google Scholar]
- Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, et al. 2014. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet 46: 1267–1273. [DOI] [PubMed] [Google Scholar]
- Wan S, Kuo N, Kryczek I, Zou W, Welling TH. 2015. Myeloid cells in hepatocellular carcinoma. Hepatology 62: 1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, Fang Z, Zhao K, Konaparthi R, Hua S, et al. 2016. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov 6: 80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, Pollard JW. 2013. Macrophage biology in development, homeostasis and disease. Nature 496: 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R, Crowley DG, et al. 2014. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514: 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Lu L, Yanger K, Wang W, Hwa Sohn B, Stanger BZ, Zhang M, Martin JF, Ajani JA, Chen J, et al. 2016. LATS1 and LATS2 regulate mouse liver progenitor cell proliferation and maturation through antagonism of the coactivators YAP and TAZ. Hepatology 64: 1757–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, Camargo FD. 2014. Hippo pathway activity influences liver cell fate. Cell 157: 1324–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimlamai D, Fowl BH, Camargo FD. 2015. Emerging evidence on the role of the Hippo/YAP pathway in liver physiology and cancer. J Hepatol 63: 1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL. 2015. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163: 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. 2015. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 17: 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Gao Y, Li P, Shi Z, Guo T, Li F, Han X, Feng Y, Zheng C, Wang Z, et al. 2014. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP–TEAD transcriptional complex. Cell Res 24: 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Qi HX, Hu ZM, Chang YN, Shi ZM, Han XH, Han YW, Zhang RX, Zhang Z, Chen T, et al. 2015. YAP and TAZ take center stage in cancer. Biochemistry 54: 6555–6566. [DOI] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. 2007. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21: 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, et al. 2008. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22: 1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. 2010. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(β-TRCP). Genes Dev 24: 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, et al. 2009. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16: 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.