

Yazinski et al. show that the functions of BRCA1 in homologous recombination and replication fork protection are sequentially bypassed during the acquisition of PARP inhibitor (PARPi) resistance. Despite the lack of BRCA1, PARPi-resistant cells regain RAD51 loading to DNA double-stranded breaks and stalled forks, enabling two distinct mechanisms of PARPi resistance.
Keywords: ATR, BRCA-deficient cancer, PARP inhibitor
Abstract
Poly-(ADP-ribose) polymerase (PARP) inhibitors (PARPis) selectively kill BRCA1/2-deficient cells, but their efficacy in BRCA-deficient patients is limited by drug resistance. Here, we used derived cell lines and cells from patients to investigate how to overcome PARPi resistance. We found that the functions of BRCA1 in homologous recombination (HR) and replication fork protection are sequentially bypassed during the acquisition of PARPi resistance. Despite the lack of BRCA1, PARPi-resistant cells regain RAD51 loading to DNA double-stranded breaks (DSBs) and stalled replication forks, enabling two distinct mechanisms of PARPi resistance. Compared with BRCA1-proficient cells, PARPi-resistant BRCA1-deficient cells are increasingly dependent on ATR for survival. ATR inhibitors (ATRis) disrupt BRCA1-independent RAD51 loading to DSBs and stalled forks in PARPi-resistant BRCA1-deficient cells, overcoming both resistance mechanisms. In tumor cells derived from patients, ATRis also overcome the bypass of BRCA1/2 in fork protection. Thus, ATR inhibition is a unique strategy to overcome the PARPi resistance of BRCA-deficient cancers.
Mutations in BRCA1 and BRCA2 genes are found in breast, ovarian, prostate, and pancreatic cancers, providing opportunities for targeted therapy (Fong et al. 2009; Audeh et al. 2010; Tutt et al. 2010; Kaufman et al. 2015; Lord et al. 2015; O'Connor 2015). Among their many functions, BRCA1 and BRCA2 proteins are important for homologous recombination (HR) and protection of stalled DNA replication forks (Prakash et al. 2015). BRCA1- and BRCA2-deficient cells are highly sensitive to inhibitors of poly-(ADP-ribose) polymerase (PARP) (Bryant et al. 2005; Farmer et al. 2005). It is believed that PARP inhibitors (PARPis) induce replication stress by trapping inactive PARP on DNA and/or inhibiting base excision repair, which creates a dependency on BRCA1 and BRCA2 for cell survival (Bryant et al. 2005; Farmer et al. 2005; Murai et al. 2012; Lord et al. 2015; Lord and Ashworth 2016). Several PARPis have shown efficacy in the treatment of BRCA-deficient cancers (O'Connor 2015). The PARPi olaparib has been approved by the FDA for the treatment of advanced ovarian cancers with BRCA1/2 mutations (Kim et al. 2015). However, as with other targeted drugs, the efficacy of PARPis is limited by drug resistance (Fojo and Bates 2013; Lord and Ashworth 2013; Sonnenblick et al. 2015). Only a fraction of BRCA1/2 mutation carriers responded to PARPis, and even those who responded subsequently developed resistance and relapsed. Thus, a strategy to overcome the PARPi resistance of BRCA-deficient cancers is much needed to improve this promising targeted therapy.
Both BRCA1 and BRCA2 are key players in HR. In the absence of BRCA1, 53BP1 inhibits HR by limiting DNA end resection, a process generating ssDNA at DNA double-stranded breaks (DSBs) (Bunting et al. 2010). BRCA1 interacts with the PALB2–BRCA2 complex and promotes its localization to DSBs, enabling PALB2–BRCA2 to load RAD51 onto ssDNA (Sy et al. 2009; Zhang et al. 2009; Orthwein et al. 2015). Independently of their HR functions, BRCA1 and BRCA2 are required for the protection of stalled replication forks (Schlacher et al. 2011, 2012; Ying et al. 2012). In BRCA1/2-deficient cells, stalled replication forks are extensively degraded by MRE11 and other nucleases (Schlacher et al. 2011; Ying et al. 2012; Chaudhuri et al. 2016). Like BRCA1 and BRCA2, RAD51 is required for the protection of stalled forks (Schlacher et al. 2011). How RAD51 is recruited to stalled forks is still unclear, but BRCA2 is needed to stabilize RAD51 on ssDNA for fork protection (Schlacher et al. 2011; Chaudhuri et al. 2016). The important functions of BRCA1/2 in HR and fork protection likely underlie the sensitivity of BRCA1/2-deficient cells to PARPis (Schlacher et al. 2011; Chaudhuri et al. 2016).
Recent genetic studies have revealed that the functions of BRCA1/2 in HR and fork protection can be bypassed by rewiring of these pathways. For example, deletion of 53BP1 suppressed the HR defects and lethality of BRCA1−/− cells (Bunting et al. 2010). Loss of the 53BP1-binding protein RIF1 and its associated protein, REV7, also suppressed the HR defects of BRCA1-deficient cells (Chapman et al. 2013; Escribano-Diaz et al. 2013; Zimmermann et al. 2013; Xu et al. 2015). In cells lacking 53BP1, RIF1, or REV7, DNA end resection is enhanced, suggesting that generation of long ssDNA at DSBs may alleviate the HR defects of BRCA1-deficient cells. Interestingly, while loss of 53BP1 suppresses the HR defects of BRCA1-deficient cells, it does not suppress the defect in fork protection (Chaudhuri et al. 2016). In contrast, loss of PTIP suppresses the fork protection defects of BRCA1- and BRCA2-deficient cells but not their HR defects (Chaudhuri et al. 2016). In the absence of PTIP, localization of the MRE11 nuclease to stalled replication forks is compromised, explaining the reduction in fork degradation in the absence of BRCA1/2 (Chaudhuri et al. 2016). Notably, loss of 53BP1 in BRCA1-deficient cells or loss of PTIP1 in BRCA1/2-deficient cells is sufficient to confer PARPi resistance, suggesting that bypasses of HR and fork protection functions of BRCA1/2 enable two distinct PARPi resistance mechanisms (Bunting et al. 2010; Chaudhuri et al. 2016). These genetic studies have provided a framework to understand how HR and fork protection pathways are rewired in the absence of BRCA1/2 and how PARPi resistance arises in BRCA1/2-deficient cells. In addition to the rewiring of HR and fork protection pathways, several other mechanisms have also been implicated in the PARPi resistance of BRCA1/2-deficient cells. These mechanisms include up-regulation of efflux pump (Rottenberg et al. 2008), loss of PARP1 (Pettitt et al. 2013), restoration of the BRCA1/2 reading frame (Edwards et al. 2008; Sakai et al. 2008), loss of KU (Patel et al. 2011; Bunting et al. 2012; Choi et al. 2016), altered DNA end processing (Wang et al. 2014), alternative splicing of BRCA1 mRNA (Wang et al. 2016), and stabilization of the BRCA1 mutant protein (Johnson et al. 2013). To what extent each of these mechanisms contributes to the PARPi resistance of BRCA-deficient tumors in patients still awaits further investigations.
In this study, we used a panel of derived cancer cell lines and tumor cells from patients to investigate how to overcome the PARPi resistance of BRCA-deficient cancers. We found that both the HR and fork protection functions of BRCA1 are commonly bypassed in PARPi-resistant cells. Interestingly, the two functions of BRCA1 are sequentially bypassed during the acquisition of PARPi resistance, suggesting that the PARPi resistance of BRCA1-deficient cancer cells arises from two distinct mechanisms through stepwise rewiring of HR and fork protection pathways. Through gene profiling and inhibitor screening, we found that the ATR kinase has a unique role in the survival of PARPi-resistant cells. In PARPi-resistant BRCA1-deficient cells, ATR controls both BRCA1-independent HR and fork protection by promoting RAD51 loading to DSBs and stalled forks. Inhibition of ATR leads to blockage of BRCA1-independent HR and fork protection, resensitizing resistant cells to PARPis. In tumor cells derived from BRCA1/2-deficient patients, PARPi resistance correlates with the bypass of BRCA1/2 in fork protection, which is also overcome by ATR inhibitors (ATRis). These results suggest that ATR inhibition is a unique strategy to overcome the PARPi resistance of BRCA-deficient cancers.
Results
A panel of PARPi-resistant BRCA1-deficient cell lines harboring distinct resistance mechanisms
To investigate how to overcome the PARPi resistance of BRCA1-deficient cancers, we used UWB1, a BRCA1-deficient ovarian cancer cell line (DelloRusso et al. 2007), to derive a panel of cell lines resistant to olaparib (Fig. 1A,B; Supplemental Fig. S1A). These cell lines were also resistant to another PARPi, ABT-888, showing that their PARPi resistance is not specific to olaparib (Supplemental Fig. S1B). Full-length BRCA1 protein was undetectable in all of the resistant lines using antibodies recognizing the N or C terminus of BRCA1 (Supplemental Fig. S1C). The BRCA1 frameshift mutation in UWB1 was retained in the resistant lines, and the secondary mutation reported to restore wild-type BRCA1 reading frame was not detected (Swisher et al. 2008; data not shown). The BRCA1Δ11q isoform and the truncated BRCA1 protein that it encodes were detected in UWB1 as well as the resistant lines (Supplemental Fig. S1D,E; Wang et al. 2016). Olaparib suppressed DNA damage-induced PARylation in the resistant lines, ruling out loss of PARP1, up-regulation of efflux pump, or compensation by other PARP family members as the cause of resistance (Supplemental Fig. S1F; Rottenberg et al. 2008; Pettitt et al. 2013). Consistently, the efflux pump protein MDR1 was not up-regulated in most of the resistant lines (Supplemental Fig. S1G). Loss of 53BP1, RIF1, REV7, PTIP, CHD4, KU70, JMJD1C, and SLFN11 has been implicated in PARPi resistance in various cellular contexts (Bunting et al. 2010; Patel et al. 2011; Callen et al. 2013; Chapman et al. 2013; Di Virgilio et al. 2013; Escribano-Diaz et al. 2013; Feng et al. 2013; Jaspers et al. 2013; Zimmermann et al. 2013; Chen et al. 2014; Boersma et al. 2015; Guillemette et al. 2015; Xu et al. 2015; Kurfurstova et al. 2016; Murai et al. 2016). Some of these proteins were expressed at reduced levels in a subset of the resistant lines compared with UWB1 (Supplemental Fig. S1C,G). Interestingly, some resistant lines displayed reductions in several proteins in this group, suggesting the presence of multiple resistance mechanisms in the same cell. However, some resistant lines did not show a reduction in any protein in this group, indicating the existence of currently unknown resistance mechanisms. Thus, this panel of PARPi-resistant BRCA1-deficient cell lines harbors a variety of genetic or epigenetic alterations leading to resistance, which likely reflects the heterogeneity of resistance mechanisms in tumors.
Figure 1.

ATRis have a unique ability to overcome the PARPi resistance of BRCA1-deficient cancer cells. (A) Schematic of derivation of PARPi-resistant cells from the parental BRCA1-deficient UWB1.289 ovarian cancer cell line. (B) Viability assay of UWB1 (parental), UWB + B1 (complemented with wild-type BRCA1), and derived PARPi-resistant cell lines after 6 d of increasing doses of PARPi (olaparib) treatment. n = 3 replicates. Error bars represent SD. (C) Gene set enrichment analysis of RNA sequencing data from UWB1, SYr12, and SYr13 cell lines. (D) Mini drug screen performed on UWB1 + B1, UWB1, and resistant lines (SYr12 and SYr13). Cells were treated with increasing doses of ATRi (VE-821, 0–0.625 µM), ATMi (KU55933, 0–0.625 µM), DNA-PKi (NU7441, 0–0.625 µM), Chk1i (MK-8776, 0–0.5 µM), or Wee1i (MK-1775, 0–0.25 µM) in the absence or presence of 2.0 µM PARPi (olaparib). Color-coding denotes the level of viability (green [100% viability] to red [0% cell viability]) relative to DMSO treatment. (E) IC50s of the indicated cell lines to PARPi (olaparib) were measured after 6 d of olaparib treatment in the presence of increasing concentrations of ATRi (VE-821). (F) The indicated cell lines were treated with ATRi (VE-821), PARPi (olaparib), or ATRi and PARPi for 7 d. Fractions of cells undergoing cell death (propidium iodide and/or annexin V-positive) were measured. n = 3 replicates. Error bars represent SD. (G) The indicated cell lines were treated with increasing doses of PARPi (0–25 µM) in combination with increasing doses of either ATRi (0–2.5 µM) or cisplatin (0–1.25 µM) in triplicate, and an overall Bliss score and P-value were calculated. Bliss score >0, synergistic; Bliss score = 0, additive; Bliss score <0, antagonistic.
ATRi has a unique ability to preferentially sensitize PARPi-resistant cells to PARPis
To understand how UWB1 acquired PARPi resistance, we performed RNA sequencing on UWB1 and two of the PARPi-resistant lines: SYr12 and SYr13. Gene set enrichment analysis (GSEA) revealed only two gene sets significantly enriched in SYr12 compared with UWB1, both of which primarily consist of checkpoint, cell cycle, and DNA repair genes (Fig. 1C; Supplemental Fig. S1H). One of these gene sets was also enriched in SYr13. Additionally, a second gene set consisting of components of the ATR checkpoint was significantly enriched in SYr13. These results suggest that the DNA damage response network is transcriptionally rewired in the PARPi-resistant cells, raising the possibility that certain checkpoint or DNA repair proteins contribute to PARPi resistance.
Prompted by the GSEA results, we used SYr12 and SYr13 to perform a “miniscreen” for checkpoint and/or DNA repair inhibitors that sensitize resistant cells to PARPis (Fig. 1D). In cell-free assays, inhibitors of ATM (ATMi: KU55933), ATR (ATRi: VE-821), and DNA-PK (DNA-PKi: NU7026) all have IC50s in the 10–100 nM range (Hollick et al. 2003; Hickson et al. 2004; Reaper et al. 2011). When used in cell cultures at submicromolar concentrations, ATMi and DNA-PKi showed little cytotoxicity in the resistant lines and did not significantly affect olaparib sensitivity (Fig. 1D). In contrast, ATRi was clearly cytotoxic to both resistant lines, and its effects were enhanced by olaparib (Fig. 1D). Inhibitors of Chk1 (Chk1i: MK-8776) and Wee1 (Wee1i: MK-1775) were also cytotoxic to the resistant lines (Fig. 1D). Compared with Chk1i and Wee1i, ATRi exhibited a greater selectivity toward the resistant lines as opposed to the BRCA1-proficient UWB1 + B1 line (UWB1 complemented with wild-type BRCA1) (Fig. 1D; Supplemental Fig. S1I,J). Furthermore, at the concentrations tested, ATRi did not sensitize untransformed RPE1-hTERT cells to PARPis (Supplemental Fig. S1J). Thus, ATRi has a unique ability to preferentially sensitize PARPi-resistant BRCA1-deficient cells, as opposed to BRCA1-proficient cells, to PARPis.
To confirm the ability of ATRi to preferentially sensitize PARPi-resistant BRCA1-deficient cells, we measured the IC50 of olaparib in UWB1, UWB1 + B1, SYr12, and SYr13 in the presence or absence of VE-821 (Fig. 1E; Supplemental Fig. S1I). Even low concentrations of VE-821 reduced the IC50 of olaparib in the resistant lines but not in UWB1 + B1. The combination of VE-821 and olaparib drastically increased the cell death of SYr12 and SYr13 but not UWB1 + B1, showing that the resistant cells were preferentially killed, and not simply arrested, by ATRi and PARPis (Fig. 1F). AZ20, a second ATRi, also preferentially sensitized SYr12 and SYr13 to olaparib compared with UWB1 + B1 (Supplemental Fig. S1K; Foote et al. 2013). To determine whether the effects of ATRi were attributed to its general cytotoxicity in S-phase cells, we compared the combinations of olaparib with VE-821 and olaparib with cisplatin in UWB1, UWB1 + B1, and SYr12 (Fig. 1G; Supplemental Fig. S1L). Olaparib and VE-821 displayed a greater synergy in SYr12 than olaparib and cisplatin according to the Bliss model (Yadav et al. 2015). Furthermore, olaparib and VE-821 were synergistic only in SYr12 but not UWB1 + B1. These results lend further support to the notion that ATRi has a unique ability to preferentially sensitize PARPi-resistant BRCA1-deficient cells and that its effects are not simply due to its cytotoxicity in S phase.
ATRi broadly overcomes pre-existing and acquired PARPi resistance in BRCA1-deficient cancer cells
We next extended our analysis to other PARPi-resistant BRCA1-deficient cell lines. Despite the heterogeneity of resistance mechanisms, VE-821 broadly overcame PARPi resistance in this panel of resistant cell lines (Fig. 2A,B). Similar to SYr12 and SYr13, other resistant lines were also preferentially sensitized to olaparib by VE-821 compared with UWB1 + B1 (Fig. 2B). In addition to the UWB1 derivative lines, a PARPi-resistant mouse cell line (BR5-R1) derived from a BRCA1Δ11/Δ11 (exon 11 deleted) mouse ovarian cancer cell line (BR5) was also sensitized to olaparib by VE-821 (Fig. 2C; Xing and Orsulic 2006). These results show that ATRi overcomes a variety of PARPi resistance mechanisms in human and mouse BRCA1-deficient cancer cells.
Figure 2.

ATRi broadly overcomes acquired and pre-existing PARPi resistance in multiple BRCA1-deficient cancer cell lines of distinct origins. (A) Colony formation assay of the indicated cell lines following 14 d of treatment with DMSO, 1 µM PARPi (olaparib), or PARPi and 0.313 µM ATRi (VE-821). Cells were stained with crystal violet. (B) Cell viability following 6 d of treatment across all cell lines using DMSO, PARPi (olaparib), ATRi (VE-821), or PARPi and ATRi. (C) Viability assay of mouse ovarian cancer cell lines ([T2] non-BRCA mutant; [BR5] BRCA1-deficient; [BR5-R1] BRCA1-deficient and PARPi-resistant) after 6 d of treatment with increasing doses of PARPi (olaparib) in the absence or presence of 1.0 µM ATRi (VE-821). n = 3 replicates. Error bars represent SD. (D) Viability assay of the BRCA1-deficient HCC1937 (de novo PARPi-resistant) and BRCA1-complemented HCC1937 + B1 cell lines after 6 d of treatment with increasing doses of PARPi (olaparib) and the indicated doses of ATRi (VE-821). n = 3 replicates. Error bars represent SD. (E) Colony formation assay of UWB1 + B1 and UWB1 cell lines after 45 d of treatment with ATMi, ATRi, PARPi, the ATMi + PARPi combination, or the ATRi + PARPi combination. Cells were stained with crystal violet.
In clinics, only a fraction of the patients with BRCA1/2 mutations responded to olaparib (Fong et al. 2009), indicating the presence of pre-existing PARPi resistance. The BRCA1-deficient breast cancer cell line HCC1937 and its BRCA1-complemented derivative, HCC1937 + B1, were similarly sensitive to olaparib, providing an example of pre-existing PARPi resistance (Fig. 2D; Supplemental Fig. S2; Johnson et al. 2013). Compared with HCC1937 + B1, HCC1937 was preferentially sensitized to olaparib by VE-821, suggesting that ATRi overcomes the pre-existing PARPi resistance (Fig. 2D; Supplemental Fig. S2). To test whether ATRi affects the emergence of PARPi resistance, we selected for olaparib-resistant UWB1 clones in the presence or absence of VE-821 (Fig. 2E). As a control, UWB1 cells were also selected in olaparib and ATMi. Strikingly, the formation of resistant clones was completely eliminated by ATRi but not ATMi. Thus, ATRi overcomes not only acquired but also pre-existing PARPi resistance of BRCA1-deficient cancer cells and suppresses the emergence of PARPi resistance when used up front with PARPis.
The HR function of BRCA1, but not PALB2–BRCA2, is partially bypassed in BRCA1-deficient cancer cells
The functional status of HR is a key determinant of PARPi sensitivity (Lord et al. 2008). To understand how ATRi overcomes PARPi resistance, we analyzed the HR status in BRCA1-deficient cancer lines and their BRCA1-complemented and PARPi-resistant derivatives. Both ionizing radiation (IR) and olaparib induced RAD51 foci in UWB1 and UWB1 + B1, indicating HR at DSBs (Fig. 3A; Supplemental Fig. S3A,B). Surprisingly, although RAD51 foci were reduced in UWB1 compared with UWB1 + B1, substantial RAD51 foci remained detectable in UWB1. Knockdown of full-length BRCA1 abolished RAD51 foci in BRCA1-proficient U2OS cells but did not reduce RAD51 foci in UWB1 (Fig. 3B; Supplemental Fig. S3C,D). Specific knockdown of the truncated BRCA1 encoded by the BRCA1Δ11q isoform in UWB1 reduced but did not eliminate RAD51 foci (Supplemental Fig. S3E,F), showing that significant amounts of RAD51 were recruited to DSBs independently of BRCA1. HCC1937 cells did not express the truncated BRCA1 but retained a substantial ability to form RAD51 foci (Fig. 3C; Supplemental Fig. S3G). Similar to UWB1, the RAD51 foci in HCC1937 were not affected by the siRNA targeting full-length BRCA1 (Fig. 3C). Knockdown of full-length BRCA1 in UWB1 + B1 and HCC1937 + B1 reduced RAD51 foci only to the levels in UWB1 and HCC1937, respectively (Fig. 3B,C; Supplemental Fig. S3D,H). Thus, while the truncated BRCA1 contributes to RAD51 localization in UWB1 (Wang et al. 2016), by and large, RAD51 is recruited to DSBs independently of BRCA1 in both UWB1 and HCC1937. The partial bypass of BRCA1 function in RAD51 recruitment in both UWB1 and HCC1937 reveals a shared feature of BRCA1-deficient cancer cells.
Figure 3.

The HR function of BRCA1, but not PALB2–BRCA2, is partially bypassed in BRCA1-deficient cancer cells. (A) Fractions of RAD51 focus-positive cells (more than five foci per cell) following 24 h of 10 µM PARPi (olaparib) treatment in the indicated cell lines. n = 3 replicates. Error bars represent SD. (B) The indicated cell lines were treated with siControl, siBRCA1, or siBRCA2 and irradiated with 10 Gy of IR, and fractions of RAD51 focus-positive cells were measured 4 h later. n = 3 replicates. Error bars represent SD. (C) The indicated cell lines were treated with siControl, siBRCA1, or siBRCA2 and irradiated with 10 Gy of IR, and fractions of RAD51 focus-positive cells were measured 4 h later. n = 3 replicates. Error bars represent SD. (D) The indicated cell lines were transfected with siControl, siPALB2, or siBRCA2 and treated with 10 µM PARPi (olaparib) for 24 h, and fractions of RAD51 focus-positive cells were determined. n = 3 replicates. Error bars represent SD. (E) The PARPi-resistant cell lines were transfected with siControl, siPALB2, or siBRCA2 and treated with increasing doses of PARPi (olaparib) for 6 d, and cell viability was measured. n = 3 replicates. Error bars represent SD.
The partial bypass of BRCA1 in UWB1 and HCC1937 suggests that the HR pathway is already rewired in BRCA1-deficient cancer cells even before they acquire PARPi resistance, which may allow cancer cells to survive the lack of BRCA1. In marked contrast to siRNAs targeting BRCA1 (including the truncated BRCA1), siPALB2 drastically reduced RAD51 foci in U2OS, UWB1, and HCC1937 (Fig. 3B,C; Supplemental Fig. S3C,D). Therefore, despite the bypass of BRCA1, PALB2 remains indispensable in the rewired HR pathway in UWB1 and HCC1937.
Compared with UWB1, the PARPi-resistant lines generally displayed similar or modestly higher levels of RAD51 foci (Fig. 3A; Supplemental Fig. S3A,B). The increase of RAD51 foci in some resistant lines suggests that HR is further restored. In other resistant lines, levels of RAD51 foci are similar to that in UWB1, suggesting that an increase in HR is not obligated for the acquisition of PARPi resistance. Knockdown of PALB2 or BRCA2 drastically reduced RAD51 foci in UWB1 and SYr12 (Fig. 3D; Supplemental Fig. S3I,J), showing that the rewired HR pathway in PARPi-resistant cells remains dependent on PALB2–BRCA2. Importantly, knockdown of PALB2 or BRCA2 significantly increased the olaparib sensitivity of SYr12 and SYr13 (Fig. 3E; Supplemental Fig. S3J), revealing that PALB2–BRCA2 remains indispensable for cell survival in PARPis even after UWB1 cells acquire resistance. These results suggest that the HR activity in UWB1 is either maintained or increased in PARPi-resistant cells, which is necessary for PARPi resistance. Furthermore, a strategy to inhibit PALB2–BRCA2 may overcome PARPi resistance.
ATRi blocks BRCA1-independent RAD51 loading to DSBs
ATR is required for HR and the formation of RAD51 foci (Wang et al. 2004; Adamson et al. 2012; Prevo et al. 2012). We and others recently showed that PALB2 is a substrate of ATR in response to DSBs and replication stress (Ahlskog et al. 2016; Buisson et al. 2017). In BRCA1-proficient U2OS cells, partial ATR inhibition reduced HR significantly (Fig. 4A; Supplemental Fig. S4A,B). VE-821 diminished BRCA2 foci but not BRCA1 foci without altering the cell cycle (Fig. 4B,C; Supplemental Fig. S4C,D), suggesting that ATR regulates BRCA2 downstream from BRCA1 in the canonical BRCA1-dependent HR pathway.
Figure 4.

ATR functions in HR downstream from BRCA1 and remains indispensable when BRCA1 is bypassed. (A) U2OS cells were treated with the indicated doses of ATMi (KU55933) or ATRi (VE821 or AZ20). The efficiency of HR was measured using the DR-GFP reporter 48 h after I-SceI transfection. n = 3 replicates. Error bars represent SD. (B) U2OS cells were treated with DMSO or ATRi and irradiated with 4 Gy of IR, and fractions of BRCA1 focus-positive cells were measured 2 h later. n = 3 replicates. Error bars represent SD. (C) Cells were treated with the indicated doses of ATRi (VE-821) and irradiated with 10 Gy of IR, and fractions of BRCA2 focus-positive cells (more than five foci per cell) were measured 4 h later. n = 3 replicates. Error bars represent SD. (D) U2OS cells were transfected with siControl, si53BP1, siBRCA1, or si53BP1 and siBRCA1. Transfected cells were treated with DMSO, 10 µM PARPi (olaparib), or PARPi and 0.3 µM ATRi (VE-821) for 24 h. Fractions of RAD51 focus-positive cells were determined. n = 3 replicates. Error bars represent SD. (*) P < 0.05; (**) P < 0.01. (E) U2OS cells or U2OS-derived 53BP1 knockout cells were transfected with siControl or siBRCA1 and treated with increasing doses of PARPi (olaparib) in the absence or presence of 62.6 nM ATRi (VE-821) for 6 d. Cell viability was measured. n = 3 replicates. Error bars represent SD.
The role of ATR in BRCA2 regulation raises the question of whether ATR controls RAD51 recruitment when BRCA1 is bypassed. Loss of 53BP1 bypasses BRCA1 in HR (Bunting et al. 2010), providing an opportunity to test ATR function in the absence of BRCA1. Knockdown of 53BP1 in cells lacking BRCA1 significantly restored the formation of RAD51 foci (Fig. 4D; Supplemental Fig. S4E). Importantly, VE-821 reduced RAD51 foci in cells lacking both BRCA1 and 53BP1, suggesting that ATR remains indispensable for RAD51 recruitment even when BRCA1 is bypassed. Furthermore, VE-821 sensitized BRCA1, 53BP1 double-deficient cells to olaparib, showing that ATRi overcomes the bypass of BRCA1 by 53BP1 loss (Fig. 4E; Supplemental Fig. S4F).
We next compared the effects of ATRi on PARPi-resistant BRCA1-deficient cell lines and BRCA1-proficient cell lines. In SYr12 cells, VE-821 drastically reduced RAD51 foci and the recruitment of BRCA2 to DNA damage stripes (Fig. 5A,B). While VE-821 also reduced BRCA2 and RAD51 localization in UWB1 + B1 cells, its effects were more pronounced in SYr12 (Fig. 5A,B). Similarly, RAD51 foci were reduced by VE-821 to a greater extent in HCC1937 than in HCC1937 + B1 (Supplemental Fig. S5). Thus, in the absence of BRCA1, the rewired HR pathway is increasingly dependent on ATR.
Figure 5.
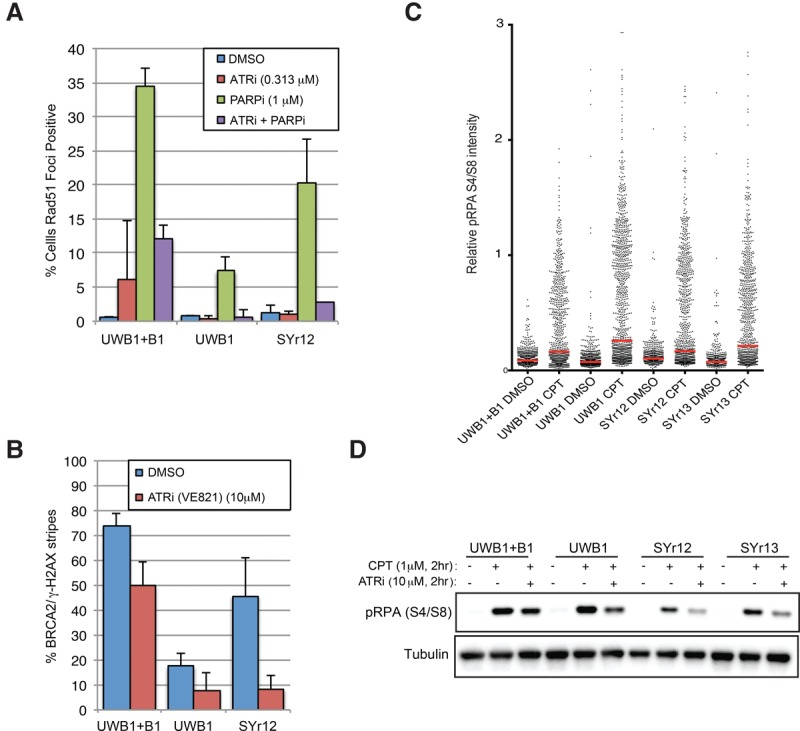
ATRi blocks BRCA1-independent HR in PARPi-resistant cells by inhibiting PALB2–BRCA2 localization to DNA breaks. (A) The indicated cell lines were treated with ATRi (VE-821), PARPi (olaparib), or ATRi and PARPi for 24 h. Fractions of RAD51 focus-positive cells were measured. n = 4 replicates for SYr12; n = 3 replicates for other samples. Error bars represent SD. (B) The indicated cell lines were treated with DMSO or 10 µM ATRi (VE-821) and irradiated with ultraviolet (UV) laser, and fractions of cells showing BRCA2 staining in γH2AX stripes were measured 1 h later. n = 4 replicates for SYr12; n = 3 replicates for other samples. Error bars represent SD. (C) The indicated cell lines were treated with 1 µM camptothecin (CPT) for 2 h. The intensity of phosphorylated RPA32 (p-RPA) (S4/S8) was measured by immunofluorescence, with each cell plotted individually. n = 550 cells for each condition. (D) The indicated cell lines were treated with 1 µM CPT or CPT and 10 µM ATRi for 2 h. Levels of p-RPA (S4/S8) were analyzed by Western blot.
In addition to BRCA1, RPA has also been implicated in the recruitment of PALB2 (Murphy et al. 2014). Phosphorylated RPA32 (p-RPA), which is generated during DSB resection, stimulates the binding of PALB2 to ssDNA in cell extracts (Murphy et al. 2014), providing a possible means to recruit PALB2–BRCA2 in the absence of BRCA1. As indicated by the BRCA1-independent HR in UWB1, SYr12, and SYr13, these BRCA1-deficient cells are able to resect DSBs. Indeed, in response to camptothecin (CPT)-induced DSBs, RPA32 was phosphorylated in UWB1, SYr12, and SYr13 (Fig. 5C). Consistent with its effects on BRCA2 and RAD51 recruitment (Fig. 5A,B), VE-821 significantly reduced the p-RPA in UWB1, SYr12, and SYr13 (Fig. 5D). These results suggest a possible mechanism by which ATRi blocks the rewired and less-efficient HR pathway in the absence of BRCA1, explaining the increased dependency of PARPi-resistant BRCA1-deficient cells on ATR.
ATRi overcomes BRCA1-independent fork protection
While BRCA1-independent HR is required for the survival of resistant cells in PARPis, RAD51 foci were not increased in several resistant lines compared with UWB1 (Supplemental Fig. S3A), suggesting that another event is necessary to confer PARPi resistance. The reduction of PTIP in some resistant lines prompted us to test whether the function of BRCA1 in fork protection is commonly bypassed (Supplemental Fig. S1C). DNA fiber analysis showed that forks stalled by hydroxyurea (HU) were degraded in UWB1 but not UWB + B1 (Fig. 6A). Remarkably, all of the PARPi-resistant lines that we tested with the fiber assay, including SYr9, SYr12, SYr13, SYr14, and SYr37, regained the ability to protect stalled forks (Fig. 6A,B; Supplemental Fig. S6A,B). The regain of fork protection was not restricted to the cell lines with reduced PTIP, indicating multiple contributing mechanisms. VE-821 significantly enhanced fork degradation in all of the resistant lines (Fig. 6B; Supplemental Fig. S6A,B). Mirin, an inhibitor of MRE11, reduced fork degradation in VE-821-treated cells (Fig. 6B). Consistent with the idea that MRE11-mediated fork degradation contributes to the PARPi sensitivity of BRCA1-deficient cells, partial knockdown of MRE11 reduced the PARPi sensitivity of UWB1 (Supplemental Fig. S6C,D). Thus, ATRi enables MRE11-mediated fork degradation in PARPi-resistant cells, reactivating a mechanism that contributes to PARPi sensitivity.
Figure 6.

ATRi reactivates degradation of stalled forks in PARPi-resistant cells. (A) DNA fiber analysis of stalled replication forks. Newly synthesized DNA was sequentially labeled with 50 µM CldU for 30 min and 100 µM IdU for 30 min. Cells were subsequently treated with 4 mM HU for 5 h, and lengths of CIdU- and IdU-labeled DNA fibers were measured. The IdU/CldU ratio was binned in increments of 0.2 and fit to a Gausian curve using Prism software. At least n = 100 fibers were measured for each condition; experiments were completed in triplicate. (B) DNA fiber analysis of stalled forks as in A, with each fiber plotted individually. The indicated cell lines were untreated or treated with 4 mM HU, HU and 10 µM ATRi (VE-821), or HU, ATRi, and 50 µM mirin for 5 h. Bars represent the median IdU/CldU ratios. n = 150 fibers for each condition; experiments were completed in triplicate. Significance was determined by Mann-Whitney test. (****) P < 0.0001. (C) The indicated cell lines were treated with 4 mM HU in the absence or presence of 10 µM ATRi (VE-821) for 5 h. The resulting genomic DNA from 5 × 105 cells for each condition was subjected to pulsed-field gel electrophoresis (PFGE) to measure DNA fragmentation. (D) The indicated PARPi-resistant cell lines were treated with 4 mM HU for 5 h in the absence or presence of 10 µM ATRi (VE-821). The levels of chromatin-bound RAD51 and RPA32 were analyzed with KU70 as a loading control. (E) The indicated cell lines were pulsed with EdU for 30 min and then collected or treated with 4 mM HU in the absence or presence of 10 µM ATRi (VE-821) for 5 h. The association of RAD51 with nascent DNA was analyzed by iPOND (isolation of proteins on nascent DNA) with histone H4 as a loading control. (F) SYr12 cells were transfected with siControl, siXRCC2, or siXRCC3 and treated with 4 mM HU for 5 h in the absence or presence of 10 µM ATRi (VE-821). The levels of chromatin-bound RAD51 were analyzed with KU70 as a loading control.
ATRi may reactivate MRE11-mediated fork degradation by inducing DSBs (Toledo et al. 2013). Using pulsed-field gel electrophoresis (PFGE), we found that similar levels of DSBs were induced by VE-821 in HU-treated UWB1 and SYr12 cells (Fig. 6C). Since VE-821 enhanced fork degradation only in SYr12 but not UWB1 (Fig. 6B), the induction of DSBs is unlikely to be the cause of increased fork degradation. In addition to HR, RAD51 is important for fork protection (Schlacher et al. 2011). RAD51 was recruited onto chromatin after HU treatment in SYr9, SYr12, SYr13, and SYr14 but not UWB1 (Fig. 6D; Supplemental Fig. S6E). The HU-induced chromatin binding of RAD51, but not RPA, was reduced by VE-821 in all of these resistant lines (Fig. 6D; Supplemental Fig. S6E). iPOND (isolation of proteins on nascent DNA) analysis showed that RAD51 was recruited to stalled forks in UWB1 + B1 and SYr12 but not UWB1 (Fig. 6E; Sirbu et al. 2011). Importantly, VE-821 reduced the RAD51 at stalled forks in SYr12. Together, these results suggest that ATRi reactivates fork degradation in PARPi-resistant cells by preventing RAD51 accumulation at stalled forks.
RAD51 paralogs are regulators of RAD51 filaments (Prakash et al. 2015). One of the RAD51 paralogs, XRCC3, is phosphorylated by ATM/ATR after DNA damage (Somyajit et al. 2013). Another paralog, XRCC2, is also a potential substrate of ATR (Matsuoka et al. 2007). Like RAD51, XRCC3 is required for the protection of stalled forks (Henry-Mowatt et al. 2003; Somyajit et al. 2015). Knockdown of XRCC3 reduced the HU-induced chromatin binding of RAD51 in SYr12 (Fig. 6F; Supplemental Fig. S6F). Depletion of XRCC2 also modestly decreased chromatin-bound RAD51. These results suggest that ATR may promote the association of RAD51 with stalled forks in resistant cells by regulating RAD51 paralogs.
ATRi overcomes fork protection in tumor cells from BRCA1/2-deficient patients
Having established that ATRi overcomes the PARPi resistance of BRCA1-deficient cancer cell lines, we asked whether this strategy is applicable to PARPi-resistant tumor cells derived from BRCA1/2-deficient patients. Primary tumor cells were isolated from a BRCA1-deficient (BRCA1-ins6kbEx13) ovarian cancer patient who had progressed after olaparib treatment and were cultured ex vivo for a few passages (Hendrickson et al. 2005). DNA fiber analyses showed that fork degradation was modest in the tumor cells but was significantly enhanced by VE-821 (Fig. 7A). Similar results were obtained using cells from patient-derived xenografts (PDXs) of the same tumor (Fig. 7B). In contrast to cells from the PARPi-resistant BRCA1-deficient patient, tumor cells and PDX cells from another ovarian cancer patient without BRCA1/2 mutations did not show fork degradation even in the presence of ATRi (Fig. 7A,B). Thus, ATRi overcomes fork protection in tumor cells from a PARPi-resistant BRCA1-deficient patient but not in BRCA-proficient tumor cells.
Figure 7.

ATRi reactivates fork degradation in tumor cells derived from PARPi-resistant BRCA-deficient patients. (A) DNA fiber analysis of tumor cells from a BRCA1-deficient PARPi-resistant ovarian cancer patient and a non-BRCA ovarian cancer patient, with each fiber plotted individually after no treatment, treatment with 4 mM HU, or treatment with HU and 10 µM ATRi (VE-821). Red bars represent the median IdU/CldU ratios. n = 125 for non-BRCA tumor cells; n = 215 for BRCA1-deficient PARPi-resistant tumor cells; experiments were performed in duplicate. Significance was determined by Mann-Whitney test. (**) P < 0.01. (B) DNA fiber analysis of PDX tumor cells derived from patients in A. Each fiber was plotted individually after no treatment, treatment with 4 mM HU, or treatment with HU and 10 µM ATRi (VE-821). Red bars represent the median IdU/CldU ratios. n = 185. Experiments were performed in duplicate. Significance was determined by Mann-Whitney test. (**) P < 0.01; (****) P < 0.0001. (C) DNA fiber analysis of circulating tumor cells (CTCs) from a BRCA2-deficient breast cancer patient and a non-BRCA breast cancer patient as in A, with each fiber plotted individually after no treatment, treatment with 4 mM HU, or treatment with HU and 10 µM ATRi (VE-821). Significance was determined by Mann-Whitney test. (****) P < 0.0001. (D) Model of how PARPi-resistant BRCA1-deficient cancer cells bypass BRCA1 in two sequential steps. In step 1, the HR function of BRCA1 is partially bypassed. This partial bypass of BRCA1 allows cancer cells to survive the lack of BRCA1 but is not sufficient to confer PARPi resistance (e.g., UWB1). In step 2, when cancer cells are under the selective pressure of PARPis, the HR function of BRCA1 is further bypassed in some cells (e.g., SY12). Furthermore, the function of BRCA1 in fork protection is commonly bypassed (e.g., SYr9, SYr12, SYr13, and SYr14). Our findings show that the bypasses of both BRCA1 functions in HR and fork protection contribute to PARPi resistance in cancer cells. This model explains why ATRi, which blocks both BRCA1-independent HR and fork protection, has a unique ability to overcome PARPi resistance.
Recent studies have suggested that ex vivo culture of circulating tumor cells (CTCs) may be used to predict treatment responses in patients (Yu et al. 2014). Brx-68, a CTC line derived from a breast cancer patient without BRCA1/2 mutations, is sensitive to the PARPi AZD7762 (Yu et al. 2014). Fork degradation occurred efficiently in Brx-68 cells (Fig. 7C), suggesting that loss of fork protection, even if it is not caused by BRCA1/2 mutations, associates with PARPi sensitivity. Consistent with the idea that fork protection is already lost in Brx-68, VE-821 did not enhance fork degradation. In contrast to Brx-68, fork degradation was not detected in Brx-50, a PARPi-resistant CTC line derived from a breast cancer patient carrying a BRCA2 frameshift mutation (Fig. 7C; Yu et al. 2014). Importantly, VE-821 enhanced fork degradation in Brx-50, lending further support to the notion that ATRi overcomes fork protection in PARPi-resistant BRCA-deficient tumor cells.
Discussion
Step-wise acquisition of PARPi resistance in BRCA-deficient cancer cells
In this study, we found that both the HR and fork protection functions of BRCA1 are commonly bypassed in PARPi-resistant cells. Previous studies have shown that loss of 53BP1, RIF1, or REV7 is sufficient to bypass the HR function of BRCA1 and confer PARPi resistance (Bunting et al. 2010; Chapman et al. 2013; Escribano-Diaz et al. 2013; Zimmermann et al. 2013; Xu et al. 2015). Loss of PTIP is also sufficient to bypass the fork protection functions of BRCA1/2 and give rise to resistance to PARPis (Chaudhuri et al. 2016). However, in a significant fraction of the PARPi-resistant cell lines that we developed, both REV7 and PTIP are reduced relative to the parental line, showing that both functions of BRCA1 are simultaneously bypassed in single-cell-derived cell populations. This observation suggests that, while the bypass of either BRCA1 function is sufficient to confer PARPi resistance in knockout or knockdown models, the acquisition of PARPi resistance in cancer cells often involves the bypass of both BRCA1 functions. The presence of multiple resistance mechanisms in individual cancer cells adds an extra layer of complexity to the heterogeneity of resistance in tumors. If one aims to identify drugs that effectively overcome PARPi resistance, these drugs would have to possess the ability to overcome the bypass of both BRCA1 functions.
Why is it necessary for BRCA1-deficient cancer cells to bypass both BRCA1 functions to acquire PARPi resistance? One possibility is that both BRCA1 functions are only partially bypassed in cancer cells, and the contributions of both bypasses are needed to confer significant PARPi resistance. Additionally, the bypasses of the two BRCA1 functions may be coordinated in some way to achieve PARPi resistance. Interestingly, the HR function of BRCA1 is already partially bypassed in UWB1 cells before the acquisition of PARPi resistance. This partial bypass of BRCA1 may help cancer cells survive the lack of BRCA1 (Fig. 7D). After UWB1 acquires PARPi resistance, RAD51 focus formation increases in a subset of resistant lines, suggesting that a further bypass of the HR function of BRCA1 may contribute to PARPi resistance (Fig. 7D). Even in a resistant line (SYr13) that does not display an increase in RAD51 foci, PALB2–BRCA2 is required for cell survival in PARPi, suggesting that the partial BRCA1 bypass inherited from UWB1 is indispensable for PARPi resistance. Importantly, the fork protection function of BRCA1 is bypassed in all of the resistant lines tested, suggesting that it is closely associated with PARPi resistance (Fig. 7D). Together, these findings suggest that the HR and fork protection functions of BRCA1 are bypassed in UWB1 cells in a sequential manner. The HR function is partially bypassed even before cells acquire PARPi resistance, priming cells for further bypass of the HR and/or fork protection functions when cells are under PARPi selection. This two-step model for acquisition of PARPi resistance may help explain how the bypasses of the two BRCA1 functions contribute to PARPi resistance, providing clues to how PARPi resistance can be prevented and overcome.
ATRi disrupts rewired HR and fork protection pathways
Through unbiased RNA profiling of PARPi-resistant cells and an inhibitor screen, we found that ATR has an important role in PARPi resistance. ATRi has a unique ability to overcome PARPi resistance because of its effects on the rewired HR and fork protection pathways in the absence of BRCA1 (Supplemental Fig. S7A). In PARPi-resistant BRCA1-deficient cells, PALB2–BRCA2 remains indispensable for the rewired HR pathway. ATR is required for the localization of PALB2–BRCA2 to DSBs in these cells, possibly due to its role in phosphorylating RPA and promoting RPA-mediated PALB2 recruitment (Supplemental Fig. S7A). When the fork protection function of BRCA1 is bypassed in PARPi-resistant cells, loading of RAD51 to stalled replication forks is restored. This rewired fork protection pathway is also dependent on ATR and its substrate, XRCC3 (Supplemental Fig. S7A). ATRi inhibits RAD51 loading to stalled forks, which reduces the protection against nucleases and leads to enhanced fork degradation. Altered expression of a number of genes could lead to rewiring of HR and fork protection pathways in BRCA1/2-deficient tumors. For example, low 53BP1 expression is associated with PARPi resistance in mouse BRCA1-deficient tumors, and low expression of 53BP1 and PTIP is associated with poor survival of human breast and ovarian cancer patients (Bouwman et al. 2010; Jaspers et al. 2013; Chaudhuri et al. 2016). ATRi broadly overcomes PARPi resistance in 53BP1-depleted cells and in a panel of resistant cell lines harboring a variety of resistance mechanisms, suggesting that ATR inhibition is an effective way to overcome the heterogeneity of resistance in tumors. In addition to overcoming acquired PARPi resistance, ATRi also overcomes pre-existing PARPi resistance and prevents the emergence of resistance when used up front with PARPi. These findings highlight the versatility of ATRi in overcoming the PARPi resistance of BRCA-deficient cancer cells.
ATRi selectively sensitizes PARPi-resistant cells to PARPis
ATR is a known regulator of HR, and ATRi is expected to sensitize cells to PARPis even in the presence of BRCA1/2 (Huntoon et al. 2013). Indeed, a synergy between ATRi and PARPi has been reported in several HR-proficient or HR-deficient contexts (Peasland et al. 2011; Huehls et al. 2012; Ogiwara et al. 2013; Abu-Sanad et al. 2015; Mohni et al. 2015; Kim et al. 2016). Here, we show that ATRi has a unique ability to preferentially sensitize PARPi-resistant BRCA1-deficient cells, as opposed to BRCA1-proficient cells, to PARPi. When used at low concentrations, ATRi displays a greater synergy with PARPis in PARPi-resistant cells than in BRCA1-proficient cells. Although ATRi preferentially kills PARPi-resistant cells, the functions of ATR in PARPi-resistant cells and BRCA-proficient cells may be the same or related. The preferential effects of ATRi on PARPi-resistant cells can be explained by several nonmutually exclusive possibilities. First, HR and fork protection pathways can function in both BRCA1-dependent and BRCA1-independent modes, and the BRCA1-independent modes of these pathways are more reliant on ATR. Second, the rewiring of HR and fork protection pathways in resistant cells may lead to increased ATR dependence because of the change of players in these pathways. Finally, the restoration of HR and fork protection in resistant cells may be partial, which renders these suboptimal pathways more sensitive to ATR inhibition compared with the fully functional pathways in BRCA-proficient cells (Supplemental Fig. S7B). Consistent with this possibility, RAD51 focus formation is generally lower in PARPi-resistant lines compared with BRCA-proficient lines (Supplemental Fig. S3A; Issaeva et al. 2010). Partial restoration of RAD51 focus formation is also prevalent in mouse PARPi-resistant BRCA1-deficient tumors (Jaspers et al. 2013). The ability of low concentrations of ATRi to preferentially sensitize PARPi-resistant cells may be important for the therapeutic window of ATRis in clinical settings.
Association of fork protection with PARPi resistance in tumor cells from patients
In addition to multiple BRCA1-deficient cancer cell lines of distinct origins and their derivative lines, we analyzed primary tumor cells and CTCs from ovarian and breast cancer patients. Our results show that it is feasible to perform DNA fiber assays using primary tumor cells and CTCs cultured ex vivo. We found that the degradation of stalled replication forks is associated with PARPi sensitivity even in tumor cells without BRCA1/2 mutations. This finding raises the possibility that the efficiency of stalled fork degradation in primary tumor cells or CTCs can be used as a biomarker to predict the PARPi responses of patients. Furthermore, fork degradation is modest or completely lost in tumor cells from PARPi-resistant patients, supporting the notion that the regain of fork protection is associated with PARPi resistance in human tumors. Finally, like in cancer cell lines, ATRi demonstrates the ability to enhance fork degradation in PARPi-resistant tumor cells. As the use of PARPis broadens in cancer clinics, primary tumor cells, CTCs, and PDXs from additional PARPi-resistant patients will become available. These reagents will allow us to further investigate the PARPi resistance mechanisms in BRCA-deficient patients and the efficacy of ATRis to overcome them, providing a new guide for ongoing and future clinical trials of ATRi–PARPi combination therapies.
Materials and methods
Cell lines
UWB1.289 and UWB1.289 + BRCA1 were obtained from American Type Culture Collection (ATCC) and maintained in RPMI1640 (ATCC) and MEGM bullet kit (1:1; Lonza) with 3% FBS and 1% penicillin/streptomycin (DelloRusso et al. 2007). SYr-resistant cell lines were derived from the parental UWB1.289 line after 45 d of selection with 1.0 µM PARPi (olaparib; SelleckChem) or following passages with incremental increases of PARPi (olaparib) from 0.025 to 1.0 µM. T2 (non-BRCA), BR5 (BRCA1Δ11/Δ11, exon 11 deleted), and resistant BR5-R1 cell lines are FVB mouse-derived ovarian tumor cell lines (Xing and Orsulic 2006) and were maintained in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% L-glutamine. BR5-R1 was derived through incremental increases of PARPi (olaparib) from 0.025 to 1.0 µM. HCC1937 and HCC1937 + BRCA1 cells were maintained in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin. RPE-hTERT cells were maintained in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% L-glutamine. Primary human ovarian tumor cells were collected from ascites or pleural fluid of ovarian cancer patients that had tested positive for malignant cells by cytological analysis. Human ovarian tumor cells were maintained in RPMI-1640 with 10% FBS, 1% L-glutamine, 1% penicillin/streptomycin, and supernatant from patient-matched ascites/pleural fluid. All cells were grown at 37°C and 5% CO2.
Inhibitors
Cells were treated with inhibitors PARPi (olaparib, ABT-888), ATRi (VE-821, VE-822, AZ20), ATMi (KU55933), Chk1i (MK-8776/SCH900776), DNA-PKi (NU7026), and Wee1i (MK-1775), all from SelleckChem, except AZ20 (custom-made).
Antibodies
Primary antibodies used for Western blots included BRCA1 (Santa Cruz Biotechnology), BRCA1 (Millipore), BRCA2 (Millipore), 53BP1 (Cell Signaling), RIF1 (Bethyl Laboratories), REV7 (BD Transduction Laboratories), PTIP (kindly provided by Dr. Junjie Chen, MD Anderson Cancer Center), KU70 (GeneTex), Tubulin (Cell Signaling), and GAPDH (EMD Millipore).
DNA fiber assay
DNA fiber assays were performed as described previously (Marechal et al. 2014). In brief, cells were pulsed with CldU for 30 min followed by a pulse of IdU for 30 min. Cells were either collected and spread or incubated with 4 mM HU, HU and 10 µM ATRi (VE-821), or HU, ATRi, and 50 µM mirin for 5 h and then collected and spread. Collected cells were resuspended in cold PBS (1 × 106 cell per milliliter), and 2.5 µL was spotted onto a glass slide. Spreading buffer (7.5 µL) (0.5% SDS, 200 mM Tris-HCl at pH 7.4, 50 mM EDTA) was added to each spot. After a brief incubation, slides were tilted ∼15°, and lysed cells were allowed to slide down and dry. Spread DNA was fixed in cold methanol:acetone (3:1). DNA was denatured in 2.5 N HCl for 30 min and blocked in 3% BSA/0.05% Tween-20 for 30 min at 37°C. Detection of CldU and IdU tracts was carried out using rat anti-BrdU (1:100; AbDSerotec, OBT0030) and mouse anti-BrdU (1:50; BD Biosciences) for 1 h at 37°C followed by Alexa-488 anti-mouse (1:100) and Cy3 anti-rat (1:100; Jackson ImmunoResearch) for 30 min at 37°C. Slides were mounted with VectaShield (Vector Laboratories). Fibers were imaged at 60× with a Nikon 90i microscope and quantified using ImageJ software.
Supplementary Material
Acknowledgments
We thank S. Cantor, J. Chen, D. Durocher, N. Dyson, L. Ellisen, S. Orsulic, and B. Xia for reagents and discussions. This work was supported by grants from the National Institutes of Health (GM076388 and CA197779 to L.Z., and CA129933 to D.A.H.), National Cancer Institute Federal Share of Program Income (to L.Z.), Jim and Ann Orr MGH Research Scholar Award (to L.Z.), Susan Komen for the Cure (KG09042 to S.M.), and the Wellcome Trust (102696 to C.H.B.). The Flow Cytometry Core is supported by National Institutes of Health instrumentation grant 1S10RR023440-01A1. S.A.Y. was supported by a fellowship from the Department of Defense (W81XWH-13-1-0027). V.C. and R.B. have received support from the Philippe Foundation. M.-M.G. is a fellow of Fonds de recherche du Québec-Santé. H.D.N. was a Medical Discovery Post-doctoral Fellow. R.B. was a Marsha Rivkin Scholar. L.Z. is the James and Patricia Poitras Endowed Chair in Cancer Research.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.290957.116.
References
- Abu-Sanad A, Wang Y, Hasheminasab F, Panasci J, Noe A, Rosca L, Davidson D, Amrein L, Sharif-Askari B, Aloyz R, et al. 2015. Simultaneous inhibition of ATR and PARP sensitizes colon cancer cell lines to irinotecan. Front Pharmacol 6: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. 2012. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol 14: 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlskog JK, Larsen BD, Achanta K, Sorensen CS. 2016. ATM/ATR-mediated phosphorylation of PALB2 promotes RAD51 function. EMBO Rep 17: 671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, et al. 2010. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376: 245–251. [DOI] [PubMed] [Google Scholar]
- Boersma V, Moatti N, Segura-Bayona S, Peuscher MH, van der Torre J, Wevers BA, Orthwein A, Durocher D, Jacobs JJ. 2015. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 521: 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 2010. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 17: 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917. [DOI] [PubMed] [Google Scholar]
- Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, Hardy EJ, Dellaire G, Haas W, Xia B, et al. 2017. Coupling of homologous recombination and the checkpoint by ATR. Mol Cell 65: 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141: 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, et al. 2012. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell 46: 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, et al. 2013. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153: 1266–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. 2013. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 49: 858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri AR, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. 2016. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535: 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang G, Niu X, Zhao J, Tan W, Wang H, Zhao L, Ge Y. 2014. Combination of AZD2281 (olaparib) and GX15-070 (obatoclax) results in synergistic antitumor activities in preclinical models of pancreatic cancer. Cancer Lett 348: 20–28. [DOI] [PubMed] [Google Scholar]
- Choi YE, Meghani K, Brault ME, Leclerc L, He YJ, Day TA, Elias KM, Drapkin R, Weinstock DM, Dao F, et al. 2016. Platinum and PARP inhibitor resistance due to overexpression of microRNA-622 in BRCA1-mutant ovarian cancer. Cell Rep 14: 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DelloRusso C, Welcsh PL, Wang W, Garcia RL, King MC, Swisher EM. 2007. Functional characterization of a novel BRCA1-null ovarian cancer cell line in response to ionizing radiation. Mol Cancer Res 5: 35–45. [DOI] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. 2013. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339: 711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. 2008. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 451: 1111–1115. [DOI] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. 2013. A cell cycle-dependent regulatory circuit composed of 53BP1–RIF1 and BRCA1–CtIP controls DNA repair pathway choice. Mol Cell 49: 872–883. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. [DOI] [PubMed] [Google Scholar]
- Feng L, Fong KW, Wang J, Wang W, Chen J. 2013. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem 288: 11135–11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fojo T, Bates S. 2013. Mechanisms of resistance to PARP inhibitors—three and counting. Cancer Discov 3: 20–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al. 2009. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361: 123–134. [DOI] [PubMed] [Google Scholar]
- Foote KM, Blades K, Cronin A, Fillery S, Guichard SS, Hassall L, Hickson I, Jacq X, Jewsbury PJ, McGuire TM, et al. 2013. Discovery of 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole (AZ20): a potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J Med Chem 56: 2125–2138. [DOI] [PubMed] [Google Scholar]
- Guillemette S, Serra RW, Peng M, Hayes JA, Konstantinopoulos PA, Green MR, Cantor SB. 2015. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev 29: 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson BC, Judkins T, Ward BD, Eliason K, Deffenbaugh AE, Burbidge LA, Pyne K, Leclair B, Ward BE, Scholl T. 2005. Prevalence of five previously reported and recurrent BRCA1 genetic rearrangement mutations in 20,000 patients from hereditary breast/ovarian cancer families. Genes Chromosomes Cancer 43: 309–313. [DOI] [PubMed] [Google Scholar]
- Henry-Mowatt J, Jackson D, Masson JY, Johnson PA, Clements PM, Benson FE, Thompson LH, Takeda S, West SC, Caldecott KW. 2003. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol Cell 11: 1109–1117. [DOI] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. 2004. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64: 9152–9159. [DOI] [PubMed] [Google Scholar]
- Hollick JJ, Golding BT, Hardcastle IR, Martin N, Richardson C, Rigoreau LJ, Smith GC, Griffin RJ. 2003. 2,6-disubstituted pyran-4-one and thiopyran-4-one inhibitors of DNA-dependent protein kinase (DNA-PK). Bioorg Med Chem Lett 13: 3083–3086. [DOI] [PubMed] [Google Scholar]
- Huehls AM, Wagner JM, Huntoon CJ, Karnitz LM. 2012. Identification of DNA repair pathways that affect the survival of ovarian cancer cells treated with a poly(ADP-ribose) polymerase inhibitor in a novel drug combination. Mol Pharmacol 82: 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntoon CJ, Flatten KS, Wahner Hendrickson AE, Huehls AM, Sutor SL, Kaufmann SH, Karnitz LM. 2013. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res 73: 3683–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issaeva N, Thomas HD, Djureinovic T, Jaspers JE, Stoimenov I, Kyle S, Pedley N, Gottipati P, Zur R, Sleeth K, et al. 2010. 6-thioguanine selectively kills BRCA2-defective tumors and overcomes PARP inhibitor resistance. Cancer Res 70: 6268–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, et al. 2013. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov 3: 68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, Wang Y, Capelletti M, Sarosiek KA, Moreau LA, et al. 2013. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci 110: 17041–17046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. 2015. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 33: 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, et al. 2015. FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res 21: 4257–4261. [DOI] [PubMed] [Google Scholar]
- Kim H, George E, Ragland RL, Rafail S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown EJ, Simpkins F. 2016. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA mutant ovarian cancer models. Clin Cancer Res 10.1158/1078-0432.CCR-16-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurfurstova D, Bartkova J, Vrtel R, Mickova A, Burdova A, Majera D, Mistrik M, Kral M, Santer FR, Bouchal J, et al. 2016. DNA damage signalling barrier, oxidative stress and treatment-relevant DNA repair factor alterations during progression of human prostate cancer. Mol Oncol 10: 879–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. 2013. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med 19: 1381–1388. [DOI] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. 2016. BRCAness revisited. Nat Rev Cancer 16: 110–120. [DOI] [PubMed] [Google Scholar]
- Lord CJ, McDonald S, Swift S, Turner NC, Ashworth A. 2008. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair (Amst) 7: 2010–2019. [DOI] [PubMed] [Google Scholar]
- Lord CJ, Tutt AN, Ashworth A. 2015. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med 66: 455–470. [DOI] [PubMed] [Google Scholar]
- Marechal A, Li JM, Ji XY, Wu CS, Yazinski SA, Nguyen HD, Liu S, Jimenez AE, Jin J, Zou L. 2014. PRP19 transforms into a sensor of RPA–ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol Cell 53: 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166. [DOI] [PubMed] [Google Scholar]
- Mohni KN, Thompson PS, Luzwick JW, Glick GG, Pendleton CS, Lehmann BD, Pietenpol JA, Cortez D. 2015. A Synthetic lethal screen identifies DNA repair pathways that sensitize cancer cells to combined ATR inhibition and cisplatin treatments. PLoS One 10: e0125482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. 2012. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 72: 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, Pommier Y. 2016. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 7: 76534–76550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AK, Fitzgerald M, Ro T, Kim JH, Rabinowitsch AI, Chowdhury D, Schildkraut CL, Borowiec JA. 2014. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol 206: 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor MJ. 2015. Targeting the DNA damage response in cancer. Mol Cell 60: 547–560. [DOI] [PubMed] [Google Scholar]
- Ogiwara H, Ui A, Shiotani B, Zou L, Yasui A, Kohno T. 2013. Curcumin suppresses multiple DNA damage response pathways and has potency as a sensitizer to PARP inhibitor. Carcinogenesis 34: 2486–2497. [DOI] [PubMed] [Google Scholar]
- Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G, et al. 2015. A mechanism for the suppression of homologous recombination in G1 cells. Nature 528: 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Patel AG, Sarkaria JN, Kaufmann SH. 2011. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci 108: 3406–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, Cliby WA, Sarkaria J, Beale G, Edmondson RJ, et al. 2011. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer 105: 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt SJ, Rehman FL, Bajrami I, Brough R, Wallberg F, Kozarewa I, Fenwick K, Assiotis I, Chen L, Campbell J, et al. 2013. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS One 8: e61520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Zhang Y, Feng W, Jasin M. 2015. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 7: a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, Muschel RJ, Brunner TB. 2012. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther 13: 1072–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JM, Pollard JR. 2011. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 7: 428–430. [DOI] [PubMed] [Google Scholar]
- Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, et al. 2008. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci 105: 17079–17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. 2008. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 451: 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51–BRCA1/2. Cancer Cell 22: 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. 2011. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25: 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somyajit K, Basavaraju S, Scully R, Nagaraju G. 2013. ATM- and ATR-mediated phosphorylation of XRCC3 regulates DNA double-strand break-induced checkpoint activation and repair. Mol Cell Biol 33: 1830–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somyajit K, Saxena S, Babu S, Mishra A, Nagaraju G. 2015. Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res 43: 9835–9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenblick A, de Azambuja E, Azim HA Jr, Piccart M. 2015. An update on PARP inhibitors—moving to the adjuvant setting. Nat Rev Clin Oncol 12: 27–41. [DOI] [PubMed] [Google Scholar]
- Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. 2008. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res 68: 2581–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sy SM, Huen MS, Chen J. 2009. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci 106: 7155–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. 2013. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103. [DOI] [PubMed] [Google Scholar]
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, et al. 2010. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376: 235–244. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang H, Powell SN, Iliakis G, Wang Y. 2004. ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res 64: 7139–7143. [DOI] [PubMed] [Google Scholar]
- Wang J, Aroumougame A, Lobrich M, Li Y, Chen D, Chen J, Gong Z. 2014. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev 28: 2693–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, Peri S, van der Gulden H, van der Heijden I, O'Brien SW, et al. 2016. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res 76: 2778–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing D, Orsulic S. 2006. A mouse model for the molecular characterization of brca1-associated ovarian carcinoma. Cancer Res 66: 8949–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, et al. 2015. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521: 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav B, Wennerberg K, Aittokallio T, Tang J. 2015. Searching for drug synergy in complex dose-response landscapes using an interaction potency model. Comput Struct Biotechnol J 13: 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, Helleday T. 2012. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 72: 2814–2821. [DOI] [PubMed] [Google Scholar]
- Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, et al. 2014. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 345: 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X. 2009. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol 19: 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 2013. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339: 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.