

Abstract
A platform has been developed to measure the activity of 23 enzymes that are involved in central carbon and nitrogen metabolism in Arabidopsis thaliana. Activities are assayed in optimized stopped assays and the product then determined using a suite of enzyme cycling assays. The platform requires inexpensive equipment, is organized in a modular manner to optimize logistics, calculates results automatically, combines high sensitivity with throughput, can be robotized, and has a throughput of three to four activities in 100 samples per person/day. Several of the assays, including those for sucrose phosphate synthase, ADP glucose pyrophosphorylase (AGPase), ferredoxin-dependent glutamate synthase, glycerokinase, and shikimate dehydrogenase, provide large advantages over previous approaches. This platform was used to analyze the diurnal changes of enzyme activities in wild-type Columbia-0 (Col-0) and the starchless plastid phosphoglucomutase (pgm) mutant, and in Col-0 during a prolongation of the night. The changes of enzyme activities were compared with the changes of transcript levels determined with the Affymetrix ATH1 array. Changes of transcript levels typically led to strongly damped changes of enzyme activity. There was no relation between the amplitudes of the diurnal changes of transcript and enzyme activity. The largest diurnal changes in activity were found for AGPase and nitrate reductase. Examination of the data and comparison with the literature indicated that these are mainly because of posttranslational regulation. The changes of enzyme activity are also strongly delayed, with the delay varying from enzyme to enzyme. It is proposed that enzyme activities provide a quasi-stable integration of regulation at several levels and provide useful data for the characterization and diagnosis of different physiological states. As an illustration, a decision tree constructed using data from Col-0 during diurnal changes and a prolonged dark treatment was used to show that, irrespective of the time of harvest during the diurnal cycle, the pgm mutant resembles a wild-type plant that has been exposed to a 3 d prolongation of the night.
INTRODUCTION
The relatively simple chemistry of nucleic acids has made it possible to develop array technologies that allow genome-wide profiling of transcript levels in sequenced organisms (DeRisi et al., 1997; Celis et al., 2000; Michaut et al., 2003), including Arabidopsis thaliana (Wang et al., 2003; Thimm et al., 2004; AtGenExpress, www.arabidopsis.org). The sensitivity and precision with which transcript levels can be quantified will be further increased by multiplexed real-time RT-PCR (Czechowski et al., 2004). Interpretation depends on assumptions about the relation between the levels of transcripts and proteins. This is difficult to predict because it will depend on how far the transcript concentration affects the rate of translation, how rapidly the protein turns over, and whether the rate of degradation is coordinately regulated. In some cases, the response may be attenuated, and in others posttranscriptional regulation may lead to exaggerated or independent changes. For example, nitrate reductase (NIA) protein increases twofold in leaves during the first part of the light period, whereas NIA transcript decreases rapidly (Scheible et al., 1997a). This occurs because illumination stimulates the synthesis and inhibits the breakdown of NIA protein (Kaiser and Huber, 1999; Weiner and Kaiser, 1999). The large number of genes involved in protein degradation in Arabidopsis (Arabidopsis Genome Initiative, 2000) underlines the potential importance of posttranscriptional regulation.
Quantification of proteins at the proteome scale poses a major technical challenge (Greenbaum et al., 2003). Although methods are being developed (Steen and Pandey, 2002), they lag behind the techniques for expression profiling and require costly and extensive infrastructure. Enzyme activity assays require inexpensive equipment and provide quantitative data. Measurements in optimized assays that allow maximum activity (ap Rees and Hill, 1994) could act as a proxy for quantitative proteomics, at least for studies of metabolism. Throughput can be increased by transferring existing protocols to microplates and using robots (Ashour et al., 1987; Kiianitsa et al., 2003). High throughput assays of individual enzymes are used routinely in drug or herbicide discovery (Lein et al., 2004). The uncommonness of each reaction and resulting diversity of assay systems will make it logistically impossible to measure an entire enzymome, but it may be possible to create efficient platforms for subsets of enzymes by grouping them in modules that share a common detection method.
Such platforms could provide quantitative data that provide insights into the relation between transcript levels and protein levels in genomics studies. Activity measurements can also provide information about posttranslational regulation in cases where it alters the kinetic properties (Kaiser and Huber, 1994, 1999). Another potential application is in the diagnosis of genotypes or physiological states. It can also be approached via machine learning, in which a training data set from a series of defined conditions is used to generate a classification key that is used as a diagnostic tool to classify unknown samples (Quinlan, 1979; Kononenko, 2001). Machine learning can be used on data sets with a relatively small number of parameters but requires good replication and reliable quantification.
This article presents a set of robotized microplate-based activity assays for 23 enzymes from central carbon and nitrogen metabolism. In addition to allowing high-throughput analyses, most of the assays are highly sensitive, allowing very low activities to be detected or very small samples to be analyzed. The platform has been used to investigate the changes of enzyme activities during the diurnal cycle in wild-type Arabidopsis and a starchless mutant where carbohydrate storage is disturbed and during a prolonged dark treatment of wild-type Arabidopsis. These treatments allow us to ask if rapid changes in transcript levels during the diurnal rhythm lead to changes in the activity of the encoded enzyme and if larger changes of transcript levels in the starchless pgm mutant or more prolonged changes in prolonged starvation lead to more marked changes in enzyme activity. Methods are presented that integrate information about transcript levels and enzyme activity, provide insights into the quantitative relation between changes of transcript levels and enzyme activities, and highlight cases where translational regulation or protein turnover may contribute. The data sets are also used to illustrate the utility of enzyme activities for diagnosis using machine learning.
RESULTS
Design of an Analytic Platform for Parallel Determination of Enzyme Activities in Central Metabolism
Enzymes were assayed using stopped assays, in which aliquots of extracts are incubated with their substrates for a fixed time before stopping the reaction and the product then determined in a second assay. Stopped assays can be performed in parallel, stored, and combined with sets of samples from the same or other enzymes before measuring the product. They can be performed in a smaller volume than is usually required for detection in a spectrophotometer or a fluorimeter, which decreases the amount of extract and reagent required. Although they require more pipetting steps than direct assays, this does not represent a major disadvantage when the assays are robotized (see Methods).
In most cases, cycling assays (Gibon et al., 2002) were used to determine the products of the enzyme reactions. The principle is that the net rate of the cycle is a pseudo zero-order reaction whose rate (d[analyte measured]/dt = −d[precursor of this analyte]/dt) depends on the initial concentration of the metabolite being determined. Quantification is achieved by comparison with a standard curve, in which different concentrations of the metabolite in question are added in the presence of pseudo-extract (Gibon et al., 2002). A combination of stopped and cycling assays can provide 10,000-fold higher sensitivity than conventional assays. The sensitivity limit for routine measurements in leaf extracts was 0.01 to 1 nmol product per assay. This allows extracts to be diluted far more strongly in the assay, which increases the reliability of stopped assays. Interference by other components in the extract is reduced, substrates can be maintained at near-constant levels throughout the assay, product inhibition is minimized because the amounts accumulated are very low (typically below 20 μM), and interference by other enzymes is decreased.
The platform uses three cycling systems (Table 1, Figure 1). Glycerol-3-P and dihydroxyacetone-P are measured by a glycerol-3-P dehydrogenase/glycerol-3-P oxidase cycle (Gibon et al., 2002), NADP+ and NADPH by a glucose-6-P dehydrogenase/phenazine methosulfate (G6PDH/PMS) cycle (Nisselbaum and Green, 1969), and NAD+ and NADH by an alcohol dehydrogenase/phenazine ethosulfate (ADH/PES) cycle (Bernofsky and Swan, 1973). Reactions were stopped using HCl, except when the direct or indirect product was a reduced coenzyme, in which case NaOH was added. Oxidized NAD(P)+ is stable at low pH and labile at high pH, and reduced NAD(P)H is labile at low pH and stable at high pH (Nisselbaum and Green, 1969). This combination allows a wide range of enzyme activities to be measured, including all NAD(P)-dependent dehydrogenases and all activities that generate ATP or UTP because the latter can be coupled to glycerol-3-P formation. In some cases, one of these metabolites is the direct product of the enzyme reaction. Otherwise, coupling enzymes are included to convert a product into one of these metabolites. The modular organization allows many different enzyme assays to be fed into a pipeline with just three downstream cycling systems, which simplifies logistics and increases throughput.
Table 1.
Assay Principles, Stability to a Freeze/Thaw Cycle, Optimal Extract Dilution, Average Activity, and Average Coefficient of Variation between Biological Replicates of Rosettes from 5-Week-Old Arabidopsis Col-0 Collected at Various Time Points throughout a 12-h-Day and 12-h-Night Cycle
| Enzyme | Method | Freeze/Thawing Stability (% Retained ± sd) | Dilution of FW | Average Activity (Arabidopsis Rosette, nmol g−1 FW min−1) | Average Coefficient of Variation (%) |
|---|---|---|---|---|---|
| AGPase | G3POX/G3PDH | 85 ± 5 | 1,000 | 1,255 | 12 |
| NAD-GAPDH | 90 ± 1 | 20,000 | 36,981 | 15 | |
| NADP-GAPDH | 86 ± 3 | 10,000 | 17,041 | 14 | |
| PFP | 26 ± 9 | 1,000 | 191 | 16 | |
| PK | 70 ± 9 | 1,000 | 952 | 18 | |
| SPS | ND | 1,000 | 540 | 8 | |
| TK | 99 ± 3 | 1,000 | 7,462 | 17 | |
| GK | 51 ± 9 | 500 | 18 | 33 | |
| cFBPase | G6PDH/(PMS + MTT) | 55 ± 26 | 500 | 105 | 23 |
| GlcK | 65 ± 13 | 1,000 | 150 | 23 | |
| FruK | 65 ± 11 | 1,000 | 179 | 18 | |
| G6PDH | 37 ± 9 | 5,000 | 849 | 13 | |
| ICDH | 97 ± 29 | 2,000 | 1,506 | 24 | |
| ShikDH | 48 ± 5 | 2,000 | 598 | 18 | |
| AlaAT | ADH/(PES + MTT) | 79 ± 21 | 20,000 | 5,462 | 20 |
| AspAT | 81 ± 30 | 20,000 | 5,229 | 19 | |
| Fumarase | 9 ± 3 | 20,000 | 4,875 | 10 | |
| GLDHam | 97 ± 7 | 1,000 | 876 | 11 | |
| PEPCase | 126 ± 35 | 1,000 | 744 | 19 | |
| Acid Inv | Glucose oxidase + dihydroxyphenoxazine | 62 ± 14 | 1,000 | 532 | 20 |
| Fd-GOGAT | GLDH | 37 ± 7 | 500 | 1,685 | 13 |
| NIA | SA + NNEDA | 175 ± 49 (Vsel) | 500 | – | 12 |
| 93 ± 18 (Vmax) | 575 | 8 | |||
| GS | PK + LDH | 129 ± 16 | 1,000 | 1,867 | 14 |
See Figure 1 for abbreviations. Acid Inv, acid invertase; cFBPase, cytosolic FBPase; GLDHam, GLDH aminating; NAD-GAPDH, NAD-dependent GAPDH; NADP-GAPDH, NADP-dependent GAPDH; MTT, methylthiazolyldiphenyl-tetrazolium bromide; NNEDA, N(1-naphtyl)ethylendiamide dihydrochloride; SA, sulfanilamide; ND, not determined; –, not determinable.
Figure 1.

Principles of Stopped Assays for Enzyme Activities That Use a Cycling Assay to Determine the Product of the Reaction.
(A) and (B) Determinations based on the glycerol-3P-cycling assay.
(C) and (D) Determinations based on the NADP-cycling assay.
(E) and (F) Determinations based on the NAD-cycling assay.
(A), (C), and (E) represent the stopped step leading to glycerol-3P or dihydroxyacetone-P, NADPH, or NAD(H), respectively, directly or via coupled reactions. The enzyme measured is in bold. For clarity, products that are not measured have been omitted as well as the cosubstrates required for the coupling reactions.
(B), (D), and (F) represent the principles of the cycling reactions for the determination of glycerol-3P or dihydroxyacetone-P, NADP(H), or NAD(H), respectively.
AGPase, glucose-1-phosphate adenyltransferase; Ald, aldolase; AlaAT, Ala aminotransferase; AspAT, Asp aminotransferase; CS, citrate synthase; FBPase, fructose-1,6-bisphosphatase; FruK, fructokinase; Fum, fumarase; G3PDH, glycerol-3P dehydrogenase; G3POX, glycerol-3P oxidase; G6PDH, glucose-6P dehydrogenase; GlcK, glucokinase; GAP DH, glyceraldehyde-3P dehydrogenase; GK, glycerokinase; GLDH, glutamate dehydrogenase; ICDH, isocitrate dehydrogenase; LDH, lactate dehydrogenase; MDH, malate dehydrogenase; PEPC, phosphoenolpyruvate carboxylase; PFP, pyrophosphate-dependent phosphofructokinase; PGI, phosphoglucose isomerase; PGK, phosphoglycerate kinase; PK, pyruvate kinase; ShikDH, shikimate dehydrogenase; SPS, sucrose-phosphate synthase; TPI, triose-P isomerase; UMPK, UMP kinase. Abbreviations for chemicals are as follows: 2OG, 2-oxoglutarate; 3PGA, 3-phosphoglycerate; 6PG, 6-phosphogluconate; ADPG, adenine-diphosphoglucose; BPGA, bisphosphoglycerate; DAP, dihydroxyacetone-P; F6P, fructose-6P; FBP, fructose-1,6-bisP; Fum, fumarate; G, glycerol; G3P, glycerol-3P; GAP, glyceraldehyde-3P; i-Cit, isocitrate; Mal, malate; MTT, methylthiazolyldiphenyl-tetrazolium bromide; OxA, oxaloacetate; PEP, phosphoenol pyruvate; Pyr, pyruvate; Shik, shikimate.
An alternative strategy to achieve high sensitivity is to use fluorigenic reactions. For example, to assay invertase, an existing stopped assay (Huber, 1984) was modified by measuring glucose via a glucose oxidase/peroxidase coupling system that oxidizes dihydroxyphenoxazin into the fluorescent dye resorufin. An analogous approach could be used for many other enzymes because a range of oxidases and fluorigenic substrates are commercially available.
A small number of enzymes required dedicated assays. We have not yet found a procedure that combines high sensitivity and high throughput for enzymes that produce ADP from ATP (e.g., Gln synthetase; GS). GS activity was high enough to use an existing assay, in which ADP is measured by coupling it to NADH formation via pyruvate kinase (PK) and lactate dehydrogenase (Scheible et al., 1997a). DTT is often required to stabilize enzymes like NIA but interferes with the G6PDH/PMS and ADH/PES cycling systems because it reduces methylthiazolyldiphenyl-tetrazolium bromide (MTT) and the peroxidase-dependent oxidation of dihydroxyphenoxazin. NIA had to be assayed using an existing protocol that measures nitrite formation (Scheible et al., 1997b, 1997c). The assay for ferredoxin-dependent glutamate synthase (Fd-GOGAT) contains a mix of reductants (DTT to stabilize Fd-GOGAT and dithionite to drive the reaction). Furthermore, the product (glutamate) has to be measured in the presence of a vast excess of 2-oxoglutarate and Gln. To achieve this, glutamate dehydrogenase (GDH) was coupled to diaphorase and MTT to displace the equilibrium toward glutamate oxidation. Before determining glutamate, an excess of N-ethylmaleimide was included to remove DTT and dithionite because they would otherwise reduce MTT to its formazan. Fortunately, GDH was not inhibited by N-ethylmaleimide.
All of the standard assays contained optimized levels of substrates and, where necessary, activators, pH, and ionic conditions to allow maximum activity. In specific cases, it is also possible to use a second assay system with suboptimal levels of specific substrates or effectors to detect posttranslational regulation. As an example, posttranscriptional regulation of NIA was monitored by comparing activity in the presence of excess Mg2+ to stabilize a complex between the phosphorylated protein and the inhibitory 14-3-3 protein and selectively assay the active nonphosphorylated protein, and in the presence of excess EDTA to trap Mg2+ and allow the phosphorylated form to be released and regain activity to determine maximal activity (Kaiser and Huber, 1999).
Optimization and Validation
This section describes the optimization and validation of the assay for fumarase. The same strategy was applied to all assays. Fumarase catalyzes the hydration of fumarate to malate. Malate dehydrogenase is included as a coupling enzyme to oxidize malate to oxaloacetate and simultaneously reduce NAD+ to NADH. Because oxaloacetate exerts strong product inhibition on malate dehydrogenase, it was removed by including citrate synthase and an excess of acetyl-CoA. The assay is stopped with an excess of NaOH, the nonreduced NAD+ destroyed by heating at 95°C for 5 min, the extract neutralized with HCl, and NADH determined using the ADH/PES cycle system. Activity was measured by comparing the signals with the complete assay buffer (Vmax) and when a substrate (here fumarate) was omitted (Vblank).
The first step in assay validation was to optimize the amount of plant material included in the assay. This exerted a dramatic effect on the apparent activity of fumarase (Figure 2A): a 20,000-fold dilution of fresh weight (w/v) in the reaction mixture was needed to reach linearity. Activity will be severely underestimated in conventional assays, which measure the accumulation of NADH via the absorbance change at 340 nm or the decrease of fumarate via the absorbance change at 280 nm (Bergmeyer, 1987). To check the reliability of the assay procedure, different amounts of pure malate were added to the extract. Recoveries of ∼90 and 100% were obtained in Vmax and Vblank conditions across a wide range of spiked malate concentrations (Figures 2B and 2C). The reaction was stopped after different times to check linearity with time. For all of the enzymes except Fd-GOGAT, the reaction was linear at least for 20 min, which was used for routine determinations (data not shown). Fd-GOGAT reaction was found to be linear for only 10 to 15 min, probably because atmospheric oxygen reoxidized the assay medium. Concentrations of substrates, ions, and pH were chosen to ensure maximal activity was detected. In cases where activators were known from the literature (not appropriate for fumarase), they were included and optimized. Stability of activity through a freeze-thawing cycle was checked by snap-freezing samples in liquid nitrogen and storing them for at least 24 h at −80°C before rethawing to assay activity. In the case of fumarase, <10% of the initial activity was recovered (Table 1). This study compares enzyme activities in comparable tissues. In cases where activities are compared between different tissues, it is strongly advised to conduct recombination experiments in which powder from the two tissues is mixed in a 50:50 ratio, extracted, and assayed to check that the activity in the mixed extract is similar to the arithmetic mean of the activities in extracts from the two tissues.
Figure 2.

Validation of the Assay for Fumarase.
(A) Relation between the dilution of the extract and the apparent activity determined. Dilutions were made with the extraction buffer before the assay.
(B) and (C) Recovery of malate standards (0, 0.1, 0.2, and 0.5 nmol) from Arabidopsis rosette extracts set to a 20,000-fold dilution of FW (w/v). The slope of the lines gives the fraction of malate recovered. Data are given in mOD min−1 ± sd (n = 3).
Overview of the Assay Systems, Activities, Variation, and Stability
Table 1 summarizes the assay principles, the extract dilution, the average activity, and standard deviation between biological replicates of rosettes from 5-week-old Arabidopsis Columbia-0 (Col-0) and the stability of the activity to a freeze/thaw cycle. The activities ranged from 20 (glycerokinase) to 40,000 (NAD-dependent GAPDH) nmol g−1 fresh weight (FW) min−1 (Table 1). The extract dilution (expressed on a FW/extract volume basis) ranged from 500 (e.g., glycerokinase, NIA, or GS) to 20,000 (e.g., fumarase, NAD-dependent glyceraldehyde-3P dehydrogenase [NAD-GAPDH], or Ala and Asp aminotransferases [AlaAT and AspAT]). Approximately half the enzymes were unaffected, and half were sensitive to a freeze-thawing cycle. The latter was always assayed directly after extract preparation, whereas for the other enzymes, aliquots could be snap-frozen and assayed later.
Sample Flow
Handling procedures and assay protocols were designed to optimize sample flow, and spreadsheets were used to allow rapid and automatic processing of the data (see Methods). Assay of one enzyme in 20 samples requires 2 h using conventional hand pipettes, including extraction, assay, and calculations, allowing one person to measure three to four different activities in 20 samples in a single working day. Throughput is increased fivefold by a four-tip pipetting robot, allowing, for example, three to four activities to be measured in 100 samples per day. We are presently transferring the platform to a 96-channel robot and expect a further twofold to 10-fold increase in throughput. The major bottleneck is the grinding and weighing of aliquots before extraction. This manual step may be difficult to automate because the sample must be kept deeply frozen to avoid loss of activity and be very finely homogenized to allow full extraction. Tissue-permeabilizing extraction media like those that are used to extract RNA and metabolites are unsuitable for enzymes because they denature proteins.
Global Comparison of Diurnal Changes of Transcript Levels and Enzyme Activities during a Diurnal Cycle in Wild-Type Arabidopsis
In a first application, 23 enzyme activities were determined in five replicate samples collected at 2-h intervals throughout a diurnal cycle in Col-0 wild-type Arabidopsis rosettes. In parallel, RNA was prepared every 4 h from three biological replicates and used for expression profiling with Affymetrix ATH1 chips. The TranscriptScavenger module from MapMan (Thimm et al., 2004) was used to identify and extract the data for genes that are members of the families for the 23 enzymes in our platform (in total 84 genes; see supplemental data online for a list). The averaged values for the diurnal changes of enzyme activities and the transcript levels for each member of the corresponding gene family are shown for NIA, fumarase, Fd-GOGAT, ADP glucose pyrophosphorylase (AGPase), and GLDH in Figure 3 and for the other 18 enzymes in the supplemental data online. The diurnal cycle in wild-type Col-0 is shown in the second panel from the left. The results will first be analyzed with respect to the overall relation between transcript levels and enzyme activities and later with respect to the response of specific enzymes.
Figure 3.

Changes of Enzyme Activity and Transcript Levels for Each Member of the Corresponding Gene Family in 5-Week-Old Arabidopsis Col-0 Wild-Type and pgm Rosettes throughout a Night and Day Cycle (12 h/12 h) and Wild-Type Rosettes after a Transfer to Continuous Darkness.
(A) Nitrate reductase.
(B) ADP-glucose pyrophosphorylase.
(C) Fumarase.
(D) Ferredoxin-dependent glutamate synthase.
(E) Glutamate dehydrogenase.
The diurnal cycle in wild-type Col-0 is shown in the second panel from left, the diurnal cycle in pgm is shown in the first panel from left, and the extended night in panels 3 and 4 from left. Enzyme activities are expressed as nmol g−1 FW min−1 ± sd (n = 5). For NIA, maximal (max) and selective (sel) activities are given. Transcript levels are expressed as robust multichip average–normalized signals ±sd (n = 3; wild type only).
Our approach assumes that the enzymatic activity in the best standardized conditions provides quantitative information about the level of the corresponding protein. An obvious problem is that small gene families encode most of the enzymes, and the assays do not distinguish the different isoforms. Fortunately, for most enzymes the signals for transcripts for one or two family members were much higher than the other members, and these major forms showed a similar diurnal response. The main exceptions are sucrose phosphate synthase (SPS), AspAT, glucose-6-phosphate dehydrogenase (G6PDH), GS, and PK (see supplemental data online). These enzymes were excluded from some of the following analyses.
The data were statistically analyzed to uncover general features of the relation between transcript levels and enzyme activities. A formula given in Methods was used to estimate a smoothness value, which has an optimal value of zero if every data point lies on a smooth curve that moves through one maximum and one minimum per diurnal cycle, rising to one as the data points become increasingly irregular (see supplemental data online for examples of the values obtained for a set of randomly generated data points). Values between 0 and 0.05 were arbitrarily chosen as indicating a good oscillation for transcripts and enzyme activities, values ranging from 0.05 to 0.1 as indicating a possible oscillation, and values >0.1 as indicating no oscillation of transcripts and enzyme. Figure 4A indicates that almost all transcripts but only 30% of the enzyme activities show a smooth oscillation (Figure 4A). A further 43% of the enzymes show a slightly irregular oscillation. The amplitude of the change is smaller for enzyme activities, making them more susceptible to experimental noise.
Figure 4.

Evaluation of the Periodic Variation for 84 Transcript Levels and the Corresponding 23 Enzyme Activities and Distribution of Their Amplitudes in Arabidopsis Col-0 Wild Type.
(A) Smoothness classes for transcripts (black bars) and activities (gray bars).
(B) Probability mass function of amplitudes for transcripts (black circles) and activities (gray circles). The calculation of the smoothness classes and the probability mass function is presented in Methods.
The amplitude of the diurnal change was estimated from the difference between the maximum and minimum values and expressed as a percentage of the maximum value (see Methods). To allow data compression, a probability mass function curve (see Methods) was calculated for both populations (Figure 4B). On average, transcript levels exhibit an almost twofold larger amplitude than enzyme activities during the 24-h diurnal cycle (Figure 4B). The same trend was obtained when the analysis was restricted to major transcripts (i.e., those arbitrarily chosen as exhibiting a maximum level >30% of the maximum level of the highest expressed transcript or when all transcripts in a given family were added together; see Supplemental Figure 2 online).
These data indicate that whereas most transcript levels exhibit a clear and marked diurnal rhythm, the response of the corresponding activities was damped, and in some cases hardly detectable. The amplitude of the diurnal changes of transcripts may still be an underestimate for low-expressed genes. It has been inferred that hybridization kinetics on expression arrays are second order (Holland, 2002), and the linear range afforded by the Affymetrix ATH1 arrays (three to four orders of magnitude) may lead to some attenuation of the signals. Comparison of the signals for >1400 transcription factors measured by real-time PCR and the ATH1 array in Arabidopsis shoot and root samples (Czechowski et al., 2004) and in Arabidopsis seedlings in differing nitrogen nutrition (Scheible et al., 2004) reveals that the ATH1 signals in the low signal range are attenuated by a factor of three. Transcription factors are low-expressed genes, and a better quantitative agreement might be expected for genes whose transcripts are present at higher levels. A well-correlated fit line with a slope close to one was found between data obtained from ATH1 probe arrays (expressed as Log2 ratios) and real-time PCR (expressed as average cycle time differences) for a small set of more strongly expressed genes (Redman et al., 2004).
Global Comparison of Diurnal Changes of Transcript Levels and Enzyme Activities during a Diurnal Cycle in the Starchless pgm Mutant
In the light, photosynthesis leads to synthesis of sucrose that is exported to support respiration and growth in the remainder of the plant. At night, the entire plant becomes a net consumer of fixed carbon. Some photosynthate is stored in leaves as starch in the light and is remobilized at night to support leaf respiration and the continued synthesis and export of sucrose. This provides a buffer against the daily alternation of the carbon budget. The starchless pgm mutant lacks plastidic phosphoglucomutase, which is essential for starch synthesis in the leaf (Caspar et al., 1985). Sugars accumulate during the day but are rapidly depleted in the first part of the night. A recurring phase of sugar starvation during the second part of the night leads to a severe growth impairment (Gibon et al., 2004). The transcriptome of pgm at the end of the day resembles wild-type Arabidopsis, whereas the transcriptome at the end of the night resembles wild-type plants after a 6-h extension of the night (Gibon et al., 2004; Thimm et al., 2004). Diurnal changes of enzyme activities were investigated in pgm because it was expected that they would be larger than in wild-type plants. It is already known that acid invertase (Caspar et al., 1985) and SPS and AGPase activities (Caspar et al., 1985) are higher in pgm plants.
For pgm, enzyme activities were measured in five replicates. Transcript profiles were only repeated at the end of the night and at the end of the day. The reproducibility found for these data points was very good (data not shown). For transcript levels, the smoothness factor moved even closer to the ideal value of zero (Figure 5A) than in wild-type plants. This trend might be explained because the larger diurnal changes make the data less susceptible to random noise. By contrast, smoothness increased only slightly for a subset of the enzyme activities (Figure 5B). There was a clear increase in the amplitude of the diurnal changes of transcript levels (the average value rose from 35% in wild-type plants to almost 60% in pgm; Figure 5C), but the amplitude of the diurnal changes of enzyme activities was not altered (Figure 5D).
Figure 5.

Comparison between Arabidopsis Col-0 Wild Type and pgm of the Periodic Variation for 84 Transcript Levels and the Corresponding 23 Enzyme Activities and for the Distribution of Their Amplitudes.
(A) Smoothness classes for transcripts levels.
(B) Smoothness classes for activities calculated after smoothing.
(C) and (D) Probability mass function of the amplitudes of transcripts levels (C) and activities (D).
Direct Comparison of the Diurnal Changes of Transcripts and Enzyme Activities
The previous statistical analysis treated transcripts and enzyme activities as populations. In a complementary approach, we compared the response for each individual enzyme. Figure 6 summarizes the amplitudes of the diurnal changes of transcripts and activities for each enzyme. To generate this plot, the signals for different transcripts for a particular enzyme were summed. Data for AspAT, G6PDH, GS, PK, and SPS were omitted because the diurnal changes of transcripts for different family members for these enzymes are markedly out of phase. Transcript levels show larger diurnal changes than enzyme activities and that the larger changes of transcripts in pgm do not lead to a larger diurnal change of enzyme activity. However, when individual enzymes are compared, there is no correlation between the amplitude of the diurnal changes of transcript and enzyme activity. A small subset of enzymes (including AGPase and NAD-GAPDH) showed larger diurnal changes of activity than of transcript levels.
Figure 6.

Comparison of the Amplitude of the Diurnal Changes of Transcripts and the Amplitudes of the Diurnal Changes of Enzyme Activities in Arabidopsis Col-0 Wild Type and pgm.
The results are calculated from the data in Figure 3 and the supplemental data online using the equation given in the text. Data for AspAT, G6PDH, PK, and SPS are omitted. The data points corresponding to AGPase and NIA are identified with arrows.
To analyze the temporal relation between changes of transcript levels and enzyme activity, the change of the transcript level and the change of the time-averaged enzyme activity was calculated for each 4-h time interval and normalized as the percentage of the average level (see Methods). This provides two local vectors that describe the change of transcript and the change of enzyme activity during a given 4-h interval. This calculation was made for each individual transcript that encodes a particular enzyme (see supplemental data online) and for the summed signal of all the transcripts for the enzyme (Figure 7). Figures 7A to 7F show the 23 vector pairs for the changes of the transcript levels and enzyme activities during each of the six time intervals. These plots allow three conclusions. First, illumination and, to a lesser extent, darkening are followed by marked changes of the levels of many transcripts and activities. This is accentuated in pgm, indicating that it is related to the provision of sugar rather than light per se. Second, transcript levels change more than enzyme activities. Two interesting exceptions are NIA and AGPase (noted by arrows in Figures 7A and 7B) during the first 4 h of the light period (see below for more details). Third, changes of activities do not correlate with changes of transcripts; indeed, in many cases, there is a negative correlation.
Figure 7.

Global Overview of the Time Lag of Changes of Enzyme Activities Compared with Transcript Levels in Arabidopsis Col-0 Wild Type and pgm Plants.
(A) to (F) The normalized changes of summed transcripts levels and enzyme activities were estimated for each 4 h. The changes of transcript levels were plotted against the change of enzyme levels in the same time interval (wild type, black circles; pgm, gray circles) and in an interval 4, 8, 12, 16, and 20 h later and the correlation coefficients calculated for each enzyme. This was repeated for each of the six time intervals.
(G) and (H) The regression coefficients for a given time delay were combined across all six time intervals and 23 enzymes and are depicted divided into seven classes (R2 ≥ 0.75; 0.50 ≤ R2 < 0.75; 0.25 ≤ R2 < 0.50; −0.25 < R2 < 0.25; −0.50 < R2 ≤ −0.25; −0.75 < R2 ≤ −0.50; R2 ≤ −0.75) for the wild type (G) and pgm (H).
(I) Time lag of the response of individual enzyme activities in wild-type (black circles) and pgm (gray circles) plants. Regression coefficients were estimated ([G] and [H]) and are plotted here for 10 selected enzymes. Each point represents the regression coefficients, calculated over six time points from a plot of the change of transcript against the change of enzyme activity after a given delay (x axis) for each of the six 4-h time intervals. Abbreviations are given in Figure 1.
One explanation for the poor correlation would be that there is a delay before changes of transcript levels lead to changes of enzyme activity. This is supported by visual inspection of the original data (Figure 3; see supplemental data online). To explore this possibility systematically, the change of transcript in a particular time interval was plotted against the change of enzyme activity in the same time interval and in a time interval 4, 8, 12, 16, or 20 h later and the regression coefficient calculated. For these analyses, all of the transcripts that encode a particular enzyme were summed. This procedure was performed six times (using the normalized change of transcript levels between 0 to 4 h, 4 to 8 h, 8 to 12 h, 12 to 16 h, 16 to 20 h, and 20 to 0 h) to provide six independent estimates of the regression coefficient between changes of transcripts and enzyme activity when a given time delay is introduced into the comparison (see supplemental data online for the original values). Figures 7G and 7H group the averaged regression coefficients for these six comparisons for 22 enzymes (SPS was omitted because data points were missing) into different classes. When changes of transcript levels are compared with changes of enzyme activities in the same 4-h time interval, most enzymes showed a very poor or even a negative correlation between the change of transcript and activity (Figures 7A to 7F). When a time delay was introduced, the proportion of positive correlations increased. The highest proportion of positive correlations was obtained when a delay of 12 to 16 h was introduced.
Figure 7I shows the results for selected enzymes. In many cases, an average regression coefficient >0.8 was obtained when changes of transcripts were plotted against the changes of enzyme activities that occurred at a later time point. Different enzymes respond with a different time delay. For example, a set of key enzymes in nitrogen metabolism (NIA, GS, Fd-GOGAT, AlaAT, and GLDH) show a lag of 12 h or longer between rapid changes in transcript levels and the corresponding changes of enzyme activity. NADP-isocitrate dehydrogenase responds similarly, whereas fumarase, pyrophosphate-dependent phosphofructokinase (PFP), and especially fructokinase respond with a shorter delay, and NADP-GAPDH shows an extremely long delay. It has been noted that AGPase shows the largest diurnal changes of all the enzymes studies. The changes are strongly delayed compared with the change of transcript levels and the regression coefficients rather poor. In most cases, a similar trend is found in wild-type plants and in pgm. Invertase showed a strikingly different response in wild-type plants and pgm. This might be because only one specific member of the invertase gene family is strongly induced in pgm (see supplemental data online).
Comparison of Time Courses of Transcripts and Activities for a Set of 23 Enzymes in a Transition to an Extended Night in Arabidopsis Col-0
The relatively short time between the maximum and minimum of transcript levels during a diurnal cycle may prevent the enzyme activity from fully adjusting to the change in transcript level. Prolongation of the night leads within hours to dramatic changes of many transcripts for enzymes involved in central metabolism (Thimm et al., 2004). This system was used to investigate the impact of prolonged changes of transcript levels on enzyme activities.
Samples were taken 2, 4, 6, 8, 24, and 48 h into the extended night to measure transcript levels and 2, 4, 8, 12, 24, 48, 72, and 148 h into the extended night to measure enzyme activities. The results were expressed relative to the value at the end of the normal night, converted to a log2 scale to linearize the distribution and used to calculate the probability mass for each time point (Figures 8A to 8G). There were detectable changes of some transcript levels by 2 h (Figure 8A) and marked changes of most by 4 h (Figure 8B). For most enzymes, there were no large changes of activity until 48 to 72 h into the extended night (Figures 8E and 8F). The only exception was NIA, which showed a rapid decrease of activity. For every enzyme activity, a P value was calculated to reveal whether the activity at each time in the prolonged night was significantly different from the activity at the end of the normal night. Figure 8G summarizes the average and se of the P values (n = 23 enzyme) at each time point and reveals that enzyme activities, considered as a whole, did not change significantly until the plants had been subjected to 2 d of carbon starvation.
Figure 8.

Comparison of the Changes in Transcript Levels with the Changes in Enzyme Activities after 2, 4, 8, 24, 48, 72, and 148 h after a Transfer to a Prolonged Night.
For each analyte, the ratios between values at t and t0 (start of the experiment) were first calculated, then converted to their basis 2 logarithm to approach the normal distribution, and the probability mass was calculated for transcripts and activities after 2 (A), 4 (B), 8 (C), 24 (D), 48 (E), 72 (F), and 148 h (G). Open circles, changes in transcript levels; closed circles, changes in enzyme activities. Transcript levels were not determined at 72 and 148 h of extended night. Significance of the changes in enzyme activities was also estimated by calculating the average P values at each point (H), obtained by calculating P values for every activity, using the t test (t0, t, two tails, and heteroscedastic). Bars = means ± se (n = 23).
Specific Case Studies of the Relation between Transcript Levels and Enzyme Activities in Diurnal Cycles and a Prolonged Night
Our data set can also be analyzed to provide specific information about the diurnal regulation of individual enzymes. Graphs displaying the changes of transcripts and the enzyme activities in Col-0 and pgm during the diurnal cycle and in Col-0 during a prolonged night are available for inspection in the supplemental data online. Two examples that show striking changes (NIA and AGPase) will be contrasted with two that show typical responses (fumarase and Fd-GOGAT).
NIA is subject to sophisticated transcriptional, posttranscriptional, and posttranslational regulation in tobacco (Nicotiana tabacum) leaves (see Introduction). Our results (Figure 3A) reveal a similarly complex network in Arabidopsis. Of the two genes that encode NIA in Arabidopsis, one is highly (At1g37130) and the other weakly (At1g77760) expressed in rosettes. Wild-type plants show large diurnal changes of transcripts and smaller changes of activity (amplitudes = 80 and 88% for NIA1 and NIA2 transcripts and 36% for NIA activity), with transcripts peaking at the end of the night and enzyme activity ∼6 h later in the middle of the light period. Compared with wild-type plants, pgm showed a slight reduction in transcript levels and a 1.5-fold to twofold reduction of activity. The diurnal changes of transcripts were less marked, but the diurnal changes of activity remained large. Prolongation of the night led to a rapid and marked decrease of NIA transcript and activity. NIA protein also decreases rapidly in response to carbon starvation in tobacco leaves (Klein et al., 2000). NIA is subject to reversible posttranslational regulation, in which phosphorylation of Ser-534 leads to binding of a 14-3-3 protein that inhibits activity and may also increase degradation of NIA protein (Kaiser and Huber, 1999; Meyer and Stitt, 2001). Posttranslational regulation was monitored using a selective assay. In wild-type plants, overall NIA activity and NIA activation are low during the night, increase rapidly after illumination, and decrease as the light period progresses. In pgm, overall activity and activation increased slowly at the start of the light period but remained high until the end of the day.
AGPase (Figure 3B) is a heterotetramer consisting of two large (AGPS) and two small (AGPB) subunits. AGPB is encoded by one gene (At5g48300; Hendriks et al., 2003) and AGPS by a four-member family, of which one is highly (At5g19220), two are weakly (At4g39210 and At1g27680), and one is very weakly (At2g21590) expressed in rosettes. Transcripts for AGPB and the major AGPS form showed weak diurnal changes, with peaks during and at the end of the light period, respectively. These results were confirmed using real-time RT-PCR (data not shown). AGPase activity was assayed in the presence of excess levels of the allosteric activator 3-phosphoglycerate and DTT to completely convert the enzyme into the active reduced form (Tiessen et al., 2002; Hendriks et al., 2003) and to ensure that the activity measurements reflected changes in AGPase protein. AGPase activity fell rapidly during the first part of the day and recovered in the last part of the light period and during the night. AGPase was the only enzyme where the changes of activity (51%) were larger than the changes in transcript levels (12 and 13% for AGPB and the major AGPS transcript). These were the largest diurnal changes of activity in the entire set of enzymes. Total AGPase activity decreased in the first part of the light period, when AGPB and AGPS transcripts were stable or rising. A similar picture emerged during diurnal changes in pgm, where the changes were even more pronounced (amplitudes of 69% for activity, 45 and 38% for transcripts for AGPB and the major AGPS family member, respectively). When wild-type plants were exposed to a prolonged night, transcripts decreased but activity stayed high.
Protein gel blots were performed to check that the changes of AGPase activity reflect changes of protein. The gels were run in the absence of DTT so that the reduced (monomer) and oxidized (dimer) forms (Figure 9A) are visible (Hendriks et al., 2003). An increase of the ratio between reduced and oxidized forms has been associated with the activation of AGPase (Tiessen et al., 2002; Hendriks et al., 2003), and the strong monomerization found in pgm has been discussed elsewhere (Hendriks et al., 2003). Total AGPB protein was measured by summing the signals from both bands. The diurnal changes of AGPB protein agree well with the changes of total AGPase activity (cf. Figures 3B and 9B). This figure also illustrates that by using suitable extraction and assay conditions, it is indeed possible to override posttranslational changes and obtain a measurement of activity that reflects total protein.
Figure 9.

Immunological Detection of AGPB Protein in Nonreducing Denaturing Gels.
Proteins were extracted from rosettes of the wild type and pgm collected at 0 (end of the night), 2, 5, and 10 h of day. Protein gel blots (A) and relative amounts of AGPB (B) calculated as the sum of monomer (50 kD) and dimer (100 kD). Levels are given as a percentage of the respective levels determined for the wild type and pgm at the end of the night ±sd (n = 3).
Fumarase (Figure 3C) is encoded by a single gene in Arabidopsis. There was a strong diurnal change of transcript and much smaller changes of activity in wild-type plants (amplitudes of 62 and 23%, respectively), with the peak of transcript preceding the peak of activity by 10 h. Compared with wild-type plants, pgm had slightly lower levels of transcript and activity, and the diurnal changes of transcript and activity were larger and smaller (73 and 11%, respectively) than in wild-type plants. During a prolonged night, transcript remained similar for at least 48 h to that found during a normal night. Activity decreased presumably as a delayed aftermath of the decrease of transcript during the normal night.
Fd-GOGAT (Figure 3D) is encoded by one major transcript (At5g04140). Wild-type plants had marked diurnal changes of the major transcript (35%), which were followed with an ∼12-h delay by damped changes of enzyme activity (18%). The average transcript and activity levels in pgm were 1.6- and 1.9-fold lower than in wild-type plants. The diurnal changes were slightly larger (51% for the transcript and 20% for activity). Prolongation of the night led to a rapid and marked 50% decrease of transcript, falling to 50% by 8 h, and 10% by 48 h, whereas activity remained unaltered for at least 7 d.
Inspection of the results for other enzymes reinforces the conclusion that changes of transcript levels are accompanied by much smaller changes of enzyme activities during the diurnal cycle and to strongly damped and slow responses in a prolonged night (AlaAT, cytosolic fructose-1,6-bisphosphatase, fructokinase, hexokinase, NAD- and NADP-GAPDH, isocitrate dehydrogenase, phosphoenolpyruvate carboxylase, PFP, and transketolase). In some cases, no relation was apparent (glycerokinase and shikimate dehydrogenase). These activities were, however, very low and more difficult to assay precisely.
Comparison of Wild-Type Arabidopsis and the pgm Mutant
Compared with Col-0, pgm has twofold lower NIA and Fd-GOGAT activity (see above), slightly lower fumarase, GS, isocitrate dehydrogenase, phosphoenol pyruvate carboxylase, PFP, and transketolase activity (see supplemental data online) and twofold higher acid invertase (Caspar et al., 1985) and GLDH activity (Figure 3E).
Transcripts for GDH1 (At5g18170) and GDH2 (At5g07440) underwent extreme diurnal changes in pgm, with a 20-fold increase toward the end of the night (Figure 7E). This contrasts with wild-type plants, where there were only small diurnal changes of transcripts. The average level of the GDH1 and GDH2 transcripts across the entire diurnal cycle was 6.7- and 3.2-fold higher, respectively, in pgm. Activity in pgm showed only a slight diurnal oscillation but was 1.8-fold higher than in wild-type plants. This indicates that recurring transient peak of GLDH transcript at the end of each night leads to a quasi-stable increase of GLDH activity throughout the entire diurnal cycle. GDH transcript levels increased dramatically during the first hour of a prolonged night before relaxing to levels that were approximately fourfold higher than those found for a normal night in wild-type rosettes. The sustained increase of GDH1 and GDH2 transcript levels in a prolonged night leads to increased GLDH activity, but the response was damped by a factor of two to three and only developed after a considerable time lag. These results indicate that the increase of the GDH transcripts in the later part of the night in pgm is a response to low sugar.
A similar response was found for acid invertase (see supplemental data online). The major transcript (At1g12240) showed much larger diurnal changes (10-fold) than in wild-type plants, falling to lower levels at the end of the day and rising to threefold higher levels at the end of the night. The 10-fold diurnal changes of transcript in pgm were not accompanied by diurnal changes of activity. However, invertase activity increased 1.7-fold, compared with wild-type plants. Transcript increased threefold at the start of a prolonged night, before relaxing to a sustained twofold higher level. This was followed over several days by a slow twofold increase of activity. In both treatments, changes of At1g12240 transcript do lead to marked changes of invertase activity but only after a considerable time lag.
The Use of Enzyme Activities as Diagnostic Markers
In machine learning, a training data set obtained in a set of defined conditions is used to generate a classification model, which can be used as a diagnostic tool to automatically classify further unknown samples. Their relative stability indicated that enzyme activities might provide a suitable data set for machine learning. A data set made of two experiments for wild-type plants, including 60 samples collected at 12 different times during the normal diurnal cycle and 55 samples collected at different times during a prolonged night were used to construct a decision tree. The decision tree separated the wild-type samples into six classes corresponding to samples harvested at any time during a normal diurnal cycle, samples harvested during the first 12 h of an extended night, and samples harvested 1, 2, 3, and 7 d into a prolonged night (Figure 10A). Four (NIA, AGPase, NAD, and NADP-GAPDH) of the 23 enzymes were excluded because their activity underwent very large changes in the normal diurnal cycle, which made the decision tree unstable (data not shown). It used information with approximately nine (GLDH, PK, shikimate dehydrogenase, G6PDH, GS, glycerokinase, fructose-1,6-bisphosphatase, fructokinase, and invertase) of the remaining 19 enzymes.
Figure 10.

Enzyme Activities as Diagnostic Markers.
(A) Six classes of wild-type samples corresponding to controls harvested throughout a normal night and day cycle or plants exposed to a prolonged night for <1, 1, 2, 3, or 7 d were used to build a decision tree that placed the sample in the correct class with a learning accuracy of 75%, which was obtained via a 15-fold cross-validation. All enzyme activities measured were used except AGPase, NAD-GAPDH, NADP-GAPDH, and NIA.
(B) The decision tree was then used to classify 60 samples of pgm plants harvested at six time points throughout a night and day cycle. The y axis shows the frequency and the weighted frequency with which a sample was assigned to a particular class.
The decision tree was then used to diagnose 60 pgm samples collected at 12 different time points during the diurnal cycle. Each sample is independently examined and classified by the decision tree. The vast majority (>90%) were classified as similar to wild-type plants that had been in the dark for 3 d (Figure 10B).
DISCUSSION
Design of a Platform for High-Throughput Determination of Multiple Enzyme Activities
Whereas high throughput has become state-of-the-art for profiling transcript and metabolite levels, techniques for assaying enzymes are still slow and cumbersome. We have developed a platform of enzyme assays that combines high sensitivity with throughput. Enzymes were assayed using stopped assays to streamline sample handling and save costs by allowing the use of small volumes. The assays were fed into a platform of three cycling assays, supplemented by a small number of dedicated tests to determine the products. Cycling assays are extremely sensitive and can be easily adapted to use with microplate readers, which are standard equipment in many laboratories. This platform, when combined with high-performance robots, has the potential to allow more than eight enzyme activities to be determined in 100 samples in a single day.
Several of the assays provide large advantages over previous approaches. Glycerokinase and shikimate dehydrogenase, which have rarely been assayed in plants, can now be routinely determined in crude extracts. The development of an assay that allows glutamate to be measured via GLDH allows hundreds of measurements of Fd-GOGAT within a day, compared with previous assays that used HPLC (Suzuki et al., 1994) or ninhydrin (Hecht et al., 1988) to measure glutamate. The new assay for AGPase is a large advance for measurements in photosynthetic tissues, where previously a cumbersome radioactive test was needed. Although a conventional coupled assay exists in which AGPase is assayed by coupling glucose-1-P formation via phosphoglucose isomerase and G6PDH to NADP+ reduction, this assay is unusable in leaves. The other product is ATP, and to obtain maximum activity, the assay must include the allosteric activator glycerate-3-P. Leaf extracts contain high activities of phosphoglycerate kinase and NADP-GAPDH, which convert ATP and glycerate-3-P to triose-phosphate and reoxidize NADPH. In the new assay, ATP reacts with glycerol in the presence of an excess of glycerokinase, and glycerate-3-P can be included in the assay without danger of interference. Previous assays for SPS also had major drawbacks. The widely used anthrone test (Huber et al., 1989) suffers from a low sensitivity and requires a desalting step and the use of concentrated sulphuric acid. In our lab, ∼40 extracts could be analyzed per day with this method. An alternative assay measures UDP via PK and lactate dehydrogenase functions (Stitt et al., 1988). However, PK has a relatively poor affinity for UDP (http://www.brenda.uni-koeln.de), which makes this assay unsuitable for many plant tissues because they contain high UDPase activity. The new assay detects UDP via UMP-kinase, glycerokinase, and a cycling assay and allows efficient recovery of UDP, especially when it is combined with high (1000-fold) dilution of the extract. It also allows >10-fold higher throughput than the anthrone assay.
Relation between Changes of Transcripts and Changes of Enzyme Activities
We have used measurements of enzyme activities in optimized conditions as a proxy for quantitative determination of proteins. The assumption that the activity in the assay reflects the amount of enzyme protein was checked by optimizing the assays with respect to all available information about the kinetic properties of the enzymes. It remains possible that some enzymes have unknown effectors or are subject to posttranslational regulation that affects their Vmax rather than kinetic properties and is not overridden by including high levels of substrates and effectors and optimizing the pH and ionic conditions. However, this would affect only the results for individual enzymes and not the general conclusions. A second limitation is that the enzyme assays do not distinguish among isoforms. For this reason, we have been cautious in using data for five enzymes, where the gene family members showed contrasting changes of their transcript levels.
The results from our study show that quantification of enzyme activities or proteins will be crucial in system biology. First, changes in transcript levels do not lead to quantitatively similar changes of enzyme activity. The diurnal changes of transcript levels were usually larger than the changes of the corresponding enzyme activities, the increased amplitude of the diurnal changes of transcripts in pgm did not lead to increased diurnal changes of enzymes, and the large and sustained changes of transcript levels in a prolonged night did not lead to correspondingly large changes in enzyme activities. In general, our results indicate that the response of enzyme activities is often damped by a factor of two or more. Second, enzyme activities typically change with a time delay. This is clearly seen in diurnal cycle in Col-0 and pgm, where the changes of activity were delayed compared with the change of transcript levels in a manner that varied from enzyme to enzyme. Most enzyme activities were also maintained for a very long time in an extended night, even though transcript levels decreased rapidly. In general, enzymes turn over more slowly than transcripts, but the relation varies on an enzyme-to-enzyme basis. Third, whereas a global analysis may provide rough guidelines about what constitutes a typical response, in some cases enzymes show very particular responses. Three examples are the large diurnal changes in AGPase activity and to a lesser extent NIA activity relative to the changes of transcript, the striking and rapid decrease of NIA activity during a prolonged night, and the unusually close correlation between the momentary changes of transcript levels and activity for fructokinase.
In cases where enzyme activity showed a large change relative to that of the encoding transcripts, closer examination of the data and comparison with the literature indicates that the change is at least partly because of posttranscriptional regulation. For example, the marked changes of AGPase activity in the diurnal cycle were accompanied by reciprocal changes of transcripts for the two subunits. Even more strikingly, whereas these transcripts rise and AGPase activity falls after illumination at the start of the day, AGPase activity stays high and the transcript levels fall if the night is prolonged. This indicates that the decrease in the first part of the normal day is because of posttranscriptional events that are triggered by a signal that is generated in the light. Jeannette and Prioul (1994) reported a similar diurnal activity change in maize (Zea mays) leaves. Further studies are needed to elucidate the biochemical mechanisms and understand their physiological function.
Comparison with published studies in tobacco indicates that the marked diurnal changes of NIA activity are also partly because of posttranscriptional regulation. Tobacco exhibits similar diurnal changes of NIA transcript and activity (Scheible et al., 1997b, 2000). In this species, overexpression of NIA under the control of a constitutive promoter abolished the diurnal changes of NIA transcript but did not alter the diurnal changes of NIA activity (for references, see Meyer and Stitt, 2001). Weiner and Kaiser (1999) have shown that synthesis of NIA protein is inhibited in the dark and stimulated in the light, whereas degradation is effected in a reciprocal manner.
The slow and damped response of enzyme activities has important implications for the analysis of multilevel genomics data sets. There is, for example, obvious interest in combining data sets from expression arrays and metabolite profiling. In the conditions we have studied, any correlations found among the levels of individual or groups of transcripts and metabolites will provide information about the role of metabolites as regulators of transcription. They cannot be because of the impact of transcription and the resulting changes of enzyme activities on metabolism because enzyme activities change too slowly and are too strongly damped to exert an effect. In other sets of biological samples, the changes in enzyme activities may be more marked, for example, when samples are compared in which a long time period is allowed for the system to stabilize or when different genotypes are compared. However, it will clearly always be important to obtain quantitative data about the response at the enzyme activity/protein level to provide an empirical base for the interpretation of the direction in which causality can operate in comparison of expression profiles and downstream phenotypic changes.
Functional Consequences of Different Dynamics for Change of Transcript Levels and Enzyme Activities
Partial uncoupling of transcription and posttranscriptional regulation generates a regulatory network that combines flexibility with robustness. Whereas transcription responds rapidly to short-term changes in the physiological status, the damped and delayed response of enzyme activities means that the changes of transcription will only affect metabolism if they are sustained for a long time. This has obvious advantages in a leaf, which is subjected every day to recurring changes of the light intensity, leaf water potential, and the levels of many metabolites every day. On the one hand, it is important to regulate transcription in response to changes in these parameters, otherwise the plant will be unable to respond to changes in its environment. On the other hand, alterations in protein levels every day would be energetically wasteful because the levels of proteins are far higher than of individual transcript. Even more crucially, changes of enzyme levels would impair metabolism and growth on the next day, until they are reversed (Gibon et al., 2004). The results in this article show that for most enzymes, changes in transcript levels have only a small impact on enzyme activities within a single day but lead to marked changes if the diurnal pattern is modified for several days. For example, the period of carbon starvation at the end of every night in pgm (Gibon et al., 2004; Thimm et al., 2004) leads to recurring changes in the levels of transcripts that generate stable changes in the levels of several enzymes, including increased activities of GLDH and invertase and decreased activities of NIA and GOGAT. In a more extreme treatment, whereas transcript levels respond to a prolonged night within 4 to 8 h, enzyme activities, with one exception, do not change markedly for 48 h.
Use of Enzyme as Diagnostic Markers
The relative stability of enzyme activities and the likelihood that they reflect long-term acclimation rather than a rapid response to a transient situation makes them ideal diagnostic markers. To illustrate this, we have used a machine-learning approach to diagnose the physiological status of the starchless pgm mutant. After several hours of acute sugar depletion during the second part of the night (Gibon et al., 2004), the transcript profile in pgm at the end of the night resembles a wild-type plant that has experienced a 4 to 6 h extension of the night (Thimm et al., 2004). The decision tree that we constructed revealed that the enzyme activity profile in pgm resembles that in a wild-type plant that has experienced 3 d of continuous darkness. This result is obtained irrespective of the time of day when pgm is harvested. This illustrates that the rapid changes of transcripts generated by the daily recurrence of a period of sugar starvation are integrated to generate a quasi-stable enzyme profile that resembles a wild-type plant that has been exposed to several days of carbon starvation. We are presently collecting a portfolio of potential diagnostic enzyme activities from plants based on data obtained across a range of growth conditions. The data will be used as training sets for machine-learning systems. We expect this approach to become useful for the characterization of ecotypes, mutants, and other genetically altered plants and for the evaluation of plant performance in different conditions.
METHODS
Plant Material
Arabidopsis thaliana var Col-0, the wild type, and a plastidic pgm (Caspar et al., 1985) were grown in an 8-h day phytotron. At least 3 weeks before their use, the plants were transferred into a small growth cabinet with a 12-h d of 160 μE and 20°C throughout the day/night cycle. Harvests of 15 plant rosettes at a time point were performed sequentially every 2 h within a day/night cycle or 0, 2, 4, 8, 24, 48, 72, and 148 h in total darkness. Each sample typically contained three rosettes, equivalent to ∼500 mg FW. The entire sample was powdered under liquid nitrogen and stored at −80°C until its use.
Reagents
Chemicals were purchased from Sigma (Taufkirchen, Germany), except NAD+, NADH, NADP+, NADPH, and phosphoenolpyruvate (Roche, Mannheim, Germany) and glucose, fructose, sucrose (Merck, Darmstadt, Germany). Xylulose-5-phosphate was purchased from W.D. Fessner (University of Darmstadt, Germany). Dihydroxyphenoxazine (Amplex red) was purchased from Molecular Probes Europe (Leiden, The Netherlands). Enzymes for analysis were purchased from Roche except phosphoglycerokinase and glycerokinase (Sigma-Aldrich). UMP-kinase was overexpressed and purified as in Serina et al. (1995). The clone encoding UMP-kinase is a generous gift from Octavian Barzu (Institut Pasteur, Paris, France).
Preparation of RNA, cDNA, and Labeled cRNA, Array Hybridization, and Data Evaluation
Extraction of RNA, cDNA synthesis and labeling, and hybridization on Affymetrix ATH1 arrays were performed as in Thimm et al. (2004) and data evaluation as in Gibon et al. (2004).
Extraction of Enzymes
For routine measurements, samples corresponding to ∼500 mg FW were collected, ground to a powder in liquid nitrogen, and stored at −80°C. Aliquots of 10 to 20 mg FW were subaliquoted at −180°C and extracted by vigorous vortexing with extraction buffer. The composition of the extraction buffer was 10% (v/v) glycerol, 0.25% (w/v) BSA, 0.1% (v/v) Triton X-100, 50 mM Hepes/KOH, pH 7.5, 10 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM benzamidine, 1 mM ɛ-aminocapronic acid, 1 mM phenylmethylsulfonyl fluoride, 10 μM leupeptin, and 1 mM DTT. Phenylmethylsulfonyl fluoride was added just before extraction. DTT was omitted when using peroxidase-based or MTT-based indicator reactions. In all cases, 500 to 1000 μL of extraction buffer was used, leading to an initial ∼50-fold (w/v) dilution. Subaliquots were diluted further to generate appropriate dilutions for the different enzymes.
Pipetting Scheme for Stopped Assays
Ninety-six–well microplates were prepared using a Multiprobe II pipetting robot equipped with a cooling block, an incubation block, a shaker, and a gripper (Perkin-Elmer, Zaventem, Belgium). When necessary, the extracts were further diluted in the extraction buffer using an automated program on the robot. Then, aliquots of 5 μL of extracts as well as standards were dispensed into a 96-well microplate held at 4°C, then 40 μL of reaction mix were added. The microplate was transferred to 25°C, gently shaken, and preincubated for 5 min. Then, 5 μL of a starter solution were added, and the microplate was shaken again. After a defined time (0, 10, or 20 min, depending on the assay), 20 μL of a stop solution (NaOH, HCl, or N-ethylmaleimide, depending on the assay) were added, and the plates were incubated at room temperature for a further 10 min. Then, 20 μL of a neutralizing solution and 50 μL of the appropriate determination mix to determine the product accumulated were added, and the reaction was immediately monitored by measuring change in absorbance or fluorescence in a Synergy HT (Bio-Tek, Denkendorf, Germany) microplate reader. The rates of reactions were calculated as the decrease of the absorbance in mOD min−1 using KC4 software (Bio-Tek). One microplate allowed the activity of one enzyme to be measured in 20 samples. Vblank and Vmax assays were performed for every sample in duplicate, and each plate included four standards. Samples were randomly distributed to avoid local artifacts.
Spreadsheets were used to track samples and allow automatic data processing. A file containing raw data from the microplate reader was pasted into a template Excel sheet, which automatically calculated the activity for each well on the microplate. Standard curves and technical error (expressed as sd for every technical duplicate) were also displayed. The time required for calculations including the quality control is <5 min per microplate. The technical error was usually <5%. Results were uploaded into standardized spreadsheets that contain information about all the samples for a given experiment, including the FW taken for extraction, to calculate activities on a FW basis.
Determinations Based on the Glycerol-3-Phosphate Cycling Assay
Step 1. Enzymatic Reaction
ADP-glucose pyrophosphorylase was assayed in the reverse direction by measuring the PPi-dependent production of ATP from ADP-glucose. The concentrations used for the substrates and for the activator 3-phosphoglycerate were chosen according to Merlo et al. (1993). Extracts, as well as ATP standards prepared in the extraction buffer and ranging from 0 to 1 nmol, were incubated in a medium containing 50 mM Hepes/KOH, pH 7.5, 5 mM MgCl2, 1 unit·mL−1 glycerokinase, 0 (blank) or 1 mM (maximal activity) ADP-glucose, 5 mM 3-phosphoglycerate, 1.5 mM sodium fluoride, and 120 mM glycerol. The reaction was started by the addition of PPi to a final concentration of 2 mM. The reaction was stopped with 20 μL of 0.5 M HCl.
Glycerokinase was assayed in conditions adapted from Sadava and Moore (1987). Extracts, as well as glycerol-3-P standards prepared in the extraction buffer and ranging from 0 to 80 μM, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 8 mM MgCl2, and 120 mM glycerol. The reaction was started by the addition of ATP to a final concentration of 0 or 0.8 mM. The reaction was stopped with 20 μL of 0.5 M HCl.
NAD-dependent GAP DH was assayed in conditions adapted from Plaxton (1990). Extracts, as well as dihydroxyacetone-P standards prepared in the extraction buffer and ranging from 0 to 500 μM, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 30 mM MgCl2, 20 mM KCl, 2 mM EDTA, 5 mM DTT, 0.4 mM NADH, 0 (blank) or 6 mM of 3-phosphoglycerate (maximal activity), 10 units·mL−1 phosphoglycerate kinase, 1 unit·mL−1 triose-P isomerase, and 1 unit·mL−1 glycerol-3-P dehydrogenase. The reaction was started by the addition of ATP to a final concentration of 2 mM. The reaction was stopped with 20 μL of 0.5 M HCl. Because the NADP-dependent GAP DH also reacts with NAD(H), its activity was retrieved.
NADP-dependent GAP DH was assayed in conditions adapted from Stitt et al. (1989). Extracts, as well as dihydroxyacetone-P standards prepared in the extraction buffer and ranging from 0 to 500 μM, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 30 mM MgCl2, 20 mM KCl, 2 mM EDTA, 5 mM DTT, 5 mM ATP, 0 (blank) or 6 mM of 3-phosphoglycerate (maximal activity), 10 units·mL−1 phosphoglycerate kinase, and 1 unit·mL−1 triose-P isomerase. The reaction was started by the addition of NADPH to a final concentration of 0.5 mM. The reaction was stopped with 20 μL of 0.5 M HCl.
PFP was assayed in conditions adapted from Scott et al. (1995). Extracts, as well as fructose-1,6-bisP standards prepared in the extraction buffer and ranging from 0 to 80 μM, were incubated in a medium containing 50 mM Hepes/KOH, pH 7.5, 2 mM MgCl2, 0.25 mM NADH, 5 mM ATP, 7.5 mM fructose-6P, 10 μM fructose-2,6-bisphosphate, 1 unit·mL−1 aldolase, 1 unit·mL−1 triose-P isomerase, and 1 unit·mL−1 glycerol-3P dehydrogenase. The reaction was started by the addition of PPi to a final concentration of 0 (blank) or 0.25 mM (maximal activity). The reaction was stopped with 20 μL of 0.5 M HCl.
PK was assayed in the reverse direction by measuring the phosphoenol pyruvate-dependent production of ATP from ADP. The concentrations used for phosphoenol pyruvate, ADP, and KCl were chosen according to Bergmeyer (1987). Extracts, as well as ATP standards prepared in the extraction buffer and ranging from 0 to 1 nmol, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 10 mM MgCl2, 1 mM EDTA, 1 unit·mL−1 glycerokinase, 200 mM KCl, 1 mM ADP, 1 mM AMP, 120 mM glycerol, and 0 (blank) or 5 mM (maximal activity) phosphoenol pyruvate. The reaction was stopped with 20 μL of 0.5 M HCl. AMP was used to inhibit myokinase activity. Preliminary tests established that AMP exerted no effect on the activity of PK.
SPS was assayed in the forward direction by measuring the fructose-6P-dependent production of UDP from UDP-glucose. The concentrations used for fructose-6P, glucose-6P, and phosphate were chosen according to Huber et al. (1989). Extracts, as well as UDP standards prepared in the extraction buffer and ranging from 0 to 1 nmol, were incubated in a medium containing 50 mM Hepes/KOH, pH 7.5, 10 mM MgCl2, 1 mM EDTA, 1 unit·mL−1 UMP-kinase, 1 unit·mL−1 glycerokinase, 10 mM UDP-glucose, 50 μM ADP, 1 mM AMP, 120 mM glycerol, 0 (blank) or 12 mM (maximal activity) fructose-6P, and 0 (blank) or 36 mM (maximal activity) glucose-6P. The reaction was stopped with 20 μL of 0.5 M HCl. Preliminary tests established that AMP exerted no effect on the activity of SPS.
Transketolase was assayed according to conditions adapted from Henkes et al. (2001). Extracts, as well as dihydroxyacetone-P standards prepared in the extraction buffer and ranging from 0 to 80 μM, were incubated in a medium containing 50 mM Hepes/KOH, pH 7.7, 10 mM MgCl2, 0.8 mM NADH, 1.25 mM thiamine pyrophosphate, 1 unit·mL−1 triose-P isomerase, and 1 unit·mL−1 glycerol-3P dehydrogenase. The reaction was started by the addition of erythrose-4P and xylulose-5P to final concentrations of 0 or 2.5 mM and 0 or 10 mM, respectively. The reaction was stopped with 20 μL of 0.5 M HCl.
Step 2
Glycerol-3-P was measured as described in Gibon et al. (2002), in the presence of 1.8 units·mL−1 glycerol-3-P oxidase, 0.7 unit·mL−1 glycerol-3-P dehydrogenase, 1 mM NADH, 1.5 mM MgCl2, and 100 mM Tricine/KOH, pH 8.0. When determining glyceraldehyde-3-phosphate dehydrogenases, 1 unit·mL−1 triose-phosphate isomerase was added. The absorbance was red at 340 nm and at 30°C until the rates were stabilized. The rates of reactions were calculated as the increase of the absorbance in mOD min−1.
Determinations Based on the NADP+ Cycling Assay
Step 1. Enzymatic Reaction
Cytosolic fructose-1,6-bisphosphatase was assayed in conditions adapted from Stitt (1989). Extracts, as well as fructose-6P standards prepared in the extraction buffer and ranging from 0 to 20 μM, were incubated in a medium containing 50 mM Hepes/KOH, pH 7.0, 2 mM MgCl2, 0.4 mM NADP+, 1 unit·mL−1 phosphoglucose isomerase, and 1 unit·mL−1 glucose-6P dehydrogenase. The reaction was started by the addition of fructose-1,6-bisphosphate to a final concentration of 0 (blank) or 20 μM (maximal activity). The reaction was stopped with 20 μL of 0.5 M NaOH.
Glucokinase activity was assayed in conditions adapted from Renz et al. (1993). Extracts, as well as glucose-6P standards prepared in the extraction buffer and ranging from 0 to 80 μM, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 5 mM MgCl2, 2 mM glucose, 0.4 mM NADP+, and 1 unit·mL−1 glucose-6P dehydrogenase. The reaction was started by the addition of ATP to a final concentration of 0 (blank) or 0.8 mM (maximal activity). The reaction was stopped with 20 μL of 0.5 M NaOH.
Fructokinase activity was assayed in conditions adapted from Renz et al. (1993). Extracts, as well as glucose-6P standards prepared in the extraction buffer and ranging from 0 to 80 μM, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 5 mM MgCl2, 2 mM fructose, 0.4 mM NADP+, 1 unit·mL−1 phosphoglucose isomerase, and 1 unit·mL−1 glucose-6P dehydrogenase. The reaction was started by the addition of ATP to a final concentration of 0 (blank) or 0.8 mM (maximal activity). The reaction was stopped with 20 μL of 0.5 M NaOH.
G6PDH was assayed in conditions adapted from Graeve et al. (1994). Extracts, as well as NADPH standards prepared freshly in the extraction buffer and ranging from 0 to 80 μM, were incubated in a medium containing 100 mM Tricine/KOH, pH 9.0, 10 mM MgCl2, and 0.5 mM NADP+. The reaction was started by the addition of glucose-6P to a final concentration of 0 (blank) or 5 mM (maximal activity). The reaction was stopped with 20 μL of 0.5 M NaOH.
NADP-dependent isocitrate dehydrogenase activity was assayed in conditions adapted from Sadka et al. (2000). Extracts, as well as NADPH standards freshly prepared in the extraction buffer and ranging from 0 to 80 μM, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 4 mM MgCl2, and 1 mM NADP+. The reaction was started by the addition of isocitrate to a final concentration of 0 (blank) or 2 mM (maximal activity). The reaction was stopped with 20 μL of 0.5 M NaOH.
Shikimate dehydrogenase activity was assayed in conditions adapted from Lourenco and Neves (1984). Extracts, as well as NADPH standards freshly prepared in the extraction buffer and ranging from 0 to 1 nmol, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 4 mM MgCl2, and 1 mM NADP+. The reaction was started by the addition of shikimate to a final concentration of 0 (blank) or 2 mM (maximal activity). The reaction was stopped with 20 μL of 0.5 M NaOH.
Step 2. Destruction of NADP+
The plates were mixed and centrifuged (2 min at 2000 rpm). They were then sealed using an adhesive aluminum foil and heated at 95°C for 5 min. After cooling down, 10 μL of HCl M and 0.2 M Tricine/KOH, pH 9.0, were added to adjust the pH to 9.
Step 3. Determination of NADPH
NADPH was measured as described by Gibon et al. (2002) in the presence of 3 units·mL−1 G6PDH grade I, 100 mM Tricine/KOH, pH 9.0, 5 mM MgCl2, 4 mM EDTA, 0.1 mM PMS, 0.6 mM MTT, and 3 mM glucose-6-phosphate. The absorbance was red at 570 nm and at 30°C until the rates were stabilized. The rates of reactions were calculated as the increase of the absorbance in mOD min−1.
Determinations Based on the NAD+ Cycling Assay
Step 1. Enzymatic Reaction
AlaAT activity was assayed in conditions adapted from Bergmeyer (1987). Extracts, as well as pyruvate standards freshly prepared in the extraction buffer and ranging from 0 to 1 nmol, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 2.5 mM Ala, 0.1 mM NADH, and 0.25 unit·mL−1 lactate dehydrogenase. Contaminating NAD+ was removed from the NADH stock solution (20 mM in 100 mM NaOH) before the assay by heating for 10 min at 95°C. The reaction was started by the addition of 2-oxoglutarate to a final concentration of 0 (blank) or 2 mM (maximal activity). The reaction was stopped with 20 μL of 0.5 M HCl.
AspAT activity was assayed in conditions adapted from Bergmeyer (1987). Extracts, as well as oxaloacetate standards freshly prepared in the extraction buffer and ranging from 0 to 1 nmol, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 2.5 mM Asp, 0.1 mM NADH, and 0.25 unit·mL−1 malate dehydrogenase. Contaminating NAD+ was removed from the NADH stock solution (20 mM in 100 mM NaOH) before the assay by heating for 10 min at 95°C. The reaction was started by the addition of 2-oxoglutarate to a final concentration of 0 (blank) or 2 mM (maximal activity). The reaction was stopped with 20 μL of 0.5 M HCl.
Fumarase was assayed in the malate-forming direction. Extracts, as well as malate standards prepared in the extraction buffer and ranging from 0 to 1 nmol, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 0.2 mM acetyl-CoA, 5 mM phosphate, 5 mM MgCl2, 0.15 mM NAD+, 0.05 (v/v) Triton X-100, 10 units·mL−1 malate dehydrogenase, and 1 unit·mL−1 citrate synthase. The reaction was started by the addition of fumarate to a final concentration of 0 (blank) or 10 mM (maximal activity). The reaction was stopped with 20 μL of 0.5 M NaOH.
GLDH activity was assayed in the aminating direction in conditions adapted from Turano et al. (1996). Extracts, as well as NAD+ standards freshly prepared in the extraction buffer and ranging from 0 to 1 nmol, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 1 mM CaCl2, 640 mM ammonium acetate, and 0.1 mM NADH. Contaminating NAD+ was removed from the NADH stock solution (20 mM in 100 mM NaOH) before the assay by heating for 10 min at 95°C. The reaction was started by the addition of 2-oxoglutarate to a final concentration of 0 (blank) or 15 mM (maximal activity). The reaction was stopped with 20 μL of 0.5 M HCl.
Phosphoenolpyruvate carboxylase activity was assayed in conditions adapted from Uedan and Sugiyama (1976). Extracts, as well as NAD+ standards freshly prepared in the extraction buffer and ranging from 0 to 1 nmol, were incubated in a medium containing 100 mM Tricine/KOH, pH 8.0, 20 mM MgCl2, 1 unit·mL−1 malate dehydrogenase, 10 mM NaHCO3, and 0.1 mM NADH. Contaminating NAD+ was removed from the NADH stock solution (20 mM in 100 mM NaOH) before the assay by heating for 10 min at 95°C. The reaction was started by the addition of phosphoenol pyruvate to a final concentration of 0 (blank) or 2 mM (maximal activity). The reaction was stopped with 20 μL of 0.5 M HCl.
Step 2. Destruction of NAD+ or NADH
The plates were mixed and centrifuged (2 min at 2000 rpm). They were then sealed using an adhesive aluminum foil and heated at 95°C for 10 min. After cooling down, 10 μL of HCl M or NaOH M, and 10 μL of 0.2 M Tricine/KOH, pH 9.0, were added to adjust the pH to 9.0.
Step 3. Determination of NADH or NAD+
NAD(H) was measured in the presence of 6 units·mL−1 ADH, 100 mM Tricine/KOH, pH 9.0, 4 mM EDTA, 0.1 mM PES, 0.6 mM MTT, and 500 mM ethanol. The absorbance was red at 570 nm and at 30°C until the rates were stabilized. The rates of reactions were calculated as the increase of the absorbance in mOD min−1.
Determination of Acid Invertase
Acid invertase was assayed in conditions adapted from Huber (1984). Sorbitol (250 mM) was added to the extraction buffer according to Greiner et al. (1999) to improve the recovery of invertase. Extracts, as well as glucose standards prepared in the extraction buffer and ranging from 0 to 1 nmol, were incubated in 50 mM acetate/KOH, pH 4.5. The reaction was started by the addition of sucrose to a final concentration of 20 mM. The reaction was stopped with 20 μL of 0.5 M NaOH after 10 and 20 min. After another 10-min incubation, the wells were neutralized with 20 μL of 0.5 M HCl and 0.1 M Hepes/KOH, pH 7.0, buffer. The determination was performed using fluorimetry (excitation 530 nm, emission 590 nm) after addition of 50 mM Hepes/KOH, pH 7.0, 1 unit·mL−1 glucose oxidase, 0.0015 unit·mL−1 horseradish peroxidase, and 0.2 mM dihydroxyphenoxazine final concentrations.
Determination of Fd-GOGAT
GOGAT activity was assayed in conditions adapted from Hecht et al. (1988). Extracts, as well as glutamate standards prepared in the extraction buffer and ranging from 0 to 10 nmol, were incubated in a medium containing 50 mM Hepes/KOH, pH 7.5, 10 mM Gln, 5 mM methyl viologen, 3 mM 2-oxoglutarate, and 1 mM aminooxyacetate. The reaction was started by the addition of sodium dithionite and NaHCO3 to respective final concentrations of 2 mg/mL. The reaction was stopped with 20 μL of 100 mM N-methylmaleimide. After heating for 10 min at 95°C, the plates were cooled and centrifuged (2 min at 2000 rpm). Then 100 μL of the determination mix were added. Final concentrations of additional reagents were 100 mM Tricine/KOH, pH 8.5, 0.6 mM MTT, 1.8 mM NAD+, 0.3% (v/v) Triton X-100, and 0.15 unit·mL−1 diaphorase. The absorbance was red at 570 nm until OD stabilized, and 1 μL of 1000 units·mL−1 GLDH was then added. Absorbance was red until the endpoint was reached (in the conditions described above, it took 30 to 40 min).
Determination of NIA
NIA was determined in conditions adapted from Scheible et al. (1997b) (1997c). Extracts, as well as nitrite standards prepared in the extraction buffer and ranging from 0 to 10 nmol, were incubated in a medium containing 50 mM Hepes/KOH, pH 7.5, 0.04% (v/v) Triton X-100, 2 mM EDTA, 10 μM Na2MoO4, 20 μM flavin adenine dinucleotide, 0.5 mM DTT, 20 μM leupeptin, 20 mM potassium nitrate, and 0 (maximal activity) or 10 mM (selective activity) MgCl2. The reaction was started by the addition of NADH to a final concentration of 0.6 mM. The reaction was stopped with 20 μL of 0.6 M zinc acetate. Then, 15 μL of 0.25 mM PMS were added, and the plates were incubated for 10 min at room temperature. Finally, 100 μL of 0.5% (w/v) sulfanilamide and 0.01% (w/v) N(1-naphtyl)ethylendiamine dihydrochloride in 2.5% (v/v) H3PO4 were added. After 5 min, the OD was red at 540 nm.
Determination of GS
GS was determined as in Scheible et al. (1997a), except that the volume was scaled down to 100 μL and the maximal activity and blank (where glutamate was omitted) rates were run simultaneously in separate wells. Extracts were incubated in a medium containing 50 mM Hepes/KOH, pH 7.5, 10% (v/v) glycerol, 10 mM MgCl2, 2 mM EDTA, 0.2 mM NaVO3, 40 μM 5′,5′-diadenosinpentaphosphate, 2 mM ammonium chloride, 5 mM ATP, 1 mM phosphoenol pyruvate, 0.6 mM NADH, 1 unit·mL−1 PK, 0.7 unit·mL−1 lactate dehydrogenase, and 0 (blank) or 45 mM (maximal activity) glutamate. The absorbance was then red at 340 nm until the rate was stabilized (20 to 30 min).
AGPase Gel Blot Analysis
Extraction of AGPase for blotting and procedures for gels and quantification were performed as in Hendriks et al. (2003).
Statistics
Standard procedures were performed using functions of the Excel program.
The smoothness value, which indicates if the variance in a set of time-related data points is because of a smooth diurnal change or random noise, was estimated as follows:
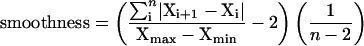 |
This equation has a value of zero if every data point lies on a smooth curve that moves through one maximum and one minimum per diurnal cycle. Increasing values reveal that the curve is becoming increasingly irregular with a maximum value of 1. The left hand term uses the data to estimate the smoothness term, and the right hand term normalizes the results so that the values vary between limits of 0 and 1. A set of illustrative sample curves with the respective smoothness value is given in the supplemental data online.
The low cost and high throughput of the enzyme determination platform allowed enzyme activities to be measured at 2-h intervals. To exploit this increased data density, moving averages were calculated throughout the night and day cycle for every two consecutive data points (see Supplemental Table 1 online). Use of these time-averaged data improved the smoothness factor for many enzyme activities and was used for statistical analyses. This transformation was not performed for the transcript data set because they were only measured every 4 h, and the smoothness factor was already close to zero for most transcripts.
To allow data compression, a probability mass function curve was calculated for both populations. The probability mass function is given by the following:
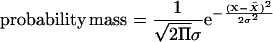 |
This calculation assumes a normal distribution. A true normal distribution gives a skewness of zero. Skewness was calculated as follows:
 |
The estimated values for transcripts and for activities were 0.7 and 1.2, respectively, which was considered acceptable.
To analyze the temporal relation between changes of transcripts and enzyme activity in more detail, the change of the transcript level and the change of the time-averaged enzyme activity was calculated for each 4-h time interval and normalized as the percentage of the average level using the following equation:
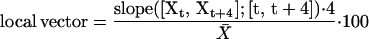 |
To generate decision trees (Quinlan, 1979), a heuristic divide-and-conquer strategy was applied that incorporated as the split criterion the normalized mutual information. Trees were generated by recursively splitting the training set according to the variable with the maximal amount of this quantity. To avoid over-fitting, trees were pruned by the removal of those subtrees that are estimated to increase the error rate minimally. The program used to generate decision trees was the J4.8 data mining software (WEKA, Waikato, New Zealand), which is a Java implementation of the C4.5 software (Quinlan, 1993). The assignment of further samples to the classes was calculated as frequencies. For each sample, an assignment probability was also calculated and used to determine weighted frequencies.
Supplementary Material
Acknowledgments
This research was supported by the Max Planck Society and by the German Ministry for Research and Technology, in the framework of the German plant genomics program Genomanalyse im Biologischen System Pflanze (0312277A). Thanks are due to Alisdair Fernie, Oliver Thimm, and John Lunn for their tips and comments. Thanks are also due to Linda Bartezko, Manuela Günther, Sonja Köhler, and Kristin Retzlaff for technical assistance.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantcell.org) is: Yves Gibon (gibon@mpimp-golm.mpg.de).
Online version contains Web-only data.
Article, publication date, and citation information can be found at www.plantcell.org/cgi/doi/10.1105/tpc.104.025973.
References
- ap Rees, T., and Hill, S.A. (1994). Metabolic control analysis of plant metabolism. Plant Cell Environ. 17, 587–599. [Google Scholar]
- Arabidopsis Genome Initiative (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815. [DOI] [PubMed] [Google Scholar]
- Ashour, M.B.A., Gee, S.J., and Hammock, B.D. (1987). Use of a 96-well microplate reader for measuring routine enzyme activities. Anal. Biochem. 166, 353–360. [DOI] [PubMed] [Google Scholar]
- Bergmeyer, H.U. (1987). Methods of Enzymatic Analysis. (Weinheim, Germany: VCH).
- Bernofsky, C., and Swan, M. (1973). Improved cycling assay for nicotinamide adenine-dinucleotide. Anal. Biochem. 53, 452–458. [DOI] [PubMed] [Google Scholar]
- Caspar, T., Huber, S.C., and Somerville, C. (1985). Alterations in growth, photosynthesis, and respiration in a starchless mutant of Arabidopsis thaliana (L) deficient in chloroplast phosphoglucomutase activity. Plant Physiol. 79, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celis, J.E., Kruhoffer, M., Gromove, I., Frederiksen, C., Ostergaard, M., Thykjaer, T., Gromova, P., Yu, J., Palsdottir, H., Mangnusson, N., and Ornoft, T.F. (2000). Gene expression profiling: Monitoring transcription and translation products using DNA microarrays and proteomics. FEBS Lett. 480, 2–16. [DOI] [PubMed] [Google Scholar]
- Czechowski, T., Bari, R.P., Stitt, M., Scheible, W.R., and Udvardi, M.K. (2004). Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: Unprecedented sensitivity reveals novel root- and shoot-specific genes. Plant J. 38, 366–379. [DOI] [PubMed] [Google Scholar]
- DeRisi, J.L., Iyer, V.R., and Brown, P.O. (1997). Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278, 680–686. [DOI] [PubMed] [Google Scholar]
- Gibon, Y., Bläsing, O.E., Palacios, N., Pankovic, D., Hendriks, J.H.M., Fisahn, J., Höhne, M., Günter, M., and Stitt, M. (2004). Adjustment of diurnal starch turnover to short days: Depletion of sugar during the night leads to a temporary inhibition of carbohydrate utilisation, accumulation of sugars and post-translational activation of ADP-glucose pyrophosphorylase in the following light period. Plant J. 39, 847–862. [DOI] [PubMed] [Google Scholar]
- Gibon, Y., Vigeolas, H., Tiessen, A., Geigenberger, P., and Stitt, M. (2002). Sensitive and high throughput metabolite assays for inorganic pyrophosphate, ADPGlc, nucleotide phosphates, and glycolytic intermediates based on a novel enzymic cycling system. Plant J. 30, 221–235. [DOI] [PubMed] [Google Scholar]
- Graeve, K., Vonschaewen, A., and Scheibe, R. (1994). Purification, characterization, and cDNA sequence of glucose-6-phosphate-dehydrogenase from potato (Solanum tuberosum L). Plant J. 5, 353–361. [DOI] [PubMed] [Google Scholar]
- Greenbaum, D., Colangelo, C., Williams, K., and Gerstein, M. (2003). Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 4, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner, S., Rausch, T., Sonnewald, U., and Herbers, K. (1999). Ectopic expression of a tobacco invertase inhibitor homolog prevents cold-induced sweetening of potato tubers. Nat. Biotechnol. 17, 708–711. [DOI] [PubMed] [Google Scholar]
- Hecht, U., Oelmueller, R., Schmidt, S., and Mohr, H. (1988). Action of light, nitrate and ammonium on the levels of NADH- and ferredoxindependent glutamate synthases in the cotyledons of mustard seedlings. Planta 175, 130–138. [DOI] [PubMed] [Google Scholar]
- Hendriks, J.H.M., Kolbe, A., Gibon, Y., Stitt, M., and Geigenberger, P. (2003). ADP-glucose pyrophosphorylase is activated by posttranslational redox-modification in response to light and to sugars in leaves of Arabidopsis and other plant species. Plant Physiol. 133, 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkes, S., Sonnewald, U., Badur, R., Flachmann, R., and Stitt, M. (2001). A small decrease of plastid transketolase activity in antisense tobacco transformants has dramatic effects on photosynthesis and phenylpropanoid metabolism. Plant Cell 13, 535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, M.J. (2002). Transcript abundance in yeast varies over six orders of magnitude. J. Biol. Chem. 277, 14363–14366. [DOI] [PubMed] [Google Scholar]
- Huber, J.L., Huber, S.C., and Hamborg-Nielsen, T. (1989). Protein phosphorylation as a mechanism for regulation of spinach leaf sucrose-phosphate synthase activity. Arch. Biochem. Biophys. 270, 681–690. [DOI] [PubMed] [Google Scholar]
- Huber, S.C. (1984). Biochemical basis for effects of K-deficiency on assimilate export rate and accumulation of soluble sugars in soybean leaves. Plant Physiol. 76, 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeannette, E., and Prioul, J.L. (1994). Variations of ADPGlucose pyrophosphorylase activity from maize leaf during day/night cycle. Plant Cell Physiol. 35, 869–878. [Google Scholar]
- Kaiser, W.M., and Huber, S.C. (1994). Posttranslational regulation of nitrate reductase in higher plants. Plant Physiol. 106, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser, W.M., and Huber, S.C. (1999). Nitrate reductase in higher plants: A case study for transduction of environmental stimuli into control of catalytic activity. Physiol. Plant. 105, 385–390. [Google Scholar]
- Kiianitsa, K., Solinger, J.A., and Heyer, W.D. (2003). NADH-coupled microplate photometric assay for kinetic studies of ATP-hydrolyzing enzymes with low and high specific activities. Anal. Biochem. 321, 266–271. [DOI] [PubMed] [Google Scholar]
- Klein, D., Krapp, A., and Stitt, M. (2000). The regulation of NIA expression by nitrate and glutamine is overridden when sugars decrease to low levels in tobacco leaves. Plant Cell Environ. 23, 863–871. [Google Scholar]
- Kononenko, I. (2001). Machine learning for medical diagnosis: History, state of the art and perspective. Artif. Intell. Med. 23, 89–109. [DOI] [PubMed] [Google Scholar]
- Lein, W., Bornke, F., Reindl, A., Ehrhardt, T., Stitt, M., and Sonnewald, U. (2004). Target-based discovery of novel herbicides. Curr. Opin. Plant Biol. 7, 219–225. [DOI] [PubMed] [Google Scholar]
- Lourenco, E.J., and Neves, V.A. (1984). Partial purification and some properties of shikimate dehydrogenase from tomatoes. Phytochemistry 23, 497–499. [Google Scholar]
- Merlo, L., Geigenberger, P., Hajirezaei, M., and Stitt, M. (1993). Changes of carbohydrates, metabolites and enzyme activities in potato tubers during development and within a single tuber along a stolon-apex gradient. J. Plant Physiol. 142, 392–402. [Google Scholar]
- Meyer, C., and Stitt, M. (2001). Nitrate reduction. In Plant Nitrogen, P.J. Lea and J.-F. Morot-Gaudry, eds (Heidelberg, Germany: Springer Verlag), pp. 37–60.
- Michaut, L., Flister, S., Neeb, M., White, K.P., Certa, U., and Gehring, W.J. (2003). Analysis of the eye developmental pathway in Drosophila using DNA microarrays. Proc. Natl. Acad. Sci. USA 100, 4024–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisselbaum, J.S., and Green, S. (1969). A simple ultramicro method for determination of pyridine nucleotides in tissues. Anal. Biochem. 27, 212–217. [DOI] [PubMed] [Google Scholar]
- Plaxton, W.C. (1990). Glycolysis. In Methods in Plant Biochemistry, Vol. 3, P.J. Lea, ed (London: Academic Press), pp. 145–173.
- Quinlan, J.R. (1979). Discovering rules from large collections of examples. In Expert Systems in the Microelectronic Age, D. Michie, ed (Edinburgh, UK: Edingburgh University Press), pp. 168–201.
- Quinlan, J.R. (1993). C4.5: Programs for Machine Learning. (San Mateo, CA: Morgan Kaufmann Publishers)
- Redman, J.C., Haas, B.J., Tanimoto, G., and Town, C.D. (2004). Development and evaluation of an Arabidopsis whole genome Affymetrix probe array. Plant J. 38, 545–561. [DOI] [PubMed] [Google Scholar]
- Renz, A., Merlo, L., and Stitt, M. (1993). Partial purification from potato tubers of 3 fructokinases and 3 hexokinases which show differing organ and developmental specificity. Planta 190, 156–165. [Google Scholar]
- Sadava, D., and Moore, K. (1987). Glycerol metabolism in higher plants: Glycerol kinase. Biochem. Biophys. Res. Commun. 143, 977–983. [DOI] [PubMed] [Google Scholar]
- Sadka, A., Dahan, E., Or, E., and Cohen, L. (2000). NADP(+)-isocitrate dehydrogenase gene expression and isozyme activity during citrus fruit development. Plant Sci. 158, 173–181. [DOI] [PubMed] [Google Scholar]
- Scheible, W.R., Gonzalez-Fontes, A., Lauerer, M., Muller-Rober, B., Caboche, M., and Stitt, M. (1997. a). Nitrate acts as a signal to induce organic acid metabolism and repress starch metabolism in tobacco. Plant Cell 9, 783–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheible, W.R., Gonzalez-Fontes, A., Morcuende, R., Lauerer, M., Geiger, M., Glaab, J., Gojon, A., Schulze, E.D., and Stitt, M. (1997. b). Tobacco mutants with a decreased number of functional nia genes compensate by modifying the diurnal regulation of transcription, post-translational modification and turnover of nitrate reductase. Planta 203, 304–319. [DOI] [PubMed] [Google Scholar]
- Scheible, W.R., Krapp, A., and Stitt, M. (2000). Reciprocal diurnal changes of phosphoenolpyruvate carboxylase expression and cytosolic pyruvate kinase, citrate synthase and NADP-isocitrate dehydrogenase expression regulate organic acid metabolism during nitrate assimilation in tobacco leaves. Plant Cell Environ. 23, 1155–1167. [Google Scholar]
- Scheible, W.R., Lauerer, M., Schulze, E.D., Caboche, M., and Stitt, M. (1997. c). Accumulation of nitrate in the shoot acts as a signal to regulate shoot-root allocation in tobacco. Plant J. 11, 671–691. [Google Scholar]
- Scheible, W.-R., Morcuende, R., Czechowski, T., Osuna, D., Fritz, C., Palacios-Rojas, N., Thimm, O., Udvardi, M.K., and Stitt, M. (2004). Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of Arabidopsis in response to nitrogen. Plant Physiol. 136, 2483–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, P., Lange, A.J., Pilkis, S.J., and Kruger, N.J. (1995). Carbon metabolism in leaves of transgenic tobacco (Nicotiana tabacum L) containing elevated fructose 2,6-bisphosphate levels. Plant J. 7, 461–469. [DOI] [PubMed] [Google Scholar]
- Serina, L., Blondin, C., Krin, E., Sismeiro, O., Danchin, A., Sakamoto, H., Gilles, A.M., and Barzu, O. (1995). Escherichia coli UMP-kinase, a member of the aspartokinase family, is a hexamer regulated by guanine nucleotides and UTP. Biochemistry 34, 5066–5074. [DOI] [PubMed] [Google Scholar]
- Steen, H., and Pandey, A. (2002). Proteomics goes quantitative: Measuring protein abundance. Trends Biotechnol. 20, 361–364. [DOI] [PubMed] [Google Scholar]
- Stitt, M. (1989). Control analysis of photosynthetic sucrose synthesis: Assignment of elasticity coefficients and flux control coefficients to the cytosolic fructose 1,6-bisphosphatase and sucrose phosphate synthase. Philos. Trans. R. Soc. Lond. B Biol. Sci. 323, 327–338. [Google Scholar]
- Stitt, M., Lilley, R.M.C., Gerhardt, R., and Heldt, H.W. (1989). Metabolite levels in specific cells and subcellular compartments in plant leaves. Methods Enzymol. 174, 518–552. [Google Scholar]
- Stitt, M., Wilke, I., Feil, R., and Heldt, H.W. (1988). Coarse control of sucrose phosphate synthase in leaves: Alterations of the kinetic properties in response to the rate of photosynthesis and the accumulation of sucrose. Planta 174, 217–230. [DOI] [PubMed] [Google Scholar]
- Suzuki, A., Vergnet, C., Morot-Gaudry, J.-F., Zehnacker, C., and Grosclaude, J. (1994). Immunological characterization of ferredoxin and methyl viologen interaction domains of glutamate synthase using monoclonal antibodies. Plant Physiol. Biochem. 32, 619–626. [Google Scholar]
- Thimm, O., Blasing, O., Gibon, Y., Nagel, A., Meyer, S., Kruger, P., Selbig, J., Muller, L.A., Rhee, S.Y., and Stitt, M. (2004). MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939. [DOI] [PubMed] [Google Scholar]
- Tiessen, A., Hendriks, J.H.M., Stitt, M., Branscheid, A., Gibon, Y., Farre, E.M., and Geigenberger, P. (2002). Starch synthesis in potato tubers is regulated by post-translational redox modification of ADP-glucose pyrophosphorylase: A novel regulatory mechanism linking starch synthesis to the sucrose supply. Plant Cell 14, 2191–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turano, F.J., Dashner, R., Upadhyaya, A., and Caldwell, C.R. (1996). Purification of mitochondrial glutamate dehydrogenase from dark-grown soybean seedlings. Plant Physiol. 112, 1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uedan, K., and Sugiyama, T. (1976). Purification and characterization of phosphoenolpyruvate carboxylase from maize leaves. Plant Physiol. 57, 906–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R., Okamoto, M., Xing, X., and Crawford, N.M. (2003). Microarray analysis of the NO3− response in Arabidopsis roots and shoots reveals over 1000 rapidly responding genes new linkages to glucose, trehalose-6-phosphate, iron, and sulfate metabolism. Plant Physiol. 132, 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner, H., and Kaiser, W. (1999). 14-3-3 proteins control proteolysis of nitrate reductase in spinach leaves. FEBS Lett. 455, 75–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.