

In this Outlook, Poulin and Haigis discuss a study from Boutin et al. that presents a new mouse model of CRC in which the expression of oncogenic K-RAS is regulated by doxycycline.
Keywords: colorectal cancer, metastasis, Kras, Apc, P53, invasion
Abstract
KRAS is the most frequently mutated oncogene in human cancer and plays a central, although poorly understood, role in colorectal cancer (CRC) progression. In this issue of Genes & Development, Boutin and colleagues (pp. 370–382) present a new mouse model of CRC in which the expression of oncogenic K-RAS is regulated by doxycycline. Using this model, they demonstrate that continued expression of oncogenic K-RAS is required for the survival of primary and metastatic colon cancers and that oncogenic K-RAS activates TGF-β signaling to promote tumor invasion and metastasis.
In 1990, Fearon and Vogelstein (1990) presented their paradigmatic model for the somatic genetics of colorectal cancer (CRC) in which mutations in tumor suppressor genes (APC and P53) and oncogenes (KRAS) cooperate to produce defined histological states during disease progression. This initial model was based on targeted sequencing studies from the 1980s but has been validated and refined by genome-wide sequencing efforts of the early 2000s. What the model does not provide is mechanistic insight into the role that each of the genetic events plays in CRC. While subsequent cellular and biochemical studies have provided some clarity (for example, APC loss activates canonical WNT signaling), details related to progression of CRC at the molecular level are still somewhat vague. This is especially true for KRAS, the most frequently mutated oncogene in cancer. More than 40% of CRCs harbor an activating missense KRAS mutation, but how mutant K-RAS contributes to CRC progression and whether aberrant K-RAS activity is required for tumor maintenance remain unclear. The study by Boutin et al. (2017) provides new insights into these important questions.
Genetically engineered mouse models (GEMMs) have been used extensively to model human disease. For example, GEMMs were used to demonstrate that K-RAS activation in the context of APC mutant colonic epithelium is sufficient to induce dysplastic adenocarcinomas, as predicted by the Fearon and Vogelstein model (Haigis et al. 2008). In their new study, Boutin et al. (2017) have created a CRC mouse model in which mutations in APC, P53, and KRAS are spatially and temporally regulated. This model improves on prior models in that expression of mutationally activated K-RASG12D is independently controlled, mosaic, and reversible, allowing the investigators to study the function of K-RAS in CRC maintenance. In the Boutin et al. (2017) model, loss of oncogenic K-RAS expression induces apoptosis in both primary and metastatic adenocarcinomas. Primary tumors do not regress completely, however, since adenomatous tissues that failed to express mutant K-RAS in the first place are spared from apoptosis (Fig. 1). Still, in essence, tumors revert from adenocarcinomas to adenomas.
Figure 1.
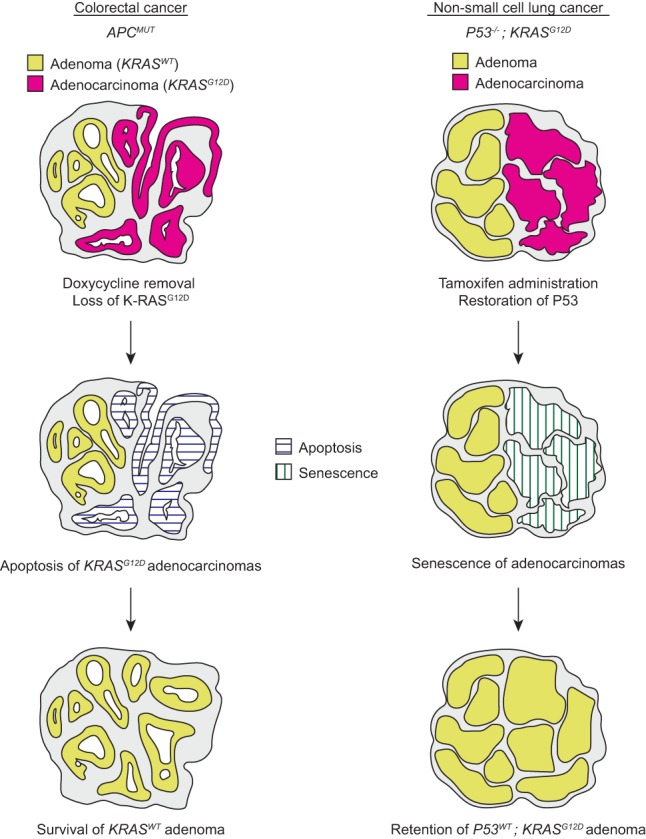
Schematic of tumor responses to reversal of progression events. In mice with CRC, expression of mutant K-RAS promotes histologic progression, invasion, and metastasis. Loss of expression of oncogenic K-RAS leads to selective apoptosis of adenocarcinoma tissue. After withdrawal of mutant K-RAS, adenomatous (KRASWT) tissues remain. In mice with non-small cell lung cancer (NSCLC), adenomas that are mutant for K-RAS and P53 spontaneously progress to adenocarcinoma. Restoration of P53 leads to senescence specifically in adenocarcinoma tissue. Adenoma tissue, although likely genotypically identical, is retained.
Doxycycline-regulated gene expression has been used to evaluate the requirement for multiple oncogenes in mouse tumor maintenance. Unlike these prior studies, which investigated initiating oncogenes, K-RAS activation in CRC is a progression event (Haigis et al. 2008). In this vein, the Boutin et al. (2017) study is more similar to a 2010 study in which Feldser et al. (2010) re-expressed P53 in K-RAS mutant, P53-null lung adenocarcinomas, selectively causing senescence in adenocarcinomas while sparing adenomas. While the Boutin et al. (2017) and Feldser et al. (2010) experiments have similar outcomes (reversion of adenocarcinomas to adenomas), the mechanisms are quite distinct. In CRC, adenocarcinoma regression is a genotype-specific phenomenon (apoptotic loss of K-RASMUT cells or retention of K-RASWT cells), while, in the lung, adenocarcinoma regression is dependent on tumor state (clearance of senescent adenocarcinoma or retention of adenoma) (Fig. 1). The most important aspect of the study by Boutin et al. (2017) is that it demonstrates for the first time that mutant K-RAS is required for tumor maintenance even in situations where K-RAS activation is not an initiating event.
Although the study by Boutin et al. (2017) firmly validates K-RAS as a therapeutic target for CRC, the fact remains that it is not presently possible to target K-RAS directly. Instead, current efforts to develop targeted therapies for K-RAS mutant cancers focus on downstream pathways. The study by Boutin et al. (2017) revealed that metastases arose only from tumors expressing mutant K-RAS and that the TGFB1/CMS4 transcriptional signature identified in human CRCs is strongly enhanced in K-RAS mutant adenocarcinomas relative to K-RAS wild-type adenomas. The investigators then demonstrated that TGF-β treatment enhanced invasion of mouse CRC cells in the absence of mutant K-RAS, while TGF-β inhibition decreased invasion of K-RAS mutant cells. Together, these data suggest that TGF-β activation by mutant K-RAS is an important step in CRC invasion and metastasis in this mouse model.
TGF-β signaling is initiated upon ligand binding to type I and type II serine/threonine kinase receptors (TGFBRI and TGFBRII), which then feed into the SMAD family of intracellular proteins that regulate contextual transcriptional responses. TGF-β signaling can have tumor-suppressive activity, including induction of cell cycle arrest and apoptosis. Paradoxically, TGF-β signaling also induces protumorigenic effects, such as epithelial-to-mesenchymal transition (EMT) and invasion, consistent with its potential role downstream from K-RAS in the Boutin et al. (2017) model. Nevertheless, the TGF-β pathway is frequently inactivated in CRC through mutation of TGFBRII or SMADs, suggesting that it is most commonly functioning as a tumor suppressor pathway in this tumor context. In mice, combined TGFBRII loss and K-RAS activation leads to intestinal tumorigenesis in the absence of an APC mutation, and these tumors metastasize to the lung (Trobridge et al. 2009). However, when TGF-β signaling remains intact, as in the Boutin et al. (2017) model, cells can take advantage of the EMT- and invasion-promoting effects. Indeed, oncogenic RAS and RAF are known to promote a TGF-β autocrine loop to stimulate EMT and tumor invasion (Oft et al. 1996; Lehmann et al. 2000; Fujimoto et al. 2001). This autocrine loop is likely functioning in the Boutin et al. (2017) model, as TGF-β1 is up-regulated by mutant K-RAS. Of note, TGF-β1 is overexpressed in a subset of human CRCs and is associated with increased metastasis and poor prognosis (Zhu et al. 2015). Nevertheless, K-RAS mutation and TGF-β1 overexpression are mutually exclusive in human CRCs (The Cancer Genome Atlas Network 2012), so the molecular mechanisms linking activated K-RAS to TGF-β signaling may be distinct in mice and humans. Regardless, the study by Boutin et al. (2017) provides an excellent example of how next-generation mouse models can be used to validate therapeutic targets and decipher molecular mechanisms of tumor progression.
Acknowledgments
We thank Alain Charest for helpful comments. The Haigis laboratory is supported by National Cancer Institute grants CA178017, CA199252, and CA195744. E.J.P. is supported by an American Cancer Society post-doctoral fellowship.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.297630.117.
References
- Boutin AT, Liao W-T, Wang M, Hwang SS, Karpinets TV, Cheung H, Chu GC, Jiang S, Hu J, Chang K, et al. 2017. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev (this issue) 10.1101/gad.293449.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network. 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. 1990. A genetic model for colorectal tumorigenesis. Cell 61: 759–767. [DOI] [PubMed] [Google Scholar]
- Feldser DM, Kostova KK, Winslow MM, Taylor SE, Cashman C, Whittaker CA, Sanchez-Rivera FJ, Resnick R, Bronson R, Hemann MT, et al. 2010. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 468: 572–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto K, Sheng H, Shao J, Beauchamp RD. 2001. Transforming growth factor-β1 promotes invasiveness after cellular transformation with activated Ras in intestinal epithelial cells. Exp Cell Res 266: 239–249. [DOI] [PubMed] [Google Scholar]
- Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, et al. 2008. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet 40: 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K, Janda E, Pierreux CE, Rytomaa M, Schulze A, McMahon M, Hill CS, Beug H, Downward J. 2000. Raf induces TGFβ production while blocking its apoptotic but not invasive responses: a mechanism leading to increased malignancy in epithelial cells. Genes Dev 14: 2610–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. 1996. TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev 10: 2462–2477. [DOI] [PubMed] [Google Scholar]
- Trobridge P, Knoblaugh S, Washington MK, Munoz NM, Tsuchiya KD, Rojas A, Song X, Ulrich CM, Sasazuki T, Shirasawa S, et al. 2009. TGF-β receptor inactivation and mutant Kras induce intestinal neoplasms in mice via a β-catenin-independent pathway. Gastroenterology 136: 1680–1688 e1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Chen X, Liao Z, He C, Hu X. 2015. TGFBI protein high expression predicts poor prognosis in colorectal cancer patients. Int J Clin Exp Pathol 8: 702–710. [PMC free article] [PubMed] [Google Scholar]