

In this review, Ng and Shyh-Chang review recent metabolomic studies of stem cell metabolism that have revealed how metabolic pathways can convey changes in the extrinsic environment or their niche to program stem cell fates.
Keywords: oxidative phosphorylation, metabolomics, stem cell
Abstract
Advances in metabolomics have deepened our understanding of the roles that specific modes of metabolism play in programming stem cell fates. Here, we review recent metabolomic studies of stem cell metabolism that have revealed how metabolic pathways can convey changes in the extrinsic environment or their niche to program stem cell fates. The metabolic programming of stem cells represents a fine balance between the intrinsic needs of a cellular state and the constraints imposed by extrinsic conditions. A more complete understanding of these needs and constraints will afford us greater mastery over our control of stem cell fates.
Stem cells are undifferentiated cells of multicellular organisms that possess long-term capacities for multipotent differentiation and self-renewal. Due to the limited life span of most somatic cells, stem cells’ capacity to replenish damaged somatic cells and maintain a self-renewing reservoir of progenitors is crucial for homeostasis in many tissues of many organisms. Thus, there is an immense interest in understanding the mechanisms for self-renewal and differentiation in stem cells, given their potential applications in regenerative medicine and studies of human development or aging.
Recent advances in metabolomics and transcriptomics analyses have increased our understanding of stem cell self-renewal and lineage specification. These novel insights have shown that, besides morphogens and growth factors, various metabolic pathways also take part in the regulation of stem cell fate. Besides their fine regulation of glycolysis and oxidative phosphorylation (OxPhos) fluxes during self-renewal and differentiation, metabolites that regulate epigenetic changes, including histone methylation and acetylation, have also been shown to be critical regulators of stem cell fates. An emerging theme is that metabolic pathways can also relay changing cues in the extrinsic environment to regulate intrinsic cell fates. Stem cell metabolism represents a combination and balance of the intrinsic metabolic needs and extrinsic metabolic constraints.
In this review, we first discuss the extensive studies of the effects of metabolism on the maintenance of embryonic (or pluripotent) stem cells. Next, we focus on the effects of metabolism on regulating the balance between quiescence and proliferation in some types of adult stem cells. Finally, we describe the roles of metabolic signaling mechanisms in aging stem cells.
Metabolism to maintain stem cell pluripotency
OxPhos in pluripotency
OxPhos refers to the mitochondrial oxygen-consuming series of reactions in the electron transport chain (ETC) and the ATP synthase complexes, which generate ATP using energy produced by the oxidation of NADH, which is in turn derived from the oxidation of nutrients through the mitochondrial Krebs cycle and other redox reactions. It is the main and most efficient source of energy for most mammalian cells, although some cell types rely on glycolysis-driven substrate-level phosphorylation as the alternative major source.
Pluripotency emerges in the epiblast during mammalian preimplantation development. The naïve state is associated with pluripotent stem cells (PSCs) in the preimplantation epiblast of embryos, and these pluripotent cells become primed during post-implantation development (Boroviak et al. 2015). In rodents, the naïve preimplantation epiblast can be captured in vitro as embryonic stem cells (ESCs) and sustained indefinitely as naïve PSCs using defined media containing LIF, GSK3β, and MEK inhibitors, also commonly known as the 2iL conditions (Ying et al. 2008). Naïve and primed PSCs display key differences in their derivation of germline-competent PSCs, epigenomic states, gene expression of naïve pluripotency markers and lineage-specific markers, signaling requirements to maintain self-renewal, and central carbon metabolism (Davidson et al. 2015). In particular, naïve PSCs use more OxPhos, whereas primed PSCs rely almost entirely on glycolysis (Zhou et al. 2012; Takashima et al. 2014; Sperber et al. 2015). Whether these changes resemble the in vivo situation in which preimplantation embryos preferentially use mitochondrial OxPhos but shift toward anaerobic glycolysis after implantation into the uterine wall (Barbehenn et al. 1978; Brinster and Troike 1979) remains unproven. In fact, a recent study suggests that the OxPhos flux in PSCs is more dependent on the medium composition and culture conditions than intrinsic changes in the pluripotency state (Zhang et al. 2016).
However, increasing evidence supports the idea that the shift in bioenergetic metabolism is intrinsically programmed by pluripotency factors and in turn regulates some epigenetic machinery that is involved in the programming of the naïve and primed pluripotency states (Takashima et al. 2014; Ware et al. 2014). For example, both LIF-induced Stat3, which can promote mitochondrial transcription (Carbognin et al. 2016), and Esrrb, which can promote transcription of mitochondrial OxPhos genes (Zhou et al. 2012), have been proposed as pluripotency factors that intrinsically promote OxPhos in naïve PSCs. OxPhos regenerates NAD+ to keep the Krebs cycle running and maintain sufficient cellular pools of α-ketoglutarate (αKG) for use as a cofactor by epigenetic modifier enzymes, including the Jumonji domain-containing (JmjC) histone demethylases and ten-eleven translocation (TET) methylcytosine dioxygenases. These Fe2+-dependent dioxygenases remove histone and DNA methylation to regulate the chromatin state and thus pluripotency.
For example, it was found that the 2i (GSK3β inhibitor + MEK inhibitor) culture conditions rewired intrinsic glucose and glutamine metabolism to control intracellular αKG (Carey et al. 2015), an intermediate in the mitochondrial Krebs cycle, which leads to the formation of succinate (Fig. 1). Because αKG is a cofactor while succinate is a competitive inhibitor for the αKG/Fe2+-dependent dioxygenases, increases in the αKG/succinate ratio were sufficient to promote αKG/Fe2+-dependent dioxygenase activities to erase multiple repressive chromatin modifications (e.g., demethylation of H3K9me3, H3K27me3, H4K20me3, and DNA) to promote naïve pluripotency (Carey et al. 2015). In vitro supplementation with αKG promoted naïve pluripotency, while succinate promoted differentiation of naïve PSCs. However, αKG is not specific to naïve pluripotency, as it can also promote primed PSC differentiation by regulating histone demethylation (Teslaa et al. 2016).
Figure 1.
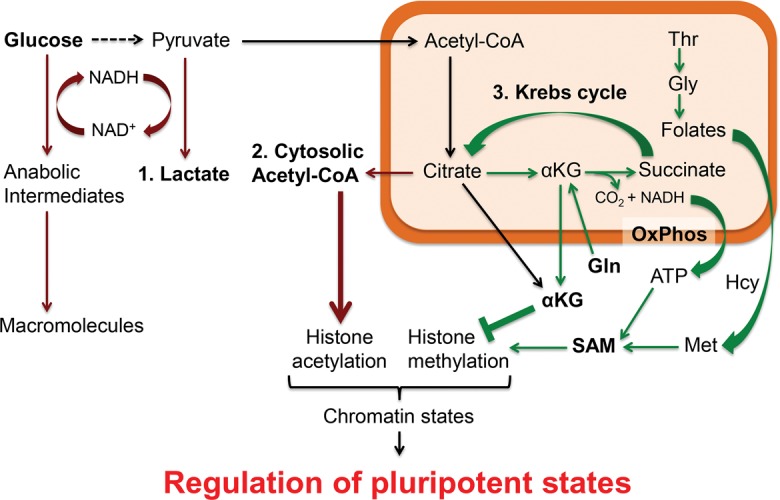
The metabolic programming of naïve and primed pluripotency states in mouse PSCs, for which the culture conditions are well established and the metabolic reactions are well characterized. Mitochondrial reactions that are more enriched in naïve (than primed) PSCs are marked in green, while cytosolic reactions that are more enriched in primed (than differentiated) PSCs are marked in red. Pyruvate essentially has three fates: (1) cytosolic reduction to lactate, (2) cytosolic conversion to acetyl-CoA via citrate, and (3) mitochondrial oxidation via the Krebs cycle. These metabolic pathways are not exclusive to PSCs but are highly prominent in PSCs to regulate histone acetylation and methylation in the chromatin, thereby influencing the epigenomic state and cell fate of PSCs.
Thus, it appears that the mitochondrial Krebs cycle intermediate αKG and the αKG/Fe2+-dependent dioxygenases are critical for altering the epigenetic states of both naïve pluripotency and differentiating PSCs. This could be one of the reasons why both naïve PSCs and differentiating PSCs activate mitochondrial OxPhos, whereas primed PSCs prefer glycolysis. However, more studies are required to fully clarify the roles of these different modes of metabolism for the different states of pluripotency. Moreover, the precise conditions for culturing human naïve PSCs remain controversial, and thus their metabolic needs are still not fully clarified.
Interestingly, another cofactor for the αKG/Fe2+-dependent dioxygenases, ascorbate (or vitamin C), has also been shown to improve reprogramming of somatic cells into induced PSCs (iPSCs) by promoting DNA 5-methyl-cytosine and H3K9me3 demethylation (Chen et al. 2013a,b). Unlike αKG, which can be synthesized and intrinsically regulated in all mammalian cells, ascorbate cannot be synthesized by human cells and must be supplemented extrinsically. Hence, the human αKG/Fe2+-dependent dioxygenases represent a node that integrates both the intrinsic metabolic needs and the extrinsic metabolic conditions to determine human stem cell fate.
Glycolysis in primed pluripotency and reprogramming
Glycolysis is the series of redox reactions in the cytosol that rapidly catabolizes each six-carbon glucose molecule to produce two three-carbon pyruvate molecules and two net ATP molecules as energy via substrate-level phosphorylation. In most cell types, the pyruvate can be shunted into two metabolic fates: as either lactate via lactate dehydrogenase (LDH) or acetyl-CoA via pyruvate dehydrogenase (PDH). The intermediates in glycolysis can also be shunted into macromolecule synthesis during rapid cell growth. Thus, while it is a less efficient source of energy, glycolysis can generate both anabolic growth intermediates and ATP very rapidly owing to the much higher speed of glycolysis reactions.
When transitioning from the naïve to the primed pluripotent state, mouse PSCs intrinsically reduce their rates of OxPhos and instead have high rates of glycolysis due to high expression of glucose transporters, which in turn leads to a high rate of glucose uptake and glycolysis (Leese 1995; Zhou et al. 2012). In addition, it has been shown that these high levels of glucose are also used in the pentose phosphate pathway in order to synthesize nucleotides in PSCs (Filosa et al. 2003; Varum et al. 2011, Manganelli et al. 2012). Therefore, it is widely thought that the high rate of glucose uptake and glycolysis is required to meet the needs of rapid proliferation of mouse PSCs, similar to the situation of cancer cells in the Warburg effect (Vander Heiden et al. 2009). The Warburg effect operates predominantly in highly proliferative cells, such as cancer cells, to accumulate glycolytic intermediates for rapid proliferation while minimizing reactive oxygen species (ROS)-induced damage. In particular, these glycolytic intermediates can be shunted into amino acid synthesis via 3-phosphoglycerate, into lipid synthesis via dihydroxyacetone phosphate and acetyl-CoA, and into nucleotide and NADPH synthesis via glucose-6-phosphate and the pentose phosphate pathway. Accumulation of these macromolecules is critical for the rapid growth of both PSCs and cancer cells.
The reprogramming of murine somatic cells into iPSCs offers another window into the intrinsic metabolic requirements of pluripotency. A switch from mitochondrial OxPhos to glycolysis is widely observed during iPSC reprogramming (Folmes et al. 2011). It is known that mitochondrial metabolism plays a fundamental role in reprogramming, and it has been shown recently that, during the course of iPSC reprogramming, an early fragmentation of mitochondria occurs. This reprogramming-induced mitochondrial fission, governed by the profission factor Drp1, was necessary for the full activation of pluripotency (Prieto et al. 2016). In studies of the role of oocyte factors in somatic cell nuclear transfer (SCNT)-mediated reprogramming, mitochondrial biogenesis was also found to be critical in regulating reprogramming and the attainment of bona fide pluripotency (Khaw et al. 2015). The oocyte-enriched factor and Oct4 target Tcl1 was found to reduce the mitochondrial localization of polynucleotide phosphorylase (PnPase). Suppression of mitochondrial PnPase led to decreased mitochondrial biogenesis, decreased OxPhos, and increased glycolysis during reprogramming. The decrease in OxPhos might be the reason behind the activation of glycolysis, as ATP is a potent allosteric inhibitor of many glycolysis enzymes (Johnson et al. 2003). Thus, the Tcl1–PnPase switch suppresses OxPhos and promotes glycolysis to remodel the metabolome and facilitate the reprogramming of somatic cells into iPSCs.
It is also important to point out that, in contrast to mouse PSCs, human naïve PSCs exhibit increased glycolysis compared with primed PSCs (Gu et al. 2016). This major difference could be due to species-specific differences, such as differences in X-chromosome dynamics and dependence on FGF signaling. The increased glycolysis is associated with high nuclear N-MYC and C-MYC in naïve PSCs. Naïve PSCs incorporate more glucose carbons into lactate, nucleotides, and serine. It appears that the presence of feeders or feeder-secreted factors affects the dependence on glucose for self-renewal of primed human PSCs. Overall, it is clear that glycolysis exerts a profound effect on the pluripotent states of human PSCs (Gu et al. 2016). Further work will be required to investigate how glycolysis regulates exit from the pluripotent states and lineage-specific differentiation.
Glycine (Gly)–methionine (Met) metabolism for self-renewal
PSCs also have a unique amino acid metabolism in order to maintain their undifferentiated, pluripotent state. Unlike other proliferative cell types, mouse PSCs are sensitive to extrinsic deprivation of threonine (Thr) (Wang et al. 2009; Shyh-Chang et al. 2013). Moreover, a remarkable up-regulation of the Thr-catabolizing enzyme Thr dehydrogenase (Tdh) is observed in mouse PSCs compared with differentiated cells. Tdh is responsible for the catabolism of Thr into Gly. Gly decarboxylase enzyme (Gldc), which is also highly up-regulated in PSCs, uses Gly to generate folate intermediates to fuel one-carbon metabolism (Zhang et al. 2012). The folate intermediates fuel nucleotide synthesis and the rapid proliferation rate of PSCs as well as neural crest progenitors (Wang et al. 2009; Tan et al. 2016). However, the specificity of Thr deprivation to PSCs and not just any proliferative cell type also suggests that the metabolites resulting from Thr–Gly degradation might be used specifically for self-renewal of the pluripotent state.
Indeed, the folate intermediates also fuel the remethylation of homocysteine to form Met and S-adenosyl-Met (SAM) in the Met salvage pathway. SAM, in turn, is required for all protein methylation reactions. Tdh and Gldc are needed to increase the synthesis of SAM via the folate intermediates, leading to a high ratio of SAM to S-adenosyl-homocysteine (SAH) and resultant histone H3 methylation states for mouse PSC self-renewal (Shyh-Chang et al. 2013). Collectively, the Thr–Gly–Met metabolism pathway conveys information on extrinsic amino acid levels to regulate the self-renewal of PSCs by feeding the folate one-carbon pool, SAM, and nucleotide synthesis to maintain the intrinsic epigenomic state of pluripotency over the long term.
A follow-up study showed that human PSCs use high levels of Met instead, in contrast to mouse PSCs, which are highly dependent on Thr (Shiraki et al. 2014). This is because Tdh had evolved into a pseudogene in humans, unlike most other mammals (Wang et al. 2009). The study showed that Met deprivation resulted in a rapid decrease in SAM, loss of H3K4me3, and reduced NANOG expression and triggered human PSCs to differentiate into any of the three embryonic germ layers. Even though human PSCs could endure short-term Met deprivation by replenishing the Met and SAM pool via the Met salvage pathway, prolonged Met deprivation led to cell cycle arrest and apoptosis in human PSCs (Shiraki et al. 2014). Further studies suggested that SAM levels are also critical for naïve human PSCs, where the epigenetic landscape was remodeled through changes in H3K27me3-repressive marks (Sperber et al. 2015). Combined, these studies have revealed the importance of Gly–Met metabolism in integrating extrinsic amino acid information with the intrinsic pluripotency state to determine the cell fate of PSCs.
Acetyl-CoA to prevent differentiation
Besides being a substrate that feeds into the Krebs cycle, acetyl-CoA also plays a role in protein acetylation. Histone H3 acetylation results in an open euchromatin state that, like H3K4me3, maintains the pluripotent epigenetic state and self-renewal of PSCs (Azuara et al. 2006; Gaspar-Maia et al. 2011). In line with this, it has been shown that chemical inhibition of histone deacetylases promotes reprogramming of somatic cells into iPSCs (Huangfu et al. 2008; Mali et al. 2010).
In a recent study performed by Moussaieff et al. (2015), metabolomic and transcriptional analyses revealed that PSCs produce cytosolic acetyl-CoA through glycolysis and the pyruvate-derived citrate flux via ATP citrate lyase (ACLY) and that this metabolic route is shut down during the course of PSC differentiation. In human and mouse PSCs, glycolysis-derived acetyl-CoA was sufficient to block histone deacetylation and stem cell differentiation. In addition, acetate, which is an alternative precursor of cytosolic acetyl-CoA, also delayed PSC differentiation by preventing histone deacetylation in a dose-dependent manner. Pharmacological perturbation experiments showed that cytosolic acetyl-CoA was both necessary and sufficient to prevent differentiation of PSCs. These data imply that a glycolytic switch controlling histone deacetylation can either maintain the pluripotency of stem cells or rapidly release them from pluripotency to start differentiation (Moussaieff et al. 2015).
These findings implicate a careful distinction between the different modes of glycolysis and the famed Warburg effect in cancer. The Warburg effect is a more specific form of glycolysis, defined as unusually rapid glycolysis followed by lactate production even under aerobic conditions. The pyruvate–lactate step in the Warburg effect is absolutely necessary to rapidly recycle the rate-limiting NAD+ coenzyme and keep the glycolysis reactions running rapidly. In stark contrast, Moussaieff et al. (2015) showed instead that it is the cytosolic pyruvate–acetyl-CoA reactions, not the pyruvate–lactate step, that is important for pluripotency. This detail represents a critical distinction because it suggests that the Warburg effect and the pyruvate–lactate step, although operational in primed PSCs (see the above discussion on glycolysis in PSCs), is not rate-limiting for pluripotency. Instead of the classic dichotomy of the Warburg effect (pyruvate–lactate) versus mitochondrial OxPhos (pyruvate–Krebs cycle), their data suggest that glycolysis makes its most significant contribution to pluripotency regulation by shunting some pyruvate through a third mode: via citrate synthase, a citrate shuttle, and, finally, cytosolic ACLY to produce cytosolic acetyl-CoA (Shyh-Chang and Daley 2015).
Metabolism to maintain stem cell quiescence or proliferation
Hypoxic niche and glycolysis
Unlike the hyperproliferative PSCs, most adult stem cells remain quiescent in their niches unless activated by stress or injury, thus resulting in rather different intrinsic metabolic needs (Fig. 2). Quiescent adult stem cells typically reside deep within hypoxic niche environments in tissues. Some examples of adult stem cells include neural stem cells (NSCs) in the brain's subventricular zone, satellite cells lying beneath skeletal muscles’ basal lamina, long-term hematopoietic stem cells (LT-HSCs), and mesenchymal stromal cells (MSCs) in the bone marrow. Adult stem cells typically maintain a quiescent state to preserve long-term self-renewal capacity for tissue maintenance (Rossi et al. 2008; Suda et al. 2011).
Figure 2.
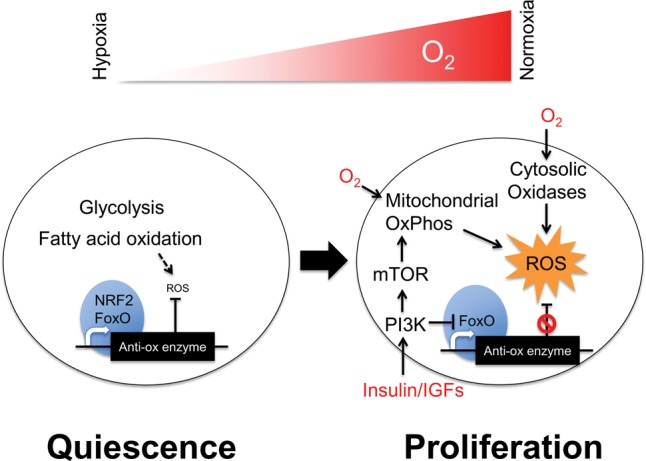
The metabolic programming of quiescent and proliferating adult stem cells relative to oxygen availability. A common theme is that quiescent adult stem cells in hypoxic niches tend to prefer glycolysis and fatty acid oxidation with high levels of nuclear NRF2- or FoxO-driven (blue) antioxidant enzyme expression to suppress ROS signaling. In contrast, proliferative adult stem cells tend to use more oxygen in a normoxic environment under the influence of growth factor kinase signaling, lower their expression of antioxidant enzymes, and activate ROS signaling (yellow). The resultant burst in proliferation also leads to irreversible commitment as the adult stem cells proliferate and differentiate into tissue-specific progeny. This model is based largely on studies in HSCs, but emerging evidence suggests that it could apply generally to many other adult stem cells.
Such a quiescent state is typically correlated with an extrinsically hypoxic niche and an intrinsically glycolytic mode of metabolism. For example, it is known that quiescent LT-HSCs adapt to the hypoxic environment of the bone marrow niche by using glycolysis as an energy source (Suda et al. 2011). Unequivocal evidence for the hypoxic environment of the LT-HSC niche was obtained directly using two-photon phosphorescence lifetime microscopy (Spencer et al. 2014). One key benefit of using anaerobic glycolysis in an hypoxic environment is the consequent minimization of mitochondrial OxPhos and ROS production. Adult LT-HSCs, like many adult stem cells, are highly sensitive to ROS. For instance, in the presence of excessive ROS, LT-HSCs are induced to activate proliferation followed by differentiation or apoptosis (Tothova et al. 2007). In the subventricular zone of the brain, adult NSCs exhibit a similar response to excessive ROS (Renault et al. 2009). The glycolytic mode of metabolism in adult LT-HSCs appears to be partly preprogrammed as well, under the influence of the transcription factor MEIS1 (Simsek et al. 2010), which up-regulates many glycolytic enzymes. The correlation between the LT-HSC state and a glycolytic program is so strong that adult LT-HSCs can be isolated from bone marrow aspirates exposed to normoxia by selecting for cells with low endogenous NADH and low mitochondrial membrane potential (Simsek et al. 2010). The resultant cells possess superior hematopoietic reconstitution capacity in vivo.
However, a low mitochondrial membrane potential at steady state could suggest either low OxPhos flux (low substrate input) or high OxPhos flux (high product output). Previous studies suggest that LT-HSCs’ low mitochondrial membrane potential is because of low OxPhos activity, since LT-HSCs express higher levels of PDK1 (PDH kinase 1) and PDK3, which inhibit PDH to shunt pyruvate into lactate production instead (Klimmeck et al. 2012). Furthermore, PDK2 and PDK4, both of which are HIF1α targets, are also required for LT-HSC self-renewal, demonstrating that the switch in pyruvate flux to lactate versus Krebs cycle is critical in toggling LT-HSC fate (Takubo et al. 2013). When the mitochondrial carrier homolog 2 (MTCH2) is conditionally deleted in the hematopoietic system, mitochondrial fusion, mitochondrial volume, and mitochondrial OxPhos are all increased, leading to HSC commitment and proliferation (Maryanovich et al. 2015). Consistent with these findings, it was found that conditional deletion of mitofusin 2 (MFN2) in the hematopoietic system, which led to mitochondrial fragmentation and presumably compromised OxPhos capacity, had no effect on primary adult LT-HSC survival and self-renewal (Luchsinger et al. 2016). However, surprisingly, MFN2 deficiency did lead to the loss of lymphoid-biased LT-HSCs in secondary transplantation assays with single LT-HSCs after lethal irradiation. This is likely due to dysfunctional Ca2+ buffering and NFAT signaling, suggesting that although LT-HSCs manifest low mitochondrial OxPhos, they still rely on mitochondria for other metabolic functions.
Interestingly, a recent follow-up study revealed that an enforcement of low mitochondrial membrane potential in LT-HSCs—by introducing an uncoupler FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone) that dissipates the mitochondrial membrane potential—could promote LT-HSCs to undergo self-renewing symmetric divisions instead of undergoing differentiation during proliferation (Vannini et al. 2016). Because FCCP typically induces maximal ETC flux in oxygen consumption studies, by dissipating the resistance from the proton gradient or membrane potential, these results paradoxically suggested that maximal ETC flux could promote LT-HSC self-renewal. Earlier results on a requirement for low OxPhos in LT-HSCs appear to contradict this finding. The investigators suggest that these results could be because FCCP induces mitophagy and reduces total OxPhos capacity over the long term even if the ETC flux is maximized for the given available OxPhos machinery over the short term. While the effects of FCCP and maximal ETC flux on ROS levels in LT-HSCs remain unclear, these interesting findings warrant further investigation.
MSCs also reside within the hypoxic environment of the bone marrow niche. MSCs intrinsically express higher levels of glycolytic enzymes and lower levels of OxPhos proteins relative to the more differentiated osteoblasts. This suggests that MSCs preferentially use glycolysis compared with osteoblasts (Chen et al. 2008a). Under normoxic conditions, MSCs can be forced into a high oxygen consumption state by using OxPhos, and, in fact, MSC proliferation is significantly increased in normoxia (Pattappa et al. 2013). However, the switch to OxPhos induces a significant increase in MSC senescence. This finding suggests that hypoxic conditions and glycolysis are necessary to prevent ROS-induced proliferation and senescence in order to ensure the long-term self-renewal capacity of bone marrow MSCs (Pattappa et al. 2013). Thus, in at least three types of adult stem cells (namely, LT-HSCs, MSCs, and NSCs), the suppression of ROS by using glycolysis within a hypoxic niche prevents entry into a more proliferative and differentiated state in normoxia. The combination and balance of intrinsic metabolic requirements and extrinsic metabolic conditions needed for quiescence as well as the tilt of balance needed to initiate proliferation may be a common mechanism for regulating adult stem cell homeostasis (Fig. 2).
ROS during proliferation
ROS is generally regarded as a stress signal even though it is also necessary for normal cellular processes in some cases. ROS can arise from many different sources, including inefficient electron transfer by mitochondrial OxPhos complexes, cytosolic oxidases, and oxygenases. ROS can also rise aberrantly due to extrinsic stress from exposure to radiation, pollutants, or drugs or intrinsic deficiencies in stress response pathways such as the FoxO or NRF2 pathways that promote the expression of antioxidant enzymes such as glutathione peroxidases or superoxide dismutases (Holmström and Finkel 2014).
As LT-HSCs proliferate, both the mitochondrial OxPhos activity and ROS levels also increase, leading to HSC differentiation. Hematopoietic cells in the Drosophila model, similar to common myeloid progenitors in mammals, also require ROS to mature into terminally differentiated blood cells (Owusu-Ansah and Banerjee 2009). In mice, hyperactivation of mitochondrial OxPhos in HSCs led to increased HSC proliferation and either myeloid or lymphoid leukemias with complete penetrance (Ueda et al. 2015). However, the targets of ROS that prime HSCs for proliferation and differentiation still remain unclear. It is plausible that the eicosanoid metabolism pathway, which responds to and generates ROS during lipid oxidation in PSCs (Yanes et al. 2010), may be involved. For example, prostaglandin E2, an eicosanoid pathway product, has been demonstrated to promote HSC proliferation in vivo by promoting Wnt signaling (Goessling et al. 2009). This suggests that eicosanoid metabolism might be critical in regulating HSC proliferation and differentiation. Another report suggested recently that p38 MAPK might be another target of ROS that activates stem cell proliferation (Karigane et al. 2016). They showed that the p38 MAPK is immediately activated in HSCs by hematological stresses, including ROS, leading to increased HSC proliferation. Conditional deletion of p38α inhibited the recovery from hematological stress and delayed the activation of HSPC proliferation. ROS-induced p38α activated the expression of IMPDH2 (inosine-5′-monophosphate dehydrogenase 2) in HSCs, which increased purine synthesis and increased cell proliferation (Karigane et al. 2016).
In NSCs, the antioxidant program driven by FoxO3 is rapidly shut off upon NSC differentiation despite the increase in mitochondrial OxPhos activity (Renault et al. 2009). This suggests that ROS is required for NSC differentiation. In fact, a deficiency in FoxO3 causes depletion of adult brain NSCs, an increase in neurogenesis in the olfactory bulb, and a significant expansion of oligodendrocytes in the corpus callosum during brain development, suggesting that ROS predisposes neural proliferation and differentiation (Renault et al. 2009; Webb et al. 2013).
In the intestines of the Drosophila model, enterocytes produce extraordinarily high levels of ROS to control the numbers of resident gut bacteria. Intestinal stem cells (ISCs) proliferate in response to these bursts of ROS released from surrounding enterocytes, as dictated by a preprogrammed intestinal regeneration response. However, as time passes during the course of aging, the cumulative oxidative stress can lead to ISC hyperproliferation, exhaustion, and, consequently, aging-induced degeneration of Drosophila intestines. This aging-induced hyperproliferation of ISCs can be blocked via activation of the NRF2 antioxidant pathway or administration of antioxidant molecules (Hochmuth et al. 2011). Deficiency in the NRF2 regulator KEAP1 also causes hyperproliferation in the mouse intestines (Wakabayashi et al. 2003), suggesting that the same ROS-based mechanism controls ISC proliferation in multiple animal models.
Optimal fatty acid oxidation (FAO)
FAO (or β-oxidation) is the series of redox reactions that catabolize fatty acid molecules in the mitochondria to generate acetyl-CoA, which enters the Krebs cycle, and NADH and FADH2, which are oxidized in the ETC to fuel OxPhos.
Interestingly, FAO is important to promote normal LT-HSC self-renewal (Ito et al. 2012). It was found that inhibition of FAO or depletion of the upstream FAO master regulator PPARδ resulted in symmetric differentiating divisions of HSCs into committed progenitor cells, whereas PPARδ activation increased asymmetric division and HSC self-renewal. A recent study found that PPARδ-FAO activation led to an increase in autophagy of mitochondria to promote LT-HSC self-renewal (Ito et al. 2016).
Similarly, NSCs also appear to use FAO for self-renewal. NSCs within the adult brain's subventricular zone express FAO enzymes and show increased oxygen consumption upon treatment with a polyunsaturated fatty acid. Conversely, NSCs demonstrate decreased oxygen consumption upon treatment with etomoxir, an inhibitor of FAO, leading to decreased NSC self-renewal (Xie et al. 2016). Lineage tracing experiments further demonstrated that FAO flux was required to prevent symmetric differentiating divisions at the expense of NSC self-renewal (Stoll et al. 2015).
In quiescent skeletal muscle stem cells (MuSCs), active SIRT1 deacetylates and activates PGC-1α to promote FAO in the mitochondria. PGC-1α transactivates OxPhos genes and represses glycolysis genes to maintain high intracellular NAD+ and keep SIRT1 active (Wu et al. 1999; Rodgers et al. 2005; Gerhart-Hines et al. 2007). During their exit from quiescence, MuSCs deactivate FAO in favor of glucose catabolism (Ryall et al. 2015). This metabolic switch decreases NAD+ and deactivates SIRT1 and its histone H4K16 deacetylation activity to activate myogenic transcription programs and muscle differentiation. Thus, FAO is necessary to maintain MuSCs in a quiescent state. However, excessive FAO in MuSCs and myocytes can also lead to excessive oxidative stress, which inhibits muscle growth and causes muscle atrophy (Fukawa et al. 2016). It appears that an optimal level of FAO, not too much or too little, is necessary for preserving self-renewal of quiescent MuSCs. Could this Goldilocks principle in FAO metabolism apply to NSCs and LT-HSCs as well? Further studies would be needed to resolve this question.
Metabolism during stem cell aging
After repeated rounds of proliferation and aging, adult stem cells such as epidermal stem cells, HSCs, ISCs, MuSCs, and NSCs gradually decline in their numbers as well as their long-term self-renewal and multipotent differentiation capacities. This progressive decline has been associated with many degenerative conditions of aging, including hair loss, immune dysfunction, colitis, sarcopenia, and neurodegenerative diseases. Numerous mechanisms have been implicated to explain this decline in numbers and function, some of which we review below.
Oxidative stress response via FoxO signaling
The insulin–PI3K–AKT signaling pathway phosphorylates and suppresses the FoxO family of transcription factors. In contrast, excessive ROS activates the FoxO family to drive the oxidative stress response. Deficiency in the FoxO transcription factors compromises the oxidative stress response and gradually depletes mouse LT-HSCs during aging (Miyamoto et al. 2007; Tothova et al. 2007). FoxO-deficient mice exhibit a pronounced increase in ROS, which ultimately leads to a decline in the HSC population and a skew toward myeloid differentiation that characterizes aging HSCs. Further studies elucidated the precise role of the FoxO-regulated oxidative stress response by showing that FoxO3 deficiency hyperactivates p53 and PI3K signaling, resulting in a vicious cycle that leads to HSC aging. This vicious cycle can be disrupted by administering antioxidant molecules (Yalcin et al. 2008, 2010). In addition to regulating ROS, FoxOs also induce mitophagy and autophagy to protect HSCs during dietary restriction (Warr et al. 2013). It has also been shown that mitochondrial metabolism can be directly regulated by FoxO3 (Rimmelé et al. 2015).
Germline stem cells in Caenorhabditis elegans and Drosophila adjust their proliferation according to nutritional conditions (Drummond-Barbosa and Spradling 2001) and especially in response to the aging regulatory insulin–PI3K–FoxO signaling pathway (Kimura et al. 1997; LaFever and Drummond-Barbosa 2005; Hsu et al. 2008; Ueishi et al. 2009). Further studies have shown that aging of the Drosophila germline stem cell niche maintained by the ovarian follicle stem cells is regulated by insulin–PI3K signaling and mitochondrial ROS production (Wang et al. 2012).
AMP/ATP and AMPK signaling
Low ATP/AMP levels during calorie restriction and exercise activate the AMPK pathway. This pathway also promotes organismal longevity by regulating mitochondrial FAO and suppressing mTOR signaling. Depletion of LKB1, an AMPK regulatory kinase, causes severe mitochondrial dysfunction, leading to LT-HSC proliferation and exhaustion and, ultimately, defects in hematopoiesis during aging (Gan et al. 2010; Gurumurthy et al. 2010; Nakada et al. 2010). During brain development and aging, AMPK signaling also regulates NSCs and their mitochondria. Depletion of AMPK leads to loss of the hippocampal dentate gyrus and severe brain atrophy (Dasgupta and Milbrandt 2009). AMPK signaling also modulates the quiescence of germline stem cells to influence nematode aging (Narbonne and Roy 2006).
Mitochondria, PGC-1α, and mTOR signaling
A variety of stem cell models have unequivocally demonstrated that mitochondrial OxPhos, regulated by PGC-1α and mTOR signaling, can regulate stem cell aging and thus overall longevity.
During aging, Drosophila ISCs undergo hyperproliferation in response to intestinal ROS, leading to stem cell exhaustion and intestinal degeneration over time (Hochmuth et al. 2011). Activation of Spargel, the Drosophila PGC-1α homolog, can significantly attenuate ISC aging by enhancing the efficiency of OxPhos in intestinal cells to reduce ROS, reducing hyperproliferation of ISCs and thus reducing misdifferentiated cells and improving intestinal integrity. This results in extended Drosophila longevity (Rera et al. 2011). Similarly, gain of function in a comaster regulatory transcription factor for mitochondrial OxPhos and antioxidant enzymes, NRF2, can enhance ISC self-renewal to extend Drosophila longevity as well (Hochmuth et al. 2011).
During NSC differentiation into neurons, PGC-1α also regulates mitochondrial biogenesis (O'Brien et al. 2015), a cellular process that often becomes dysfunctional during neurodegenerative diseases. For example, deficiency in Parkin is widely thought to induce Parkinson's disease by reducing PGC-1α to reduce mitochondrial biogenesis and reducing mitophagy to reduce mitochondrial turnover, thus causing the accumulation of faulty mitochondria in dopaminergic neurons during development and aging. These faulty mitochondria then gradually become unmasked during aging, leading to neuronal apoptosis later in life (Shaltouki et al. 2015; Stevens et al. 2015). In Drosophila models of neurogenesis and neurodegeneration, deficiencies due to mutations in the Parkin pathway have been shown to dysregulate NSC self-renewal to cause Parkinsonism (Goh et al. 2013).
Downstream from growth factor and amino acid-sensing pathways, the mTOR signaling pathway also plays a major role in regulating mitophagy (Egan et al. 2011) and mitochondrial activity to control stem cell aging. By phosphorylating mitochondrial proteins and activating PGC-1α, mTOR has been shown to boost mitochondrial OxPhos (Schieke et al. 2006; Cunningham et al. 2007; Ramanathan and Schreiber 2009). In LT-HSCs, activation of mTOR signaling by conditionally deleting TSC1 increases mitochondrial oxidative stress, thus driving the LT-HSC population out of quiescence and ultimately into exhaustion to impair hematopoiesis during aging (Chen et al. 2008b).
Conversely, rapamycin-mediated inhibition of mTOR signaling can delay hematopoietic aging by preserving the long-term self-renewal and hematopoietic capacity of LT-HSCs (Chen et al. 2009). This molecular mechanism is well-conserved in Drosophila too, as insulin–PI3K–TOR signaling has been shown to regulate the long-term self-renewal capacity of hematopoietic progenitors (Shim et al. 2012). Excessive mTOR signaling can also cause exhaustion of epidermal stem cells and progressive alopecia in adult mice, whereas rapamycin can delay this aging phenotype (Castilho et al. 2009). Most importantly, rapamycin treatment late in adulthood has been shown to extend longevity in mice (Harrison et al. 2009).
Conclusion
Many metabolic pathways are emerging as important regulatory mechanisms for programming stem cell fates. While many of the metabolic changes in stem cells are undoubtedly a response to changes in their niche or environment, it does not follow that they have no effect on the intrinsic programming of stem cell fates. Like growth factor signaling pathways, metabolic pathways can also convey changes in the extrinsic environment to reprogram stem cell fates via changes in epigenetics, proliferation, and differentiation (Fig. 3). Moreover, it is becoming clear that many metabolic pathways are also intrinsically programmed by stem cell factors to facilitate the metabolic needs of that particular cellular state. Thus, in retrospect, the recent flurry of studies on stem cell metabolism supports the notion that the metabolic programming of stem cells represents a fine balance between the intrinsic needs of a cellular state and the constraints imposed by extrinsic nutrient and oxygen levels. A more complete understanding of these needs and constraints and the effects of manipulating them will afford us greater mastery and control over the fates of stem cells for both tissue engineering in vitro and regenerative medicine in vivo.
Figure 3.
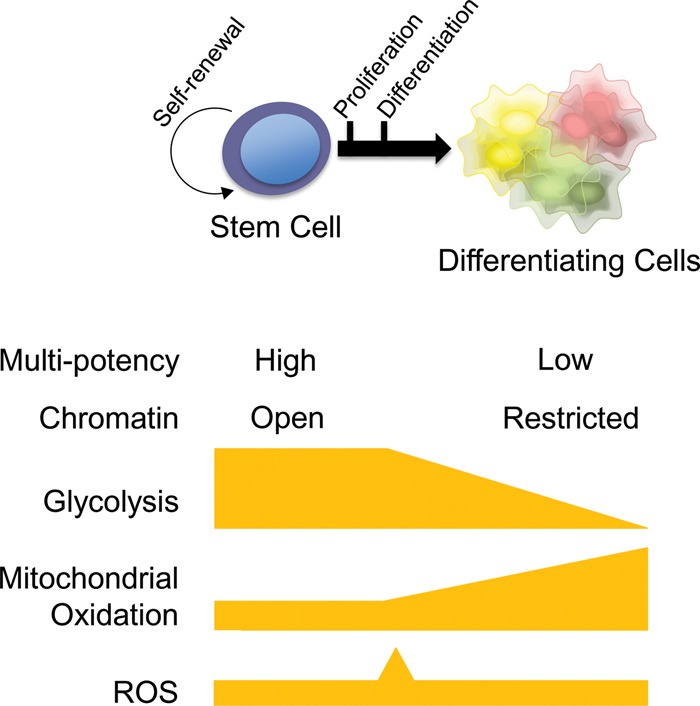
Unified summary of common metabolic features often observed during both PSC and adult stem cell differentiation. During self-renewal, both PSCs and adult stem cells maintain their multipotent capacity and a relatively open chromatin state. In conjunction with their epigenomic status, glycolysis and mitochondrial (glutamine or fatty acid) oxidation tend to be used, but total OxPhos flux and ROS levels typically stay low in a hypoxic environment. Proliferation of committed progenitors typically coincides with high glycolysis and a burst of ROS in a normoxic environment (see Fig. 2), while mitochondrial oxidation begins to rise. Further differentiation often leads to a decline in glycolysis and a further increase in mitochondrial oxidation.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.293167.116.
References
- Azuara V, Perry P, Sauer S, Spivakov M, Jørgensen HF, John RM, Gouti M, Casanova M, Warnes G, Merkenschlager M, et al. 2006. Chromatin signatures of pluripotent cell lines. Nat Cell Biol 8: 532–538. [DOI] [PubMed] [Google Scholar]
- Barbehenn EK, Wales RG, Lowry OH. 1978. Measurement of metabolites in single preimplantation embryos; a new means to study metabolic control in early embryos. J Embryol Exp Morphol 43: 29–46. [PubMed] [Google Scholar]
- Boroviak T, Loos R, Lombard P, Okahara J, Behr R, Sasaki E, Nichols J, Smith A, Bertone P. 2015. Lineage-specific profiling delineates the emergence and progression of naive pluripotency in mammalian embryogenesis. Dev Cell 35: 366–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Troike DE. 1979. Requirements for blastocyst development in vitro. J Anim Sci 49(Suppl 2): 26–34. [DOI] [PubMed] [Google Scholar]
- Carbognin E, Betto RM, Soriano ME, Smith AG, Martello G. 2016. Stat3 promotes mitochondrial transcription and oxidative respiration during maintenance and induction of naive pluripotency. EMBO J 35: 618–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LWS, Cross JR, Allis CD, Thompson CB. 2015. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518: 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS. 2009. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell 5: 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-T, Shih Y-RV, Kuo TK, Lee OK, Wei Y-H. 2008a. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 26: 960–968. [DOI] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, Zheng P. 2008b. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med 205: 2397–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu Y, Zheng P. 2009. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal 2: ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Guo L, Zhang L, Wu H, Yang J, Liu H, Wang X, Hu X, Gu T, Zhou Z, et al. 2013a. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat Genet 12: 1504–1509. [DOI] [PubMed] [Google Scholar]
- Chen J, Liu H, Liu J, Qi J, Wei B, Yang J, Liang H, Chen Y, Chen J, Wu Y, et al. 2013b. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat Genet 45: 34–42. [DOI] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. 2007. mTOR controls mitochondrial oxidative function through a YY1–PGC-1α riptional complex. Nature 450: 736–740. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Milbrandt J. 2009. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev Cell 16: 256–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson K, Mason EA, Pera MF. 2015. The pluripotent state in mouse and human. Development 142: 3090–3099. [DOI] [PubMed] [Google Scholar]
- Drummond-Barbosa D, Spradling AC. 2001. Stem cells and their progeny respond to nutritional changes during Drosophila oogenesis. Dev Biol 231: 265–278. [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. 2011. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filosa S, Fico A, Paglialunga F, Balestrieri M, Crooke A, Verde P, Abrescia P, Bautista JM, Martini G. 2003. Failure to increase glucose consumption through the pentose-phosphate pathway results in the death of glucose-6-phosphate dehydrogenase gene-deleted mouse embryonic stem cells subjected to oxidative stress. Biochem J 370: 935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CDL, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A. 2011. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 14: 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukawa T, Yan-Jiang BC, Min-Wen JC, Jun-Hao ET, Huang D, Qian CN, Ong P, Li Z, Chen S, Mak SY, et al. 2016. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat Med 22: 666–671. [DOI] [PubMed] [Google Scholar]
- Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, Fletcher-Sananikone E, Colla S, Wang YA, Chin L, et al. 2010. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 468: 701–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar-Maia A, Alajem A, Meshorer E, Ramalho-Santos M. 2011. Open chromatin in pluripotency and reprogramming. Nat Rev Mol Cell Biol 12: 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. 2007. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J 26: 1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, et al. 2009. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 136: 1136–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh LH, Zhou X, Lee MC, Lin S, Wang H, Luo Y, Yang X. 2013. Clueless regulates aPKC activity and promotes self-renewal cell fate in Drosophila lgl mutant larval brains. Dev Biol 381: 353–364. [DOI] [PubMed] [Google Scholar]
- Gu W, Gaeta X, Sahakyan A, Chan AB, Hong CS, Kim R, Braas D, Plath K, Lowry WE, Christofk HR. 2016. Glycolytic metabolism plays a functional role in regulating human pluripotent stem cell state. Cell Stem Cell 19: 476–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurumurthy S, Xie SZ, Alagesan B, Kim J, Yusuf RZ, Saez B, Tzatsos A, Ozsolak F, Milos P, Ferrari F, et al. 2010. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature 468: 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. 2009. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460: 392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochmuth CE, Biteau B, Bohmann D, Jasper H. 2011. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell 8: 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmström KM, Finkel T. 2014. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15: 411–421. [DOI] [PubMed] [Google Scholar]
- Hsu H-J, LaFever L, Drummond-Barbosa D. 2008. Diet controls normal and tumorous germline stem cells via insulin-dependent and -independent mechanisms in Drosophila. Dev Biol 313: 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, Melton DA. 2008. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol 26: 795–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT, Evans RM, Suda T, Lee C-H, et al. 2012. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med 18: 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Turcotte R, Cui J, Zimmerman SE, Pinho S, Mizoguchi T, Arai F, Runnels JM, Alt C, Teruya-Feldstein J, et al. 2016. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 354: 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MT, Mahmood S, Patel MS. 2003. Intermediary metabolism and energetics during murine early embryogenesis. J Biol Chem 278: 31457–31460. [DOI] [PubMed] [Google Scholar]
- Karigane D, Kobayashi H, Morikawa T, Ootomo Y, Sakai M, Nagamatsu G, Kubota Y, Goda N, Matsumoto M, Nishimura EK, et al. 2016. p38α activates purine metabolism to initiate hematopoietic stem/progenitor cell cycling in response to stress. Cell Stem Cell 19: 192–204. [DOI] [PubMed] [Google Scholar]
- Khaw SL, Min-Wen C, Koh CG, Lim B, Shyh-Chang N. 2015. Oocyte factors suppress mitochondrial polynucleotide phosphorylase to remodel the metabolome and enhance reprogramming. Cell Rep 12: 1080–1088. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. 1997. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277: 942–946. [DOI] [PubMed] [Google Scholar]
- Klimmeck D, Hansson J, Raffel S, Vakhrushev SY, Trumpp A, Krijgsveld J. 2012. Proteomic cornerstones of hematopoietic stem cell differentiation: distinct signatures of multipotent progenitors and myeloid committed cells. Mol Cell Proteomics 11: 286–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFever L, Drummond-Barbosa D. 2005. Direct control of germline stem cell division and cyst growth by neural insulin in Drosophila. Science 309: 1071–1073. [DOI] [PubMed] [Google Scholar]
- Leese HJ. 1995. Metabolic control during preimplantation mammalian development. Hum Reprod Update 1: 63–72. [DOI] [PubMed] [Google Scholar]
- Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M, Snoeck HW. 2016. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 529: 528–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Chou BK, Yen J, Ye Z, Zou J, Dowey S, Brodsky RA, Ohm JE, Yu W, et al. 2010. Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells 28: 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganelli G, Fico A, Masullo U, Pizzolongo F, Cimmino A, Filosa S. 2012. Modulation of the pentose phosphate pathway induces endodermal differentiation in embryonic stem cells. PLoS One 7: e29321. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Maryanovich M, Zaltsman Y, Ruggiero A, Goldman A, Shachnai L, Zaidman SL, Porat Z, Golan K, Lapidot T, Gross A. 2015. An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat Commun 6: 7901. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, Matsuoka S, Miyamoto T, Ito K, Ohmura M, et al. 2007. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 1: 101–112. [DOI] [PubMed] [Google Scholar]
- Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, et al. 2015. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab 21: 392–402. [DOI] [PubMed] [Google Scholar]
- Nakada D, Saunders TL, Morrison SJ. 2010. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature 468: 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narbonne P, Roy R. 2006. Inhibition of germline proliferation during C. elegans dauer development requires PTEN, LKB1 and AMPK signalling. Development 133: 611–619. [DOI] [PubMed] [Google Scholar]
- O'Brien LC, Keeney PM, Bennett JP Jr. 2015. Differentiation of human neural stem cells into motor neurons stimulates mitochondrial biogenesis and decreases glycolytic flux. Stem Cells Dev 24: 1984–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E, Banerjee U. 2009. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 461: 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattappa G, Thorpe SD, Jegard NC, Heywood HK, de Bruijn JD, Lee DA. 2013. Continuous and uninterrupted oxygen tension influences the colony formation and oxidative metabolism of human mesenchymal stem cells. Tissue Eng Part C Methods 19: 68–79. [DOI] [PubMed] [Google Scholar]
- Prieto J, León XP, Sendra R, Bort R, Ferrer-Lorente R, Raya A, López-García C, Torres J. 2016. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat Commun 7: 11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan A, Schreiber SL. 2009. Direct control of mitochondrial function by mTOR. Proc Natl Acad Sci 106: 22229–22232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault VM, Rafalski VA, Morgan AA, Salih DA, Brett JO, Webb AE, Villeda SA, Thekkat PU, Guillerey C, Denko NC, et al. 2009. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 5: 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rera M, Bahadorani S, Cho J, Koehler CL, Ulgherait M, Hur JH, Ansari WS, Lo T Jr, Jones DL, Walker DW. 2011. Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell Metab 14: 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmelé P, Liang R, Bigarella CL, Kocabas F, Xie J, Serasinghe MN, Chipuk J, Sadek H, Zhang CC, Ghaffari S. 2015. Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO3. EMBO Rep 16: 1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. 2005. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434: 113–118. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Jamieson CH, Weissman IL. 2008. Stems cells and the pathways to aging and cancer. Cell 132: 681–696. [DOI] [PubMed] [Google Scholar]
- Ryall JG, Dell'Orso S, Derfoul A, Juan A, Zare H, Feng X, Clermont D, Koulnis M, Gutierrez-Cruz G, Fulco M, et al. 2015. The NAD+-dependent SIRT deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 16: 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieke SM, Phillips D, McCoy JP Jr, Aponte AM, Shen RF, Balaban RS, Finkel T. 2006. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem 281: 27643–27652. [DOI] [PubMed] [Google Scholar]
- Shaltouki A, Sivapatham R, Pei Y, Gerencser AA, Momčilović O, Rao MS, Zeng X. 2015. Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem Cell Rep 4: 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J, Mukherjee T, Banerjee U. 2012. Direct sensing of systemic and nutritional signals by haematopoietic progenitors in Drosophila. Nat Cell Biol 14: 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, Kume S. 2014. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab 19: 780–794. [DOI] [PubMed] [Google Scholar]
- Shyh-Chang N, Daley GQ. 2015. Metabolic switches linked to pluripotency and embryonic stem cell differentiation. Cell Metab 21: 349–350. [DOI] [PubMed] [Google Scholar]
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, et al. 2013. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339: 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC, Sadek HA. 2010. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 7: 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, Zaher W, Mortensen LJ, Alt C, Turcotte R, et al. 2014. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 508: 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperber H, Mathieu J, Wang Y, Ferreccio A, Hesson J, Xu Z, Fischer KA, Devi A, Detraux D, Gu H, et al. 2015. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat Cell Biol 17: 1523–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A, Jiang H, Kang SU, Andrabi SA, Dawson VL, et al. 2015. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci 112: 11696–11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll EA, Makin R, Sweet IR, Trevelyan AJ, Miwa S, Horner PJ, Turnbull DM. 2015. Neural stem cells in the adult subventricular zone oxidize fatty acids to produce energy and support neurogenic activity. Stem Cells 33: 2306–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takubo K, Semenza GL. 2011. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9: 298–310. [DOI] [PubMed] [Google Scholar]
- Takashima Y, Guo G, Loos R, Nichols J, Ficz G, Krueger F, Oxley D, Santos F, Clarke J, Mansfield W, et al. 2014. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell 158: 1254–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson RS, Soga T, et al. 2013. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12: 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JL, Fogley RD, Flynn RA, Ablain J, Yang S, Saint-André V, Fan ZP, Do BT, Laga AC, Fujinaga K, et al. 2016. Stress from nucleotide depletion activates the transcriptional regulator HEXIM1 to suppress melanoma. Mol Cell 62: 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TeSlaa T, Chaikovsky AC, Lipchina I, Escobar SL, Hochedlinger K, Huang J, Graeber TG, Braas D, Teitell MA. 2016. α-Ketoglutarate accelerates the initial differentiation of primed human pluripotent stem cells. Cell Metab 24: 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, et al. 2007. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128: 325–339. [DOI] [PubMed] [Google Scholar]
- Ueda T, Nagamachi A, Takubo K, Yamasaki N, Matsui H, Kanai A, Nakata Y, Ikeda K, Konuma T, Oda H, et al. 2015. Fbxl10 overexpression in murine hematopoietic stem cells induces leukemia involving metabolic activation and upregulation of Nsg2. Blood 125: 3437–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueishi S, Shimizu H, H Inoue Y. 2009. Male germline stem cell division and spermatocyte growth require insulin signaling in Drosophila. Cell Struct Funct 34: 61–69. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini N, Girotra M, Naveiras O, Nikitin G, Campos V, Giger S, Roch A, Auwerx J, Lutolf MP. 2016. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat Commun 7: 13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA IV, Ramalho-Santos J, Van Houten B, Schatten G. 2011. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE 6: e20914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, et al. 2003. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 35: 238–245. [DOI] [PubMed] [Google Scholar]
- Wang J, Alexander P, Wu L, Hammer R, Cleaver O, McKnight SL. 2009. Dependence of mouse embryonic stem cells on threonine catabolism. Science 325: 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA, Huang J, Kalderon D. 2012. Drosophila follicle stem cells are regulated by proliferation and niche adhesion as well as mitochondria and ROS. Nat Commun 3: 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CB, Nelson AM, Mecham B, et al. 2014. Derivation of naive human embryonic stem cells. Proc Natl Acad Sci 111: 4484–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegué E. 2013. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494: 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AE, Pollina EA, Vierbuchen T, Urbán N, Ucar D, Leeman DS, Martynoga B, Sewak M, Rando TA, Guillemot F, et al. 2013. FOXO3 shares common targets with ASCL1 genome-wide and inhibits ASCL1-dependent neurogenesis. Cell Rep 4: 477–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. 1999. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124. [DOI] [PubMed] [Google Scholar]
- Xie Z, Jones A, Deeney JT, Hur SK, Bankaitis VA. 2016. Inborn errors of long-chain fatty acid β-oxidation link neural stem cell self-renewal to autism. Cell Rep 14: 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin S, Zhang X, Luciano JP, Mungamuri SK, Marinkovic D, Vercherat C, Sarkar A, Grisotto M, Taneja R, Ghaffari S. 2008. Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J Biol Chem 283: 25692–25705. [DOI] [PubMed] [Google Scholar]
- Yalcin S, Marinkovic D, Mungamuri SK, Zhang X, Tong W, Sellers R, Ghaffari S. 2010. ROS-mediated amplification of AKT/mTOR signalling pathway leads to myeloproliferative syndrome in Foxo3−/− mice. EMBO J 29: 4118–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanes O, Clark J, Wong DM, Patti GJ, Sánchez-Ruiz A, Benton HP, Trauger SA, Desponts C, Ding S, Siuzdak G. 2010. Metabolic oxidation regulates embryonic stem cell differentiation. Nat Chem Biol 6: 411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. 2008. The ground state of embryonic stem cell self-renewal. Nature 453: 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S, Soh BS, Sun LL, Tai BC, Nga ME, et al. 2012. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 148: 259–272. [DOI] [PubMed] [Google Scholar]
- Zhang H, Badur MG, Divakaruni AS, Parker SJ, Jäger C, Hiller K, Murphy AN, Metallo CM. 2016. Distinct metabolic states can support self-renewal and lipogenesis in human pluripotent stem cells under different culture conditions. Cell Rep 16: 1536–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, et al. 2012. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J 31: 2103–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]