

Abstract
Elucidation of the driving forces that govern interactions between nanoparticles and intrinsically disordered proteins (IDP) is important for the understanding of the effect of nanoparticles in living systems and for the design of new nanoparticle-based assays to monitor health and combat disease. The quantitative interaction profile of the intrinsically disordered transactivation domain of p53 and its mutants with anionic silica nanoparticles is reported at atomic resolution using nuclear magnetic spin relaxation experiments. These profiles are analyzed with a novel interaction model that is based on a quantitative nanoparticle affinity scale separately derived for the 20 natural amino acids. The results demonstrate how the interplay of attractive and repulsive Coulomb interactions with hydrophobic effects is responsible for the sequence-dependent binding of a disordered protein to nanoparticles.
TOC image
INTRODUCTION
The emergence of nanoscience applications in biology calls for a detailed understanding of the interactions between synthetic nanoparticles and biological molecules.1–3 Nanoparticles can be used in biomedicine, for example, as drugs, drug carriers, or imaging agents.4,5 In all these situations, they are inevitably exposed to in vivo environments, such as biofluids, cells, or tissues, where a vast array of diverse native biomolecules exist, encompassing proteins, nucleic acids, and metabolites. The complex nature of such nanoparticle-biomolecule interactions is well recognized6,7. Nanoparticles have differential binding propensities to small molecules. For example, anionic and cationic silica nanoparticles (SNPs) bind to a host of metabolites as evidenced by NMR peak broadening.8 Many synthetic nanomaterials are covered by small molecules via covalent or non-covalent interactions supporting their structural integrity, solubility, and function, but quantitative information about binding strengths is limited or missing.9 Nanoparticles can also interact with polypeptides, whereby most studies have focused on globular, well-structured proteins. Only few studies reported nanoparticle-protein interactions at atomic resolution.10–15
Intrinsically disordered proteins (IDPs) form a separate class of polypeptides that is linked to a wide range of human diseases.16–18 Due to the flexible nature of IDPs and the heterogeneous distribution of amino-acid types along the primary sequence with their different physical chemical properties, the interaction strength with nanoparticles can considerably vary from residue to residue. It thereby induces differential mobility or rigidification along the protein sequence. Nanoparticle-IDP interactions can be potentially utilized to target specific IDPs in vitro or in vivo for interventional therapies in ways that are not possible using small molecules.19–21 As important progress is being made,20 a critical bottleneck remains the lack of a fundamental, quantitative understanding of IDP-nanoparticle interactions based on the surface properties of nanoparticles and IDP amino-acid sequence information.
The transactivation domain of the human tumor suppressor p53 (p53TAD), which contains 73 amino-acid residues, is a well-known IDP.22–24 In this work, p53TAD serves as a model system to study its interactions with anionic silica nanoparticles at atomic resolution. For this purpose, transverse 15N backbone NMR spin relaxation experiments were performed for both the wild type (WT) and mutants of p53TAD as they sensitively reflect the populations of the bound state at the sites of individual residues. Based on experimentally determined binding propensities of isolated amino acids to SNPs a general analytical model is developed that quantitatively accounts for the binding properties of p53TAD and several of its mutants.
RESULTS AND DISCUSSION
Electrostatic and size properties
The SNPs used in this study are negatively charged with a ζ-potential −23.0 ± 7.4 mV due to the partial deprotonation of silanol group at neutral pH. Since p53TAD has also a negative net charge (pI = 3.77), its interactions with SNPs are expected to be relatively weak and transient due to Coulomb repulsion. The size distribution of the SNPs was measured to be 19.5 ± 5.3 nm by transmission electron microscopy and 25.3 ± 8.9 nm by dynamic light scattering. In the presence of p53TAD with a p53TAD:SNP concentration ratio of 20:1, no significant shift in the hydrodynamic diameter distribution was observed (Figure S7), which confirms that the SNPs remain monodisperse with p53TAD present. Sucrose cushion separation followed by SDS-PAGE analysis confirmed the dynamic and reversible nature of p53TAD-SNP association and dissociation, which is fully consistent with the NMR results (vide infra).
Differential NMR line broadening due to SNP presence
Two-dimensional (2D) heteronuclear NMR spectroscopy allows one to study nanoparticle-protein interactions at a spatial resolution of individual amino acids without the need for non-native chemical tags. To study the effect of SNPs on p53TAD, we collected 2D 1H-15H heteronuclear single quantum correlation (HSQC) NMR spectra of WT p53TAD samples in the absence and presence of SNPs (Figure 1). Since HSQC cross-peaks correspond to directly bonded 1H-15H spin pairs, the individual behavior of each p53TAD residue (with the exception of the 13 Pro residues) can be monitored by its own cross-peak belonging to the amide bond. While no apparent chemical shift changes were observed, the relative peak intensities decreased noticeably and non-uniformly upon the titration of SNPs to the protein sample whereby some cross-peaks completely disappeared (Figures 1a–e and S4). For p53TAD samples (at 200 μM concentration), cross-peaks that were most affected belong to residues V10-Q16, D21-A39, and G59-V73 experiencing more than 80% intensity loss as a result of line broadening upon the addition of SNPs (4.7% by weight or 9.5 μM), whereas residues M1-D7 and M40-D57 experienced much less cross-peak attenuation (Figure 1f).
Figure 1.
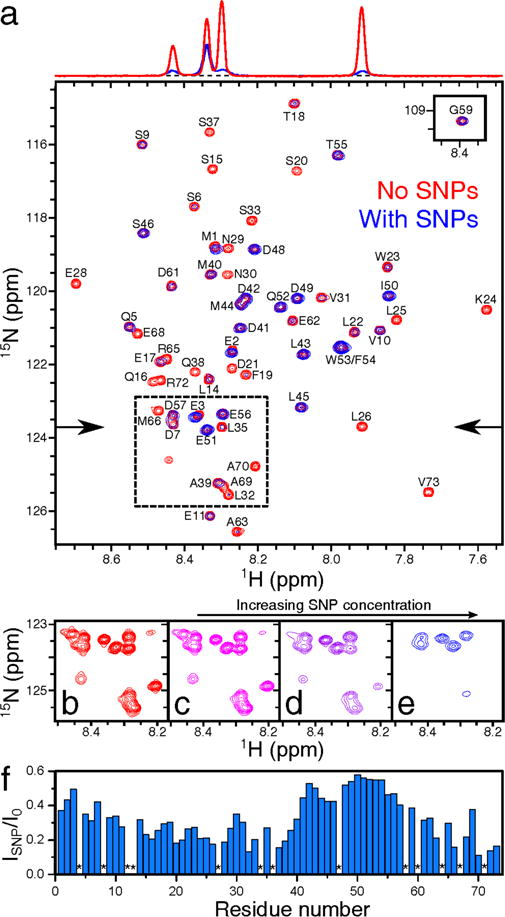
Effect of interactions of WT p53TAD with silica nanoparticles on 1H-15H HSQC NMR spectrum. (a) Superimposed spectra of 200 μM protein samples in the absence (red) and presence (blue) of 4.7% [9.5 μM] SNPs by weight. The 1H cross-sections of the two HSQC spectra, indicated by the arrows, are displayed at the top, showing the effect of SNPs on the relative cross-peak intensities. (b) – (e) Representative spectral region (dashed box in (a)) showing a differential intensity decrease of some but not all cross-peaks with increasing SNP concentration from (b) 0.0% [0 μM], (c) 0.6% [1.2 μM], (d) 2.3% [4.7 μM], (e) 7.0% [14 μM] by weight in 250 μM protein samples. (f) Residue-intensity ratios are plotted as blue bars vs the primary sequence, except for prolines (*).
To quantify SNP-induced differential peak attenuation, transverse backbone 15H spin relaxation rates (R2)25 were measured for all (non-proline) amino-acid residues along the backbone of p53TAD for 300 μM protein samples in the absence and presence of 1.3% anionic SNPs by weight (2.5 μM). The red symbols in Figure 2a depict the residue-specific differences of measured R2, ΔR2, in the presence and absence of SNPs for p53TAD. The profile reveals a large dynamic range with up to a 5-fold difference in ΔR2 between the TAD2 region (D41-P60), showing the smallest effect, and the C-terminal residues displaying the largest effect. Several portions of the profile reflect a distinct fine structure with multiple local minima and maxima, which are experimentally well reproducible. By contrast, the longitudinal 15H spin relaxation rates (R1) remained essentially unchanged by the addition of the SNPs (Figure S5).
Figure 2.
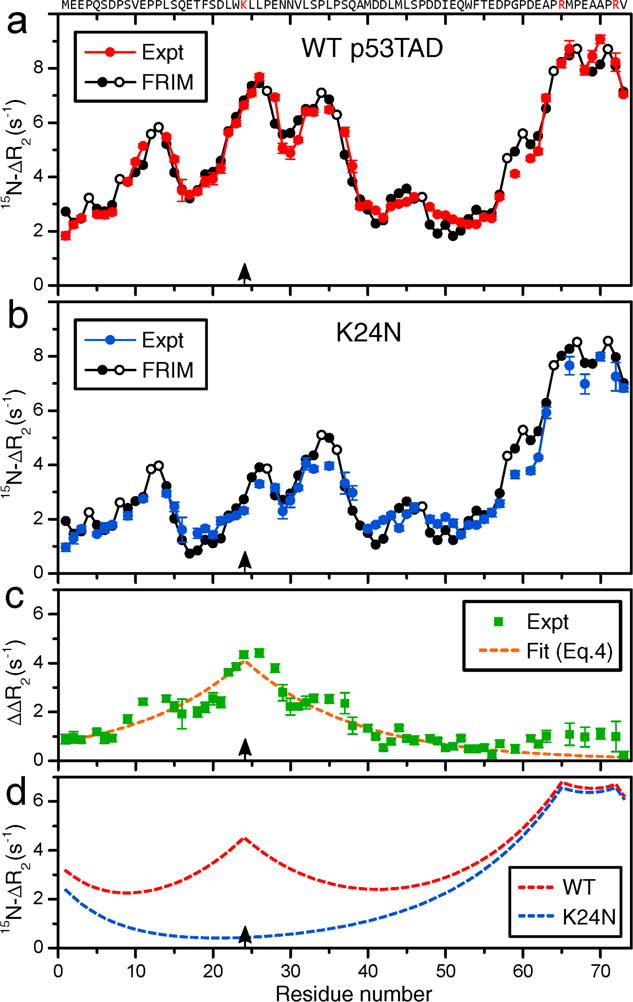
Residue-specific differences of NMR 15H backbone transverse relaxation rates of 300 μM (a) WT p53TAD (red solid circles) and (b) K24N mutant (blue solid circles) in the presence (2.5 μM) and absence of SNPs, i.e. , were plotted as a function of residue number. (a),(b) Predicted ΔR2 values by the free residue interaction model (FRIM) (Eq. (5)) are shown as black solid circles (except black open circles for Pro residues). (c) Difference of experimental ΔR2 profiles of (a) and (b) (green squares), ΔΔR2, depicting the effect of Lys24 (black arrows) on ΔR2. The dashed orange line is a fit using Eq. (4). (d) Cumulative effect of positively charged residues on global ΔR2 profiles using Eq. (4) for WT (red) and K24N mutant (blue).
Transverse spin relaxation analysis
Experimental transverse NMR relaxation data were interpreted by a two-site exchange process where each amino-acid residue i exists either in a free or an SNP-bound state with normalized populations and . The experimentally observed effective R2 relaxation rate in the presence of SNPs, , is then a population-weighted average
| (1) |
of the transverse relaxation rate of nuclear spin of residue i of the free protein and of its SNP-bound state, , provided that the bound and free states are in fast exchange, i.e. they have millisecond lifetimes or faster. The populations can be directly determined from
| (2) |
where . The term can be determined directly from experiment by measuring R2 in the presence and absence of SNPs. is according to Redfield relaxation theory (see, e.g., Ref. [26] and references therein) directly proportional to the overall tumbling correlation time τc of the nanoparticles, which can be estimated based on the Stokes-Einstein-Debye relationship
| (3) |
where V is the hydrodynamic volume of a single SNP, η is the solvent (water) viscosity, kB is the Boltzmann constant, and T is the absolute temperature (see the Supporting Information for details). Therefore, is constant for all residues. In this way, the experimental determination of ΔR2,i offers direct insights into the effective bound population at the site of each residue. is a quantitative measure of the restriction of the reorientational motion of the N-H internuclear vector of residue i due to the interaction between the molecule and the SNPs. For a well-structured globular protein, is the same for all residues. For an IDP, by contrast, may vary from residue to residue due to differences in the interaction strengths with SNPs between different residues and due to the SNP interactions of neighboring residues j whose effect on residue i is attenuated because of the intrinsically flexible nature of the polypeptide chain that separates the two residues. A value indicates that residue i is rigidly attached to the surface of the SNP whereas indicates that residue i does not interact with SNPs. For the weak binding conditions that apply for the present study, are in the sub-percentage range. In the following we report ΔR2,i, which is directly proportional to according to Eq. (2). Working under conditions where the for p53TAD, as well as of individual amino acids, is in the sub-percentage range, allows the use of high-resolution NMR to quantitatively measure the interactions in a site-specific manner.
WT p53TAD vs. K24N mutant
To interpret the experimental ΔR2 profile (red symbols in Figure 2a) in terms of the physical-chemical properties of individual residues, we first study the effect of the attractive electrostatic interactions between the positively charged residues and the anionic SNPs. For this purpose, we also performed 15H-R2 spin relaxation measurements of the K24N mutant of p53TAD where the positively charged Lys24 has been replaced by a neutral Asn residue. While the R2 profile of free K24N closely resembles the one of WT p53TAD, indicating that K24N is also intrinsically disordered, the ΔR2 profile of K24N (blue symbols in Figure 2b) is substantially lowered for many residues on both sides of the mutation site. According to Eq. (2), the site-specific bound populations of the mutant in this region are systematically lower than those of the WT. The difference of ΔR2,i between the WT and K24N mutant, , reflects a remarkably simple profile displaying a symmetric cusp that is centered at the mutation site (Figure 2c). It follows in very good approximation the mono-exponentially decaying function:
| (4) |
where prefactor Aj = 4.1 s−1, i is the primary sequence position of the residue of interest, j (= 24) is the position of the mutated residue, and λ = 14.0 reflects the interaction range (measured in terms of residue distance along the primary sequence). Assuming that the presence of all other positively charged parts of p53TAD, namely Arg65 and Arg72 as well as the N-terminus (with the interaction length reduced by half), have an analogous effect that is additive, overall ΔR2 profiles were predicted shown in Figure 2d. Although they reproduce dominant features of the experimental ΔR2 profiles of both WT and K24N mutant (Figure 2a,b), they fail to explain the local variations that determine the fine structure.
SNP-binding affinities of all 20 natural amino acids
With its intrinsically disordered nature, all amino acid residues of p53TAD are exposed to the solvent, which allows each of them to closely interact with the SNP. In order to determine whether the local variations in the experimental ΔR2 profiles (colored symbols in Figure 2a,b) can be explained by binding properties of individual amino-acid residues to SNPs, we determined the SNP-binding affinities of all 20 standard amino acids each measured in a separate sample. We measured the 13Cα R2 and R1 relaxation rates of individual amino acids at natural 13C abundance both in the absence and presence of SNPs to evaluate their differential interaction propensities. The Cα sites are the most suitable probes, since they are the only protonated carbons common to all 20 amino acids and due to their vicinity to the amino groups used as NMR spin relaxation probes in p53TAD. In the presence of SNPs, the R2 relaxation rates increased in an amino-acid specific manner whereby the differences in R2 between SNP-containing and SNP-free samples, ΔR2, reflect according to Eq. (2) the relative binding affinities (populations pb) of the different amino acids to the SNP surface (Figure 3). Three of the five amino acids with the strongest effects (largest ΔR2) are Arg, Lys, and His. These amino acids are the only ones with a net positive charge at neutral pH, indicating that electrostatic attraction makes a major contribution to the binding affinity to the SNP surface. Interestingly, Pro possesses the second largest ΔR2 among all amino acids despite its neutral net charge. Ile, Leu, and Val, all with hydrophobic side-chains, also experience significantly accelerated transverse relaxation in the presence of SNPs. This suggests that hydrophobic interactions also make an important contribution to SNP-binding. In contrast, Asp, Glu, Ser, Thr, Ala, Gly, Asn, and Gln showed negligible elevation in ΔR2 and, hence, negligible binding propensities. The observed variations of ΔR2 are depicted in Figure 3 providing a quantitative scale for the relative affinities of amino acids with anionic SNPs. These results show that SNPs interact with amino acids with widely variable binding affinities. An analogous behavior is expected for other small molecules, including metabolites, with potential therapeutic applications and implications for nanotoxicity.
Figure 3.
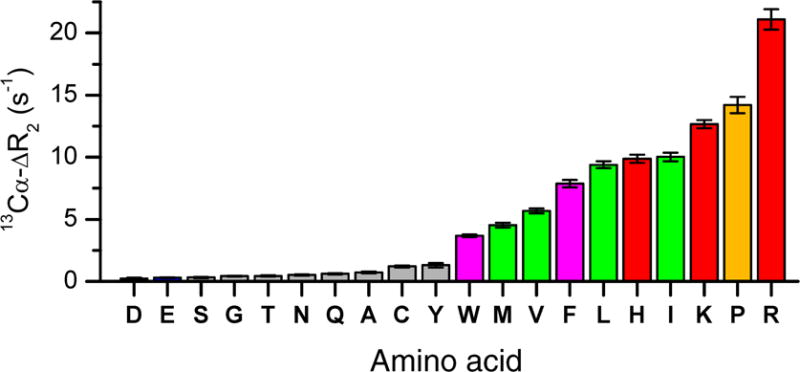
Experimental 13Cα values of individual amino acids in the presence and absence of silica nanoparticles (SNPs). These values served as residue-specific affinities to explain the nanoparticle-IDP interactions (Table S2 and Fig. S2). The color-coded amino acids have the highest binding affinities, which include the positively charged amino acids (red), proline (orange), methyl-containing (green), and aromatic amino acids (magenta).
Free residue interaction model
Based on these amino-acid specific affinities the model of Eq. (4) can be generalized to
| (5) |
where the sum extends over all residues as well as the N- and C-terminal charges (Table S2). All positively charged residues were treated with the parameters of Eq. (4). For the negatively charged residues Aj = −1.0 s−1 and λj = 6.4, and the interaction range of both terminal charges was reduced by half. This accounts for the repulsive electrostatic force between these residues and the anionic SNP surface. For the neutral residues, Aj is proportional to the experimentally determined amino-acid specific SNP affinity (Figure 3) and their interaction range λj = 2.8 was obtained by fitting. The sum reflects the weighted contributions from neighboring amino acids that are within λj of residue i along the primary sequence. We term this model the Free Residue Interaction Model (FRIM).
The ΔR2 profiles were back-calculated by FRIM for WT p53TAD and K24N mutant (black curves in Figure 2a,b). They are in excellent agreement with the experimentally measured ΔR2 profiles with a Pearson correlation coefficient r of 0.975 and 0.970, respectively. The predicted curves do not only reproduce the overall shape of the affinity profile along the polypeptide chain, but they now also explain its fine structure. This level of agreement between the prediction by FRIM and experiment is remarkable considering the very small number of fit parameters entering Eq. (5) (see also Supporting Information).
The overall shape of the ΔR2 profile is dominated by electrostatic attraction between the positively charged residues and the anionic SNPs. These interactions have an unexpectedly long range over approximately 14 sequential residues on either side of a charged amino acid side-chain as was directly measured for Lys24 (Figure 2c). This interaction range significantly exceeds the characteristic persistence length of unfolded proteins, which is only about 7 residues.27,28 Hydrophobic residues, such as Ile, Leu, Phe, and Val, also exert attractive forces on the SNPs. In fact, many of these interactions are responsible for the fine details of the ΔR2 profile of p53TAD. Although the SNPs used in this work have an anionic surface, this does not exclude the presence of hydrophobic patches on the surface of the SNPs that preferentially bind to hydrophobic side chains. It was previously found that certain hydrophobic metabolites (e.g. Val and dimethylglycine) display significant interactions with anionic SNPs.8 In a computational study, it was found that various forms of DNA can bind to SNPs by a combination of electrostatic and hydrophobic effects.29 In this work, the distance range of the hydrophobic interactions was determined to be 2.8 residues, which is much smaller than that of the electrostatic interactions. This was further corroborated by the relaxation profiles measured for the double mutant F19A/W23A (Figure S6). A larger interaction range is inconsistent with experiment, as it would eventually cause the disappearance of the fine structure in the ΔR2 profiles. p53TAD contains a total of 18 negatively charged sites comprising all Asp and Glu residues as well as the C-terminus. They experience a repulsive Coulomb force from the anionic SNP surface, which is reflected in a negative affinity. Direct measurement of the negative affinities of individual Asp and Glu amino acids is a challenge, because their bound population is nearly zero rendering their effective R2 rates essentially identical to those of the SNP-free sample (see also Eq. (2)). This leads to very small 13Cα R2 rates of Asp and Glu in Figure 3, which are non-zero as a consequence of increased viscosity due to the presence of SNPs. However, the negative affinities of Asp and Glu residues are manifested in the ΔR2 profiles of IDPs as they reduce the binding affinities of nearby residues according to the FRIM model (Eq. (5)). It assumes the same SNP affinity value and interaction range for Asp and Glu residues. Since these affinities could not be directly determined for the free amino acids, they are less accurately defined compared to the ones of the other residues.
The proposed interaction model (FRIM) offers a predictive understanding of the location and strengths of interactions between IDPs and silica nanoparticles. The positively charged residues increase the binding propensities of other residues over a strikingly large distance range (14 residues along the primary sequence on both sides). This may be caused by a rise in the kinetic on-rates kon as the average distance to the SNP surface is shortened due to the attractive Coulomb interaction between the nearby positively charged residue and the SNP surface (Figure 4). Changes in conditions, such as the ζ-potential, pH, or ionic strength, can affect the protein-nanoparticle interactions and, hence, the ΔR2 profile and might require adjustment of the FRIM parameters.
Figure 4.
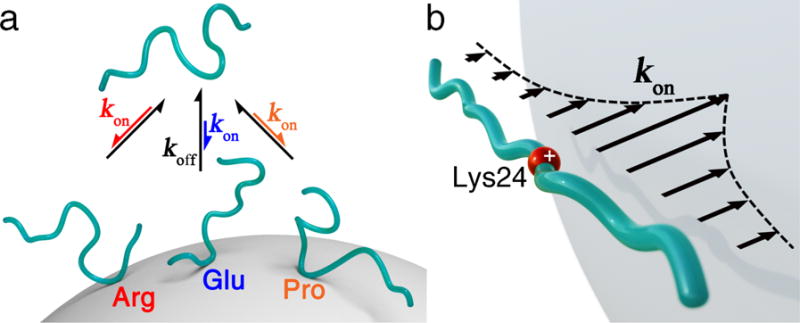
(a) Schematic representation of multiple conformational substates of p53TAD that interact with a SNP with differential binding propensities determined by the primary sequence. The lengths of the arrows reflect variable kon for different p53TAD regions. (b) Long-range effect of positively charged Lys24 on the association rate constant kon of p53TAD with anionic SNP as a function of residue position along the primary sequence.
CONCLUSIONS
The molecular driving forces for the interaction between intrinsically disordered p53TAD and silica nanoparticles could be quantitatively monitored by NMR spin relaxation in solution. They report on the combined effect of both long-range electrostatic and short-range hydrophobic interactions, which considerably vary along the protein amino-acid sequence and which are well captured by the proposed FRIM model. This model is transferrable to other intrinsically disordered proteins and should be adaptable to other types of nanoparticles. It will help guide the design of nanoparticles with altered binding properties to target, for example, specific IDP regions, and the calibration of computational approaches for the atomistic modeling of these systems. This study also demonstrates that free amino acids display a wide range of binding affinities to nanoparticles, which were quantitatively measured and converted to a binding propensity scale. Since in vivo, amino acids are metabolites, this could prove useful to control biochemical pathways or understand nanotoxicity.
MATERIALS AND METHODS
SNP preparation
Bindzil 2040 SNPs were extensively dialyzed in the same buffers used for amino acids and protein constructs, respectively. These amorphous, near-spherical SNPs have a relatively narrow size distribution and are negatively charged at the surface (ζ-potential −23.0 ± 7.4 mV) at neutral pH, as characterized previously8. In all SNP-doped samples for relaxation experiments, either 2.5% w/w (2.1 μM) for amino acid samples or 1.3% w/w (1.1 μM) for protein samples SNPs were used unless otherwise specified.
p53TAD and its mutants
The DNA fragment corresponding to WT p53TAD (1–73 residues) was amplified with polymerase chain reaction and was cloned into a pTBSG ligation independent cloning vector derivative as reported previously.24 Plasmid cDNAs of the K24N and F19A/W23A mutants of p53TAD were constructed using the QuikChange Lightening site-directed mutagenesis protocol. 15H-labeled proteins were overexpressed in Escherichia coli with 1 L M9 minimal media containing 1 g 15HH4Cl and 5 g D-Glucose, and were purified with Ni-NTA agarose affinity column. The final protein concentration of NMR samples was 300 μM in 50 mM HEPES buffer (containing 100 mM NaCl, pH 7.0) unless otherwise specified.
Amino acid samples
Solutions of the twenty common amino acids (natural 13C abundance) were individually prepared in 20 mM phosphate buffer (D2O, containing 100 mM NaCl, pD 7.0) to achieve 16 mM (except for Try c.a. 1.3 mM) final concentration.
Dynamic light scattering
DLS data were collected of the SNP suspension in the absence and presence of p53TAD (see Supporting Information). These data demonstrate that the SNPs remain monodispersed under both conditions.
NMR spin relaxation experiments
13C and 15H spin relaxation experiments were performed on Bruker Avance III HD Ascend 850 MHz spectrometers equipped with TCI cryoprobes. For protein samples, either without or with SNPs, the backbone amide 15N spin relaxation parameters were obtained in a pseudo-3D fashion using conventional R1 and R1ρ experiments by extracting the 2D peak intensities from 1H-15H cross-peaks of enhanced-HSQC-type experiments recorded with eight delay intervals at 298 K, followed by fitting the intensity-delay data with a single exponential decay curve. R2 relaxation rates were thereafter obtained using the equation:
| (S1) |
where θ = arctan(ν/Ω) is the tilt angle in the rotating frame. Here, ν denotes 15H spin-lock field strength (Hz), which was set to 2047 Hz and was calibrated as reported previously25, and Ω is the resonance offset from the spin-lock carrier (Hz) for each cross-peak. To estimate statistical errors of R1 and R2, 200 iterations of Monte Carlo error analysis were used. The peak assignments of WT p53TAD and F19A/W23A mutant were previously done by Shan et al.24
Similarly, the effective R1 and R2 (CPMG) for the 20 amino acids, without or with SNPs, were obtained in a pseudo-2D fashion based on the 1D peak intensities from natural abundance 1Hα-13Cα cross-peaks of enhanced-HSQC-type experiments vs delay intervals. The proton-decoupled CPMG pulse sequence with νCPMG = 109 Hz and rf field strength = 3.125 kHz for DIPSI-2 decoupling was used, which allows a longer observation window (> 500 ms) for R2 relaxation with better precision compared to conventional R1ρ-type of experiments for molecules in the extreme narrowing limit, hence is favorably applicable to amino acids. Due to the large variation in R2 rates for different amino acids in the presence of SNPs, the delay intervals were adjusted so that the peak intensity corresponding to the longest interval was sufficiently decreased (approximately 2.7-fold less) compared to the peak intensity without any delay.
Supplementary Material
Acknowledgments
We thank AkzoNobel and Eka Chemicals for a gift of Bindzil 2040 silica nanoparticles. We also thank Dr. L. Bruschweiler-Li for expert assistance with p53TAD sample preparation and Dr. G. W. Daughdrill for providing K24N assignments. This work was supported by National Science Foundation (grant MCB-1360966) and the National Institutes of Health (grant R01GM066041). All NMR experiments were conducted at the CCIC NMR facility at the Ohio State University.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Experimental details about protein expression and purification, amino-acid sample preparation; silica nanoparticle treatment, NMR spin relaxation experiments, R1 and R2 relaxation data, and their analysis (tables and figures); prediction of spin relaxation properties of nanoparticle-bound molecule; amino-acid affinity scale (table and figure), parametrization of the FRIM model (PDF)
Notes
The authors declare no competing financial interests.
References
- 1.Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M. Understanding Biophysicochemical Interactions at the Nano-Bio Interface. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 2.Kim ST, Saha K, Kim C, Rotello VM. The Role of Surface Functionality in Determining Nanoparticle Cytotoxicity. Acc Chem Res. 2013;46:681–691. doi: 10.1021/ar3000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelly PM, Åberg C, Polo E, O’Connell A, Cookman J, Fallon J, Željka K, Dawson KA. Mapping Protein Binding Sites on the Biomolecular Corona of Nanoparticle. Nat Nanotechol. 2015;10:472–479. doi: 10.1038/nnano.2015.47. [DOI] [PubMed] [Google Scholar]
- 4.Li Z, Barnes JC, Bosoy A, Stoddart JF, Zink JI. Mesoporous Silica Nanoparticles in Biomedical Applications. Chem Soc Rev. 2012;41:2590–2605. doi: 10.1039/c1cs15246g. [DOI] [PubMed] [Google Scholar]
- 5.Mitragotri S, Anderson DG, Chen X, Chow EK, Ho D, Kabanov AV, Karp JM, Kataoka K, Mirkin CA, Petrosko SH, et al. Accelerating the Translation of Nanomaterials in Biomedicine. ACS Nano. 2015;9:6644–6654. doi: 10.1021/acsnano.5b03569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monopoli MP, Aberg C, Salvati A, Dawson KA. Biomolecular Coronas Provide the Biological Identity of Nanosized Materials. Nat Nanotechnol. 2012;7:779–786. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- 7.Docter D, Westmeier D, Markiewicz M, Stolte S, Knauer S, Stauber R. The Nanoparticle Biomolecule Corona: Lessons Learned–Challenge Accepted? Chem Soc Rev. 2015;44:6094–6121. doi: 10.1039/c5cs00217f. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B, Xie M, Bruschweiler-Li L, Bingol K, Brüschweiler R. Use of Charged Nanoparticles in NMR-Based Metabolomics for Spectral Simplification and Improved Metabolite Identification. Anal Chem. 2015;87:7211–7217. doi: 10.1021/acs.analchem.5b01142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rimola A, Costa D, Sodupe M, Lambert J-F, Ugliengo P. Silica Surface Features and Their Role in the Adsorption of Biomolecules: Computational Modeling and Experiments. Chem Rev. 2013;113:4216–4313. doi: 10.1021/cr3003054. [DOI] [PubMed] [Google Scholar]
- 10.Engel MFM, Visser AJWG, van Mierlo CPM. Conformation and Orientation of a Protein Folding Intermediate Trapped by Adsorption. Proc Natl Acad Sci USA. 2004;101:11316–11321. doi: 10.1073/pnas.0401603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lundqvist M, Sethson I, Jonsson B-H. Protein Adsorption onto Silica Nanoparticles: Conformational Changes Depend on the Particles’ Curvature the Protein Stability. Langmuir. 2004;20:10639–10647. doi: 10.1021/la0484725. [DOI] [PubMed] [Google Scholar]
- 12.Shrivastava S, McCallum SA, Nuffer JH, Qian X, Siegel RW, Dordick JS. Identifying Specific Protein Residues that Guide Surface Interactions and Orientation on Silica Nanoparticles. Langmuir. 2013;29:10841–10849. doi: 10.1021/la401985d. [DOI] [PubMed] [Google Scholar]
- 13.Calvaresi M, Arnesano F, Bonacchi S, Bottoni A, Calò V, Conte S, Falini G, Fermani S, Losacco M, Montalti M, et al. C60@Lysozyme: Direct Observation by Nuclear Magnetic Resonance of a 1:1 Fullerene Protein Adduct. ACS Nano. 2014;8:1871–1877. doi: 10.1021/nn4063374. [DOI] [PubMed] [Google Scholar]
- 14.Zanzoni S, Pedroni M, D’Onofrio M, Speghini A, Assfalg M. Paramagnetic Nanoparticles Leave Their Mark on Nuclear Spins of Transiently Adsorbed Proteins. J Am Chem Soc. 2016;138:72–75. doi: 10.1021/jacs.5b11582. [DOI] [PubMed] [Google Scholar]
- 15.Ceccon A, Tugarinov V, Bax A, Clore GM. Global Dynamics and Exchange Kinetics of a Protein on the Surface of Nanoparticles Revealed by Relaxation-Based Solution NMR Spectroscopy. J Am Chem Soc. 2016;138:5789–5792. doi: 10.1021/jacs.6b02654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 17.Wright PE, Dyson HJ. Intrinsically Disordered Proteins in Cellular Signalling and Regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larion M, Hansen AL, Zhang F, Bruschweiler-Li L, Tugarinov V, Miller BG, Brüschweiler R. Kinetic Cooperativity in Human Pancreatic Glucokinase Originates from Millisecond Dynamics of the Small Domain. Angew Chem Int Ed. 2015;127:8247–8250. doi: 10.1002/anie.201501204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uversky VN. Proteins without Unique 3D Structures: Biotechnological Applications of Intrinsically Unstable/Disordered Proteins. Biotechnol J. 2015;10:356–366. doi: 10.1002/biot.201400374. [DOI] [PubMed] [Google Scholar]
- 20.Lin W, Insley T, Tuttle MD, Zhu L, Berthold DA, Král P, Rienstra CM, Murphy CJ. Control of Protein Orientation on Gold Nanoparticles. J Phys Chem C. 2015;119:21035–21043. doi: 10.1021/acs.jpcc.5b07701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pregent S, Lichtenstein A, Avinery R, Laser-Azogui A, Patolsky F, Beck R. Probing the Interactions of Intrinsically Disordered Proteins Using Nanoparticle Tags. Nano Lett. 2015;15:3080–3087. doi: 10.1021/acs.nanolett.5b00073. [DOI] [PubMed] [Google Scholar]
- 22.Lee H, Mok KH, Muhandiram R, Park K-H, Suk J-E, Kim D-H, Chang J, Sung YC, Choi KY, Han K-H. Local Structural Elements in the Mostly Unstructured Transcriptional Activation Domain of Human p53. J Biol Chem. 2000;275:29426–29432. doi: 10.1074/jbc.M003107200. [DOI] [PubMed] [Google Scholar]
- 23.Wells M, Tidow H, Rutherford TJ, Markwick P, Jensen MR, Mylonas E, Svergun DI, Blackledge M, Fersht AR. Structure of Tumor Suppressor p53 and Its Intrinsically Disordered N-Terminal Transactivation Domain. Proc Natl Acad Sci USA. 2008;105:5762–5767. doi: 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shan B, Li D-W, Bruschweiler-Li L, Brüschweiler R. Competitive Binding between Dynamic p53 Transactivation Subdomains to Human MDM2 Protein: Implications for Regulating the p53·MDM2/MDMX Interaction. J Biol Chem. 2012;287:30376–30384. doi: 10.1074/jbc.M112.369793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmer AG, III, Kroenke CD, Loria JP. Nuclear Magnetic Resonance Methods for Quantifying Microsecond-to-Millisecond Motions in Biological Macromolecules. Methods Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 26.Gu Y, Li D-W, Brüschweiler R. NMR Order Parameter Determination from Long Molecular Dynamics Trajectories for Objective Comparison with Experiment. J Chem Theory Comput. 2014;10:2599–2607. doi: 10.1021/ct500181v. [DOI] [PubMed] [Google Scholar]
- 27.Klein-Seetharaman J, Oikawa M, Grimshaw SB, Wirmer J, Duchardt E, Ueda T, Imoto T, Smith LJ, Dobson CM, Schwalbe H. Long-Range Interactions within a Nonnative Protein. Science. 2002;295:1719–1722. doi: 10.1126/science.1067680. [DOI] [PubMed] [Google Scholar]
- 28.Schwarzinger S, Wright PE, Dyson HJ. Molecular Hinges in Protein Folding: the Urea-Denatured State of Apomyoglobin. Biochemistry. 2002;41:12681–12686. doi: 10.1021/bi020381o. [DOI] [PubMed] [Google Scholar]
- 29.Shi B, Shin YK, Hassanali AA, Singer SJ. DNA Binding to the Silica Surface. J Phys Chem B. 2015;119:11030–11040. doi: 10.1021/acs.jpcb.5b01983. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.