

Hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP [MIM#615704]) is an extremely rare syndromic form of autosomal dominant poikiloderma. This genetic disorder was first identified in a South African family in 2006.1 To date, 3 families and 9 independent sporadic cases have been reported.2, 3, 4 Here we report an additional family of POIKTMP and expand the clinical spectrum. We describe, for the first time to our knowledge, a pancreatic cancer in the clinical course in 1 patient. We also address the differential diagnosis of inherited poikiloderma and related disorders.
Case series
In 2007, at the Strasbourg University Hospital, the department of medical genetics referred a family to the dermatology department with a diverse clinical skin picture, dominated by poikiloderma. The father, a white 64 year old, was the sixth (I: 6) of sibship born from nonconsanguineous parents. Very soon after birth, his grandmother observed that “he was not like the others,” and he was described to have red cheeks and heat (but not sun) intolerance since early childhood. Clinical evaluation found a very severe case of poikiloderma, predominant in the sun-exposed areas, resulting from the combination of skin atrophy, mottled pigmentation with hyperpigmented and hypopigmented lesions, and telangiectasia (Fig 1, A). He had a distinct intolerance for heat with marked hypohidrosis. Diffuse xerosis was combined with multiple depigmented macules on the trunk and limbs. The patient reported lymphedema of the lower limbs (Fig 1, B) starting in adolescence, complicated by recurrent erysipelas. His feet and hands were small, both affected by tendon contractures (Fig 1, B). His nails and teeth were normal. He did not have a history of cataract formation or pulmonary disease. He also had recent-onset alopecia, which was subsequent to chemotherapy. The patient had pancreatic cancer at the time of referral. The appearance of an obstructive jaundice had led, several months earlier, to the diagnosis of pancreatic cancer. A thoraco-abdominal pelvic computed tomography scan found a diffuse infiltration of the pancreas, with peritoneal carcinosis. Magnetic resonance imaging of the pancreas found a tumor in the pancreatic isthmus, with numerous cystic lesions, most likely caused by an intraductal papillary mucinous neoplasm (IPMN). There was no fatty infiltration of the pancreas on magnetic resonance imaging. The diagnosis of invasive adenocarcinoma originating from an IPMN was established by scan-guided pancreatic biopsy. Because of locally advanced stage, only palliative chemotherapy with gemcitabine was administered. The patient had none of the known risk factors of pancreatic cancer such as type 2 diabetes, obesity, pancreatitis, or smoking.
Fig 1.

A, Father. Poikiloderma, predominant in the sun-exposed areas but sparing in some zones the forehead and scalp. B, Father. Chronic lymphedema of the lower limbs with tendon contractures.
His son (II: 1), age 30, had similar skin changes—red cheeks since 6 months old, developing into an essentially facial poikiloderma; hypohidrosis with heat intolerance; lymphedema of the lower limbs starting in adolescence; guttate leukoderma; and stiffness of the fingers. Teeth, hair, and nails were normal. There was no pulmonary impairment.
His 27-year-old daughter (II: 2) had erythematous cheeks since she was 1 year old. Identical lesions to those of her father were observed: poikiloderma mainly localized to the face; xerosis and innumerable achromic or hypochromic macules, measuring between 1 and 2 mm, of the trunk and the limbs (Fig 2, A); marked hypohidrosis; and lower-limb lymphedema. Her hands and feet were small, with atrophy of both thenar and hypothenar eminences (Fig 2, B). A biopsy of the palm found marked reduction in eccrine glands, and a biopsy of an achromic macule found a clear decrease in melanin pigment in the basal layer of the epidermis without loss of melanocytes. She had mildly elevated liver transaminases on repeat blood samples, but the search for an etiology was negative. Pulmonary function tests and thoracic computed tomography scan were normal.
Fig 2.

A, Daughter. Multiple, discrete round or oval, porcelain-white macules. B, Daughter. Atrophy of both thenar and hypothenar eminences with inability to extend the fingers fully. No loss of dermal ridges.
Her daughter (III: 3) had facial telangiectatic erythema since the age of 6 months. This family's phenotype, characterized by poikiloderma, hypohidrosis, small feet and hands with tendon contractures or atrophy of the thenar and hypothenar eminences, was reminiscent of the case of a South African family reported in 20061 and recently found to have a mutation in the FAM111B gene.5 Sanger sequencing of the family reported herein identified also a mutation in FAM111B: p.[Ser628Arg]; [=] c.[1884T>A] (Fig 3) that segregated with the disease confirming that the diagnosis belonged to the POIKTMP spectrum.
Fig 3.
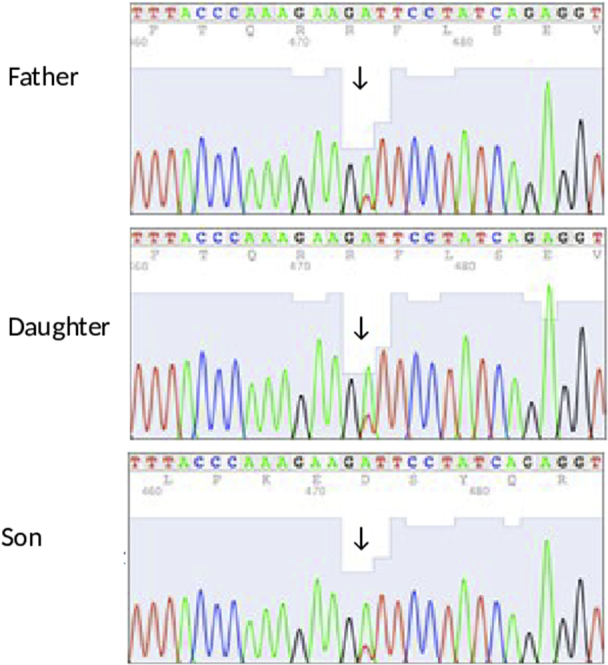
Chromatograms of Sanger sequencing of the FAM111B gene in the patients. The arrows indicate mutation sites.
Discussion
We report an additional family of POIKTMP (Fig 4) in which a pancreatic cancer was discovered in one of the members. POIKTMP is a recently described entity of syndromic inherited poikiloderma.2, 5, 6 In 2013, whole exome sequencing enabled identification of causative mutations in FAM111B-encoding a protein of still unknown function.5 This is a rare disorder; only 26 patients belonging to 12 independent families have been reported.1, 2, 3, 4 These families were of South African, Algerian, Northern European, French, Irish, Italian, Moroccan, Dominican Republic, and Kuwaiti origins.1, 2, 3, 4 Congenital poikiloderma is a constant skin condition.2 The condition begins in early childhood, typically in the first 6 months of age (Table I). It affects sun-exposed areas. It is a true poikiloderma and not a simple reticulated pigmentation disorder or a mottled pigmentation with atrophy. Poikiloderma is almost invariably combined with alopecia and hypohidrosis with heat intolerance.2 We found that hypohidrosis could be explained by a rarefaction of sweat glands. As in the family reported here, lymphedema of the extremities, complicated by recurring erysipelas, is often observed (Table I). The variability of dermatologic characteristics is found in our cases: no alopecia, but diffuse xerosis and depigmented macules, which do not usually occur in this syndrome. To our knowledge, this is the first case of guttate hypomelanosis described in POIKTMP. The skin lesions improve with age, whereas the extracutaneous manifestations become worse.2 Musculo-tendinous impairments begin during the first 10 years of life5 consisting of muscle contractures, progressive muscular weakness, and amyotrophy (Table I). Interstitial pulmonary fibrosis is found in adulthood, and was found to be fatal in a few individuals.1, 2 No respiratory abnormalities were observed in our patients. Liver impairment,2 reported in one of our patients with transaminases fluctuating between normal and abnormal, and exocrine pancreatic dysfunction3 are also described. An extensive fatty infiltration of the pancreas is reported.1
Fig 4.

Pedigree of the family consistent with autosomal dominant inheritance. The proband is indicated by arrow. Asterisk indicates sequenced patients.
Table I.
Clinical and molecular data of the different types of syndromic inherited poikiloderma
| OMIM number | RTS #268400 | PN #604173 | DKC #127550 #224230 #305000 #613987 #613988 #613989 #613990 #615190 #616353 | KS #173650 | WHSK 173700 | XP #278700 #278720 #278730 #278740 #278760 #278780 #610651 | BS #210900 | BGS #218600 | FA #227645 #227646 #227650 #300514 #600901 #603467 #609053 #609054 #610832 #613390 #613951 #614082 #614083 #615272 #616435 | POIKTMP #615704 |
|---|---|---|---|---|---|---|---|---|---|---|
| Inheritance | AR | AR | AD AR X-linked | AR | AD | AR | AR | AR | AR X-linked | AD |
| Onset of poikiloderma | 3-6 mo | 6 mo Prevalent acral distribution | 3-5 y Lacy reticular pigmentation rather than true poikiloderma | 2-3 y | 2-4 y Accentuated in flexural areas | Late childhood Abnormal freckling on the face before age 2 y ; real poikiloderma is rare | 1-2 y Butterfly distribution of face; initially telangiectatic erythema and not poikiloderma | Late onset (not in early infancy) | Variable Poikilodermoidbrownish pigmentation and hypo pigmentation | 6 mo |
| Palmoplantar keratoderma | About 30% of patients | Frequent | May be present | 65% | May be present | Absent | Absent | Absent | Absent | May be present |
| Blister | In early childhood; in sun-exposed areas | May be present | In areas of trauma | Acral; after trauma or sun exposure present at birth: fourth type of inherited epidermolysis bullosa | Absent | In neonate; in sun-exposed areas | Blister around the mouth | In infancy (face buttocks extremities) | Absent | May be present |
| Nail abnormalities | Pachyonychia is common | Pachyonychia | Major feature (lichen planus-like changes) | Frequent | Absent | Absent | Absent | Absent | Absent | May be present |
| Dental defects | Frequent (27%–59%); wide variety of malformations | Dental eruption delay; fragile carious teeth | Poor dentition; early dental loss | Periodontal disease | Absent | Absent | Occasional absence of lateral incisors | Delayed eruption of teeth | Severe generalized periodontitis | Absent (rarely poor dentition; recurrent gingivitis) |
| Mucous membrane lesions | Absent | Absent | Major feature: leukoplakia, stenosis | Orogenital leukokeratosis; mucous stenosis | Absent | Tongue leukoplakia | Absent | Anus anteposition; imperforated anus | Oral leukoplakia | No leukoplakia |
| Other dermatologic signs | Hypotrichosis/ alopecia photosensitivity; café-au-lait spots | Photosensitivity; sparse eyelash-eyebrows | No photosensitivity; absent fingerprints; alopecia; canities | Photosensitivity; skin fragility; skin atrophy; loss of dermal ridges; pseudosyndactyly pseudoainhum; phimosis ; anhidrosis | Sclerosis of palms and soles; linear hyperkeratosis and sclerotic bands in skin folds; calcinosis; clubbing fingers; Raynaud's phenomenon | Photosensitivity; xerosis | Photosensitivity; paucity of subcutaneous fat; loss of the lower eyelashes; café-au-lait spots | Swelling of the extremities | Café-au-lait spots | Hypohidrosis/heat intolerance; hypotrichosis/alopecia; lymphoedema of extremities; xerosis; eczema or psoriasis-like lesions; sclerosis of the digits; guttate hypo- melanosis |
| Ocular abnormalities | Cataract | Absent | Epiphora blepharitis retinopathy | Conjunctivitis; corneal erosion; ectropion of the lower eyelids | Absent | Photophobia; severe keratitis | Wide variety | Ocular proptosis; hypertelorism | Microphthalmia | May be present (cataract) |
| Skeletal defects Malformations |
Radial ray defects; patellar hypoplasia; frontal bossing; saddle nose | Craniofacial dysmorphism; hypermobile fingers with beak of swan appearence | Microcephaly; osteoporosis | Skull or mandibular abnormalities | Mandibuloacral dysplasia; maxillary bossing; micrognathia | Absent | Microcephaly; dolichocephaly; prominence of the nose and ears | Coronal cranio-synostosis; radial ray defects; patellar hypoplasia | Microcephaly; absent or abnormal thumbs and radii | May be present (scoliosis) small feet |
| Respiratory system | Bronchiectasis | Recurrent pulmonary infections | Diffuse interstitial pulmonary fibrosis | Normal | No pulmonary involvement | Normal | Pneumonia bronchiectasis chronic pulmonary disease | Normal | Pulmonary infections | Interstitial pulmonary fibrosis |
| Hematologic features | Leukopenia anemia | Permanent neutropenia | High frequency of bone marrow failure; pancytopenia | Absent | Absent | Absent | Myelodysplasia; low immunoglobulin counts | Absent | Major feature (bone marrow failure); myelodysplasia | Eosinophilia |
| Other visceral abnormalities | Chronic diarrhea in early childhood | Recurrent otitis media; splenomegaly | Liver cirrhosis | Severe colitis | Cardiac involvement; cardiac valvular diseases; aortic stenosis | Acquired microcephaly; neuro-degeneration (30%) in some variants | Immunodeficiency with recurrent infections; diabetes mellitus; gastroesophageal reflux; diarrhea; lower urinary tract obstruction | Heart-hand syndrome: radial abnormalities and defects in the heart | Type 2 diabetes (adults); ear abnormalities; hearing loss | Muscle contractures (triceps surae); muscle atrophy; weakness of proximal and distal muscles; liver involvement; pancreatic exocrine insufficiency |
| Physical development | Pre- and postnatal growth deficiency | Short stature | Short stature | Normal | Growth retardation | Normal | Pre- and postnatal growth deficiency | Short stature | Pre- and postnatal growth retardation | Growth retardation |
| Mental development | Normal | Normal or slightly delayed | Mental retardation | Normal | Normal | Intellectual deficiency (30%) | Normal or limited | Mental retardation in some patients | May be delayed | Normal |
| Malignancy | Osteosarcomas (childhood); skin carcinomas (adults); myelodysplasia; leukemia; lymphoma | Myelodysplasia; acute myeloid leukemia | Squamous cell carcinoma in and outside the areas of leukoplakia; Myelodysplasia ; acute myeloid leukemia | Mucocutaneous squamous cell carcinomas | Absent | Skin carcinomas, melanomas in early childhood; ocular and tongue neoplasms; leukemia | Types and sites of cancer very broad; early frequent (46%) | Osteosarcomas; skin carcinomas; lymphoma | Acute myeloid leukemia; squamous cell carcinomas of the head and neck, esophagus, and vulva; liver tumors | Absent (pancreatic cancer?) |
| Genetic defect | RECQL4 on 8q24.3∗ | C16orf57 on 16q21 |
TERC on 3q26.2 NOLA3 on 15q14 DKC1 on Xq28 NOLA2 on 5q35.3 WRAP53 on 17p13.1 TERT on 5p15.33 TINF2 on 14q12 RTEL1 on 20q13.33 PARN on 16p13.12 |
KIND1 on 20p12.3 | Unknown |
XPA on 9q22.33 XPC on 3p25.1 ERCC2 on 19q13.32 DDB2 on 11p11.2 ERCC4 on 16p13.12 ERCC5 on 13q33.1 ERCC3 on 2q14.3 |
BLM/RECQL3 on 15q26.1 | RECQL4 on 8q24.3∗ |
FANCC on 9q22.32 FANCD2 on 3p25.3 FANCA on 16q24.3 FAAP95 on Xp22.2 FANCE on 6p21.31 FANCF on 11p14.3 FANCI on 15q26 BRIP1 on 17q23.2 PALB2 on 16p12.2 RAD51C on 17q22 SLX4 X on 16p13.3 XRCC9 on 9p13.3 PHF9 on 2p16.1 ERCC4 on 16p13.12 UBE2T on 1q32.1 |
FAM111B on 11q12.1 |
Some of the discussed entities rather display a mottled disorder of pigmentation than true poikiloderma, which associates a reticulate hyperpigmentation, atrophy and telangiectases, but distinction between those 2 dermatologic phenotypes is not always done in published literature. Werner syndrome (OMIM #277700) is not mentioned because the symptoms appear in adulthood. It is associated with increased risk of malignancies (thyroid carcinoma, melanoma, soft-tissue sarcoma, meningioma).
AD, Autosomal dominant; AR, autosomal recessive; BGS, Baller-Gerold syndrome; BS, Bloom syndrome; DKC, dyskeratosis congenita; FA, Fanconi anemia; KS, Kindler syndrome; PN, poikiloderma with neutropenia; POIKTMP, hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis; WHSK, Weary hereditary sclerosing poikiloderma; XP, xeroderma pigmentosum.
Mutations in RECQL4 are also associated with RAPADILINO syndrome, which overlaps clinically with RTS but without poikiloderma.
In 2013, Mercier et al5 identified the causative gene, FAM111B (NM_198947.3), on chromosome 11 (11q12.1). The c.1884T>A heterozygous mutation of the FAM111B gene found in our 4 cases yields a p.Ser628Arg amino acid substitution at position 628 (Fig 3). A mutation at the same position (serine: p.Ser628Asn) was previously reported.2, 5 This mutation segregated with the disease in this large pedigree (Fig 4).
Autosomal dominant POIKTMP has overlapping clinical findings with Rothmund-Thomson syndrome (RTS), which has a recessive inheritance pattern. Several patients were initially misdiagnosed with RTS in childhood,2, 6 although the mode of inheritance is different. Differential diagnoses that must be considered in a child with poikiloderma are reviewed in Table I.2, 7, 8, 9, 10, 11
Although the son (II: 1), the daughter (II: 2), and the granddaughter (III: 3) had no personal history of cancer, the proband died of pancreatic cancer. No cancer has previously been described in POIKTMP2, 6; however, considering the relative young age of most patients reported to date, this does not rule out any cancer-predisposing effect of FAM111B deleterious variants occurring later in life.6 The length bias may also have played a role, as the first case of cancer was reported in the oldest patient with a FAM111B mutation. Although the pancreatic adenocarcinoma could have been an incidental finding in this patient, to report a pancreatic cancer in a syndrome in which pancreatic manifestations do occur3 is intriguing. Most congenital poikiloderma syndromes are inherited cancer-prone diseases (Table I).8, 9, 10, 11 Furthermore, 2 studies imply FAM111B as a cancer predisposition gene. The first12 using a genomewide association study, identified several susceptibility loci for prostate cancer. One locus was on chromosome 11q12 and FAM111B is a candidate gene. In the second study,13 transcriptomic analysis on a pancreatic cancer cell line previously treated by antiproliferative drugs found downregulation of FAM111B, raising the question as to the oncogenic activity of this gene. Further functional studies and the analysis of a broader cross-section of patients are requested to address this question and the putative cancer-promoting effects of FAM111B mutations.
Congenital poikiloderma presents a diagnostic challenge for dermatologists.4 The differential diagnosis begins with a complete dermatologic examination.9 Definite diagnosis relies on identification of a pathogenic mutation in specific genes implicated in the broad group of the inherited poikiloderma, as summarized in Table I.6 This report shows the variability of the clinical spectrum of POIKTMP.2 The malignant transformation of IPMN in the index case raises the question as to whether FAM111B could be a new predisposition gene for cancer or precancerous lesions in the pancreas.
Footnotes
Funding sources: None.
Conflicts of interest: None declared.
References
- 1.Khumalo N.P., Pillay K., Beighton P. Poikiloderma, tendon contracture and pulmonary fibrosis: a new autosomal dominant syndrome? Br J Dermatol. 2006;155:1057–1061. doi: 10.1111/j.1365-2133.2006.07473.x. [DOI] [PubMed] [Google Scholar]
- 2.Mercier S., Küry S., Salort-Campana E. Expanding the clinical spectrum of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis due to FAM111B mutations. Orphanet J Rare Dis. 2015;10:135. doi: 10.1186/s13023-015-0352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seo A., Walsh T., Lee M.K. FAM111B Mutation is associated with inherited exocrine pancreatic dysfunction. Pancreas. 2016;45:858–862. doi: 10.1097/MPA.0000000000000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeichi T., Nanda A., Yang H.S. Syndromic inherited poikiloderma due to a de novo mutation in FAM111B. Br J Dermatol. 2017;176:534–536. doi: 10.1111/bjd.14845. [DOI] [PubMed] [Google Scholar]
- 5.Mercier S., Küry S., Shaboodien G. Mutations in FAM111B cause hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Am J Hum Genet. 2013;93:1100–1107. doi: 10.1016/j.ajhg.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Küry S., Mercier S., Shaboodien G. CUGC for hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP) Eur J Hum Genet. 2016;24(5) doi: 10.1038/ejhg.2015.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fradin M., Merklen-Djafri C., Perrigouard C. Long-term follow-up and molecular characterization of a patient with a RECQL4 mutation spectrum disorder. Dermatology. 2013;226:353–357. doi: 10.1159/000351311. [DOI] [PubMed] [Google Scholar]
- 8.Jaju P.D., Ransohoff K.J., Tang J.Y., Sarin K.Y. Familial skin cancer syndromes: Increased risk of nonmelanotic skin cancers and extracutaneous tumors. J Am Acad Dermatol. 2016;74:437–451. doi: 10.1016/j.jaad.2015.08.073. [DOI] [PubMed] [Google Scholar]
- 9.Larizza L., Roversi G., Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2. doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colombo E.A., Bazan J.F., Negri G. Novel C16orf57 mutations in patients with Poikiloderma with Neutropenia: bioinformatic analysis of the protein and predicted effects of all reported mutations. Orphanet J Rare Dis. 2012;7:7. doi: 10.1186/1750-1172-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Renty C., Ellis N.A. Bloom's syndrome: Why not premature aging?: A comparison of the BLM and WRN helicases. Ageing Res Rev. 2017;33:36–51. doi: 10.1016/j.arr.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akamatsu S., Takata R., Haiman C.A. Common variants at 11q12, 10q26 and 3p11.2 are associated with prostate cancer susceptibility in Japanese. Nat Genet. 2012;44:426–429. doi: 10.1038/ng.1104. [DOI] [PubMed] [Google Scholar]
- 13.Yue W., Wang T., Zachariah E. Transcriptomic analysis of pancreatic cancer cells in response to metformin and aspirin: an implication of synergy. Sci Rep. 2015;5:13390. doi: 10.1038/srep13390. [DOI] [PMC free article] [PubMed] [Google Scholar]