

Abstract
Deep brain stimulation (DBS) has revolutionized the clinical care of late stage Parkinson’s disease and shows promise for improving the treatment of intractable neuropsychiatric disorders. However, after over 25 years of clinical experience, numerous questions still remain on the neurophysiological basis for the therapeutic mechanisms of action. At their fundamental core, the general purpose of electrical stimulation therapies in the nervous system are to use the applied electric field to manipulate the opening and closing of voltage-gated sodium channels on neurons, generate stimulation induced action potentials, and subsequently control the release of neurotransmitters in targeted pathways. Historically, DBS mechanisms research has focused on characterizing the effects of stimulation on neurons and the resulting impact on neuronal network activity. However, when electrodes are placed within the central nervous system, glia are also being directly (and indirectly) influenced by the stimulation. Mounting evidence shows that non-neuronal tissue can play an important role in modulating the neurochemistry changes induced by DBS. The goal of this review is to evaluate how DBS effects on both neuronal and non-neuronal tissue can potentially work together to suppress oscillatory activity (and/or information transfer) between brain regions. These resulting effects of ~100 Hz electrical stimulation help explain how DBS can disrupt pathological network activity in the brain and generate therapeutic effects in patients.
Keywords: Neuromodulation, Electrode, Synapse
I. Introduction
The goal of this review is to integrate results on the effects of deep brain stimulation (DBS) on both neuronal and non-neuronal tissue to develop a more holistic understanding of possible therapeutic mechanisms for ~100 Hz focal stimulation in the brain for the treatment of Parkinson’s disease (PD). The premise of this review is rooted in the prevailing hypothesis that disorders treated with DBS are fundamentally the result of dysfunctional brain circuit activity arising from pathological synaptic interactions between multiple nuclei. This general hypothesis of a “circuitopathy” has existed for decades, with roots in epilepsy [Papez, 1937; MacLean, 1949] and PD [Alexander et al., 1986]. Successful application of DBS therapy has since provided additional motivation to better understand the origin of circuitopathies, as well as their modulation and/or cessation with surgical intervention [Benabid et al., 1991; Lozano et al., 2002]. In parallel, recent systems-level theories on brain network communication deterioration in PD are providing new kinds of circuitopathy metrics that can be directly acquired from human patients [de Hemptinne et al., 2015; Cagnan et al., 2015]. However, bridging the gap between cellular-level details and systems-level theories remains a formidable challenge.
After more than 25 years of scientific debate, a generalized consensus statement on the basic mechanisms of DBS would be that it overrides the underlying neural activity near the implanted electrode and replaces it with an alternative activity pattern that is less deleterious to brain function. Unfortunately, this generalized statement is not especially specific or satisfying. The reality is that the effects of DBS on the nervous system are generated at the ionic level, protein level, cellular level, and network level to then produce changes in behavior. Given our current data collection and analysis methods, it has not been possible to truly link these various effects into a single unifying hypothesis that completely explains the overall system.
Early hypotheses on the mechanisms of DBS focused on the parallels between the behavioral outcomes achieved with either ~100 Hz stimulation or lesioning a focal target region in the brain [Benabid et al., 1998]. Prior to DBS, lesioning globus pallidus (GP) was the surgical method of choice for treating late stage PD [Laitinen et al., 1992]. So when early electrophysiological recordings of neural activity at the site of ~100 Hz stimulation showed blocked or reduced somatic spiking [e.g. Benazzouz et al., 2000; Dostrovsky et al., 2000], the logical conclusion was that therapeutic DBS generated a neural activity lesion. However, theoretical models [e.g. McIntyre et al., 2004a], experimental recordings from nuclei downstream of the site of stimulation [e.g. Hashimoto et al., 2003], and functional imaging results [e.g. Perlmutter et al., 2002] did not support the blocking hypothesis, but instead suggested increased neural activity. In turn, recent hypotheses on the mechanisms of DBS have migrated toward network-based theories, forgoing a direct link between lesioning and stimulation, and focusing instead on stimulation induced disruption of pathological network oscillations [McIntyre and Hahn, 2010].
An interesting caveat in attempting to define the mechanisms of DBS is that it modulates different symptoms with different time courses (Fig. 1). The temporal details of these effects are also dependent on the focal target region being directly stimulated [Johnson et al., 2008]. Such observations have prompted hypotheses that the different parkinsonian symptoms are generated by different oscillatory patterns in the basal-ganglia-thalamo-cortical network [Blumenfeld and Bronte-Stewart, 2015]. For example, ~4 Hz bursting activity in thalamus has been linked to tremor [Zirh et al., 1998] and ~20 Hz oscillations in the basal ganglia have been linked with rigidity [Kuhn et al., 2009]. Further elucidation of these kinds of hypotheses are being directly facilitated by new DBS systems that enable chronic local field potential recording in freely moving patients [Rouse et al., 2011]. Early results from these systems suggest that specific PD phenotypes may even further segregate the pertinent oscillatory patterns [Quinn et al., 2015]. In addition, there is strong interest in dissecting the role of individual pathways (or connections) within the basal-ganglia-thalamo-cortical network as key in the generation of specific oscillations and/or the associated PD symptoms. For example, direct stimulation of the dentatothalamic pathway has been linked with tremor control [Groppa et al., 2014] and direct stimulation of the hyperdirect pathway has been linked with rigidity improvements [Xu et al., 2011]. These trends highlight the need to better understand the network connection patterns and network activity patterns of both the normal and pathological state, likely representing major focuses for the field of DBS mechanisms research over the next decade. However, the fundamentals of electrical stimulation still occur at the cellular-level to modulate the neurochemistry of the brain and generate the subsequent network-level changes. In turn, the need to characterize and understand the effects of DBS on individual neurons and glia remains as important as ever.
Figure 1.
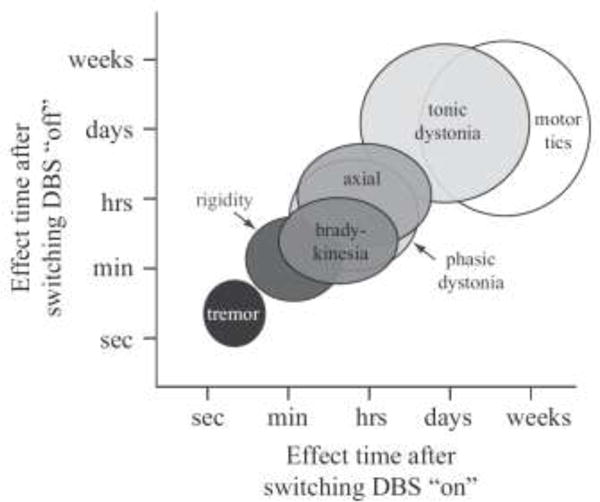
Time course of DBS symptom control. DBS has a nearly immediate effect on tremor, while other symptoms require longer periods of time to observe relief. The return of symptoms following cessation of stimulation follows a similar pattern. Adapted from Johnson et al. [2008].
II. Fundamentals of Electrical Stimulation
A key step in characterizing brain circuit modulation by DBS is to first define the direct neural response to the applied electric field near the site of electrode implantation (Fig. 2A). This is commonly performed using DBS electric field models coupled to models of individual neurons (or axons) [McNeal, 1976]. The DBS electric field is a three-dimensionally complex phenomenon that is distributed throughout the brain [McIntyre et al., 2004b]. The field is generated by the redistribution of charged ions in the extracellular space, resulting from the electrode polarization [Plonsey and Barr, 2007]. Modern DBS computer models typically simulate DBS electric fields using finite element methods that have evolved over the last decade to be able to match in vivo experimental recordings of the voltage distribution in the brain with impressive fidelity [Miocinovic et al., 2009].
Figure 2.
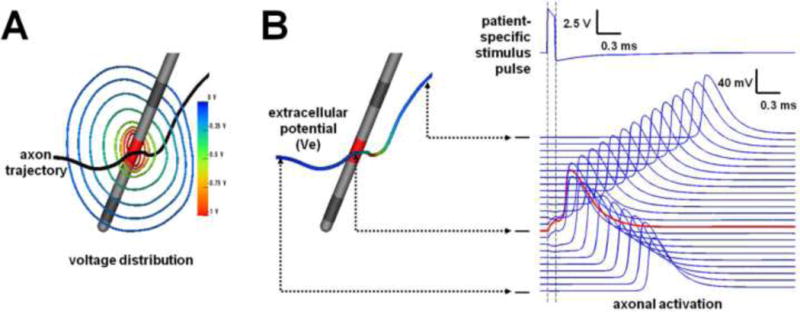
Neural response to stimulation. A) DBS electrodes generate a nonlinear voltage gradient (radiant isolines depict the voltage distribution in 2D) in the brain. B) Each neural process (or axon) surrounding the electrode will be polarized by the stimulus, but each different axon will experience different magnitudes of polarization depending on the stimulus parameters and its relative location to the electrode. If the stimulus pulse is strong enough, an action potential will be generated and propagate in both directions. Adapted from Lujan et al. [2012].
Experimentalists from 60’s and 70’s established that the primary effect of electrical stimulation in the central nervous system was the generation of action potentials (APs) in axons [Ranck, 1976]. Theoreticians then quantified that the response of an individual neural process (or axon) to the applied field was related to the second derivative of the extracellular voltage distribution along its trajectory [McNeal, 1976] (Fig. 2B). Therefore, modern methods enable the use a tractography streamline derived from diffusion-weighted imaging data to represent the trajectory of a simulated axon and them impose the DBS voltage distribution on that trajectory (aka - tractography-activation model) [e.g. Chaturvedi et al., 2010]. Such models subsequently enable prediction of which axonal pathways are directly activated by DBS pulses within the context of the patient-specific anatomy.
The fundamental purpose of the applied electric field on the neuronal process is to induce a transmembrane voltage change that can open voltage-gated sodium channels on the axon. The cathodic phase of the stimulus pulse generates a membrane depolarization in the nodes of Ranvier closest to the electrode contact. The magnitude of that depolarization is dependent on the shape of the axon trajectory and its proximity to the electrode contact. If the stimulus amplitude is sufficient to generate a depolarization that crosses the activation threshold for that axon, an AP will be generated and propagate in both directions (i.e. antidromically and orthodromically) (Fig 2).
Action potential initiation is a binary phenomenon, so once the stimulus amplitude is greater than the threshold for that individual axon, it is often capable of following stimulus frequencies up to and over 100 Hz with very high fidelity [Bucher and Goaillard, 2011]. However, an interesting caveat of stimulating neural tissue for long periods of time at high frequencies is that the robustness of AP generation and propagation can waver, especially for small diameter axons [e.g. Jensen and Durand, 2009]. Nonetheless, in vivo electrophysiological recordings from the most clinically relevant DBS experiments strongly support the initiation and propagation of axonal APs in response to therapeutic DBS settings [e.g. Hashimoto et al., 2003].
III. DBS is a Chemical Therapy
Once action potentials get generated they typically propagate to their axon terminals and induce neurotransmitter release. Simulations suggests that DBS-induced APs are equally effective at propagating in either the orthodromic or antidromic directions [Grill et al., 2008]. In turn, each AP, in each directly stimulated neuron, can result in hundreds of synaptic events throughout the complex axonal arbor of that neuron. For example, anatomical reconstructions of the axonal arbors of non-human primate subthalamic nucleus (STN) neurons show ~500 synapses in globus pallidus (GP) [Sato et al., 2000]. The human STN is estimated to have ~400,000 neurons [Hardman et al., 2002]. Patient-specific DBS models estimate that ~30% of the STN projection neurons are directly stimulated with typical therapeutic settings [Chaturvedi et al., 2012]. Therefore, a single DBS pulse can theoretically generate ~60 million (400,000 × 500 × 0.30) synaptic events in GP via direct stimulation of STN projection neurons. In addition, the subthalamopallidal projection is only one of many pathways surrounding DBS electrodes implanted in the subthalamic region. Other key pathways include the lenticular fasiculus, dentatothalamic tract, and internal capsule. It is highly likely that at least some of these other pathways are also directly stimulated during therapeutic DBS [Chaturvedi et al., 2012], generating large numbers of additional synaptic events throughout the brain.
All of those DBS-induced synaptic events can alter the balance of neurotransmitters within the stimulated brain network. The effects of electric fields on axons are nondiscriminatory to the type of neurotransmitter used by any particular pathway. So depending on the specific pathway being stimulated, the effect could be either inhibitory via modulated GABA release or excitatory via modulated glutamate release. Within the simplified context of the STN-GP microcircuit, therapeutic subthalamic DBS should theoretically generate both glutamate and GABA modulation simultaneously [Hahn and McIntyre, 2010]. In this example, directly stimulated STN neuron projection axons generate glutamate release in the GP (GPe and GPi), while direct stimulation of the GPe axonal afferents generate GABA release in the STN.
Attempts to directly measure the neurochemistry modulation induced by DBS, typically via intracerebral microdiaylsis, have generated important insights into the mechanisms of DBS. Studies in human PD patients undergoing DBS surgery have measured significant increases in cGMP, which is indicative of increased glutamatergic transmission, in the GPi [Stefani et al., 2005; 2011] and substantia nigra (SN) [Galati et al., 2006] during subthalamic DBS. In addition, they measured a decrease in GABA concentration in the thalamus [Stefani et al., 2011]. A key aspect of those studies was the tight correlation between the measured biochemical changes and the therapeutic benefit observed in the patients.
Numerous studies in rodent models have also investigated the impact of subthalamic DBS on glutamate and/or GABA tone in the basal ganglia [e.g. Windels et al., 2000; Lee et al., 2004; Windels et al., 2005; Boulet et al., 2006; Walker et al. 2009]. Taken together, most of the available microdialysis results on DBS-induced neurochemistry modulation support the theory that ~100 Hz stimulation generates APs in the axonal processes near the electrode, enhancing neurotransmitter release within the connected circuitry. For example, early work studying normal rats with 1 hour of subthalamic DBS demonstrated significant increases in extracellular glutamate in GP [Windels et al., 2000]. However, when similar experiments were performed in parkinsonian rats they found that basal levels of glutamate in GP were dramatically higher than normal rats and subsequent subthalamic DBS did not generate any additional increase in extracellular glutamate [Windels et al., 2005]. This lead to the hypothesis that the hyperactivity of STN neurons in parkinsonian rats resulted in a basal level of released glutamate that was already too high to measure additional increases under stimulation. However, the SN exhibited a significant increase in GABA, which was associated with activation of pallidal fibers of passage [Windels et al., 2005].
Recent studies of rodent neurochemistry modulation with DBS have turned to in vivo proton magnetic resonance spectroscopy and begun to address key questions on the effects of chronic stimulation [Melon et al., 2015; Chassain et al., 2016]. Results from these studies show that chronic (5 week) continuous subthalamic DBS efficiently counteracts the metabolic and synaptic defects that arise from dopaminergic lesion, which include significant increases in both glutamate and GABA in the striatum and SN. Interestingly, the long-term effects of DBS in this model were an overall decrease in neurotransmitter levels, relative to the parkinsonian state, and this was seen throughout the basal ganglia. While these chronic stimulation results might appear in conflict with the theory of DBS-induced AP generation, the overall effect on synaptic transmission during long-term high frequency driving of axon terminals could potentially reconcile this disconnect (see below).
IV. DBS Creates an Informational Lesion
While the direct neurophysiological connections between destructive lesioning and DBS are not strong, the underlying concept of disrupting signaling between basal-ganglia-thalamo-cortical nuclei remains the key premise of nearly every current hypothesis on DBS mechanisms. The underlying concept is that the pathological activity of the microcircuit directly affected by DBS can be overridden by the new stimulation induced activity. When the stimulation frequency approaches a level that is at least ~twice as high as the underlying average firing frequency of the neurons being stimulated, the stimulation induced APs begin to take over control the synaptic communication of the neurons. These directly stimulated neurons have effectively been “captured” by the stimulation and lose the ability to transmit information associated with the brain circuit to which they are attached. This occurs because the stimulation induced APs outnumber the intrinsically generated APs and the stimulation induced APs traveling antidromically collision block most of the intrinsically generated APs traveling othodromically [McIntyre et al., 2004a]. Since the stimulation frequency remains constant (i.e. 100 Hz) the information content of the stimulation signal is effectively zero, generating what is known as an “informational lesion” in the circuit [Grill et al., 2004]. Therefore, if the underlying circuitopathy is rooted in pathological brain network oscillatory activity, as prevailing hypotheses in PD suggest, then creating a DBS-induced informational lesion anywhere in the affected circuit should eliminate the oscillation and generate therapeutic effects.
The concepts of an informational lesion provide a relatively simple answer to the complex questions of DBS mechanisms and they work perfectly well in idealized simulations. However, the realities of high frequency neurotransmitter release and synaptic modulation are likely much more complicated than assumed by informational lesion theories and models. Early efforts at dissecting DBS modulated synaptic transfer focused on simple stimulation induced neurotransmitter depletion as the key mechanism of action [Urbano et al., 2002]. Yet, neurons within the basal ganglia commonly fire at high rates under normal conditions, implying that synaptic vesicle recycling in these neurons is still capable of operating under DBS conditions and generating a range of plasticity effects [Shen et al., 2003]. Therefore, more recently attention has turned to understanding the role of synaptic filtering induced by DBS [Anderson et al., 2006; Rosenbaum et al., 2014] (Fig. 3). The concept is that short-term depression can preferentially suppress the synaptic transfer of low frequency oscillatory activity during periods of elevated presynaptic spiking. In the context of PD, beta oscillations (~20 Hz) are commonly assumed to be a hallmark electrophysiological biomarker of the circuitopathy [e.g. Little and Brown, 2014]. Therefore, introducing ~100 Hz stimulation into a pathway also associated with transmitting the lower frequency “pathological” oscillation can result in a selective synaptic suppression of the lower frequency content transmission. This can occur via DBS-induced short-term depression, where after a variable time course of equilibration, the total synaptic drive from the input pathway to the output pathway is only moderately increased (or possibly even decreased) from baseline during DBS, but the transfer of low frequency synchrony/oscillation is substantially suppressed (Fig. 3).
Figure 3.
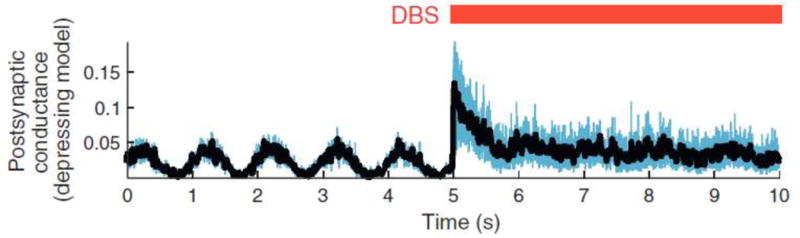
Synaptic filtering. Theoretical model demonstrating the post-synaptic conductance of a neuron driven by a low frequency oscillation. Application of high frequency DBS to the pre-synaptic drive abolishes the low frequency oscillation. Adapted from Rosenbaum et al. [2014].
V. Role of Non-Neuronal Tissue
The evolving concepts of DBS-induced synaptic filtering appear to represent a promising platform for analyzing the basic mechanisms of DBS at the molecular level. Key to such analyses are intimate understanding of the basic synaptic unit and its molecular control under high frequency driving. Unfortunately, experiments and data specifically focused on DBS questions at this level are limited. In addition, the control and regulation of the synapse is really an interplay between neuronal and non-neuronal tissue [Perea et al., 2009]. Therefore, it is likely that DBS-induced synaptic events effect both the pre- and post-synaptic terminals, as well as astrocytes and blood vessels associated with the stimulated brain circuitry (Fig. 4). In non-neuronal tissue these effects can range from modulating the release of gliotransmitters from astrocytes to changing the permeability of blood vessels. The specific molecular signaling mechanisms for the tools of synaptic modification are beyond the scope of this review, but likely represent important aspects of the overall mechanisms of DBS [Vedam-Mai et al., 2012].
Figure 4.
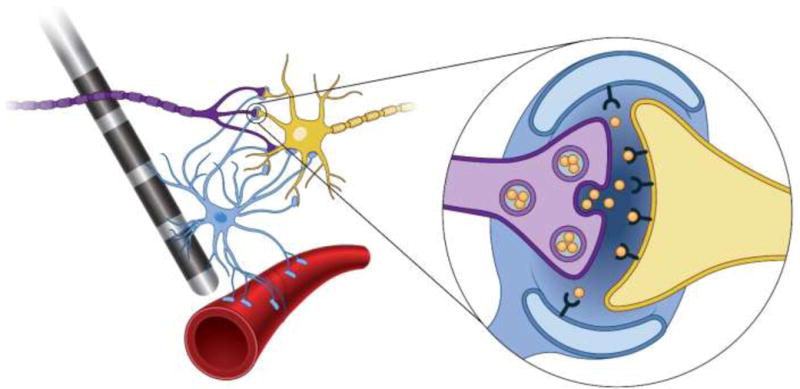
Neuronal and non-neuronal DBS effects. Direct modulation of the axonal firing of the purple neuron generates synaptic action on the yellow neuron. Stimulation induced release of neurotransmitters (inset – orange molecules) generate calcium waves in the blue astrocyte, which can then generate the release of gliotransmitters, modulating synaptic plasticity, and dilating the arteriole (red tube), increasing blood flow to the region.
Astrocytes far outnumber neurons and play important roles in neuronal metabolism and homeostasis of the extracellular medium [Kettenmann and Ransom, 2005]. They can also integrate synaptic information and regulate synaptic plasticity. As part of a tripartite synapse, astrocytes respond to neurotransmitter release with their own calcium elevations that generate calcium-dependent release of gliotransmitters, which can include glutamate and/or adenosine [Perea et al., 2009]. Basal ganglia synapses are known to display the structural features of tripartite synapses [Villalba and Smith, 2011]. However, the parkinsonian state is associated with significant pathology of glutamatergic synapses that may impact the interactions between neurons and astrocytes [Villalba et al., 2015]. Nonetheless, while astrocytes operate on slower time scales than synaptic neurotransmission, they are known to transiently control synaptic strength, as well as contribute to long-term plasticity [Perea and Araque, 2005].
While work specifically focused on the effects of DBS on astrocytes is still in its infancy, some details on their direct modulation during stimulation are beginning to emerge. Key questions on the role of astrocytes in DBS revolve around their response to prolonged high frequency synaptic activity. It has been suggested that astrocytic calcium, and subsequent gliotransmitter release, is depressed during high stimulation frequencies and enhanced during low frequency stimulation [Perea and Araque, 2005]. However, detailed experiments in rodents specifically focused on studying the role of astrocytes in DBS have clearly demonstrated the release of gliotransmitters in response to ~100 Hz stimulation [Bekar et al., 2008; Tawfik et al., 2010]. Of particular interest, in vivo experiments showed that cortical astrocytes exhibited a rapid calcium increase in response to thalamic stimulation in both a frequency and amplitude dependent manner [Bekar et al., 2008 sup. mat.]. Given that the dilation of cortical arterioles triggered by neuronal activity is dependent on glutamate-mediated calcium oscillations in astrocytes [Zonta et al., 2003], these findings also correspond well with recent in vivo optical measurements of increased cortical perfusion during high frequency thalamic stimulation in rats [Noor et al., 2015]. Therefore, axonal projections far from the site of stimulation appear to exhibit robust synaptic activity that can influence not only post-synaptic neural spiking, but also non-neuronal tissue of that distant nucleus (Fig. 4). These neuronal and non-neuronal responses appear to be interlinked and could likely work in concert to generate the synaptic filtering effects described above (Fig. 3).
Another interesting role of astrocytes in the mechanisms of DBS could be their control of the time course of synaptic filtering. Understanding the rate at which DBS modulated synapses recover from the initial onslaught of high frequency neurotransmitter release and then reach a new operating equilibrium during tonic stimulation, is of great importance for drawing links between the possible “molecular” mechanisms of DBS and the possible “network” mechanisms of DBS. Such linkages could also expand our understanding of the subsequent behavioral effects observed in patients. For example, the variable time courses of DBS-induced symptom control in patients cover a wide range that spans from seconds to days (Fig. 1). Not surprisingly, synaptic plasticity has been proposed to play a key role in the time course of modulation for various symptoms [e.g. Cooper et al., 2011]. In addition, recent results from chronic DBS in rodents have demonstrated the ability to restore both forms of corticostriatal synaptic plasticity, long-term depression and potentiation, which were impaired in parkinsonian rats [Chassain et al., 2016]. Astrocytes likely play an important role in DBS-induced synaptic plasticity changes, especially at the longer time scales, and understanding their effect represents an important direction for the future.
VI. Conclusions
Developing a conceptual understanding of the direct neural and non-neural responses to DBS is necessary for characterizing the circuit modulation induced by stimulation. In addition, interesting phenomena that remain to be fully characterized are what happens to DBS-induced APs once they reach their axon terminals (locally and distally, in both the antidromic and orthodromic directions). Do they faithfully trigger synaptic vesicle release in perpetuity? At what fidelity? Does high frequency driving of the axon terminal deplete the readily releasable pool of neurotransmitters and/or overwhelm the synaptic vesicle machinery? How do the astrocytes maintain the homeostasis of the extracellular medium during DBS? And, how do these effects (whatever they are) impact information transfer between the directly connected nuclei, as well as information transfer throughout the entire circuit?
Available experimental data can provides some clues toward answering these questions at the terminal level [e.g. Shen et al., 2003], at the pathway level [e.g. Gradinaru et al., 2009], at the microcircuit level [e.g. Hashimoto et al., 2003], at the brain circuit level [e.g. Fox et al., 2014], as well as the underlying neurochemistry changes [e.g. Melon et al., 2015]. In addition, computational models are beginning to integrate these types of experimental results into unifying hypotheses [e.g. Rosenbaum et al., 2014]. However, an enormous systems neuroscience challenge remains to map the diverse array of directly stimulated pathways, identify their synaptic targets, and evaluate the functional connectivity between the nuclei that make up the networks responsible for the underlying circuitopathies treated by DBS.
While many of the key next steps in defining the mechanisms of DBS appear to be biased toward system-level or network analyses, the root of any electrical stimulation therapy is to control the release of neurotransmitters in directly stimulated neurons. In addition, new tools are becoming available to measure neurochemistry modulation in humans during DBS [e.g. Grahn et al., 2014; Bennet et al., 2016] and these results will extend the foundational data on neurotransmitter fluctuations that were generated in rodent DBS models. Together with knowledge from the molecular mechanisms of synaptic transmission and plasticity, a neurochemical basis should facilitate detailed explanation of the network mechanisms of DBS. This will require new neurochemistry experiments specifically designed to address DBS questions and elucidate the modulation of neurotransmitters during therapeutic DBS conditions.
Acknowledgments
This work was supported by grant R01 NS086100 from the National Institutes of Health.
ABBREVIATIONS
- DBS
Deep Brain Stimulation
- PD
Parkinson’s Disease
- AP
Action Potential
- STN
Subthalamic Nucleus
- GP
Globus Pallidus
- SN
Substantia Nigra
Footnotes
Conflict of Interest Statement: CCM is a paid consultant for Boston Scientific Neuromodulation, and is a shareholder in the following companies: Surgical Information Sciences, Inc.; Autonomic Technologies, Inc.; Cardionomic, Inc.; Enspire DBS, Inc.; Neuros Medical, Inc.
References
- Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–81. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- Anderson TR, Hu B, Iremonger K, Kiss ZH. Selective attenuation of afferent synaptic transmission as a mechanism of thalamic deep brain stimulation-induced tremor arrest. J Neurosci. 2006 Jan 18;26(3):841–50. doi: 10.1523/JNEUROSCI.3523-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekar L, Libionka W, Tian GF, Xu Q, Torres A, Wang X, Lovatt D, Williams E, Takano T, Schnermann J, Bakos R, Nedergaard M. Adenosine is crucial for deep brain stimulation-mediated attenuation of tremor. Nat Med. 2008 Jan;14(1):75–80. doi: 10.1038/nm1693. [DOI] [PubMed] [Google Scholar]
- Benabid AL, Pollak P, Gervason C, Hoffmann D, Gao DM, Hommel M, Perret JE, de Rougemont J. Long-term suppression of tremor by chronic stimulation of the ventral intermediate thalamic nucleus. Lancet. 1991 Feb 16;337(8738):403–6. doi: 10.1016/0140-6736(91)91175-t. [DOI] [PubMed] [Google Scholar]
- Benabid AL, Benazzouz A, Hoffmann D, Limousin P, Krack P, Pollak P. Long-term electrical inhibition of deep brain targets in movement disorders. Mov Disord. 1998;13(Suppl 3):119–25. doi: 10.1002/mds.870131321. [DOI] [PubMed] [Google Scholar]
- Benazzouz A, Gao DM, Ni ZG, Piallat B, Bouali-Benazzouz R, Benabid AL. Effect of high-frequency stimulation of the subthalamic nucleus on the neuronal activities of the substantia nigra pars reticulata and ventrolateral nucleus of the thalamus in the rat. Neuroscience. 2000;99(2):289–95. doi: 10.1016/s0306-4522(00)00199-8. [DOI] [PubMed] [Google Scholar]
- Bennet KE, Tomshine JR, Min HK, Manciu FS, Marsh MP, Paek SB, Settell ML, Nicolai EN, Blaha CD, Kouzani AZ, Chang SY, Lee KH. A Diamond-Based Electrode for Detection of Neurochemicals in the Human Brain. Front Hum Neurosci. 2016 Mar 15;10:102. doi: 10.3389/fnhum.2016.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld Z, Brontë-Stewart H. High Frequency Deep Brain Stimulation and Neural Rhythms in Parkinson’s Disease. Neuropsychol Rev. 2015 Dec;25(4):384–97. doi: 10.1007/s11065-015-9308-7. [DOI] [PubMed] [Google Scholar]
- Boulet S, Lacombe E, Carcenac C, Feuerstein C, Sgambato-Faure V, Poupard A, Savasta M. Subthalamic stimulation-induced forelimb dyskinesias are linked to an increase in glutamate levels in the substantia nigra pars reticulata. J Neurosci. 2006 Oct 18;26(42):10768–76. doi: 10.1523/JNEUROSCI.3065-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher D, Goaillard JM. Beyond faithful conduction: short-term dynamics, neuromodulation, and long-term regulation of spike propagation in the axon. Prog Neurobiol. 2011 Sep 1;94(4):307–46. doi: 10.1016/j.pneurobio.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnan H, Duff EP, Brown P. The relative phases of basal ganglia activities dynamically shape effective connectivity in Parkinson’s disease. Brain. 2015 Jun;138(Pt 6):1667–78. doi: 10.1093/brain/awv093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassain C, Melon C, Salin P, Vitale F, Couraud S, Durif F, Kerkerian-Le Goff L, Gubellini P. Metabolic, synaptic and behavioral impact of 5-week chronic deep brain stimulation in hemiparkinsonian rats. J Neurochem. 2016 Mar;136(5):1004–16. doi: 10.1111/jnc.13438. [DOI] [PubMed] [Google Scholar]
- Chaturvedi A, Butson CR, Lempka SF, Cooper SE, McIntyre CC. Patient-specific models of deep brain stimulation: influence of field model complexity on neural activation predictions. Brain Stimul. 2010;3(2):65–7. doi: 10.1016/j.brs.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi A, Foutz TJ, McIntyre CC. Current steering to activate targeted neural pathways during deep brain stimulation of the subthalamic region. Brain Stimul. 2012 Jul;5(3):369–77. doi: 10.1016/j.brs.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SE, Noecker AM, Abboud H, Vitek JL, McIntyre CC. Return of bradykinesia after subthalamic stimulation ceases: relationship to electrode location. Exp Neurol. 2011 Oct;231(2):207–13. doi: 10.1016/j.expneurol.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hemptinne C, Swann NC, Ostrem JL, Ryapolova-Webb ES, San Luciano M, Galifianakis NB, Starr PA. Therapeutic deep brain stimulation reduces cortical phase-amplitude coupling in Parkinson’s disease. Nat Neurosci. 2015 May;18(5):779–86. doi: 10.1038/nn.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MD, Buckner RL, Liu H, Chakravarty MM, Lozano AM, Pascual-Leone A. Resting-state networks link invasive and noninvasive brain stimulation across diverse psychiatric and neurological diseases. Proc Natl Acad Sci. 2014;111(41):E4367–75. doi: 10.1073/pnas.1405003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostrovsky JO, Levy R, Wu JP, Hutchison WD, Tasker RR, Lozano AM. Microstimulation-induced inhibition of neuronal firing in human globus pallidus. J Neurophysiol. 2000 Jul;84(1):570–4. doi: 10.1152/jn.2000.84.1.570. [DOI] [PubMed] [Google Scholar]
- Galati S, Mazzone P, Fedele E, Pisani A, Peppe A, Pierantozzi M, Brusa L, Tropepi D, Moschella V, Raiteri M, Stanzione P, Bernardi G, Stefani A. Biochemical and electrophysiological changes of substantia nigra pars reticulata driven by subthalamic stimulation in patients with Parkinson’s disease. Eur J Neurosci. 2006 Jun;23(11):2923–8. doi: 10.1111/j.1460-9568.2006.04816.x. [DOI] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324(5925):354–9. doi: 10.1126/science.1167093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahn PJ, Mallory GW, Khurram OU, Berry BM, Hachmann JT, Bieber AJ, Bennet KE, Min HK, Chang SY, Lee KH, Lujan JL. A neurochemical closed-loop controller for deep brain stimulation: toward individualized smart neuromodulation therapies. Front Neurosci. 2014 Jun 25;8:169. doi: 10.3389/fnins.2014.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill WM, Snyder AN, Miocinovic S. Deep brain stimulation creates an informational lesion of the stimulated nucleus. Neuroreport. 2004 May 19;15(7):1137–40. doi: 10.1097/00001756-200405190-00011. [DOI] [PubMed] [Google Scholar]
- Grill WM, Cantrell MB, Robertson MS. Antidromic propagation of action potentials in branched axons: implications for the mechanisms of action of deep brain stimulation. J Comput Neurosci. 2008 Feb;24(1):81–93. doi: 10.1007/s10827-007-0043-9. [DOI] [PubMed] [Google Scholar]
- Groppa S, Herzog J, Falk D, Riedel C, Deuschl G, Volkmann J. Physiological and anatomical decomposition of subthalamic neurostimulation effects in essential tremor. Brain. 2014 Jan;137(Pt 1):109–21. doi: 10.1093/brain/awt304. [DOI] [PubMed] [Google Scholar]
- Hahn PJ, McIntyre CC. Modeling shifts in the rate and pattern of subthalamopallidal network activity during deep brain stimulation. J Comput Neurosci. 2010 Jun;28(3):425–41. doi: 10.1007/s10827-010-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman CD, Henderson JM, Finkelstein DI, Horne MK, Paxinos G, Halliday GM. Comparison of the basal ganglia in rats, marmosets, macaques, baboons, and humans: volume and neuronal number for the output, internal relay, and striatal modulating nuclei. J Comp Neurol. 2002 Apr 8;445(3):238–55. doi: 10.1002/cne.10165. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Elder CM, Okun MS, Patrick SK, Vitek JL. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J Neurosci. 2003;23(5):1916–23. doi: 10.1523/JNEUROSCI.23-05-01916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AL, Durand DM. High frequency stimulation can block axonal conduction. Exp Neurol. 2009 Nov;220(1):57–70. doi: 10.1016/j.expneurol.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MD, Miocinovic S, McIntyre CC, Vitek JL. Mechanisms and targets of deep brain stimulation in movement disorders. Neurotherapeutics. 2008 Apr;5(2):294–308. doi: 10.1016/j.nurt.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Ransom BR. Neuroglia. 2nd. Oxford University Press; 2005. [Google Scholar]
- Kühn AA, Tsui A, Aziz T, Ray N, Brücke C, Kupsch A, Schneider GH, Brown P. Pathological synchronisation in the subthalamic nucleus of patients with Parkinson’s disease relates to both bradykinesia and rigidity. Exp Neurol. 2009 Feb;215(2):380–7. doi: 10.1016/j.expneurol.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Laitinen LV, Bergenheim AT, Hariz MI. Leksell’s posteroventral pallidotomy in the treatment of Parkinson’s disease. J Neurosurg. 1992 Jan;76(1):53–61. doi: 10.3171/jns.1992.76.1.0053. [DOI] [PubMed] [Google Scholar]
- Lee KH, Chang SY, Roberts DW, Kim U. Neurotransmitter release from high-frequency stimulation of the subthalamic nucleus. J Neurosurg. 2004 Sep;101(3):511–7. doi: 10.3171/jns.2004.101.3.0511. [DOI] [PubMed] [Google Scholar]
- Little S, Brown P. The functional role of beta oscillations in Parkinson’s disease. Parkinsonism Relat Disord. 2014 Jan;20(Suppl 1):S44–8. doi: 10.1016/S1353-8020(13)70013-0. [DOI] [PubMed] [Google Scholar]
- Lozano AM, Dostrovsky J, Chen R, Ashby P. Deep brain stimulation for Parkinson’s disease: disrupting the disruption. Lancet Neurol. 2002 Aug;1(4):225–31. doi: 10.1016/s1474-4422(02)00101-1. [DOI] [PubMed] [Google Scholar]
- Lujan JL, Chaturvedi A, Malone DA, Rezai AR, Machado AG, McIntyre CC. Axonal pathways linked to therapeutic and nontherapeutic outcomes during psychiatric deep brain stimulation. Hum Brain Mapp. 2012;33(4):958–68. doi: 10.1002/hbm.21262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean PD. Psychosomatic disease and the visceral brain; recent developments bearing on the Papez theory of emotion. Psychosom Med. 1949;11(6):338–53. doi: 10.1097/00006842-194911000-00003. [DOI] [PubMed] [Google Scholar]
- McIntyre CC, Hahn PJ. Network perspectives on the mechanisms of deep brain stimulation. Neurobiol Dis. 2010 Jun;38(3):329–37. doi: 10.1016/j.nbd.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre CC, Grill WM, Sherman DL, Thakor NV. Cellular effects of deep brain stimulation: model-based analysis of activation and inhibition. J Neurophysiol. 2004a;91(4):1457–69. doi: 10.1152/jn.00989.2003. [DOI] [PubMed] [Google Scholar]
- McIntyre CC, Mori S, Sherman DL, Thakor NV, Vitek JL. Electric field and stimulating influence generated by deep brain stimulation of the subthalamic nucleus. Clin Neurophysiol. 2004b;115(3):589–95. doi: 10.1016/j.clinph.2003.10.033. [DOI] [PubMed] [Google Scholar]
- McNeal DR. Analysis of a model for excitation of myelinated nerve. IEEE Trans Biomed Eng. 1976;23(4):329–337. doi: 10.1109/tbme.1976.324593. [DOI] [PubMed] [Google Scholar]
- Melon C, Chassain C, Bielicki G, Renou JP, Kerkerian-Le Goff L, Salin P, Durif F. Progressive brain metabolic changes under deep brain stimulation of subthalamic nucleus in parkinsonian rats. J Neurochem. 2015 Mar;132(6):703–12. doi: 10.1111/jnc.13015. [DOI] [PubMed] [Google Scholar]
- Miocinovic S, Lempka SF, Russo GS, Maks CB, Butson CR, Sakaie KE, Vitek JL, McIntyre CC. Experimental and theoretical characterization of the voltage distribution generated by deep brain stimulation. Exp Neurol. 2009;216(1):166–76. doi: 10.1016/j.expneurol.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor MS, Murari K, McCracken CB, Kiss ZH. Spatiotemporal dynamics of cortical perfusion in response to thalamic deep brain stimulation. Neuroimage. 2015 Nov 11;126:131–139. doi: 10.1016/j.neuroimage.2015.11.017. [DOI] [PubMed] [Google Scholar]
- Papez JW. A proposed mechanism of emotion. Arch Neurol Psychiatry. 1937;38:725–43. [Google Scholar]
- Perea G, Araque A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci. 2005 Mar 2;25(9):2192–203. doi: 10.1523/JNEUROSCI.3965-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009 Aug;32(8):421–31. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Perlmutter JS, Mink JW, Bastian AJ, Zackowski K, Hershey T, Miyawaki E, Koller W, Videen TO. Blood flow responses to deep brain stimulation of thalamus. Neurology. 2002 May 14;58(9):1388–94. doi: 10.1212/wnl.58.9.1388. [DOI] [PubMed] [Google Scholar]
- Plonsey R, Barr RC. Bioelectricity, a quantitative approach. 3rd. Springer; 2007. [Google Scholar]
- Quinn EJ, Blumenfeld Z, Velisar A, Koop MM, Shreve LA, Trager MH, Hill BC, Kilbane C, Henderson JM, Brontë-Stewart H. Beta oscillations in freely moving Parkinson’s subjects are attenuated during deep brain stimulation. Mov Disord. 2015 Nov;30(13):1750–8. doi: 10.1002/mds.26376. [DOI] [PubMed] [Google Scholar]
- Ranck JB. Which elements are excited in electrical stimulation of mammalian central nervous system: a review. Brain Res. 1975;98(3):417–40. doi: 10.1016/0006-8993(75)90364-9. [DOI] [PubMed] [Google Scholar]
- Rosenbaum R, Zimnik A, Zheng F, Turner RS, Alzheimer C, Doiron B, Rubin JE. Axonal and synaptic failure suppress the transfer of firing rate oscillations, synchrony and information during high frequency deep brain stimulation. Neurobiol Dis. 2014 Feb;62:86–99. doi: 10.1016/j.nbd.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse AG, Stanslaski SR, Cong P, Jensen RM, Afshar P, Ullestad D, Gupta R, Molnar GF, Moran DW, Denison TJ. A chronic generalized bi-directional brain-machine interface. J Neural Eng. 2011 Jun;8(3):036018. doi: 10.1088/1741-2560/8/3/036018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato F, Parent M, Levesque M, Parent A. Axonal branching pattern of neurons of the subthalamic nucleus in primates. J Comp Neurol. 2000 Aug 14;424(1):142–52. doi: 10.1002/1096-9861(20000814)424:1<142::aid-cne10>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Shen KZ, Zhu ZT, Munhall A, Johnson SW. Synaptic plasticity in rat subthalamic nucleus induced by high-frequency stimulation. Synapse. 2003;50(4):314–9. doi: 10.1002/syn.10274. [DOI] [PubMed] [Google Scholar]
- Stefani A, Fedele E, Galati S, Pepicelli O, Frasca S, Pierantozzi M, Peppe A, Brusa L, Orlacchio A, Hainsworth AH, Gattoni G, Stanzione P, Bernardi G, Raiteri M, Mazzone P. Subthalamic stimulation activates internal pallidus: evidence from cGMP microdialysis in PD patients. Ann Neurol. 2005 Mar;57(3):448–52. doi: 10.1002/ana.20402. [DOI] [PubMed] [Google Scholar]
- Stefani A, Fedele E, Pierantozzi M, Galati S, Marzetti F, Peppe A, Pastore FS, Bernardi G, Stanzione P. Reduced GABA Content in the Motor Thalamus during Effective Deep Brain Stimulation of the Subthalamic Nucleus. Front Syst Neurosci. 2011 Apr 5;5:17. doi: 10.3389/fnsys.2011.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik VL, Chang SY, Hitti FL, Roberts DW, Leiter JC, Jovanovic S, Lee KH. Deep brain stimulation results in local glutamate and adenosine release: investigation into the role of astrocytes. Neurosurgery. 2010 Aug;67(2):367–75. doi: 10.1227/01.NEU.0000371988.73620.4C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbano FJ, Leznik E, Llinas RR. Cortical activation patterns evoked by afferent axons stimuli at different frequencies: an in vitro voltage-sensitive dye imaging study. Thalamus Rel Syst. 2002;1:371–378. [Google Scholar]
- Vedam-Mai V, van Battum EY, Kamphuis W, Feenstra MG, Denys D, Reynolds BA, Okun MS, Hol EM. Deep brain stimulation and the role of astrocytes. Mol Psychiatry. 2012 Feb;17(2):124–31. doi: 10.1038/mp.2011.61. [DOI] [PubMed] [Google Scholar]
- Villalba RM, Smith Y. Neuroglial plasticity at striatal glutamatergic synapses in Parkinson’s disease. Front Syst Neurosci. 2011 Aug 23;5:68. doi: 10.3389/fnsys.2011.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Mathai A, Smith Y. Morphological changes of glutamatergic synapses in animal models of Parkinson’s disease. Front Neuroanat. 2015 Sep 25;9:117. doi: 10.3389/fnana.2015.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RH, Koch RJ, Sweeney JE, Moore C, Meshul CK. Effects of subthalamic nucleus lesions and stimulation upon glutamate levels in the dopamine-depleted rat striatum. Neuroreport. 2009 May 27;20(8):770–5. doi: 10.1097/WNR.0b013e32832ad556. [DOI] [PubMed] [Google Scholar]
- Windels F, Bruet N, Poupard A, Urbain N, Chouvet G, Feuerstein C, Savasta M. Effects of high frequency stimulation of subthalamic nucleus on extracellular glutamate and GABA in substantia nigra and globus pallidus in the normal rat. Eur J Neurosci. 2000 Nov;12(11):4141–6. doi: 10.1046/j.1460-9568.2000.00296.x. [DOI] [PubMed] [Google Scholar]
- Windels F, Carcenac C, Poupard A, Savasta M. Pallidal origin of GABA release within the substantia nigra pars reticulata during high-frequency stimulation of the subthalamic nucleus. J Neurosci. 2005 May 18;25(20):5079–86. doi: 10.1523/JNEUROSCI.0360-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Miocinovic S, Zhang J, Baker KB, McIntyre CC, Vitek JL. Dissociation of motor symptoms during deep brain stimulation of the subthalamic nucleus in the region of the internal capsule. Exp Neurol. 2011 Apr;228(2):294–7. doi: 10.1016/j.expneurol.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirh TA, Lenz FA, Reich SG, Dougherty PM. Patterns of bursting occurring in thalamic cells during parkinsonian tremor. Neuroscience. 1998 Mar;83(1):107–21. doi: 10.1016/s0306-4522(97)00295-9. [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003 Jan;6(1):43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]