

Abstract
PURPOSE: Although targeting angiogenesis with tyrosine kinase inhibitors (TKIs) has become standard of care in the treatment of clear cell renal cell carcinoma (RCC), resistance mechanism are not fully understood, and there is a need to develop new therapeutic options overcoming them. METHODS AND MATERIALS: To develop a preclinical model that predicts clinical activity of novel agents in 19 RCC patients, we established patient-derived cell (PDC) and xenograft (PDX) models derived from malignant effusions or surgical specimen. RESULTS: Successful PDCs, defined as cells that maintained growth following two passages, were established in 5 of 15 malignant effusions and 1 of 4 surgical specimens. One PDC, clinically refractory to TKIs, was implanted and engrafted in mice, resulting in a comparable histology to the primary tumor. The PDC-PDX model also showed similar genomic features when tested using targeted sequencing of cancer-related genes. When we examined the drug effects of the PDX model, the tumor cells showed resistance to TKIs and everolimus in vitro. CONCLUSION: The results suggest that the PDC-PDX preclinical model we developed using malignant effusions can be a useful preclinical model to interrogate sensitivity to targeted agents based on genomic alterations.
Introduction
Clear cell renal cell carcinoma (RCC) represents a unique clinical setting for the application of antiangiogenic therapy. Targeting angiogenesis via the vascular endothelial growth factor receptor (VEGFR) or mammalian target of rapamycin (mTOR) pathways has produced robust clinical effects and revolutionized the treatment of metastatic RCC (mRCC) [1]. Multitargeted tyrosine kinase inhibitors (TKIs) against VEGFR such as sunitinib [2], sorafenib [3], and pazopanib [4] have demonstrated improved progression-free survival and/or overall survival compared with interferon and/or supportive care. However, some patients with clear cell mRCC who received these TKIs do not achieve response. Also, in most responders, resistance to therapy will eventually develop. While the mechanisms of resistance to VEGFR TKIs are not yet well understood, there is a need to develop new therapeutic options overcoming TKI resistance. The goal should be met through preclinical models that reliably predict clinical activity of novel antiangiogenic compounds in patients.
It becomes increasingly clear that novel preclinical models that more closely simulate the heterogeneity of human cancers are needed for more efficient oncology drug development. Until recently, drug screening of cancer has emphasized xenograft models derived from the established, conventional cell lines and [5], in some cases, from patient samples [6]. As the limitations of current xenograft models derived from previously established cell lines have been well described [7], patient-derived xenograft (PDX) models may provide more accurate depiction of the human cancers they are derived from than cell line–derived xenografts. As patient-derived models might reflect a clinical response better [8] and the ability to obtain metastatic tumor samples is not always possible, we already have established disease-specific panels of patient-derived cell (PDC) models directly from malignant effusions [9]. Both PDC lines and patient-derived xenografts (PDX) made from malignant effusions are alternative models that may overcome sample challenges.
So far, numerous tumor-specific PDX models have been established, and importantly, they are biologically mostly stable when passaged in mice in terms of gene expression patterns, mutational status, metastatic potential, and drug responsiveness [10]. However, the practical relevance of PDX models for application in clinical oncology is limited owing to the time required for PDX establishment (~4 months) since mRCC patients with refractory disease live less than 1 year [11]. Despite an obvious advantage of PDX over xenografts from cell lines [12], their application has been criticized by the fact that many PDXs are established from the primary tumors or, in some cases, from metastatic sites of previously untreated patients. Thereby, they fail to reproduce the refractory patient population in whom most novel therapeutics will undergo their initial trials [13]. In addition, tumor take rates may be higher for metastases with more aggressive phenotypes than primary tumors.
In an effort to develop a novel PDX model with PDCs, we established a large collection of RCC PDC models directly derived from malignant effusions or ascites collected after TKI failure. This model could be used to develop new therapeutic targets, to better understand the basis of sensitivity of tumors from individual patients, and potentially to help the stratification of patients according to molecular characteristics. TKI-resistant PDCs were selected and tested further using PDX cells.
Methods
This prospective, pilot study is a part of the Samsung Medical Center Oncology Biomarker study (ClinicalTrials.gov identifier: NCT01831609). The Institutional Review Board at Samsung Medical Center (Seoul, Korea) approved the study. After obtaining a written informed consent, we collected effusions or ascites from 15 adult (>18 years), clear cell mRCC patients who failed treatment with sunitinib or pazopanib as a first-line TKI therapy. Effusions were obtained for a therapeutic purpose and in accordance with the Declaration of Helsinki. In addition, four RCC samples from surgical resection of metastases were collected for primary culture. Details of patient characteristics are listed in Table 1. Patients had either intrinsic (i.e., primary; n = 14) or developed secondary (n = 5) resistance to TKIs. We defined patients with intrinsic resistance as those who had progressive disease (PD) as their best response during the first 8 weeks after starting TKI treatment. In accordance with the Response Evaluation Criteria in Solid tumors, PD was defined as a >20% increase in the sum of the diameters of the target lesions, and the sum must also demonstrate an absolute increase of at least 5 mm. Patients who showed complete response (CR), partial response (PR), or stable disease (SD) as their best response to TKIs but eventually progressed were classified as those with secondary resistance.
Table 1.
Patient Characteristics
| No. | Age/Sex | Therapy | Resistance | Survival | Origin of PDC | Cell Number | Passage |
|---|---|---|---|---|---|---|---|
| 123 | 48/M | Sunitinib | Intrinsic | 6 mo | Surgery | 0 | |
| 161 | 55/F | Sunitinib | Intrinsic | 8 mo | Ascites | P1 | |
| 162 | 66/M | Pazopanib | Intrinsic | 6 mo | Surgery | 0 | |
| 166 | 71/M | Sunitinib | Secondary | 14 mo | Surgery | P2 | |
| 189 | 59/F | Sunitinib | Secondary | 21 mo | Surgery | 0 | |
| 192 | 52/F | Sunitinib | Secondary | 16 mo | Ascites | P1 | |
| 391 | 53/M | Sunitinib | Secondary | 13 mo | Ascites | 1.26E+ 07/ml | P1 |
| 395 | 39/M | Sunitinib | Intrinsic | 4 mo | Pleural effusion | 1.20E+ 07/ml | P2 |
| 413 | 67/M | Sunitinib | Secondary | 27 mo | Pericardial effusion | 1.73E+ 07/ml | 0 |
| 426 | 55/M | Sunitinib | Intrinsic | 9 mo | Pleural effusion | 2.00E+ 06/ml | 0 |
| 483 | 53/M | Sunitinib | Intrinsic | 5 mo | Pleural effusion | 2.30E+ 06/ml | 0 |
| 491 | 62/M | Pazopanib | Intrinsic | 11 mo | Pleural effusion | 1.22E+ 07/ml | P1 |
| 518 | 65/M | Pazopanib | Intrinsic | 19 mo | Pleural effusion | 1.20E+ 06/ml | 0 |
| 539 | 63/F | Sunitinib | Secondary | 26 mo | Ascites | 5.00E+ 06/ml | P3 |
| 573 | 36/M | Pazopanib | Intrinsic | 8 mo | Pleural effusion | 3.40E+ 07/ml | P3 |
| 575 | 44/M | Pazopanib | Intrinsic | 13 mo | Pleural effusion | 5.00E+ 05/ml | 0 |
| 590 | 42/F | Sunitinib | Intrinsic | 6 mo | Ascites | 6.08E+ 06/ml | P2 |
| 603 | 61/M | Sunitinib | Secondary | 37 mo | Pleural effusion | 8.83E+ 06/ml | P2 |
| 624 | 54/F | Sunitinib | Intrinsic | 9 mo | Ascites | 6.39E+ 06/ml | 0 |
Primary cultures of human effusions and metastatic tumors were conducted according to a method described previously [9]. In brief, collected effusions (1 to 5 l) were divided into 50-ml tubes, centrifuged at 1500 rpm for 10 minutes, and washed twice with PBS. Cell pellets were resuspended in culture medium and divided into 75-cm2 culture flasks. Cells were grown in RPMI 1640 supplemented with 10% FBS and 1% antibiotic-antimycotic solution (Gibco BRL, Paisley, UK). The media were changed every 3 days, and cells were maintained at 37°C in a humidified 5% CO2 incubator. PDCs were passaged using TrypLE Express (Gibco BRL) to detach cells when the culture was at 80% to 90% confluence. A similar process of tumor primary culture was conducted on snap-frozen tumor samples collected immediately after surgery.
Once the cells reached 80% to 90% confluence, they were washed and detached using TrypLE Express, and incubated for 3 minutes at 37°C with 5% CO2. Following detachment, 4 ml of complete culture media was added to stop the activity of trypsin, and cells were transferred to a 15-ml sterile centrifuge tube. After centrifugation, cells were resuspended in 1 ml of freezing medium (Cellbanker, Zenoaq, Japan), transferred into cryovials (Nalge Nunc, Naperville, IL), and slowly frozen in a −80°C freezer overnight.
PDCs were received frozen and were transferred to Oncotest GmbH (Germany) for the establishment of PDX models [9]. On site, cells were thawed, the freezing media removed, and the cells resuspended and transferred into T75 flasks. Cells were grown for 3 to 7 days in RPMI/10% FBS until the culture reached around 80% confluence. Cells were collected and counted, and 5 × 106 cells were injected into the hind flanks of NOD scid gamma (NSG) mice (Jackson Laboratories). Tumors developed within 25 to 85 days postinjection. These tumors were explanted, and viable portions of the tumors were cut into pieces and implanted subcutaneously into female NMRI nu/nu mice (Harlan Laboratories). This process was repeated to serially passage the respective models. From each passage, formalin-fixed, paraffin-embedded blocks were prepared, and tumor slices were stained with hematoxylin and eosin. Slides were scanned with a Hamamatsu slide scanner, and images were extracted using the Nanozoomer program from Hamamatsu. All animal handling and experiments with animals were in accordance with the guidelines set by the Samsung Biological Research Institute (Seoul, Korea).
To genomically compare the final PDX model to the primary tumor, we performed targeted deep sequencing using Illumina HiSeq 2500. Genomic DNA was extracted from a single cell suspension of PDX cells and sequenced for capturing cancer-related genes (Supplement file 1) [9]. Concurrently, copy number variations were analyzed using the nCounter Copy Number Variation Assay Kit (NanoString Technologies, Seattle, WA). Average count numbers of greater than 3 were called and confirmed by immunohistochemistry, fluorescent in situ hybridization, or real-time polymerase chain reaction.
For exploratory purposes, we examined the drug effects on cell viability with single cell suspension using the PDX tumor at passage 5. The overall process of cell proliferation inhibition assay and the high-throughput screening (Samsung Electro-Mechanics CO [SEMCO], Suwon, Korea) is summarized in Supplement file 2 [14], and cell viability was determined with the CellTiter 96 Aqueous One Solution assay (Promega, Madison, WI) according to the manufacturer's protocol. Agents including sunitinib, pazopanib, lapatinib, and everolimus were purchased from Selleck Chemicals (Houston, TX).
Results
PDCs were derived from malignant effusions (n = 15) and surgical specimens (n = 4) of metastatic disease. All patients had clear cell carcinoma in either alveolar or tubular configurations. These tumors were aggressive in clinical behavior; 12 patients experienced intrinsic resistance to first-line sunitinib or pazopanib, whereas 7 patients had secondary resistant tumors (Table 1). Cells from all patients were stained with either hematoxylin and eosin or Papanicolaou, and micrographs were prospectively stored in our internal database. Successful PDCs, defined as cells that maintained growth following 2 passages, were established in 5 of 15 effusions and 1 of 4 surgical specimens.
After successful PDC establishment from malignant effusions, we implanted one PDC (no. 395) in NSG mice to evaluate whether PDCs could successfully be converted to a PDX model. The PDX was serially passaged for five times before it was used for experiments. The PDC-PDX model exhibited histological features and immunohistochemistry findings similar to those of the primary tumor (Figure 1). In the primary tumor, clear cells are arranged in an alveolar pattern, while granular cells are in a solid pattern. PDX tumor showed similar findings where the cytoplasm was eosinophilic and the nuclei had prominent nucleoli and coarse chromatin. The patient (no. 395, 39-year-old male) presented with a diagnosis of clear cell carcinoma of the right kidney. Fifteen months after his surgical resection, the patient developed multiple lung and lymph node metastases (Figure 2A). He had been treated with sunitinib at 50 mg per day for 4 weeks followed by 2 weeks of rest. Six weeks after starting the sunitinib therapy, metastatic lesions showed progression, and large amount of bilateral pleural effusions developed (Figure 2B). After thoracentesis for a therapeutic purpose, at which a PDC was established, everolimus had been administered but failed to achieve any clinical benefit. The clinical condition of the patient deteriorated rapidly after 1 month of everolimus therapy that only supportive care was given until death.
Figure 1.

Morphology of primary tumor (A and B) and xenograft (P1, C; P2, D; P3, E; P4, F).
Figure 2.
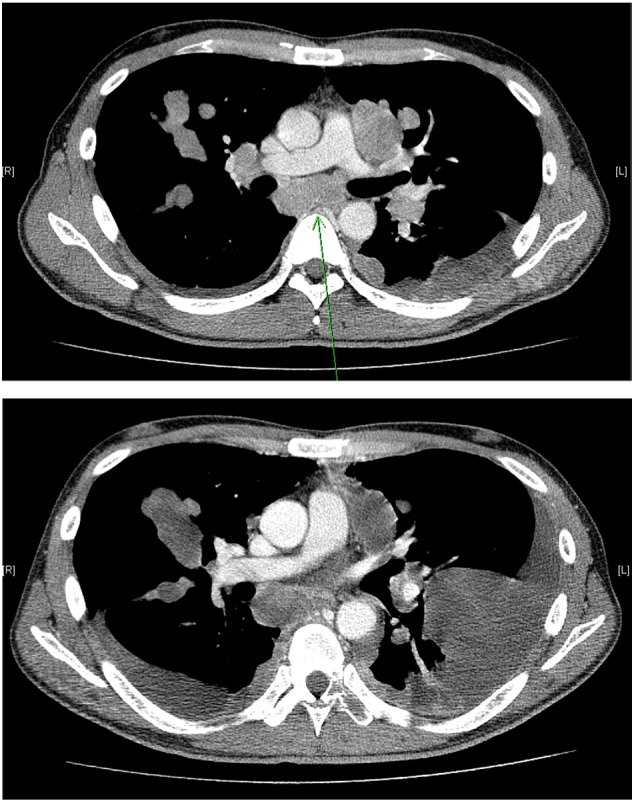
Chest computed tomography scans of the patient no. 395 with refractory RCC. (A) Baseline scan showing multiple lung and lymph node metastases. (B) Subsequent scan after 6 weeks of sunitinib therapy showing disease progression and the development of large amount of bilateral pleural effusions.
To confirm the similarity of genomic features between the primary tumor and PDX, we compared variant allele frequency results of the primary tumor and single cell suspensions from the PDX tumor at passage 5 (Table 2). With targeted sequencing for 381 cancer-related genes, these 2 samples showed surprisingly similar genomic alterations. Sequencing of primary tumor and PDX revealed mutual deletions in TP53, as well as cyclin-dependent kinase inhibitor 2A (CDKN2A) and von Hippel–Lindau (VHL), both of which are thought to be related with RCC carcinogenesis.
Table 2.
Copy Number Variations Seen in Patient (No. 395)-Derived Xenograft
| Type | Gene Name | Alteration | Sig. Ratio | Copy Number | Sig. Exon |
|---|---|---|---|---|---|
| Known | CDKN2A | Deletion | −9.8149 | 0.00222 | 5 |
| Known | TP53 | Deletion | −1.0027 | 0.998127 | 13 |
| Known | VHL | Deletion | −1.07139 | 0.951723 | 3 |
| Unknown | DDR2 | Amplification | 0.900786 | 3.734165 | 15 |
| Unknown | SRC | Amplification | 0.931351 | 3.814123 | 11 |
| Unknown | TOP1 | Amplification | 0.987788 | 3.966284 | 21 |
| Unknown | AURKB | Deletion | −0.83248 | 1.12313 | 8 |
| Unknown | FLT3 | Deletion | −0.88982 | 1.079362 | 25 |
| Unknown | MTOR | Deletion | −0.80689 | 1.143227 | 45 |
| Unknown | PTCH2 | Deletion | −0.83586 | 1.120501 | 19 |
We then examined the drug effects on cell viability as described in the Methods section. Antitumor activity of 18 drugs (pazopanib, sunitinib, everolimus, lapatinib, BEZ235, BGJ-398, and others) was tested against no. 395 PDCs (Table 3). In accordance to his clinical resistance to VEGFR TKIs and everolimus, we confirmed that cells were resistant to most VEGFR TKIs tested, with an IC50 of more than 10 μM for everolimus, pazopanib, sorafenib, and sunitinib. The drugs with IC50 values below 4.0 μM included neratinib, BEZ235, and BGJ-398. To define the role of these drugs in TKI-resistant clear cell RCC, cells were treated again to assess cell viability using CellTiter-Glo reagents (Supplement file 2). As expected, BEZ235 and BGJ-398 inhibited cell proliferation with IC50 values of 0.015 μM and 0.79 μM, respectively (Figure 3B). A subsequent immunofluorescence assay showed mTOR protein expression in the cytoplasm and FGFR2 in the nuclei (Figure 3A). Western blot confirmed the expression of mTOR/PI3K and FGFR2 proteins in the cells (Figure 3C). We observed that the inhibition of mTOR/PI3K pathway with BEZ235 resulted in the downregulation of phosphorylation of the pathway. BGJ-398 also inhibited FGFR and downstream phosphorylation of AKT, MEK, and ERK1/2. However, the results with BEZ235 and BGJ-398 were not reproduced when we tested with an in vivo PDX model (Figure 4).
Table 3.
Drug Sensitivity Profiles Seen in Patient (No. 395)-Derived Xenograft
| Drug | Mechanism | IC50 (μM) |
|---|---|---|
| AZD1775 | Wee1 kinase inhibitor | 5.1 |
| Everolimus | MTOR inhibitor | >10 |
| Crizotinib | ALK and ROS1 inhibitor | >10 |
| Pazopanib | VEGFR kinase inhibitor | >10 |
| Sorafenib | VEGFR kinase inhibitor | >10 |
| Sunitinib | VEGFR kinase inhibitor | >10 |
| Vemurafenib | Anti-BRAF antibody | >10 |
| Ceruximab | Anti-EGFR antibody | >10 |
| Trastuzumab | Anti-HER2 antibody | >10 |
| Gefitinib | EGFR inhibitor | >10 |
| Dacomitinib (PF-0299804) | EGFR inhibitor | >10 |
| Lapatinib | Pan-HER inhibitor | >10 |
| BEZ235 | MTOR/PI3K inhibitor | 3.3 |
| AZD2014 | MTOR inhibitor | >10 |
| LEE011 | CDK4/6 inhibitor | >10 |
| 5-Fluorouracil | Cytotoxic | >10 |
| Neratinib | Pan-HER inhibitor | 1.0 |
| BGJ-398 | FGFR inhibitor | 1.2 |
Figure 3.

Antitumor activity of BEZ235 and BGJ-398.
(A) mTOR and FGFR2 protein expression by immunofluorescence.
(B) The viability of RCC PDCs was measured by CellTiter-Glo assay after treatment with various concentrations of BEZ235 and BGJ-398 for 5 days. The cell viability (%) represents the percent growth as compared to the control (no treatment), and IC50 values are 0.015 μM and 0.79 μM, respectively.
(C) The western blot for mTOR and FGFR phosphorylation and targeted downstream pathways. Cells were treated with 1 μM BEZ235 and 1 μM BGJ-398 for 5 days, respectively. Control cell was treated with DMSO.
Figure 4.

Tumor responses to BEZ235 (A), BGJ-398 (B), and temsirolimus in a PDX model.
Discussion
Treatment strategies targeting angiogenesis via the VEGFR TKIs or mTOR inhibitors have dramatically improved the treatment outcomes for patients with clear cell mRCC [1]. While most patients would develop adaptive or secondary resistance to these targeted agents after months or even after years of clinical benefit, a substantial number of patients exhibit intrinsic resistance to VEGFR TKIs. Once this has happened, limited treatment options remain. Furthermore, this subset of patients with intrinsic resistance rarely responds to subsequent therapy such as mTOR inhibitors or other VEGFR TKIs [15], [16], [17]. This raises a need for the development of novel therapeutics to overcome VEGFR TKI resistance. Historically, in cancer research, the development of new agents is predominantly based on preclinical studies of drug activity, typically tested with in vitro cell lines and in vivo murine model systems. While conventional cell lines are convenient and easily accessible, their predictive power has been poor [18], [19]. The ability to establish preclinical models that closely resemble actual tumor in vivo in terms of molecular profiles and clinical behaviors holds enormous promise in oncology drug development.
In this study, we developed PDC and PDX models using cancer cells derived from malignant effusions that exhibited morphologic features and genomic profiles similar to those of tumor tissue samples in patients with clear cell mRCC. PDC from malignant effusions has more than a few advantages over conventional cell lines [9]. First, PDC models faithfully recapitulated primary patient tumors, with the molecular and gross phenotypic characteristics of the primary tumor retained. Second, the median time from specimen collection to PDC passage 1 takes only 3 weeks, which is more feasible for clinical application than xenograft models. Third, we found that the success rate is as high as >70% of attempted cases. Fourth, although only tested in a limited setting so far, PDC could be successfully engrafted into immunocompromised mice; thus, PDCs can be used both in vitro and as a cell source for further in vivo analyses. Although tested for exploratory purposes, we assessed the sensitivity of tumor to a number of targeted drugs including sunitinib and pazopanib. As a result, the tumor cells were resistant to VEGFR TKIs as well as to everolimus, concordant with the actual clinical responses to sunitinib and everolimus.
As stated above, the limitations of current preclinical models have been well described [12]. In contrast, PDX may represent more meaningful models for the development of novel therapeutic agents. As they normally display similar heterogeneity to the tumors they are originally derived from, they can predict the subsequent clinical outcomes more accurately. This allows for mechanistic studies of clinical agents that cannot be done in the patient themselves. Establishing PDX models from surgical specimens faces certain limitations. There is a need for a sufficient amount of fresh tumor tissue, and unless the patient is scheduled for surgery, available tumor material might not be sufficient. Moreover, depending on the originating histology, the failure rate in PDX establishment can be substantial. In addition, the use of “personalized” PDX models (Avatar trials) is most likely limited due to the need for sufficient time to generate in vivo study results. Very often, oncologists need to decide much quicker which second-line or third-line therapy the patient has to be switched to. The time required for generating a PDX model, as well as the success rate, directly correlates with tumor aggressiveness, invasiveness, and the quality and quantity of malignant tissue received [20]. Malignant effusions in mRCC are known to be seen only in patients with multiple metastatic disease [21]. Therefore, PDCs from malignant effusions can be preferred to surgical specimens in PDX establishment.
One may argue that passages and the establishment of a PDX may allow additional DNA alterations to occur within the tumor. In addition, malignant body fluids such as effusions or ascites could only be collected from patients with far-advanced disease, and most of them already acquired clinical resistant to targeted agents. While the mechanisms of resistance to TKIs are not fully understood, it is generally acknowledged that secondary genetic alterations after repeated exposure to targeted agents may be responsible for the development of resistance [22]. In the present study, the results from the PDC drug test with BEZ235 and BGJ-398 were not reproduced in subsequent PDX model. Further investigations are needed if it may be due to a different tumor microenvironment or secondary mutations. However, we observed the similarity of genomic features between the primary tumor and single cell suspensions from the PDX tumor at passage 5. Still, it also is advantageous having not only PDX models of primary tumors at hand but also models generated from tumors after therapy. It is of increasing interest to pharmaceutical companies getting their hands on models that have arisen from tumor material that has received initial chemotherapy. This way, second-line and third-line therapeutic agents can be developed. Taken together, our study demonstrated that the preclinical model we developed can be a useful preclinical model to interrogate sensitivity to targeted agents based on genomic alterations [9]. Although the results are based on an observation of a single case, the PDC model from malignant effusions of patients was successfully converted to a PDX model. While larger studies are required to further evaluate and confirm the usefulness of the PDC-PDX model, this pilot study suggests that our novel patient-derived preclinical model from malignant effusions represents an important and feasible platform for future cancer research.
The following are the supplementary data related to this article.
Panel Designed to Enrich Exons of 83 Cancer-Related Genes
Footnotes
All authors read and approved the final manuscript. No authors declared conflicts of interest.
Contributor Information
Hyun Moo Lee, Email: besthml@skku.edu.
Se Hoon Park, Email: hematoma@skku.edu.
References
- 1.Hutson TE. Targeted therapies for the treatment of metastatic renal cell carcinoma: clinical evidence. Oncologist. 2011;16(Suppl. 2):14–22. doi: 10.1634/theoncologist.2011-S2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Motzer RJ, Hutson TE, Tomczak P. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 3.Escudier B, Eisen T, Stadler WM. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 4.Sternberg CN, Davis ID, Mardiak J. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 5.Troiani T, Schettino C, Martinelli E, Morgillo F, Tortora G, Ciardiello F. The use of xenograft models for the selection of cancer treatments with the EGFR as an example. Crit Rev Oncol Hematol. 2008;65:200–211. doi: 10.1016/j.critrevonc.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Cree IA, Glaysher S, Harvey AL. Efficacy of anti-cancer agents in cell lines versus human primary tumour tissue. Curr Opin Pharmacol. 2010;10:375–379. doi: 10.1016/j.coph.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Ellis LM, Fidler IJ. Finding the tumor copycat. Therapy fails, patients don't. Nat Med. 2010;16:974–975. doi: 10.1038/nm0910-974. [DOI] [PubMed] [Google Scholar]
- 8.DeRose YS, Wang G, Lin YC. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17:1514–1520. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JY, Kim SY, Park C. Patient-derived cell models as preclinical tools for genome-directed targeted therapy. Oncotarget. 2015;6:25619–25630. doi: 10.18632/oncotarget.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tentler JJ, Tan AC, Weekes CD. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JH, Won YW, Kim ST. Clinical features of metastatic renal cell carcinoma patients with intrinsic resistance to sunitinib: A Korean multicenter study. J Clin Oncol. 2015;33(Suppl. 7) [Abstr 462] [Google Scholar]
- 12.Kopetz S, Lemos R, Powis G. The promise of patient-derived xenografts: the best laid plans of mice and men. Clin Cancer Res. 2012;18:5160–5162. doi: 10.1158/1078-0432.CCR-12-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim MP, Truty MJ, Choi W. Molecular profiling of direct xenograft tumors established from human pancreatic adenocarcinoma after neoadjuvant therapy. Ann Surg Oncol. 2012;19(Suppl. 3):S395–S403. doi: 10.1245/s10434-011-1839-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee DW, Choi YS, Seo YJ. High-throughput screening (HTS) of anticancer drug efficacy on a micropillar/microwell chip platform. Anal Chem. 2014;86:535–542. doi: 10.1021/ac402546b. [DOI] [PubMed] [Google Scholar]
- 15.Seidel C, Busch J, Weikert S. Progression free survival of first line vascular endothelial growth factor-targeted therapy is an important prognostic parameter in patients with metastatic renal cell carcinoma. Eur J Cancer. 2012;48:1023–1030. doi: 10.1016/j.ejca.2012.02.048. [DOI] [PubMed] [Google Scholar]
- 16.Vickers MM, Choueiri TK, Rogers M. Clinical outcome in metastatic renal cell carcinoma patients after failure of initial vascular endothelial growth factor-targeted therapy. Urology. 2010;76:430–434. doi: 10.1016/j.urology.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 17.Heng DY, Mackenzie MJ, Vaishampayan UN. Primary anti-vascular endothelial growth factor (VEGF)-refractory metastatic renal cell carcinoma: clinical characteristics, risk factors, and subsequent therapy. Ann Oncol. 2012;23:1549–1555. doi: 10.1093/annonc/mdr533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Voskoglou-Nomikos T, Pater JL, Seymour L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin Cancer Res. 2003;9:4227–4239. [PubMed] [Google Scholar]
- 19.Johnson JI, Decker S, Zaharevitz D. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84:1424–1431. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stebbing J, Paz K, Schwartz GK. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer. 2014;120:2006–2015. doi: 10.1002/cncr.28696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kutty K, Varkey B. Incidence and distribution of intrathoracic metastases from renal cell carcinoma. Arch Intern Med. 1984;144:273–276. [PubMed] [Google Scholar]
- 22.Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009;10:992–1000. doi: 10.1016/S1470-2045(09)70240-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Panel Designed to Enrich Exons of 83 Cancer-Related Genes