

Abstract
Somatic or germline mutations in the tuberous sclerosis complex (TSC) tumor suppressor genes are associated closely with the pathogenesis of lymphangioleiomyomatosis (LAM), a rare and progressive neoplastic disease that predominantly affects women in their childbearing years. Serum levels of the lymphangiogeneic growth factor VEGF-D are elevated significantly in LAM. However, there are gaps in knowledge regarding VEGF-D dysregulation and its cellular origin in LAM. Here we show that increased expression and activation of the tyrosine kinase Syk in TSC2-deficient cells and pulmonary nodules from LAM patients contributes to tumor growth. Syk kinase inhibitors blocked Syk signaling and exhibited potent anti-proliferative activities in TSC2-deficient cells and an immunodeficient mouse xenograft model of LAM. In TSC2-deficient cells, Syk signaling increased expression of monocyte chemoattractant protein MCP-1, which in peripheral blood mononuclear cells (PBMC) stimulated production of VEGF-D. In clinical isolates of PBMC from LAM patients, VEGF-D expression was elevated. Further, levels of VEGF-D and MCP-1 in patient sera correlated positively with each other. Our results illuminate the basis for LAM growth and demonstrate the therapeutic potential of targeting Syk in this and other settings driven by TSC genetic mutation.
Keywords: Syk Kinase, Lymphangioleiomyomatosis, Macrophages, MCP-1, Vascular Endothelial Growth Factor D
Introduction
Tuberous sclerosis complex (TSC), an inheritable disorder resulting from mutations in either the TSC1 or TSC2 gene, is associated with benign tumors in multiple tissues, cognitive impairment, seizures and skin abnormalities (1). TSC deficiency causes aberrant activation of the mammalian target of rapamycin complex 1 (mTORC1) signaling and thereby results in excessive protein synthesis and uncontrolled cell proliferation (2). Up to 30% of women with TSC develop lymphangioleiomyomatosis (LAM), a rare and progressive neoplastic disease that predominantly affects women in their childbearing years and ranks as the third leading cause of TSC-related death (3,4). Other than TSC-associated LAM (TSC-LAM), there is a separate form of the disease with a distinct clinical entity called sporadic LAM (S-LAM), which is caused by somatic rather than germ line mutations in TSC genes (5). Lung lesions from LAM patients are characterized by excessive growth of smooth muscle-like cells (LAM cells) and cyst formation, leading to gradual airflow obstruction and potentially death from respiratory failure within two decades (4).
It is estimated that up to 3,500 patients worldwide and 1,400 patients in the US have been diagnosed with LAM and that up to a quarter million women may have LAM (6). Significant efforts have been made over the past two decades to identify, develop and implement effective therapeutic strategies to combat this potentially fatal disease. Based on its ability to inhibit mTORC1 activation of downstream kinases, sirolimus (rapamycin) has become an established treatment for LAM (7,8). Rapamycin has been shown to suppress TSC2-null xenograft tumor growth in animal models (9,10). Correspondingly, rapamycin treatment leads to shrinkage of angiomyolipomas (AMLs) (11) and lymphangioleiomyomas (12), reduction of chylous effusions (13) and preservation of lung function in patients with LAM (7). Rapamycin decreases serum levels of vascular endothelial growth factor (VEGF)-D, which is elevated in the majority of LAM patients and has been widely used as a diagnostic biomarker (14,15). However, the mechanism for its upregulation is not fully known. While LAM cells are generally thought to be the source of VEGF-D (16), we have previously shown that silencing TSC2 in human lung fibroblasts does not lead to increases in VEGF-D levels (17), suggesting that TSC2-deficient cells may not be the direct source of excessive VEGF-D in LAM.
It should be noted, however, that more than half of LAM patients enrolled in the Multicenter International Lymphangioleiomyomatosis Efficacy and Safety of Sirolimus (MILES) clinical trial did not exhibit enhanced or stabilized lung function during the rapamycin treatment phase (7), and as such, it is absolutely imperative to evaluate new therapies that may expand options for patients with LAM and other malignancies characterized by activation of the mTORC1 pathway who are unresponsive or intolerant to rapamycin treatment. The identification of elevated Src kinase activity in LAM cells has led to an ongoing clinical trial evaluating the therapeutic efficacy of a Src inhibitor (Saracatinib) in LAM patients (18). Similar to Src kinase, spleen tyrosine kinase (Syk) is a non-receptor tyrosine kinase and a key component of both innate and adaptive immunity. Syk also contributes to the initiation and metastatic progression of multiple types of solid tumors. For instance, Syk plays a role in breast cancer invasive cell growth (19) and acts as an oncogenic driver in small cell lung cancer (20). In addition, upregulated Syk expression corresponds with metastasis of both prostate cancer (21) and squamous cell carcinomas of the head and neck (22). More intriguingly, studies have shown that Syk regulates mTORC1 activation in acute myeloid leukemia and lymphoma (23,24). Taken together, the aberrant expression of Syk in various malignancies and the association between Syk and mTORC1 prompt us to hypothesize that Syk may be tightly involved in LAM pathogenesis.
In the present study, we found deregulated Syk expression and activation in TSC2- deficient cells and in LAM lung lesions. We also determined the therapeutic efficacy of clinically relevant Syk inhibitors in curbing TSC2-null tumor progression. Furthermore, we identified a unique cascade of Syk-dependent signals between TSC2-deficient cells and peripheral blood mononuclear cells (PBMCs) that eventually induced VEGF-D expression. Given these collective findings, Syk could be a therapeutic target for the treatment of LAM and other diseases associated with mutations in the TSC genes.
Materials and Methods
Human studies
All human subjects gave their written informed consent. Human samples were acquired under protocols approved by the National Heart, Lung, and Blood Institute Institutional Review Board (Protocol 95-H-0186)
Lung tissues
Pulmonary parenchyma from subjects with LAM (n=6) was fixed in 10% neutral buffered formalin and subsequently embedded with paraffin.
Clinical features of patient cohort
Serum was collected from 27 LAM patients for a total of 200 visits before (80 visits) and after (120 visits) sirolimus treatment. The diagnosis of LAM was confirmed by histopathology or the presence of cystic disease by computed tomography scan plus TSC, AMLs, high vascular endothelial growth factor D (VEGF-D) levels, and/or LLMs. Three of the patients have LAM-TSC; the rest have sporadic LAM. There are 2 Asian, 1 white Hispanic/Latino, and 24 white not Hispanic/Latino patients. Age and visit information are in Supplementary Table S1.
Luminex analysis of serum samples
MCP-1 and VEGF-D were measured in serum (dilution 1:2) using magnetic bead-based multiplex screening assays from R&D Systems (Minneapolis, MN) on a Luminex IS100 instrument (Luminex Corp, Austin, TX). Data were analyzed using Bio-Plex Manager Pro 6.1 analysis software (Bio-Rad, Hercules, CA). If a measurement exceeded or fell below the limit of detection, either the upper or lower limit of detection was substituted.
Peripheral blood mononuclear cells
20 ml of blood samples were collected from healthy volunteers (n=16) and subjects with LAM (n=22). Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Paque Plus (GE Healthcare, Pittsburgh, PA) gradient centrifugation. Characteristics of healthy volunteers and LAM subjects whose PBMCs were studied are summarized in Supplementary Table S2.
Cell culture
Cells were incubated at 37°C in a humidified 5% CO2 atmosphere. TSC2-null ELT3 cells, first isolated in 1995, were derived from the Eker rat uterine leiomyoma as previously described (25). Cells were generously provided by Dr. Cheryl Walker, and authenticated by the study team by Western blotting for Tuberin. ELT3-V cells (vector) or ELT3-T (TSC2 addback) cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). TSC2-null ELT3-V cells and TSC2-reexpressing ELT3-T cells are denoted as TSC2- and TSC2+, respectively. Cells were serum-starved overnight before they were treated with R406 (1uM) (Selleckchem, Houston, TX), rapamycin (20 nM) (LC Laboratories, Woburn, MA) or Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO) as control vehicle for the indicated times. PBMCs were isolated, seeded and incubated with recombinant human MCP-1 (10 ng/ml, R&D Systems) for 24h.
RNA Interference
Sequences for rat Syk and Stat3 siRNA were designed using an online siRNA selection program (26) and were synthesized by Sigma-Aldrich. Please refer to Supplementary Table S3 for siRNA sequences. TSC2- cells were transfected with siRNA for targeted gene suppression or with MISSION siRNA Universal Negative Control (Sigma-Aldrich). Cells were transfected with 1 nmol/L siRNA using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Cambridge, MA).
Animal studies
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee at Brigham and Women’s Hospital. The mouse xenograft tumor model was established as previously described (9,10). 2.5 × 106 TSC2- cells (ELT3-V) were subcutaneously injected bilaterally into the flank and shoulder of female immunodeficient C.B17 scid mice (Taconic, Hudson, NY). When tumor surface area reached 40 mm2, mice were randomly assigned to one of the following treatments through intraperitoneal injection: 1) Vehicle control (n = 5 mice, three divided doses at three-hour intervals per day); 2) R788 (n = 5 mice, 80mg/kg, three divided doses at three-hour intervals per day) (Selleckchem); and 3) Rapamycin (n = 4 mice, 3 mg/kg, three times per week). The dose, frequency and route of drug administration were chosen on the basis of published studies (9,27). Mice underwent 12 days of designated treatment and were then sacrificed. Prior to the treatment phase, tumors were measured with a Vernier caliper three times per week, while during the treatment phase these measurements were recorded daily. Tumor surface area was calculated as follows: π × (Major diameter/2) × (Minor diameter/2). After tumors were excised, final tumor volume was calculated as follows: (4/3) × π × (Length/2) × (Width/2) × (Height/2).
Immunohistochemistry and immunofluorescence
Formalin-fixed, paraffin-embedded tissue sections were deparaffinized in xylene and rehydrated through a graded series of ethanol, followed by antigen retrieval with citrate buffer (pH 6.0) for 20 min at 95°C. Sections were incubated with a primary antibody or an isotype control in a humidified chamber overnight at 4°C. Please refer to Supplementary Table S4 for a complete list of antibodies. 1) For immunohistochemistry: Sections were incubated with appropriate secondary antibodies using a HRP-DAB Cell & Tissue Staining Kit (R&D Systems) according to manufacturer’s instructions; 2) For immunofluorescence: Sections were incubated in Alexa Fluor conjugated secondary antibodies (Thermo Fisher Scientific) for 1 h at room temperature in the dark. Slides were then mounted using mounting medium containing 4,6-diamidino-2-phenylindole-2-HCl (DAPI) (Vector Laboratories Inc., Burlingame, CA). To detect in vivo cell apoptosis, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using the In Situ Cell Death Detection Kit, TMR red (Roche Applied Science, Branford, CT) according to the manufacturer’s recommendations.
RNA Extraction and Real-Time Polymerase Chain Reaction (RT-PCR)
RT-PCR was performed as described in supplementary Material and Methods. Please refer to Table S5 for a list of primer sequences.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Two-tailed Student t-test was used to compare two groups. Variables with more than two factors were evaluated by one-way analysis of variance (ANOVA) followed by the Tukey post hoc test. Mixed effects models were used to incorporate the within and between subject variability as well as the repeated measurements assessing the effect of rapamycin before and after treatment. Correlation coefficients between two variables were calculated with Pearson correlation test. Analyses were performed by using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA) and SAS (Information Technology Services, Iowa City, IA). P < 0.05 was considered significant.
Results
Deregulated Syk expression and activation are identified in TSC2-deficient cells
Since we hypothesize that Syk is a critical kinase involved in LAM pathogenesis, we first sought to determine whether Syk expression is TSC2-dependent. We used the TSC2-deficient ELT-3 cell line derived from a uterine leiomyoma in an Eker rat with a germ line mutation in TSC2 gene (28). Real-time PCR analysis showed that Syk expression was upregulated almost four-fold in TSC2- cells (ELT3-V) compared to TSC2+ cells (ELT3-T) (Fig. 1A). Western blot analysis verified the elevated Syk expression in the absence of tuberin (the protein encoded by TSC2 gene) and revealed that TSC2- cells exhibited increased phosphorylation of Syk at Tyr525/526 (Fig. 1B), which is pivotal for Syk activation and its downstream signal transduction (29). As expected, TSC2 deficiency led to mTORC1 hyperactivation characterized by increased presence of phospho-p70S6 kinase, which was almost completely abolished by rapamycin treatment. Notably, we found that treatment with rapamycin yielded a concomitant reduction of both phospho-Syk and total Syk expression (Fig. 1B), suggesting that Syk is mTORC1-dependent.
Fig. 1. TSC2-deficient cells display deregulated Syk expression and activation.
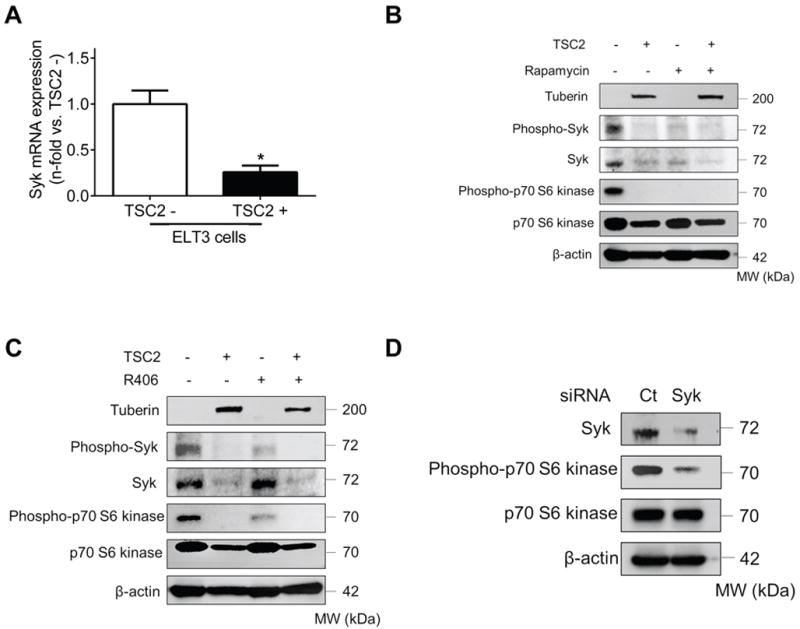
(A) Real-time PCR analysis of rat Syk mRNA in TSC2- cells compared to TSC2+ cells. Results were expressed as the fold change relative to TSC2- cells. Data represent means ± SEM of three independent experiments. * P < 0.05, by Student’s t test. (B) TSC2- and TSC2+ cells were treated with either control vehicle (DMSO) or rapamycin (20 nM) for 24 h. (C) TSC2- and TSC2+ cells were treated with either control vehicle (DMSO) or R406 (1μM) for 24 h. (D) TSC2- cells were transfected with control siRNA (Ct) or Syk siRNA for 48 h. (B to D) Equal amounts of protein from whole cell lysates were analyzed by Western blot using antibodies against Tuberin, Phospho-Syk, Syk, Phospho-p70 S6 Kinase and p70 S6 Kinase. β-actin was used as a loading control. All experiments were repeated at least three times.
Consistent with published data (30), we confirmed that treatment with R406 (a Syk inhibitor) effectively blocked Syk phosphorylation in TSC2-deficient cells, but did not modulate total Syk expression (Fig.1C). Since Syk has been reported to play a significant role in mTORC1 activation in hematopoietic malignancies (23,24), we queried whether Syk inhibition could affect mTOR signaling in TSC2-deficient cells as well. As anticipated, we found that R406 decreased the levels of phospho-p70S6 kinase in TSC2- cells (Fig.1C). To corroborate these findings, we transfected TSC2- cells with Syk siRNA and found that knocking down Syk expression dampened mTORC1 activity, similar to the effects of R406 (Fig. 1D). Taken together, our data suggest that the Syk and mTORC1 pathways are reciprocally regulated.
Syk inhibition reduces proliferation of TSC2-deficient cells in vitro
TSC2 deficiency and the constitutive activation of mTORC1 signaling induce uncontrolled cell growth, a distinctive characteristic in LAM (2). To determine whether the abnormal proliferation of TSC2- cells is Syk-dependent, we treated these cells with either R406 or rapamycin and monitored cell density at different time points. R406 was as effective as rapamycin in diminishing cell proliferation (Fig.2A). Correspondingly, TSC2- cells treated with either R406 or rapamycin exhibited a substantial increase in the percentage of G1-phase cells accompanied by a reduced fraction of cells in S phase, indicating that R406 and rapamycin induced G1 cell cycle arrest (Fig. 2B and Supplementary Fig.S1). The anti-proliferative effects of R406 and rapamycin were further corroborated by Western blot analysis, which revealed that both treatments were associated with decreased proliferating cell nuclear antigen (PCNA) expression in TSC2- cells, although neither treatment activated caspase-3 (Fig. 2C).
Fig.2. Inhibition of Syk suppresses growth of TSC2-deficient cells through induction of cell cycle arrest.

TSC2- cells were treated with DMSO (vehicle control), R406 (1μM) or rapamycin (20 nM). (A) Effects of R406 and rapamycin on cell proliferative capacity. Cell proliferation was determined at the indicated time points after treatment. (B) Effects of R406 and rapamycin on cell cycle distribution. Cells were harvested 24h after treatment. Results were expressed as a percentage of cells in G1, S and G2/M phases. Data represent mean ± SEM of at least three independent experiments. NS, not significant, *P < 0.05, **P < 0.01, ***P < 0.001 (versus DMSO), by one-way ANOVA. (C) Cells were collected 24 h after treatment. Equal amounts of protein from whole cell lysates were analyzed by Western blot using antibodies against PCNA, cleaved caspase-3, and caspase 3. β-actin was used as a loading control. All experiments were repeated at least three times.
Syk inhibition suppresses TSC2-null xenograft tumor development in vivo
Based on the anti-proliferative effects of R406 in vitro, we evaluated the activity of R788, the prodrug of R406, in an immunodeficient mouse model bearing subcutaneous TSC2-null ELT-3 xenograft tumors (9,10). As expected, TSC2-null xenograft tumors treated with control grew progressively. In contrast, both R788 and rapamycin treatment resulted in a striking and persistent decrease in tumor area (Fig. 3A). After twelve days of treatment, R788 significantly reduced xenograft tumor burden to a similar degree as rapamycin, evidenced by the volume of the excised tumors (Fig.3B and C). In agreement with our observations in TSC2- cells in vitro, Western blot analysis of tumor homogenates showed that expressions of phospho-Syk and phospho-p70S6 kinase were similarly decreased in R788- and rapamycin-treated groups compared to controls, while total Syk protein expression was only reduced in the rapamycin-treated group (Fig. 3D and Supplementary Fig.S2). In addition, we found reduced immunostaining of PCNA in the tumors of R788- and rapamycin-treated animals (Fig 3E). Nonetheless, quantification of TUNEL staining showed no difference in the percentage of positively labeled cells among all experimental conditions (Fig. 3F), indicating that tumor shrinkage after treatment was not caused by induction of apoptosis. Thus, these data demonstrate that TSC2-null xenograft tumor growth is Syk-dependent in vivo and provide further rationale to test the efficacy of Syk inhibitors in future clinical trials.
Fig.3. Syk inhibition impairs TSC2-null xenograft tumor development.

Female CB17-SCID mice were inoculated with TSC2- cells subcutaneously. Mice were treated with control vehicle (n=5), R788 (n=5), or rapamycin (n=4) through intraperitoneal injection after tumor surface area reached 40 mm2. (A) Tumor surface area was measured daily with a caliper and tumor growth curve was plotted. Results were expressed as a percentage of baseline tumor surface area before treatment. (B) Mice were sacrificed 12 days after the initial treatment. Representative gross appearance of mice bearing xenograft tumors and excised tumors is displayed. Scale bars: 1 cm. (C) Volume of the excised tumors. (D) Equal amounts of protein from tumor homogenates were analyzed by Western blot using antibodies against Phospho-Syk, Syk, Phospho-p70 S6 Kinase and p70 S6 Kinase. β-actin was used as a loading control. (E) Left panel: representative images of PCNA (red) immunofluorescence staining in tumor tissue. Right panel: percentage of PCNA-positive cells. (F) Left panels: representative images of TUNEL (red) staining in tumor tissue. Right panel: percentage of TUNEL-positive cells. Nuclei were counterstained with DAPI (blue). Scale bars: 50 μm (E and F). Data are presented as the mean ± SEM. NS, not significant, *P < 0.05, **P < 0.01, ***P < 0.001 (versus control), by one-way ANOVA.
Syk regulates MCP-1 expression via Stat3 signaling in TSC2-deficient cells
Loss of functional TSC2 in embryonic fibroblasts has been described to mediate increased monocyte chemoattractant protein (MCP)-1 production (31). In the current study, we made a similar observation that TSC2 deficiency led to upregulation of MCP-1 gene expression (Supplementary Fig. S3). In order to evaluate whether Syk signaling is involved in this pathological process, we treated TSC2- and TSC2+ cells with R406 and analyzed the levels of MCP-1 in cell culture supernatants. We found that increased MCP-1 secretion from TSC2- cells was markedly attenuated by pharmacological blockade of Syk kinase activity (Fig. 4A). To further confirm the role of Syk in the induction of MCP-1, we silenced Syk using siRNA in TSC2- cells and observed a dramatic reduction in MCP-1 production compared to control siRNA (Fig. 4B).
Fig.4. Aberrant Syk activation induces MCP-1 overexpression via Stat3 signaling in TSC2-deficient cells.

(A) TSC2- and TSC2+ cells were treated with either control vehicle (DMSO) or R406 (1μM) for 24 h. (B) TSC2- cells were transfected with control siRNA (Ct) or Syk siRNA for 48 h. (A and B) Levels of MCP-1 in cell culture supernatants were determined by enzyme-linked immunosorbent assay (ELISA). Data represent means ± SEM of three independent experiments. ***P < 0.001 (versus TSC2- cells treated with DMSO), by one-way ANOVA (A); * P < 0.05, by Student’s t test (B). (C) TSC2- and TSC2+ cells were treated with either control vehicle (DMSO) or R406 (1μM) for 24 h. (D) TSC2- cells were transfected with control siRNA (Ct) or Stat3 siRNA for 48 h. (C and D) Equal amounts of protein from whole cell lysates were analyzed by Western blot using antibodies against Phospho-Stat3 and Stat3. β-actin was used as a loading control. (E) TSC2- cells were transfected with control siRNA (Ct) or Stat3 siRNA for 48 h. Levels of MCP-1 in cell culture supernatants were determined by ELISA. Data represent means ± SEM of three independent experiments. * P < 0.05, by Student’s t test.
Considering that Syk regulates signal transducer and activator of transcription (Stat) 3 and Stat3-dependent signaling transduction is prominently associated with the induction of MCP-1(32,33), we next set out to explore whether the Stat3 pathway interconnects aberrant Syk activation and the eventual MCP-1 overexpression. Consistent with previous findings (34), we observed that TSC2- cells displayed elevated phosphorylation of Stat3 (Fig.4C). Treatment with R406 abrogated Stat3 activation but did not affect total Stat3 expression. Furthermore, siRNA-mediated silencing of Stat3 resulted in decreased MCP-1 levels in cell culture supernatants from TSC2- cells (Fig. 4D and E). These data collectively indicate that Syk regulates MCP-1 production through Stat3 signaling in TSC2-deficient cells.
MCP-1 upregulates VEGF-D expression in peripheral blood mononuclear cells
VEGF-D is elevated in the sera of ~70% of patients with LAM (14,15). The prevailing view holds that LAM cells are the fundamental source of increased VEGF-D production (15), although the exact cellular origin of increased VEGF-D has not been completely established. Interestingly, we found that levels of VEGF-D gene expression were comparable between TSC2- and TSC2+ ELT-3 cells (Fig. 5A), suggesting that TSC2 deficiency might not directly upregulate VEGF-D. Since MCP-1 is overexpressed in TSC2- cells, we hypothesized that increased amounts of MCP-1 recruit PBMCs, culminating in excessive VEGF-D production. Indeed, we found that incubation of LAM patient-derived PBMCs with exogenous MCP-1 resulted in a ~2.5 fold increase in VEGF-D gene expression (Fig. 5B). Considering that PBMCs can differentiate into tumor-associated macrophages (TAMs), which constitute a predominant source of cellular infiltrates around solid tumors (35), we next examined the presence of CD68+ TAMs in TSC2-null xenograft tumors. TSC2-null xenografts were associated with accumulation of CD68+ macrophages near the tumor edges. Treatment with R788 or rapamycin resulted in a remarkable reduction in CD68+ macrophage density (Fig. 5C). In addition, we found a significant decrease in rat MCP-1 expression in tumors treated with R788 or rapamycin (Fig. 5D, left panel). Consistent with our in vitro data, we did not observe changes in rat VEGF-D expression among all groups (Fig.5D, middle panel) but found that R788 and rapamycin led to downregulation of mouse VEGF-D (Fig 5D, right panel) in tumors. Further statistical analysis showed that rat MCP-1 levels correlated with the presence of CD68+ cells (r=0.76, p=0.0017) (Fig. 5E, left panel), which in turn correlated with mouse VEGF-D levels (r=0.9, p<0.0001) (Fig. 5E, middle panel). A positive correlation also existed when rat MCP-1 levels were plotted against mouse VEGF-D levels (r=0.76, p=0.0015) (Fig. 5E, right panel). Taken together, these data suggest an association between MCP-1, TAM infiltration, and VEGF-D production.
Fig.5. MCP-1 stimulates VEGF-D expression in peripheral blood mononuclear cells.

(A) Real-time PCR analysis of rat VEGF-D mRNA in TSC2- cells compared to TSC2+ cells. Results were expressed as the fold change relative to TSC2- cells. (B) Real-time PCR analysis of human VEGF-D mRNA in human peripheral blood mononuclear cells (PBMCs). PBMCs from four LAM subjects were isolated, seeded and stimulated with recombinant human MCP-1 (10 ng/ml) for 24 h. Results were expressed as the fold change relative to PBMCs treated with control vehicle. (A and B) Data represent means ± SEM of three independent experiments. NS, not significant, * P < 0.05, by Student’s t test. (C) Left panels: Representative images of CD68 (green) immunofluorescence staining on TSC2-null tumor edges. Nuclei were counterstained with DAPI (blue). Scale bars: 50 μm. Right panel: percentage of CD68-positive cells. (D) Real-time PCR analysis of rat MCP-1(left panel), rat VEGF-D (middle panel) and mouse VEGF-D (right panel) mRNA in tumor homogenates. (C and D) Data are presented as the mean ± SEM. NS, not significant, **P < 0.01, ***P < 0.001 (versus control), by one-way ANOVA. (E) Correlation between percentage of CD68-positive cells, rat MCP-1 mRNA expression and mouse VEGF-D mRNA expression in tumors. Correlation coefficients between two variables were calculated with Pearson correlation test.
Aberrant Syk signaling is evident in LAM patients
To determine whether our in vitro and in vivo results were consistent with findings in LAM patients, we first performed immunohistochemical staining for Syk expression and activation in LAM lungs. While unaffected areas in LAM lungs exhibited minimal phospho- and total Syk immunoreactivity, abundant phospho- and total Syk staining was detected in LAM nodules (Fig. 6A).
Fig. 6. Aberrant increase of Syk activation and expression is identified in LAM.
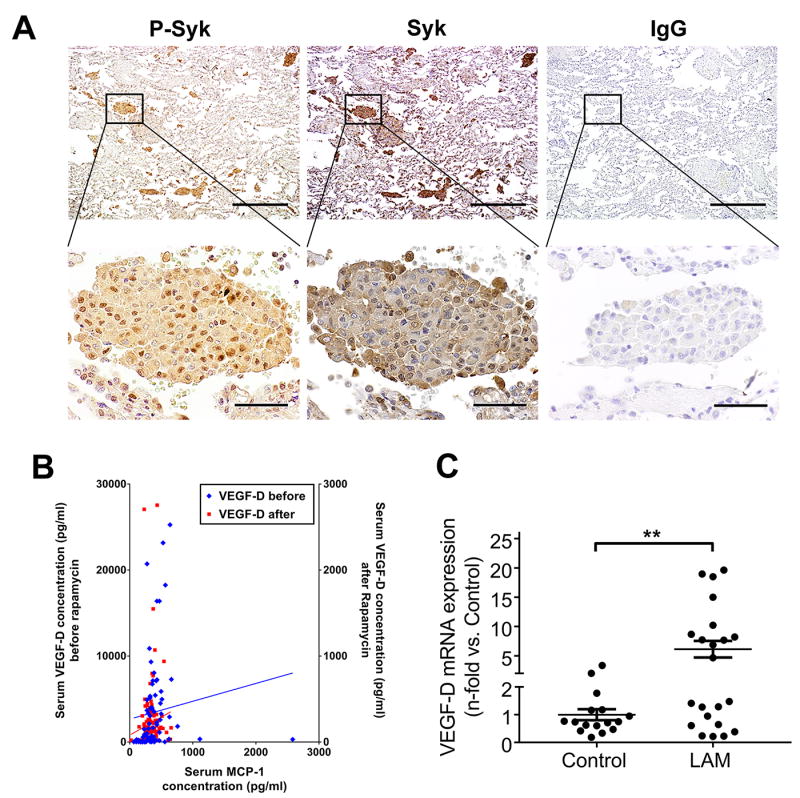
(A) Representative immunohistochemical staining of LAM lungs. From left to right: immunoreactivity for phospho-Syk and total Syk visualized with diaminobenzidine (DAB) is observed in LAM lung nodules. Absence of immunoreactivity with nonimmune IgG in the LAM lung. (n=6 LAM patients). Scale bars: 500 μm (upper panels), 50 μm (lower panels). (B) Serum was collected from 27 LAM patients for a total of 200 visits before (80 visits, Y-axis on the left) and after (120 visits, Y-axis on the right) sirolimus treatment. MCP-1 and VEGF-D were measured in serum using magnetic bead-based multiplex screening assays. Correlation coefficients between levels of MCP-1 and VEGF-D were calculated with Pearson correlation test. (C) Real-time PCR analysis of human VEGF-D mRNA in human peripheral blood mononuclear cells (PBMCs) from healthy volunteers as control (n=16) and LAM patients (n=22). Results were expressed as the fold change relative to healthy control subjects. Data represent means ± SEM. **P < 0.01, by Student’s t test.
To validate the downstream effects of Syk activation, we measured levels of MCP-1 and VEGF-D in sera from 27 LAM patients before and after treatment with sirolimus. Patients had an average follow-up of 3.8 years before sirolimus therapy (covering 80 visits) and 3.4 years after sirolimus therapy (120 visits). We found that when we corrected for factors previously shown to influence rate of decline (36), rapamycin reduced the rate of change in the forced expiratory volume in 1 second (FEV1) (p<0.001) and in diffusion capacity in carbon monoxide (p<0.001). MCP-1 levels were significantly different before and after sirolimus treatment (p=0.010), as were VEGF-D levels (p<0.001). Notably, we found that MCP-1 was a significant predictor of VEGF-D regardless of sirolimus treatment (p<0.05; correlation r=0.346) (Fig. 6B).
Based on the strong positive correlation between density of TAMs and mouse VEGF-D expressions in xenograft tumors, we investigated if PBMCs could be a potential source of VEGF-D in LAM. We collected PBMCs from women with LAM and healthy controls (table S1), and we detected a six-fold increase in VEGF-D mRNA expression in the PBMCs of LAM subjects compared to those of healthy controls (Fig. 6C). Similar to our findings in vitro and in xenograft tumors, these data collectively suggest that Syk activation led to increased MCP-1 production, which triggered overexpression of VEGF-D from PBMCs. Our results also demonstrated that PBMCs could potentially contribute to increased VEGF-D levels in LAM.
Discussion
After its original discovery in 1988, Syk was initially investigated almost exclusively in hematopoietic cells because of its key functions in immunoreceptor signaling. Subsequent studies show that Syk can also be detected in non-hematopoietic cells, such as bronchial epithelial cells (37) and vascular smooth muscle cells (38). In the present study, we demonstrate for the first time that there is an aberrant increase in the expression and activation of Syk in TSC2-deficient cells as well as in pulmonary nodules from LAM patients. Furthermore, treatment with rapamycin reduces Syk expression and activity, indicating that Syk signaling is mTORC1-dependent. We also provide compelling evidence that Syk inhibitors (R406 and R788) effectively block Syk signaling and elicit potent anti-proliferative activities in TSC2-deficient cells in vitro and in an immunodeficient mouse xenograft tumor model, indicating a pivotal role of Syk signaling in TSC2-deficient tumor growth and suggesting the therapeutic potential of targeting Syk in LAM. The beneficial effects of R788 (fostamatinib disodium), a prodrug of the biologically active compound R406, have been demonstrated in experimental models of lymphoma (39) and rheumatoid arthritis (40). The overwhelming success in the preclinical phase has laid the foundation for several clinical trials evaluating R788’s effects and safety profile in autoimmune diseases (41,42). Collectively, these properties render Syk inhibition an attractive alternative in LAM treatment and an ideal candidate for personalized therapy.
Interestingly, protein expression analysis shows that Syk inhibitors R406 and R788 reduced the level of phospho-p70S6 kinase, a hallmark of mTORC1 activation, in TSC2-deficient cells in vitro and in xenograft tumors. Correspondingly, siRNA-mediated knockdown of Syk diminished phospho-p70S6 kinase in TSC2-deficient cells, indicating that the modulation of mTORC1 signaling by Syk inhibitors is less likely due to an off-target effect attributed to nearly all tyrosine kinase inhibitors. Our findings suggest that the Syk pathway is not only a downstream target of mTORC1 signaling, but also engages to regulate mTORC1 activity in a reciprocal manner, which is consistent with previous studies describing the indispensable role of Syk in mTORC1 signaling in lymphoma and acute myeloid leukemia (23,24). This feedback loop -with Syk both upstream and downstream of the mTORC1 pathway- makes it challenging to separate the effects of Syk on TSC2 null cells independent of its effects on mTORC1. Nevertheless, it is important to acknowledge that Syk kinase activity is the principal but not the only target for Syk inhibitors. An in vitro pharmacological profiling reveals that R406 inhibits crucial kinases in vascular endothelial growth factor receptor (VEGFR) signaling, including VEGFR1 and VEGFR2 (43). Interestingly, it was reported that an inhibitor of VEGFR signaling (axitinib) restrained TSC2-null tumor development in a mouse model of LAM (44), indicating that signaling pathways besides mTORC1 could also contribute to LAM pathogenesis. In light of the multifactorial aspects of LAM, we see tremendous potential in utilizing a multi-target compound such as a Syk inhibitor for LAM treatment.
LAM has been labeled as “a low grade, destructive, metastasizing neoplasm” (45). Similar to cancer, LAM generates a proinflammatory environment (46). In fact, TSC2-deficiency has been directly associated with elevated levels of MCP-1 (31), a potent chemotactic factor for monocytes, although the exact underlying mechanisms of this phenomenon remain to be elucidated. Here, we employed a combination of pharmacological and genetic manipulations to define the signaling pathways and discovered that Syk mediates increased production of MCP-1 in TSC2-deficient cells through a Stat3-dependent mechanism. Our findings further extend previous reports that Syk directly activates Stat3 (32) and Stat3 mediates MCP-1 production (33).
Serum VEGF-D concentration is elevated in most LAM patients and has been confirmed to be a reliable diagnostic and prognostic biomarker of LAM (14,15). Previous investigations have also shown that VEGF-D contributes to LAM cell proliferation in vitro (47) and correlates with lymphatic involvement (14). Despite intense research efforts and major advances in the understanding of VEGF-D biology, the origin of excessive VEGF-D in LAM patients’ sera remains enigmatic. Traditionally, LAM cells are considered as the major source for elevated VEGF-D levels based on the evidence that VEGF-D immunostaining is present in LAM cells (15). Notably, LAM cells also express VEGFR3 (47), a receptor for VEGF-D, and therefore it is difficult to ascertain whether VEGF-D originates within LAM cells or if the observed VEGF-D immunostaining may be in part due to ligand-receptor complex internalization. Here, we provide evidence that endogenous VEGF-D is not directly modulated by TSC2 deficiency and mTORC1 activation in vitro and in mouse xenograft tumors. These data suggest the possibility that cellular sources of VEGF-D other than LAM cells may exist in the LAM microenvironment and be sensitive to the effect of rapamycin. Remarkably, we also found that VEGF-D expression is elevated in PBMCs from LAM patients and could be further enhanced by exogenous MCP-1 stimulation. These findings suggest that PBMCs could be the missing link between aberrant MCP-1 production from TSC-2 deficient cells and increased VEGF-D concentration in peripheral blood. The direct role of PBMCs in VEGF-D production was further substantiated in the mouse xenograft tumor model. Quantification of TAM immunostaining and real-time PCR analysis of tumor homogenates suggest that TAM infiltration correlates with gene expressions of rat MCP-1 and mouse VEGF-D. Furthermore, we found that the gene expression of rat MCP-1 also correlates with that of mouse VEGF-D. Considering that TAMs predominantly differentiate from PBMC-derived monocytes and produce an abundant amount of VEGF-D (48), our findings support a two-step mechanism: 1) In TSC2-deficient cells (primary LAM lesions), activation of mTORC1 and Syk signaling induces MCP-1 overproduction via a Stat3-dependent manner; and 2) MCP-1 recruits and activates PBMCs, leading to increased VEGF-D expression (Fig. 7). Taken together, our study extends a recent report that proposes alternative sources of VEGF-D in LAM (47), and we demonstrate for the first time that PBMCs indeed participate in VEGF-D production.
Fig.7. Schematic illustration depicts the roles of Syk signaling in VEGF-D overexpression and TSC2-null tumor growth.

In TSC2-deficient cells, activation of mTORC1 and Syk signaling induces MCP-1 overproduction via a Stat3-dependent manner. Subsequently, MCP-1 recruits and activates peripheral blood mononuclear cells (PBMCs), leading to increased VEGF-D expression. In contrast, treatment with Syk inhibitors impairs proliferation of TSC2-deficient cells and decreases production of MCP-1. As a result, VEGF-D expression is reduced in PBMCs.
Consistent with results from the MILES trial (7), the present study demonstrates that rapamycin treatment effectively preserved lung function and decreased levels of serum VEGF-D in LAM patients. By distinction, we also found that rapamycin led to a reduction in serum MCP-1 levels, which positively correlated with the decline in serum VEGF-D concentrations. Of particular relevance to the current findings, elevated levels of MCP-1 were previously detected in the bronchoalveolar lavage fluid (BALF) from LAM patients (49). The cumulative findings of the current and the prior study provide an additional argument for the crucial role of MCP-1 in LAM pathogenesis. Moreover, the changes of MCP-1 in LAM suggest that it could also be a feasible target for therapeutic modulation and could be utilized to predict the course of the disease. Future studies are required to determine the sensitivity and specificity of MCP-1 as a LAM biomarker.
In conclusion, we delineate a novel cascade of Syk-dependent signals and demonstrate that Syk inhibition is a potential therapy for patients with LAM and perhaps other manifestations of TSC who are intolerant, allergic or unresponsive to conventional mTOR inhibitors. Syk inhibitors may also be particularly useful for LAM patients awaiting lung transplant, since most transplant programs require patients to stop taking mTOR inhibitors due to concerns regarding impaired wound healing (50). Furthermore, we provide mechanistic insight of how cells of the immune system are linked to the aberrant regulation of VEGF-D in LAM. In view of the favorable safety profile of Syk inhibitors, evaluating their therapeutic efficacy in patients with LAM, other manifestations of TSC and malignancies characterized by activation of the mTORC1 pathway seems well warranted.
Supplementary Material
Acknowledgments
The authors are grateful to LAM patients and their families for their participation in this research. We would also like to thank Leigh Samsel, Venina Dominical, and J. Philip McCoy of the Flow Cytometry Core at NIH/NHLBI for their excellent technical assistance.
Financial support: Supported in part by the Department of Defense (TS130031) (to S.E-C), NIH grant R01 HL130275 (to S.E-C) and the Anne Levine LAM research fund (to S.E-C); BRI microgrant (to Y.C.); the Division of Intramural Research NIH/NHLBI.
Footnotes
Potential Conflicts of Interest: The authors declare that they have no conflicts of interest.
References
- 1.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345–56. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 2.Glasgow CG, Steagall WK, Taveira-Dasilva A, Pacheco-Rodriguez G, Cai X, El-Chemaly S, et al. Lymphangioleiomyomatosis (LAM): molecular insights lead to targeted therapies. Respir Med. 2010;104(Suppl 1):S45–58. doi: 10.1016/j.rmed.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc. 1991;66(8):792–6. doi: 10.1016/s0025-6196(12)61196-3. [DOI] [PubMed] [Google Scholar]
- 4.Henske EP, McCormack FX. Lymphangioleiomyomatosis - a wolf in sheep’s clothing. The Journal of clinical investigation. 2012;122(11):3807–16. doi: 10.1172/JCI58709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(11):6085–90. doi: 10.1073/pnas.97.11.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCormack FX, Panos RJ, Trapnell BC, editors. Molecular basis of pulmonary disease: insights from rare lung disorders. New York: Humana Press; 2010. [Google Scholar]
- 7.McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364(17):1595–606. doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCormack FX, Gupta N, Finlay GR, Young LR, Taveira-DaSilva AM, Glasgow CG, et al. Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines: Lymphangioleiomyomatosis Diagnosis and Management. American journal of respiratory and critical care medicine. 2016;194(6):748–61. doi: 10.1164/rccm.201607-1384ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parkhitko A, Myachina F, Morrison TA, Hindi KM, Auricchio N, Karbowniczek M, et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(30):12455–60. doi: 10.1073/pnas.1104361108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Shin S, Sun Y, Yoon SO, Li C, Zhang E, et al. mTORC1-Driven Tumor Cells Are Highly Sensitive to Therapeutic Targeting by Antagonists of Oxidative Stress. Cancer research. 2016;76(16):4816–27. doi: 10.1158/0008-5472.CAN-15-2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. The New England journal of medicine. 2008;358(2):140–51. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohammadieh AM, Bowler SD, Silverstone EJ, Glanville AR, Yates DH. Everolimus treatment of abdominal lymphangioleiomyoma in five women with sporadic lymphangioleiomyomatosis. The Medical journal of Australia. 2013;199(2):121–3. doi: 10.5694/mja12.11567. [DOI] [PubMed] [Google Scholar]
- 13.Taveira-DaSilva AM, Hathaway O, Stylianou M, Moss J. Changes in lung function and chylous effusions in patients with lymphangioleiomyomatosis treated with sirolimus. Annals of internal medicine. 2011;154(12):797–805. W-292-3. doi: 10.7326/0003-4819-154-12-201106210-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glasgow CG, Avila NA, Lin JP, Stylianou MP, Moss J. Serum vascular endothelial growth factor-D levels in patients with lymphangioleiomyomatosis reflect lymphatic involvement. Chest. 2009;135(5):1293–300. doi: 10.1378/chest.08-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seyama K, Kumasaka T, Souma S, Sato T, Kurihara M, Mitani K, et al. Vascular endothelial growth factor-D is increased in serum of patients with lymphangioleiomyomatosis. Lymphatic research and biology. 2006;4(3):143–52. doi: 10.1089/lrb.2006.4.143. [DOI] [PubMed] [Google Scholar]
- 16.Seyama K, Kumasaka T, Souma S, Sato T, Kurihara M, Mitani K, et al. Vascular endothelial growth factor-D is increased in serum of patients with lymphangioleiomyomatosis. Lymphat Res Biol. 2006;4(3):143–52. doi: 10.1089/lrb.2006.4.143. [DOI] [PubMed] [Google Scholar]
- 17.Cui Y, Osorio JC, Risquez C, Wang H, Shi Y, Gochuico BR, et al. Transforming growth factor-β1 downregulates vascular endothelial growth factor-D expression in human lung fibroblasts via the Jun NH2-terminal kinase signaling pathway. Mol Med. 2014;20(1):120–34. doi: 10.2119/molmed.2013.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tyryshkin A, Bhattacharya A, Eissa NT. SRC kinase is a novel therapeutic target in lymphangioleiomyomatosis. Cancer research. 2014;74(7):1996–2005. doi: 10.1158/0008-5472.CAN-13-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katz E, Dubois-Marshall S, Sims AH, Faratian D, Li J, Smith ES, et al. A gene on the HER2 amplicon, C35, is an oncogene in breast cancer whose actions are prevented by inhibition of Syk. Brit J Cancer. 2010;103:401–10. doi: 10.1038/sj.bjc.6605763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Udyavar AR, Hoeksema MD, Clark JE, Zou Y, Tang Z, Li Z, et al. Co-expression network analysis identifies Spleen Tyrosine Kinase (SYK) as a candidate oncogenic driver in a subset of small-cell lung cancer. BMC Syst Biol. 2013;7(Suppl 5):S1. doi: 10.1186/1752-0509-7-S5-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghotra VP, He S, van der Horst G, Nijhoff S, de Bont H, Lekkerkerker A, et al. SYK is a candidate kinase target for the treatment of advanced prostate cancer. Cancer Res. 2015;75(1):230–40. doi: 10.1158/0008-5472.CAN-14-0629. [DOI] [PubMed] [Google Scholar]
- 22.Luangdilok S, Box C, Patterson L, Court W, Harrington K, Pitkin L, et al. Syk tyrosine kinase is linked to cell motility and progression in squamous cell carcinomas of the head and neck. Cancer Res. 2007;67:7907–16. doi: 10.1158/0008-5472.CAN-07-0331. [DOI] [PubMed] [Google Scholar]
- 23.Leseux L, Hamdi SM, Al Saati T, Capilla F, Recher C, Laurent G, et al. Syk-dependent mTOR activation in follicular lymphoma cells. Blood. 2006;108(13):4156–62. doi: 10.1182/blood-2006-05-026203. [DOI] [PubMed] [Google Scholar]
- 24.Carnevale J, Ross L, Puissant A, Banerji V, Stone RM, DeAngelo DJ, et al. SYK regulates mTOR signaling in AML. Leukemia. 2013;27(11):2118–28. doi: 10.1038/leu.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howe SR, Gottardis MM, Everitt JI, Goldsworthy TL, Wolf DC, Walker C. Rodent model of reproductive tract leiomyomata. Establishment and characterization of tumor-derived cell lines. The American journal of pathology. 1995;146(6):1568–79. [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan B, Latek R, Hossbach M, Tuschl T, Lewitter F. siRNA Selection Server: an automated siRNA oligonucleotide prediction server. Nucleic acids research. 2004;32(Web Server issue):W130–4. doi: 10.1093/nar/gkh366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suljagic M, Longo PG, Bennardo S, Perlas E, Leone G, Laurenti L, et al. The Syk inhibitor fostamatinib disodium (R788) inhibits tumor growth in the Emu- TCL1 transgenic mouse model of CLL by blocking antigen-dependent B-cell receptor signaling. Blood. 2010;116(23):4894–905. doi: 10.1182/blood-2010-03-275180. [DOI] [PubMed] [Google Scholar]
- 28.Kobayashi T, Hirayama Y, Kobayashi E, Kubo Y, Hino O. A germline insertion in the tuberous sclerosis (Tsc2) gene gives rise to the Eker rat model of dominantly inherited cancer. Nature genetics. 1995;9(1):70–4. doi: 10.1038/ng0195-70. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Billingsley ML, Kincaid RL, Siraganian RP. Phosphorylation of Syk activation loop tyrosines is essential for Syk function. An in vivo study using a specific anti-Syk activation loop phosphotyrosine antibody. J Biol Chem. 2000;275(45):35442–7. doi: 10.1074/jbc.M004549200. [DOI] [PubMed] [Google Scholar]
- 30.Kuiatse I, Baladandayuthapani V, Lin HY, Thomas SK, Bjorklund CC, Weber DM, et al. Targeting the Spleen Tyrosine Kinase with Fostamatinib as a Strategy against Waldenstrom Macroglobulinemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(11):2538–45. doi: 10.1158/1078-0432.CCR-14-1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li S, Takeuchi F, Wang JA, Fuller C, Pacheco-Rodriguez G, Moss J, et al. MCP-1 overexpressed in tuberous sclerosis lesions acts as a paracrine factor for tumor development. The Journal of experimental medicine. 2005;202(5):617–24. doi: 10.1084/jem.20042469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uckun FM, Qazi S, Ma H, Tuel-Ahlgren L, Ozer Z. STAT3 is a substrate of SYK tyrosine kinase in B-lineage leukemia/lymphoma cells exposed to oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):2902–7. doi: 10.1073/pnas.0909086107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schroer N, Pahne J, Walch B, Wickenhauser C, Smola S. Molecular pathobiology of human cervical high-grade lesions: paracrine STAT3 activation in tumor-instructed myeloid cells drives local MMP-9 expression. Cancer research. 2011;71(1):87–97. doi: 10.1158/0008-5472.CAN-10-2193. [DOI] [PubMed] [Google Scholar]
- 34.Goncharova EA, Goncharov DA, Damera G, Tliba O, Amrani Y, Panettieri RA, Jr, et al. Signal transducer and activator of transcription 3 is required for abnormal proliferation and survival of TSC2-deficient cells: relevance to pulmonary lymphangioleiomyomatosis. Molecular pharmacology. 2009;76(4):766–77. doi: 10.1124/mol.109.057042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka M, Shimamura S, Kuriyama S, Maeda D, Goto A, Aiba N. SKAP2 Promotes Podosome Formation to Facilitate Tumor-Associated Macrophage Infiltration and Metastatic Progression. Cancer research. 2016;76(2):358–69. doi: 10.1158/0008-5472.CAN-15-1879. [DOI] [PubMed] [Google Scholar]
- 36.Taveira-DaSilva AM, Pacheco-Rodriguez G, Moss J. The natural history of lymphangioleiomyomatosis: markers of severity, rate of progression and prognosis. Lymphatic research and biology. 2010;8(1):9–19. doi: 10.1089/lrb.2009.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ulanova M, Puttagunta L, Marcet-Palacios M, Duszyk M, Steinhoff U, Duta F, et al. Syk tyrosine kinase participates in beta1-integrin signaling and inflammatory responses in airway epithelial cells. American journal of physiology Lung cellular and molecular physiology. 2005;288(3):L497–507. doi: 10.1152/ajplung.00246.2004. [DOI] [PubMed] [Google Scholar]
- 38.Yaghini FA, Li F, Malik KU. Expression and mechanism of spleen tyrosine kinase activation by angiotensin II and its implication in protein synthesis in rat vascular smooth muscle cells. The Journal of biological chemistry. 2007;282(23):16878–90. doi: 10.1074/jbc.M610494200. [DOI] [PubMed] [Google Scholar]
- 39.Young RM, Hardy IR, Clarke RL, Lundy N, Pine P, Turner BC, et al. Mouse models of non-Hodgkin lymphoma reveal Syk as an important therapeutic target. Blood. 2009;113(11):2508–16. doi: 10.1182/blood-2008-05-158618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pine PR, Chang B, Schoettler N, Banquerigo ML, Wang S, Lau A, et al. Inflammation and bone erosion are suppressed in models of rheumatoid arthritis following treatment with a novel Syk inhibitor. Clinical immunology. 2007;124(3):244–57. doi: 10.1016/j.clim.2007.03.543. [DOI] [PubMed] [Google Scholar]
- 41.Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. The New England journal of medicine. 2010;363(14):1303–12. doi: 10.1056/NEJMoa1000500. [DOI] [PubMed] [Google Scholar]
- 42.Genovese MC, Kavanaugh A, Weinblatt ME, Peterfy C, DiCarlo J, White ML, et al. An oral Syk kinase inhibitor in the treatment of rheumatoid arthritis: a three-month randomized, placebo-controlled, phase II study in patients with active rheumatoid arthritis that did not respond to biologic agents. Arthritis and rheumatism. 2011;63(2):337–45. doi: 10.1002/art.30114. [DOI] [PubMed] [Google Scholar]
- 43.Rolf MG, Curwen JO, Veldman-Jones M, Eberlein C, Wang J, Harmer A, et al. In vitro pharmacological profiling of R406 identifies molecular targets underlying the clinical effects of fostamatinib. Pharmacology research & perspectives. 2015;3(5):e00175. doi: 10.1002/prp2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Atochina-Vasserman EN, Abramova E, James ML, Rue R, Liu AY, Ersumo NT, et al. Pharmacological targeting of VEGFR signaling with axitinib inhibits Tsc2-null lesion growth in the mouse model of lymphangioleiomyomatosis. American journal of physiology Lung cellular and molecular physiology. 2015;309(12):L1447–54. doi: 10.1152/ajplung.00262.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCormack FX, Travis WD, Colby TV, Henske EP, Moss J. Lymphangioleiomyomatosis: calling it what it is: a low-grade, destructive, metastasizing neoplasm. American journal of respiratory and critical care medicine. 2012;186(12):1210–2. doi: 10.1164/rccm.201205-0848OE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pacheco-Rodriguez G, Moss J. The role of chemokines in migration of metastatic-like lymphangioleiomyomatosis cells. Critical reviews in immunology. 2010;30(4):387–94. doi: 10.1615/critrevimmunol.v30.i4.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Issaka RB, Oommen S, Gupta SK, Liu G, Myers JL, Ryu JH, et al. Vascular endothelial growth factors C and D induces proliferation of lymphangioleiomyomatosis cells through autocrine crosstalk with endothelium. The American journal of pathology. 2009;175(4):1410–20. doi: 10.2353/ajpath.2009.080830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schoppmann SF, Birner P, Stockl J, Kalt R, Ullrich R, Caucig C, et al. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. The American journal of pathology. 2002;161(3):947–56. doi: 10.1016/S0002-9440(10)64255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pacheco-Rodriguez G, Kumaki F, Steagall WK, Zhang Y, Ikeda Y, Lin JP, et al. Chemokine-enhanced chemotaxis of lymphangioleiomyomatosis cells with mutations in the tumor suppressor TSC2 gene. Journal of immunology. 2009;182(3):1270–7. doi: 10.4049/jimmunol.182.3.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.King-Biggs MB, Dunitz JM, Park SJ, Kay Savik S, Hertz MI. Airway anastomotic dehiscence associated with use of sirolimus immediately after lung transplantation. Transplantation. 2003;75(9):1437–43. doi: 10.1097/01.TP.0000064083.02120.2C. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.