

Abstract
Historically, the syndrome of primary paroxysmal dyskinesias was considered a group of disorders due to ion channel dysfunction. This proposition was primarily based on the discovery of mutations in ion channels, which caused other episodic neurological disorders such as epilepsy and migraine and also supported by the frequent association between paroxysmal dyskinesias and epilepsy. However, the discovery of the genes responsible for the three classic forms of paroxysmal dyskinesias disproved this ion channel theory. On the other hand, novel gene mutations implicating ion channels have been recently reported to produce episodic movement disorders clinically similar to the classical paroxysmal dyskinesias. Here, we review the clinical and pathophysiological aspects of the paroxysmal dyskinesias, further proposing a pathophysiological framework according to which they can be classified as synaptopathies (PRRT2 and MR1), channelopathies (KCNMA1 and SCN8A) or transportopathies (SLC2A1). This proposal might serve to explain similarities and differences among the various paroxysmal dyskinesias in terms of clinical features, treatment response, and natural history.
Keywords: paroxysmal dyskinesias, pathophysiology, PPRT2, GLUT1, MR1
1. Introduction
The syndrome of paroxysmal dyskinesias (PxDs) encompasses a rubric of disorders characterized by recurrent episodes of abnormal movements, frequently co-occurring with epilepsy or other episodic neurological symptoms (i.e., migraine and/or ataxia) [1]. Historically, PxDs and other episodic neurological disorders were mainly considered as disorders representing ion channel dysfunction (channelopathies) [2]. This proposition has been supported by the discovery of mutations in genes encoding for ion channels and responsible for epilepsy, periodic paralyses and episodic ataxias among other episodic neurological disorders, and linkage studies showed close vicinity to ion channels in cohorts including PxDs [2–5]. The ‘channelopathy hypothesis’ would tie in with the observed sensitivity of some PxD subtypes (i.e., the kinesigenic variant) to anticonvulsant drugs, which modulate the activity of ion channels [6]. Moreover, the PxDs-epilepsy association [6,7] suggested the concept that PxDs represented a form of “basal ganglia epilepsy” related to altered ion channel function at both cortical and subcortical levels [8]. Thus, in one patient with epilepsy and PxDs, the epileptogenic source was identified within the supplementary sensory motor cortex, the ictal discharge rapidly spreading to the basal ganglia [9].
However, the discovery of the three major PxDs-associated genes (PRRT2, MR-1 and SLC2A1), none of which encode for ion channels, lessened the strength of the channelopathy hypothesis [6]. On the other hand, PxDs and epilepsy co-occur in the setting of recently described novel mutations in KCNMA1 and SCN8A genes, implicated in ion channel function. Despite the significant advances in the molecular understanding of the PxDs, their pathophysiological mechanisms (with the exception of SLC2A1, which encodes for glucose transporter type 1) remain largely unclear.
Here, we review the clinical aspects of PxDs and accumulating evidence regarding their pathomechanisms, discuss novel genetic disorders, and propose a pathophysiological framework that may serve to explain similarities and differences among the various PxDs in terms of clinical features, treatment response, and natural history.
2. Clinical overview
PxDs are characterised by recurrent attacks of abnormal involuntary movements, typically chorea, dystonia or a combination thereof [1,6]. During the attacks, consciousness is preserved; between attacks most patients are normal [1,6].
The first clear description of PxDs probably dates back to 1940 when Mount and Reback described several family members over 5 generations who had attacks of choreo-dystonia triggered by alcohol and coffee and lasting from minutes to several hours [10]. Over the decades, it became clear that the PxDs were quite heterogeneous in terms of clinical characteristics and two further variants have been recognized [11,12]. Accordingly, a classification was proposed based on triggers: paroxysmal kinesigenic dyskinesias (PKD); paroxysmal non-kinesigenic dyskinesias (PNKD); and paroxysmal exercise-induced dyskinesias (PED) [13]. Despite the pragmatic usefulness of such a classification [13], the elucidation of the genetic basis of PxDs suggests there might be some heterogeneity within each type and clinical overlap between them [6,14]. Moreover, novel genetic disorders (i.e., KCNMA1 and SCN8A) have been described to produce episodes similar to the “classical” PxDs. As such, the following clinical overview will be structured according to the main genetic disorders generating PxDs, with or without epilepsy (figure 1).
Figure 1.
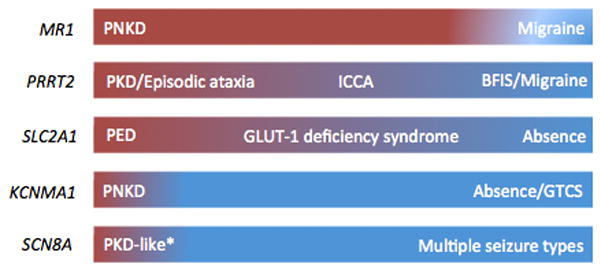
Spectrum of episodic neurological disorders occurring with mutations in the genes associated with paroxysmal dyskinesias. The underlying colour reflects the presumed cortical (blue) vs subcortical (red) origin of the clinical manifestations. PNKD; paroxysmal non-kinesigenic dyskinesias; PKD: paroxysmal kinesigenic dyskinesias; ICCA: infantile convulsions with choreoathetosis; BFIS: benign familial infantile seizures; PED: paroxysmal exercise-induced dyskinesias; GTCS: generalized tonic-clonic seizures. * The nature of the movement disorder occurring with SCN8A mutation has been questioned (see text for details).
2.1 PRRT2 (PKD, ICCA, and BFIS)
Since 2011 [15], mutations in the proline-rich transmembrane protein 2 (PRRT2) gene have been found in autosomal dominant early-onset neurological disorders such as PKD, benign familial infantile seizures (BFIS), with their association known as infantile convulsions plus choreoathetosis (ICCA), and rarely in some patients with PED, PNKD, hemiplegic migraine, episodic ataxia, childhood absence epilepsy, paroxysmal torticollis, and febrile seizures [7].
PKD, ICCA and BFIS have long been thought to be allelic disorders because they co-occur in some families and have been linked to the same region on chromosome 16p11.2-q12.1 [16,17]. By whole exome sequencing technology, Chen and colleagues first identified mutations in PRRT2 on chromosome 16p11.2 in eight Chinese PKD families [15], opening the way to the identification of PRRT2 mutations in its related clinical syndromes, ICCA and BFIS [7]. The PRRT2 mutations identified thus far include a considerable number of loss-of-function and missense amino-acid changing mutations [14]. The most common mutation is the frameshift single-nucleotide duplication c.649dupC (p.R217fsX224), which was found in over 60% of PKD, ICCA and BFIS families. This mutation likely arose independently in some of the families, given their diverse ethnic and geographic origins [18,19].
PRRT2-associated PKD
Currently, PRRT2 is the major gene responsible for PKD, with a frequency ranging from about 40% to over 90%, depending on case ascertainment [6,7,14,17,20–23]. Almost all PRRT2 cases have a clear kinesigenic trigger, and in up to 40% anxiety or prolonged exercise can also induce them [6]. The attacks are very short, classically lasting less than 1 minute, feature both chorea and dystonia, and tend to generalize [6]. Often patients have many attacks per day, but their frequency usually decreases with advancing age after puberty, regardless of any treatments [6,7].
PRRT2-associated BFIS
PRRT2 mutations accounts for about 80% of patients with BFIS, an autosomal dominant disorder occuring in infancy with onset between 3 and 12 months of age. BFIS is characterized by brief seizures featuring motor arrest, cyanosis, hypertonia, and limb jerks. There is a good response to antiepileptic drugs and seizures eventually remit before 2 years of age [24,25].
PRRT2-associated ICCA
PRRT2 mutations are also found in a large percentage (>90%) of ICCA families of different ancestry [18]. The ICCA syndrome is characterized by the combination of infantile seizures and paroxysmal dyskinesias, co-inherited as a single trait [18]. The PxDs start after the onset of epilepsy, usually after age 5, but some patients might exhibit epileptic seizures at a much later age [7].
2.2 MR1 (PNKD)
In 2004, missense mutations in the myofibrillogenesis regulator gene (MR1) were for the first time discovered in two unrelated families with PNKD [26]. Subsequently, several additional families have been reported on and this has allowed delineating the clinical characteristics of patients carrying MR1 mutations as compared to those with a similar phenotype but who do not carry such mutations [6,27]. Thus, onset is usually in the first decade and very rarely after age 18 [6,27]. There is always a dominant family history for similar problems (no sporadic cases have been described so far) [6]. Paroxysmal attacks frequently encompass both dystonic and choreic features and are generalized in about half of the cases [6,27]. In rare instances, attacks can be complicated by dysarthria, dysphagia, oculogyric crises, inability to move and/or pain and might be also fatal [6,27,28]. The duration of the attacks is variable but usually tends to last for up to hours [1,6,27]. Attacks are generally induced by coffee and alcohol intake, even though other precipitants, including stress, tiredness and prolonged exercise, have been rarely described [6]. Patients with MR1 mutations do not have epilepsy (and this makes the phenotype associated with this gene different from that associated with KCNMA1 mutations, as discussed below) [6]. However, a substantial proportion of patients with MR1 mutations can have migraine, thus linking this gene to another episodic neurological disorder [14]. Many patients seem to respond, at least partially, to benzodiazepines. However, there is a general tendency for the attacks to decrease with aging, regardless of any treatments.
2.3 SLC2A1 (PED)
The genetic overlap between epilepsy and movement disorders has also been widely recognized in glucose-transporter-1 (GLUT1) deficiency syndrome (OMIM 138140), caused by mutations in SLC2A1 encoding for the glucose transporter type 1 (GLUT1) protein of the blood-brain barrier. Both epilepsy, particularly early-onset absence, and PED co-occur in families and individuals [29–31]. The estimated frequency of SLC2A1 mutations is approximately 1:80,000 [32].
SLCA2A1-associated PED
The attacks usually consist of choreoathetosis and dystonia affecting mainly the lower limbs and are typically triggered by sustained exercise [33]. Notably, this disturbance might be misdiagnosed as epileptic myoclonic seizures [34]. The combination of epilepsy with a possible family history and PED in the setting of an unremarkable neurological examination, along with low CSF glucose concentration, represents an important clinical clue to raise the correct diagnostic suspicion [33]. An early diagnosis of GLUT-1 deficiency is crucial given that the syndrome can be well managed with a ketogenic diet (see below).
2.4 Other genes implicated in PxDs (KCNMA1 and SCN8A)
Two genetic mutations have been reported to include in their phenotype episodes resembling the “classic” PxDs, namely KCNMA1 and SCN8A [35,36].
KNCMA1-associated PNKD-like episodes
In 2005, Du and colleagues reported on a family with an autosomal dominant form of generalized epilepsy and PxDs carrying a mutation in the KCNMA1 gene, which encodes for a subunit of a calcium-activated potassium channel [36]. Clinically, the PxDs were described to resemble the non-kinesigenic variant with alcohol being a possible trigger, whereas the epileptic attacks could be either in the form of absence or GTCS [36]. The presence of epilepsy makes patients with KCNMA1 mutations different from those carrying MR1 mutations, which has never been associated with epilepsy. While two additional PNKD patients with KCNMA1 mutations have been subsequently reported to lack epilepsy [37], they presented with neurodevelopmental delay, also unlike MR1-associated PNKD [37].
SCN8A-associated PKD-like episodes
PKD, BFIS, and their combination, known as ICCA, a clinical syndrome classic of PRRT2 mutations, have been reported in patients with SCN8A mutations [14,21]. Gardella et al. reported 3 families with 16 members manifesting either with afebrile focal or GTCS during the first two years of life [38]. Additionally, 5 of these 16 patients had at older age choreo-dystonic or “shivering” attacks that were brought on by stretching, movement initiation and/or emotional stimuli [38]. All patients but one had normal developmental milestones, uninformative neuroimaging, and unremarkable neurological examination between attacks. Moreover, they were mostly seizure-free without medications in their adulthood [38]. These three families carried the same heterozygous mutation (c.4447G>A; p.E1483K) in the SCN8A gene. The phenomenology of the attacks suggests but does not resemble classical PKD [39]. Moreover, in one case a typical PKD spell was recorded by video-EEG-polygraphy and a cortical involvement was documented, suggesting that such attacks might in fact be epileptic in nature [39]. Nonetheless, this report highlights the difficulty in making the diagnosis of PxDs based on clinical features alone. Notably, other reports on the phenotypic spectrum of SCN8A-related epileptic encephalopathies have described the presence of “choreo-dystonia” and “dystonic dyskinesias” in some cases [40], thus supporting the idea that episodic movement disorders (arguably resembling PxDs) can occur with SCN8A mutations. Despite the clinical similarities with patients carrying PRRT2 mutations, there are several differences that might help the differential diagnosis [40]. Besides the families reported by Gardella et al. [39], other SCN8A cases have been reported with a variety of seizure types encompassing focal, tonic, clonic, myoclonic and absence seizures, all of which are reported to be usually refractory to antiepileptic drugs. Eventually, SCN8A patients develop moderate to severe intellectual disability, regardless of whether early development was normal [40]. Moreover, these might co-occur with non-episodic movement disorders, including dystonia and ataxia [40]. Finally, most SCN8A mutations are de-novo, although a single case of somatic mosaicism in an unaffected parent has been observed [40]. All these features make the phenotype(s) associated with SCN8A different from that associated with PRRT2.
3. A pathophysiological framework
3.1 PxD as synaptopathies
PRRT2 and presynaptic vesicles
PRRT2 encodes for a protein that is highly conserved across species [41]. The encoded 340-amino acid protein, the proline-rich transmembrane protein 2 (PRRT2), is highly expressed in the cerebral cortex, cerebellum and basal ganglia (figure 2), which matches with the localization expected based on the human PRRT2 phenotype. Yet, it physiologic function has not been entirely elucidated.
Figure 2.
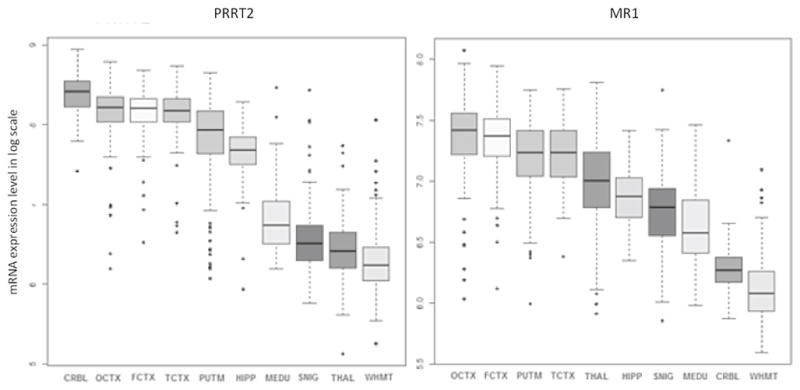
Regional expression of mRNA from PRRT2 (left panel) and MR1 (right panel) in the normal human brain. Brain regions included in the analysis: cerebellum (CRBL); occipital cortex (OCTX); frontal cortex (FCTX); temporal cortex (TCTX); putamen (PUTM); hippocampus (HPP); medulla (MEDU); substantia nigra (SNIG); thalamus (THAL); white matter (WHMT). [Source: UK Brain Expression Consortium]
PRRT2 consists of two transmembrane helices in the C-terminal region, a long N-terminal intra-cytoplasmic region that includes a proline-rich domain, an intracellular loop linking the two transmembrane helices, and a very short C-terminal end [17]. Subsequent to alternative splicing at the 3′ terminus, there are recognized six splice variants and three different isoforms (www.ensemble.org; ENSG00000167371). However, since the large majority of pathogenic mutations in PRRT2 are localized in a conserved region shared by all splice variants, the contribution of the different isoforms to PRRT2-associated diseases appears elusive [14,21].
There is evidence that PRRT2 localizes mostly in axons of glutamatergic synapses [41,42]. However, previous works have suggested that it interacts with the SNARE protein SNAP-25 as well as the GluA1 subunit of the AMPA-type glutamate receptor complex, leaving the question open of whether PRRT2 localizes at the presynaptic terminal, postsynaptic terminal, or both. Recent evidence suggests that PRRT2 main localization is at the presynaptic terminal, where it interacts with a number of other proteins involved in synaptic vesicle docking and neurotransmitter release. Indeed, Valente and colleagues recently demonstrated that PRRT2 is highly distributed in presynaptic terminals and interacts with SNAP-25, VAMP2 and synaptotagmin1/2 (Syt1/2), intimately implicated in the Ca2+-sensing machinery involved in the final steps of neurotransmitter release [43]. PRRT2-silenced neurons have an abnormal asynchronous/synchronous neurotransmitter release ratio indicating that the fusion mechanism per se is not altered and that a specific defect in coupling Ca2+ influx to exocytosis is the most likely pathomechanism [43]. Interestingly, similar results were obtained in a mouse strain carrying a Syt2 mutation and manifesting with ataxia [44], one of the clinical phenotypes of PRRT2 in humans. PRRT2 may also have other functions. Its temporal expression increases during the development and in fact PRRT2 silencing in primary neurons decreases synaptic density and alters nerve terminal ultrastructure. Using RNA interference, it has been shown that knocking PRRT2 in mouse embryos leads to a delay in neuronal migration and defects in synaptic development [45]. Therefore, it is not surprising that homo- or heterozygous biallelic PRRT2 mutations can lead to neurodevelopmental disorders [46–48]. Finally, proteomic studies demonstrated an interaction of PRRT2 with the AMPA receptor complex [49]. After infection with shRNA-Prrt2 lentivirus, there is an increase in glutamate levels in the culture medium of neurons as well as in the distribution of GluA1 on the cell membrane [42]. One of the reasons why carbamazepine is very effective in patients with PRRT2 mutations might be owing to its ability to enhance the activity of the glutamate transporter 3 at the post-synaptic terminal [50], thus allowing glutamate to be pumped into postsynaptic terminals without activating its receptor.
MR1 and synaptic regulation
The MR1 or PNKD gene encodes for the Myofibrillogenesis Regulator 1, a protein with 3 isoforms that have distinct cellular distribution and, likely, biological functions. The brain specific isoform MR1L consists of 385 amino acids [26] and is highly expressed in the basal ganglia (figure 2). At the cellular level, MR1L localizes on the membrane [51]. However, its function is not yet entirely clear. MR1 shares up to 40% identity with the human hydroxyacylglutathione hydrolase (HAGH), especially for the beta-lactamase domain [52]. HAGH is involved in the pathway to detoxify methylglyoxal, a compound present in both caffeinated and alcoholic drinks that is produced as a by-product of oxidative stress [52]. This would suggest a putative mechanism whereby alcohol, coffee, and stress could induce attacks in patients with MR1 mutations. However, it was found in cultured cells and transgenic animals that MR1L could not effectively restore absent HAGH activity, suggesting that it does not intervene in glutathione metabolism at appreciable levels in vivo [51]. More recently, in vivo data from the MR1−/− mice and the identification of RIM1 and RIM2 as MR1-binding proteins hinted at the possibility that MR1 could be involved in synaptic regulation [53]. Rab3-interacting molecules (RIMs) are a group of proteins of the active zone that interact among others with vesicle proteins and presynaptic membrane proteins, playing therefore a crucial role in vesicle priming and calcium-dependent neurotransmitter release [53]. Thus, MR1-knockout animals have significantly reduced RIM levels and, in turn, display an abnormal neurotransmitter release [53]. MR1 might be hence involved in stabilizing RIM proteins at the active zone and inhibiting RIM-dependent pathway. Consistent with this, overexpressed MR1 inhibits exocytosis [53]. Given that caffeine acts as a ryanodine receptor agonist at presynaptic terminals and stimulates calcium efflux from the endoplasmic reticulum, the hypothesis has been raised that neurons might be more vulnerable to high concentration of calcium and hence might become hyperexcitable when challenged with caffeine [53]. Striatal neurons are more susceptible to this pathomechanism, as also demonstrated by a proportional increase of dopamine release after caffeine or ethanol challenge in mutated transgenic MR1 mice that recapitulate the human MR1 phenotype [54].
In summary, PRRT2 and MR1 play important roles in synapse formation and/or functioning and should be hence classified, from a pathophysiological standpoint, as synaptopathies, an emerging rubric of brain diseases including neurodevelopmental disorders as well as other forms of epilepsy [55], all of which share aberrant synapse physiology as the main pathomechanism.
3.2 PxD as channelopaties
Because electrical signals are critical to the function of neurons, proteins that regulate neuronal electrical signaling are obvious sites where abnormalities might lead to episodic neurological diseases [56].
KCNMA1 encodes for a subunit of a calcium-activated potassium channel, belonging to the family of large conductance for potassium ion channels (BK channels) [36,56]. BK channels are involved in many physiological processes such as neuronal excitability, synaptic transmission as well as regulation of myogenic tone [57]. BK channels are mainly activated by internal Ca2+ concentration but an alternative mechanism of activation relies on the voltage sensitivity of the S4 domain [58]. Thus, calcium opens BK channels when all the voltage sensors are in their resting configuration and, vice versa, voltage can activate channels when Ca2+ is absent [58]. It was demonstrated that the original mutation enhances channel activity at the same voltage and intracytoplasmic Ca2+, so that more BK channels are open than the wild-type (WT) channels at physiological conditions [36]. However, this evidence stemmed form models where β subunits were absent. In addition to the α subunit of the BK channel, 4 β subunits have been recognized [57]. The most expressed β subunits in the brain are β2, 3, and 4 [57]. Interestingly, a mutation on the β3 subunit has been linked to idiopathic epilepsy [59] and β4 knock-out mice show temporal lobe seizures [60]. Especially with the β4, BK channels regulate neuronal excitability at both cortical and sub-cortical levels [61–63]. Therefore, it is conceivable that mutations altering these processes can lead to both epilepsy and PxDs.
SCN8A mutations are also predicted to alter neuronal excitability. Most SCN8A mutations are missense mutations causing increased NaV1.6 channel activity, which provides the molecular basis for the initiation and propagation of action potentials [64]. NaV1.6 is widely expressed both in the cerebral cortex and subcortical structures [65]. Functional analyses have suggested that SCN8A mutations can lead to either gain-of-function (i.e., by an increased persistent sodium current) or loss-of-function effects (i.e., by an unstable protein) [64]. In fact, both hypoactivity and hyperactivity of NaV1.6 are suggested to be pathogenic, yet with different outcomes: haploinsufficiency would be more likely associated with cognitive dysfunction and hyperactivity within an epileptic encephalopathy [64]. However, due to the relatively small number of mutations reported on, no strict genotype-phenotype correlation can be definitely suggested.
In summary, both KCNMA1 and SCN8A mutation alter the function of ion channels, neuronal excitability with or without a net impairment in neurotransmitter release, and should be hence classified, from a pathophysiological standpoint, as channelopathies.
3.3 PxD as transportopathies
SLC2A1 encodes the glucose transporter type 1 protein (GLUT1) that is responsible for transporting glucose across the blood-brain barrier. Haploinsufficiency of SLC2A1 results in a hypoglycemic state of the brain, which can cause a wide spectrum of neurological conditions. Functional studies revealed that a chronic hypoglycemic state is responsible for intermittent generalized epileptiform activity. In particular, positron emission tomography (PET) findings indicated that there is a permanent hypometabolism in the frontal lobes of patients with epilepsy that matches with EEG data showing epileptic activity with anterior predominance [34]. Moreover, there was a positive correlation between PED frequency and the FDG uptake in the left putamen [34]. Additionally, ictal SPECT analysis showed hyperperfusion in the aforementioned regions during an episode of PED [34]. Altogether, these layers of evidence suggest that disordered glucose metabolism in the corticostriate pathways is crucial in the pathophysiology of PED [34]. An open issue remains the broad phenotypic variability of the GLUT1 defects. Genotype–phenotype correlations show that deletions, nonsense, frameshift and splice-site mutations resulting in complete loss of one allele typically lead to the classic GLUT1 deficiency syndrome, whereas heterozygous missense mutations where a residual function is predicted are more often found in milder phenotypes, including isolated PED [31,67,68]. Finally, the reduced penetrance and the variable clinical expressivity of SLC2A1 mutations suggest that the other modifying genetic and/or acquired environmental factors may influence the underlying pathophysiology and clinical expressivity of this disorder.
Although the term transportopathies has been coined for disorders due to deficient neurotransmitter transporters [69] and glucose is not strictly a neurotransmitter, we thought reasonable to consider it a transportopathy. In fact, bypassing the transport defects by means of a ketogenic diet represents the first therapeutic choice.
4. Knowledge gaps and future perspectives
The similarities among epilepsy and PxDs, including their episodic nature, triggering factors, and the therapeutic response to the same drugs, are striking. Historically, epilepsy has been considered a disorder originating from the cerebral cortex whereas PxDs have mostly been considered to reflect a subcortical dysfunction. However, increased understanding of the genetics underpinnings of epilepsy syndromes and PxDs have provided insights into the shared mechanisms of these two conditions, revealing the role of ion channels, and of proteins associated to the vesical synaptic cycle or involved in energy metabolism (figure 3) [14,31,36,38,43,53,70–72]. Nevertheless, there remains controversy about the extent to which paroxysmal movement disorder may have epileptic components, as variably measured by video-EEG monitoring [9,73–75], recognizing that abnormally organized electrical activity of subcortical structures (“subcortical seizures”) may not be readily recorded with scalp electrodes. In general, a clear differentiation between the pathophysiology of purely subcortical and cortical events risks to be an oversimplification due to the strong reciprocal interconnectivity between basal ganglia and cerebral cortex, particularly within the fronto-striatal network, as well as the differential age-related expression of dysfunction in these structures. For instance, ictal SPECT demonstrated that a network including both basal ganglia and cortical motor areas was involved in patients with SLC2A1 mutations manifesting with epilepsy and PxD, [34]. The age-dependent expression of clinical features in patients with PRRT2 mutations is likely to be explained by the particular temporal pattern of the protein at both cerebral and spinal level [17], which is in accordance with the observation that mutations in this gene more likely lead to epilepsy in infancy and PKD in childhood or adolescence, with a tendency to spontaneous remission later in life. Moreover, this temporal pattern implicates PRRT2 in neurodevelopment, as also suggested by the fact that biallelic PRRT2 mutations cause mental retardation [76]. However, it is still difficult to explain how the same mutation in a certain gene can cause both epilepsy and movement disorders either in a given patient or family, or in separate families. Future studies should address this issue by exploring the putative role of modifier genes or environmental factors on phenotypic variability. In fact, the brain localization of the specific mutated protein does not seem to entirely explain the clinical phenotype. For instance, MR1 is also expressed at the cortical level (figure 2), yet without causing any “cortical” paroxysms with the exception of migraine [14]. It is therefore likely that modifier genes and interacting proteins play a role in modulating the clinical phenotype.
Figure 3.
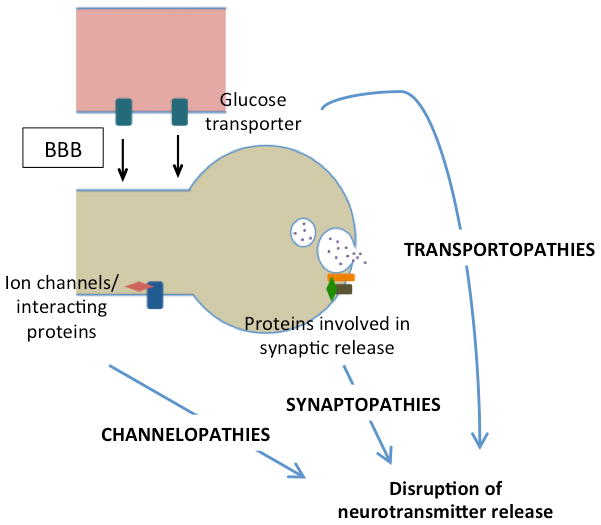
Schematic diagram of the paroxysmal dyskinesia pathomechanisms. (BBB: brain blood barrier)
On the other hand, the main objective for future research should be shifting from the identification of the underlying molecular cause to the development of pharmacogenomics and related tailored therapeutic strategies [77]. Indeed, the advent of high-throughput genomic sequencing and related tools in molecular diagnosis further offers a great occasion to develop specific therapies by directly targeting the pathophysiological mechanisms that produce the outspread effects of these disorders [78].
Footnotes
Financial disclosures related to the manuscript: none
Full financial disclosure:
RE received consultancies from Zambon.
KPB has received grant support from Welcome/MRC, NIHR, Parkinsons’s UK and EU Horizon 2020. He has received royalties from publication of the Oxford Specialist Handbook Parkinson’s Disease and Other Movement Disorders (Oxford University Press, 2008, 2016) and of Marsden’s Book of Movement Disorders (Oxford University Press, 2013). He has received honoraria/personal compensation for participating as consultant/scientific board member from Ipsen, Allergan, Merz and honoraria for speaking at meetings and from Allergan, Ipsen, Merz, Sun Pharma, Teva, UCBPharmaceuticals and from the American Academy of Neurology and Movement Disorders Society.
AJE has received grant support from the NIH, CleveMed/Great Lakes Neurotechnologies, Davis Phinney Foundation, and Michael J Fox Foundation; personal compensation as a consultant/scientific advisory board member for Solvay, Abbott, Chelsea Therapeutics, TEVA, Impax, Merz, Lundbeck, and Eli Lilly; honoraria from TEVA, UCB, the American Academy of Neurology, and the Movement Disorders Society; and publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer.
PS received grant support from the Telethon Foundation and Italian MoH; personal compensation as a consultant/scientific advisory board member for Eisai pharma, Kolfarma, Shire; and publishing royalties from Springer and Ambrosiana Editrice.
References
- 1.Bhatia KP. Paroxysmal dyskinesias. Mov Disord. 2011;26:1157–1165. doi: 10.1002/mds.23765. [DOI] [PubMed] [Google Scholar]
- 2.Bhatia KP, Griggs RC, Ptáček LJ. Episodic movement disorders as channelopathies. Mov Disord. 2000 May;15(3):429–33. [PubMed] [Google Scholar]
- 3.Fouad GT, Servidei S, Durcan S, Bertini E, Ptáček LJ. A gene for familial paroxysmal dyskinesia (FPD1) maps to chromosome 2q. Am J Hum Genet. 1996;59:135–139. [PMC free article] [PubMed] [Google Scholar]
- 4.Fink JK, Rainier S, Wilkowski J, et al. Paroxysmal dystonic choreoathetosis: tight linkage to chromosome 2q. Am J Hum Genet. 1996;59:140–145. [PMC free article] [PubMed] [Google Scholar]
- 5.Ptáček LJ. Ion channel diseases: episodic disorders of the nervous system. Semin Neurol. 1999;19(4):363–9. [PubMed] [Google Scholar]
- 6.Erro R, Sheerin UM, Bhatia KP. Paroxysmal dyskinesias revisited: a review of 500 genetically proven cases and a new classification. Mov Disord. 2014 Aug;29(9):1108–16. doi: 10.1002/mds.25933. [DOI] [PubMed] [Google Scholar]
- 7.Nobile C, Striano P. PRRT2: a major cause of infantile epilepsy and other paroxysmal disorders of childhood. Prog Brain Res. 2014;213:141–58. doi: 10.1016/B978-0-444-63326-2.00008-9. [DOI] [PubMed] [Google Scholar]
- 8.Singh R, Macdonell RA, Scheffer IE, Crossland KM, Berkovic SF. Epilepsy and paroxysmal movement disorders in families: evidence for shared mechanisms. Epileptic Disord. 1999 Jun;1(2):93–9. [PubMed] [Google Scholar]
- 9.Lombroso CT. Paroxysmal kinesigenic choreoathetosis: an epileptic or non-epileptic disorder? Ital J Neurosci. 1995;16: 271–7. doi: 10.1007/BF02249102. [DOI] [PubMed] [Google Scholar]
- 10.Mount L, Reback S. Familial paroxysmal choreoathetosis. Arch Neurol. 1940;44:841–847. [Google Scholar]
- 11.Kertesz A. Paroxysmal kinesigenic choreoathetosis: an entity within the paroxysmal choreoathetosis syndrome. Description of 10 cases, including 1 autopsied. Neurology. 1967;17:680–690. doi: 10.1212/wnl.17.7.680. [DOI] [PubMed] [Google Scholar]
- 12.Lance JW. Familial paroxysmal dystonic choreoathetosis and its differentiation from related syndromes. Ann Neurol. 1977;2:285–293. doi: 10.1002/ana.410020405. [DOI] [PubMed] [Google Scholar]
- 13.Demirkiran M, Jankovic J. Paroxysmal dyskinesias: clinical features and classification. Ann Neurol. 1995;38:571–579. doi: 10.1002/ana.410380405. [DOI] [PubMed] [Google Scholar]
- 14.Gardiner AR, Jaffer F, Dale RC, et al. The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain. 2015 Dec;138(Pt 12):3567–80. doi: 10.1093/brain/awv310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan GH, Guo SL, He J, Chen YF, Zhang QJ, Li HF, Lin Y, Murong SX, Xu J, Wang N, Wu ZY. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet. 2011;43:1252–5. doi: 10.1038/ng.1008. [DOI] [PubMed] [Google Scholar]
- 16.Lee WL, Tay A, Ong HT, Goh LM, Monaco AP, Szepetowski P. Association of infantile convulsions with paroxysmal dyskinesias (ICCA syndrome): confirmation of linkage to human chromosome 16p12-q12 in a Chinese family. Hum Genet. 1998 Nov;103(5):608–12. doi: 10.1007/s004390050876. [DOI] [PubMed] [Google Scholar]
- 17.Striano P, Lispi ML, Gennaro E, et al. Linkage analysis and disease models in benign familial infantile seizures: a study of 16 families. Epilepsia. 2006 Jun;47(6):1029–34. doi: 10.1111/j.1528-1167.2006.00521.x. [DOI] [PubMed] [Google Scholar]
- 18.Szepetowski P, Rochette J, Berquin P, Piussan C, Lathrop GM, Monaco AP. Familial infantile convulsions and paroxysmal choreoathetosis: a new neurological syndrome linked to the pericentromeric region of human chromosome 16. Am J Hum Genet. 1997;61:889–98. doi: 10.1086/514877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker F, Schubert J, Striano P, et al. PRRT2-related disorders: further PKD and ICCA cases and review of the literature. J Neurol. 2013;260(5):1234–44. doi: 10.1007/s00415-012-6777-y. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Zhu X, Wang X, et al. Targeted genomic sequencing identifies PRRT2 mutations as a cause of paroxysmal kinesigenic choreoathetosis. J Med Genet. 2012 Feb;49(2):76–8. doi: 10.1136/jmedgenet-2011-100635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Zhao X, Du Y, He F, Peng G, Luo B. Phenotypic overlap among paroxysmal dyskinesia subtypes: Lesson from a family with PRRT2 gene mutation. Brain Dev. 2013 Aug;35(7):664–6. doi: 10.1016/j.braindev.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 22.Méneret A, Grabli D, Depienne C, et al. PRRT2 mutations: a major cause of paroxysmal kinesigenic dyskinesia in the European population. Neurology. 2012 Jul 10;79(2):170–4. doi: 10.1212/WNL.0b013e31825f06c3. [DOI] [PubMed] [Google Scholar]
- 23.Ebrahimi-Fakhari D, Saffari A, Westenberger A, Klein C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain. 2015 Dec;138(Pt 12):3476–95. doi: 10.1093/brain/awv317. [DOI] [PubMed] [Google Scholar]
- 24.Heron SE, Grinton BE, Kivity S, et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am J Hum Genet. 2012;90:152–160. doi: 10.1016/j.ajhg.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zara F, Specchio N, Striano P, et al. Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia. 2013 Mar;54(3):425–36. doi: 10.1111/epi.12089. [DOI] [PubMed] [Google Scholar]
- 26.Rainier S, Thomas D, Tokarz D, et al. Myofibrillogenesis regulator 1 gene mutations cause paroxysmal dystonic choreoathetosis. Arch Neurol. 2004;61:1025–1029. doi: 10.1001/archneur.61.7.1025. [DOI] [PubMed] [Google Scholar]
- 27.Bruno MK, Lee HY, Auburger GWJ, et al. Genotype-phenotype correlation of paroxysmal nonkinesigenic dyskinesia. Neurology. 2007;68:1782–9. doi: 10.1212/01.wnl.0000262029.91552.e0. [DOI] [PubMed] [Google Scholar]
- 28.Zittel S, Ganos C, Münchau A. Fatal paroxysmal non-kinesigenic dyskinesia. Eur J Neurol. 2015;22(2):e30–1. doi: 10.1111/ene.12574. [DOI] [PubMed] [Google Scholar]
- 29.Mullen SA, Marini C, Suls A, et al. Glucose transporter 1 deficiency as a treatable cause of myoclonic astatic epilepsy. Arch Neurol. 2011;68:1152–1155. doi: 10.1001/archneurol.2011.102. [DOI] [PubMed] [Google Scholar]
- 30.Agostinelli S, Traverso M, Accorsi P, et al. Early-onset absence epilepsy: SLC2A1 gene analysis and treatment evolution. Eur J Neurol. 2013;20(5):856–9. doi: 10.1111/j.1468-1331.2012.03871.x. [DOI] [PubMed] [Google Scholar]
- 31.De Giorgis V, Veggiotti P. GLUT1 deficiency syndrome 2013: current state of the art. Seizure. 2013;22:803–811. doi: 10.1016/j.seizure.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 32.Larsen J, Johannesen KM, Ek J, Tang S, et al. The role of SLC2A1 mutations in myoclonic astatic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome. Epilepsia. 2015 Dec;56(12):e203–8. doi: 10.1111/epi.13222. [DOI] [PubMed] [Google Scholar]
- 33.Erro R, Stamelou M, Ganos C, et al. The clinical syndrome of paroxysmal exercise-induced dystonia: diagnostic outcomes and an algorithm. Mov Disord Clin Pract. 2014;1:57–61. doi: 10.1002/mdc3.12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suls A, Dedeken P, Goffin K, et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain. 2008 Jul;131(Pt 7):1831–44. doi: 10.1093/brain/awn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erro R. Expanding the genetic spectrum of paroxysmal dyskinesias. Mov Disord. 2016;31(7):936. doi: 10.1002/mds.26646. [DOI] [PubMed] [Google Scholar]
- 36.Du W, Bautista JF, Yang H, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733–738. doi: 10.1038/ng1585. [DOI] [PubMed] [Google Scholar]
- 37.Zhang ZB, Tian MQ, Gao K, Jiang YW, Wu Y. De novo KCNMA1 mutations in children with early-onset paroxysmal dyskinesia and developmental delay. Mov Disord. 2015;30(9):1290–2. doi: 10.1002/mds.26216. [DOI] [PubMed] [Google Scholar]
- 38.Gardella E, Becker F, Møller RS, et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol. 2016 Mar;79(3):428–36. doi: 10.1002/ana.24580. [DOI] [PubMed] [Google Scholar]
- 39.Balint B, Erro R, Salpietro V, Houlden H, Bhatia KP. PKD or Not PKD: That is the question. Ann Neurol. 2016 Jul;80(1):167–8. doi: 10.1002/ana.24668. [DOI] [PubMed] [Google Scholar]
- 40.Larsen J, Carvill GL, Gardella E, et al. EuroEPINOMICS RES Consortium CRP. The phenotypic spectrum of SCN8A encephalopathy. Neurology. 84(5):480–9. doi: 10.1212/WNL.0000000000001211. 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee HY, Huang Y, Bruneau N, et al. Mutations in the gene PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions. Cell Rep. 2012;1:2–12. doi: 10.1016/j.celrep.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li M, Niu F, Zhu X, Wu X, Shen N, Peng X, Liu Y. PRRT2 Mutant Leads to Dysfunction of Glutamate Signaling. Int J Mol Sci. 2015 Apr 23;16(5):9134–51. doi: 10.3390/ijms16059134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valente P, Castroflorio E, Rossi P, et al. PRRT2 Is a Key Component of the Ca(2+)-Dependent Neurotransmitter Release Machinery. Cell Rep. 2016 Apr 5;15(1):117–31. doi: 10.1016/j.celrep.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pang ZP, Sun J, Rizo J, Maximov A, Seudhof TC. Genetic analysis of synaptotagmin 2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J. 2006;25:2039–2050. doi: 10.1038/sj.emboj.7601103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu YT, Nian FS, Chou WJ, et al. PRRT2 mutations lead to neuronal dysfunction and neurodevelopmental defects. Oncotarget. 2016 May 9; doi: 10.18632/oncotarget.9258. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delcourt M, Riant F, Mancini J, et al. Severe phenotypic spectrum of biallelic mutations in PRRT2 gene. Journal of neurology, neurosurgery, and psychiatry. 2015;86:782–785. doi: 10.1136/jnnp-2014-309025. [DOI] [PubMed] [Google Scholar]
- 47.Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 48.Labate A, Tarantino P, Palamara G, et al. Mutations in PRRT2 result in familial infantile seizures with heterogeneous phenotypes including febrile convulsions and probable SUDEP. Epilepsy Res. 2013;104:280–284. doi: 10.1016/j.eplepsyres.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 49.Schwenk J, Harmel N, Brechet A, et al. High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron. 2012;74:621–633. doi: 10.1016/j.neuron.2012.03.034. [DOI] [PubMed] [Google Scholar]
- 50.Lee G, Huang Y, Washington JM, Briggs NW, Zuo Z. Carbamazepine enhances the activity of glutamate transporter type 3 via phosphatidylinositol 3-kinase. Epilepsy Res. 2005 Aug-Sep;66(1–3):145–53. doi: 10.1016/j.eplepsyres.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 51.Shen Y, Lee HY, Rawson J, Ojha S, Babbitt P, Fu YH, Ptacek LJ. Mutations in PNKD causing paroxysmal dyskinesia alters protein cleavage and stability. Hum Molec Genet. 2011;20:2322–2332. doi: 10.1093/hmg/ddr125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee HY, Xu Y, Huang Y, Ahn AH, et al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum Mol Genet. 2004 Dec 15;13(24):3161–70. doi: 10.1093/hmg/ddh330. [DOI] [PubMed] [Google Scholar]
- 53.Shen Y, Ge WP, Li Y, et al. Protein mutated in paroxysmal dyskinesia interacts with the active zone protein RIM and suppresses synaptic vesicle exocytosis. Proc Natl Acad Sci U S A. 2015 Mar 10;112(10):2935–41. doi: 10.1073/pnas.1501364112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee HY, Nakayama J, Xu Y, et al. Dopamine dysregulation in a mouse model of paroxysmal nonkinesigenic dyskinesia. J Clin Invest. 2012 Feb;122(2):507–18. doi: 10.1172/JCI58470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grant SG. Synaptopathies: diseases of the synaptome. Curr Opin Neurobiol. 2012;22:522–9. doi: 10.1016/j.conb.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 56.Ptácek LJ. Channelopathies: ion channel disorders of muscle as a paradigm for paroxysmal disorders of the nervous system. Neuromuscul Disord. 1997 Jun;7(4):250–5. doi: 10.1016/s0960-8966(97)00046-1. [DOI] [PubMed] [Google Scholar]
- 57.Lee US, Cui J. {beta} subunit-specific modulations of BK channel function by a mutation associated with epilepsy and dyskinesia. J Physiol. 2009 Apr 1;587(Pt 7):1481–98. doi: 10.1113/jphysiol.2009.169243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Latorre R, Brauchi S. Large conductance Ca2+-activated K+ (BK) channel: activation by Ca2+ and voltage. Biol Res. 2006;39(3):385–401. doi: 10.4067/s0716-97602006000300003. [DOI] [PubMed] [Google Scholar]
- 59.Lorenz S, Heils A, Kasper JM, Sander T. Allelic association of a truncation mutation of the KCNMB3 gene with idiopathic generalized epilepsy. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:10–13. doi: 10.1002/ajmg.b.30369. [DOI] [PubMed] [Google Scholar]
- 60.Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. 2005;8:1752–1759. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- 61.Faber ES, Sah P. Calcium-activated potassium channels: multiple contributions to neuronal function. Neuroscientist. 2003;9:181–194. doi: 10.1177/1073858403009003011. [DOI] [PubMed] [Google Scholar]
- 62.Hu H, Shao LR, Chavoshy S, Gu N, Trieb M, Behrens R, Laake P, Pongs O, Knaus HG, Ottersen OP, Storm JF. Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci. 2001;21:9585–9597. doi: 10.1523/JNEUROSCI.21-24-09585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bentzen BH, Olesen SP, Rønn LC, Grunnet M. BK channel activators and their therapeutic perspectives. Front Physiol. 2014 Oct 9;5:389. doi: 10.3389/fphys.2014.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meisler MH, Helman G, Hammer MF, et al. SCN8A encephalopathy: Research progress and prospects. Epilepsia. 2016 Jun 8; doi: 10.1111/epi.13422. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Caldwell JH, Schaller KL, Lasher RS, et al. Sodium channel Na(v)1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A. 2000;97:5616–5620. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Striano P, de Jonghe P, Zara F. Genetic epileptic encephalopathies: is all written into the DNA? Epilepsia. 2013 Nov;54(Suppl 8):22–6. doi: 10.1111/epi.12419. [DOI] [PubMed] [Google Scholar]
- 67.Schneider SA, Paisan-Ruiz C, Garcia-Gorostiaga I, et al. GLUT1 gene mutations cause sporadic paroxysmal exercise-induced dyskinesias. Mov Disord. 2009 Aug 15;24(11):1684–8. doi: 10.1002/mds.22507. [DOI] [PubMed] [Google Scholar]
- 68.Erro R, Stamelou M, Bhatia KP. Paroxysmal dyskinesias. In: Jancovic J, Tolosa E, editors. Parkinson’s Disease and Movement Disorders. 6. Philadelphia, PA: Wolters Kluwer; 2015. [Google Scholar]
- 69.Blackstone C. Infantile parkinsonism-dystonia: a dopamine “transportopathy”. J Clin Invest. 2009;119:1455–8. doi: 10.1172/JCI39632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee HY, Fu YH, Ptáček LJ. Episodic and electrical nervous system disorders caused by nonchannel genes. Annu Rev Physiol. 2015;77:525–41. doi: 10.1146/annurev-physiol-021014-071814. [DOI] [PubMed] [Google Scholar]
- 71.Ptáček LJ. Episodic disorders: channelopathies and beyond. Annu Rev Physiol. 2015;77:475–9. doi: 10.1146/annurev-physiol-021014-071922. [DOI] [PubMed] [Google Scholar]
- 72.Rossi P, Sterlini B, Castroflorio E, et al. A Novel Topology of Proline-rich Transmembrane Protein 2 (PRRT2): HINTS FOR AN INTRACELLULAR FUNCTION AT THE SYNAPSE. J Biol Chem. 2016 Mar 18;291(12):6111–23. doi: 10.1074/jbc.M115.683888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sadamatsu M, Masui A, Sakai T, et al. Familial paroxysmal kinesigenic choreoathetosis: an electrophysiologic and genotypic analysis. Epilepsia. 1999;40:942–949. doi: 10.1111/j.1528-1157.1999.tb00801.x. [DOI] [PubMed] [Google Scholar]
- 74.Akiyama T, Ohtsuka Y, Kobayashi K, Oka E. Kinesigenic attacks with ictal electroencephalographic abnormalities. Pediatr Neurol. 2004;3:357–359. doi: 10.1016/j.pediatrneurol.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 75.Ohmori I, Ohtsuka Y, Ogino T, et al. The relationship between paroxysmal kinesigenic choreoathetosis and epilepsy. Neuropediatrics. 2002;33:15–20. doi: 10.1055/s-2002-23594. [DOI] [PubMed] [Google Scholar]
- 76.Labate A, Tarantino P, Viri M, Mumoli L, Gagliardi M, Romeo A, Zara F, Annesi G, Gambardella A. Homozygous c.649dupC mutation in PRRT2 worsens the BFIS/PKD phenotype with mental retardation, episodic ataxia, and absences. Epilepsia. 2012 Dec;53(12):e196–9. doi: 10.1111/epi.12009. [DOI] [PubMed] [Google Scholar]
- 77.Tan L, Jiang T, Tan L, Yu JT. Toward precision medicine in neurological diseases. Ann Transl Med. 2016 Mar;4(6):104. doi: 10.21037/atm.2016.03.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Striano P, Vari MS, Mazzocchetti C, Verrotti A, Zara F. Management of genetic epilepsies: From empirical treatment to precision medicine. Pharmacol Res. 2016;107:426–9. doi: 10.1016/j.phrs.2016.04.006. [DOI] [PubMed] [Google Scholar]