

ABSTRACT
The β2-integrin lymphocyte function-associated antigen 1 (LFA-1) is needed for the T cell receptor (TCR)-induced activation of LFA-1 to promote T cell adhesion and interaction with antigen-presenting cells (APCs). LFA-1-mediated cell-cell interactions are critical for proper T cell differentiation and proliferation. The Src kinase-associated phosphoprotein of 55 kDa (SKAP55) is a key regulator of TCR-mediated LFA-1 signaling (inside-out/outside-in signaling). To gain an understanding of how SKAP55 controls TCR-mediated LFA-1 activation, we assessed the functional role of its pleckstrin homology (PH) domain. We identified two critical amino acid residues within the PH domain of SKAP55, aspartic acid 120 (D120) and lysine 152 (K152). D120 facilitates the retention of SKAP55 in the cytoplasm of nonstimulated T cells, while K152 promotes SKAP55 membrane recruitment via actin binding upon TCR triggering. Importantly, the K152-dependent interaction of the PH domain with actin promotes the binding of talin to LFA-1, thus facilitating LFA-1 activation. These data suggest that K152 and D120 within the PH domain of SKAP55 regulate plasma membrane targeting and TCR-mediated activation of LFA-1.
KEYWORDS: LFA-1, SKAP55, adhesion
INTRODUCTION
For proper lymphocyte function, T cells interact with antigen-presenting cells (APCs) that display foreign peptide antigen/major histocompatibility complexes (Ag/MHCs) that are recognized by the clonotypic T cell receptor (TCR). The individual interactions between T cells and APCs strongly depend on the activation of the β2-integrin lymphocyte function-associated antigen 1 (LFA-1). On nonactivated T cells, LFA-1 is expressed in an inactive conformation and exhibits a low affinity for its ligand intercellular adhesion molecule 1 (ICAM-1). Triggering of the TCR by Ag/MHCs or monoclonal antibodies (MAbs) induces a conformational change of LFA-1 that increases its affinity for ICAM-1 (affinity modulation) and facilitates the clustering of LFA-1 (avidity regulation), a process termed “inside-out signaling.” Subsequently, the interaction between ICAM-1 and LFA-1 transmits a costimulatory signal into T cells, thereby driving their activation, differentiation, and proliferation, a process termed “outside-in signaling” (1, 2).
Several molecules have been shown to play critical roles for the TCR-mediated activation of LFA-1. Among these molecules are LFA-1-associated talin, the GTPase Rap1, and its binding partners regulator for cell adhesion and polarization enriched in lymphoid tissues (RAPL) and Rap1-interacting adapter molecule (RIAM). The loss of these molecules attenuates TCR-mediated adhesion, interactions with APCs, and affinity/avidity regulation of LFA-1 (2–7). Besides these molecules, additional adapter proteins are involved in TCR-mediated LFA-1 activation. These proteins include the transmembrane adapter protein linker of activation of T cells (LAT) and cytosolic adapter proteins such as Src homology 2 (SH2) domain-containing leukocyte phosphoprotein of 76 kDa (SLP-76), adhesion- and degranulation-promoting adapter protein (ADAP), and Src kinase-associated phosphoprotein of 55 kDa (SKAP55) (8).
Analysis of ADAP−/− T cells revealed defects in LFA-1-mediated adhesion, avidity modulation, and, consequently, an impaired interaction of T cells with APCs in vitro and in vivo (9–13). ADAP possesses a central proline-rich region, two helical SH3 domains, and one Ena/VASP homology 1 (EVH1) binding domain (8). Via its proline-rich region, ADAP directly and constitutively interacts with another adapter protein, SKAP55 (14). This constitutive interaction with ADAP protects SKAP55 from degradation (15, 16). Consequently, ADAP−/− T cells are also deficient for SKAP55 (11, 16). Similar to ADAP, the loss of SKAP55 in T cells leads to defective TCR-mediated LFA-1 function and attenuated T cell/APC interactions and is termed here the ADAP/SKAP55 module (11, 12, 16, 17).
SKAP55 possesses a dimerization (DM) domain followed by a pleckstrin homology (PH) domain and a C-terminal SH3 domain (interaction site with ADAP) (14, 18). Via the DM domain, SKAP55 constitutively interacts with RAPL and RIAM (18–21). The loss of SKAP55 or deletion of the DM domain abrogates membrane targeting of RAPL, RIAM, and talin and also their interaction with LFA-1 (18–22). This indicates that the interaction of RAPL and RIAM with the DM domain of SKAP55 is crucial for TCR-mediated LFA-1 activation.
In contrast to the DM domain, the role of the PH domain of SKAP55 for TCR-mediated LFA-1 activation is still controversial. Two studies reported that a deletion of the PH domain or mutation of arginine 131 (R131) within the PH domain of SKAP55 impairs adhesion and conjugate formation of T cells with APCs (12, 21). In contrast, two other reports showed that neither the deletion of the PH domain within full-length SKAP55 nor the overexpression of the isolated PH domain of SKAP55 alters TCR-mediated adhesion (16, 18).
Here we investigated the functional role of the PH domain within SKAP55 for TCR-mediated LFA-1 activation. We show that the isolated PH domain of SKAP55 has a preference for phosphatidylinositol (3,4,5)-trisphosphate [PI(3,4,5)P3] over phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] binding in vitro. Interestingly, however, in T cells, plasma membrane (PM) recruitment of the PH domain does not depend on PI(3,4,5)P3 formation. Rather, the isolated PH domain of SKAP55 interacts with actin via lysine 152 (K152). We show here that this interaction is critically involved in TCR-mediated adhesion to ICAM-1 and affinity modulation of LFA-1. Besides K152, we identified aspartic acid 120 (D120) within the PH domain of SKAP55 as a critical negative regulator of SKAP55 function. Mutation of D120 to lysine (D120K mutant) induces the constitutive membrane association of SKAP55 and LFA-1 activation in the absence of TCR-mediated stimuli. Importantly, however, an additional mutation of K152 to glutamic acid (K152E) overrides the functional properties of the D120K mutant and completely blocks inside-out signaling, leading to LFA-1 activation by attenuating the interaction of the ADAP/SKAP55 module with actin/talin and LFA-1. Thus, our data suggest that two critical amino acid residues within the phosphoinositide phosphate (PIP) binding site of the PH domain of SKAP55 regulate PM targeting of the ADAP/SKAP55 module and TCR-mediated activation of LFA-1.
RESULTS
In vitro lipid binding properties of the isolated PH domain of SKAP55.
We used a purified recombinant His-tagged fusion protein to assess the lipid head group specificity of the isolated PH domain of SKAP55 by nuclear magnetic resonance (NMR) spectroscopy. Because PH domains typically recognize PI(3,4,5)P3 or PI(4,5)P2, we investigated the interaction of the isolated PH domain of SKAP55 with the corresponding head groups inositol 1,3,4,5-tetrakisphosphate (IP4) and inositol 1,4,5-triphosphate (IP3), respectively. Exemplarily, the heteronuclear single quantum coherence (HSQC) spectra of the 15N-labeled PH domain in the presence of increasing amounts of IP4 are shown in Fig. 1A. Amide group resonances of residues that experience large changes in their chemical shifts are indicated in the spectra, and the titration curves for some of these resonances are shown in Fig. 1B. A mean equilibrium dissociation constant (KD) value of 119 ± 59 μM was determined for IP4, while IP3 displayed a KD of 641 ± 276 μM. Comparable results were obtained when we used the short lipid chain variants C4-PI(3,4,5)P3 and C4-PI(4,5)P2 as ligands (74 ± 12 μM versus 604 ± 202 μM). Several charged residues in the vicinity of the anticipated IP4 binding pocket (Fig. 1C) displayed significant chemical shift changes and were therefore mutated in order to obtain potential non-lipid binding variants of the domain. For the R131M mutant, we observed that IP4 binding was significantly reduced in NMR experiments, while no binding was observed for the K152E variant. Based on these results, R131M, K152E, and, additionally, K116M mutants were generated for cellular experiments.
FIG 1.

In vitro lipid binding properties of the isolated PH domain of SKAP55. (A) 1H-15N HSQC titration of 270 μM the wild-type SKAP55 PH domain with increasing concentrations (50, 150, 300, 600, 1,130, and 2,466 μM) of IP4, the head group of PI(3,4,5)P3. Some of the shifts are indicated. (B) Curve fits of combined 1H-15N HSQC chemical shift changes with increasing IP4 concentrations for significantly shifting residues E115, K117, R131, Y142, T154, and R185. The individual KD values derived from the fits are given. (C) Significant chemical shift differences in the isolated PH domain of wild-type SKAP55 upon the addition of a 10-fold molar excess of IP4 are highlighted on the structure (PDB accession number 1U5D) overlaid with IP4 cocrystallized with the PH domain of AKT (PDB accession number 1UNQ). Residues K116, R131, and K152, involved in IP4 binding and suggested for mutation, are drawn as sticks. (D) Chemical shifts using 270 μM wild-type (WT) SKAP55 and R131M and K152E mutants of the PH domain of SKAP55 upon incubation with 2,466 μM IP4. While large chemical shifts are seen for certain NH resonances of the wild-type protein, shifts are very small for the R131M variant and are totally abolished for the K152E mutant protein.
PI(3,4,5)P3-independent recruitment of the isolated PH domain of SKAP55 to the PM.
Given its PI(3,4,5)P3 binding properties, we next investigated whether the PH domain of SKAP55 is recruited to the PM in T cells. Confocal microscopy analysis was performed by using nonstimulated Jurkat T cells expressing either green fluorescent protein (GFP) alone or a GFP-tagged PH domain of SKAP55 (PH-SKAP55-GFP) to determine the ratios of fluorescence intensity at the PM of GFP fusion proteins (see Fig. S1 in the supplemental material). The isolated GFP-tagged PH domains of phospholipase C delta (GFP-PH-PLCδ), which possesses a large preference for PI(4,5)P2 binding (23), and of AKT (protein kinase B) (GFP-PH-AKT), displaying a high preference for PI(3,4,5)P3 binding (24), served as controls.
Similarly to GFP-PH-AKT and GFP-PH-PLCδ, PH-SKAP55-GFP was targeted to the PM in Jurkat T cells (Fig. 2A; see also Fig. S1 in the supplemental material). To assess whether binding to PI(3,4,5)P3 is responsible for the constitutive PM localization of PH-SKAP55-GFP, Jurkat T cells were treated with the phosphatidylinositol 3-kinase (PI3K) inhibitor wortmannin, which induced a clear relocalization of GFP-PH-AKT to the cytoplasm (Fig. 2A). In contrast, and as expected, the PM localization of GFP-PH-PLCδ was not influenced by wortmannin treatment. Unexpectedly, the same was true for PH-SKAP55-GFP, whose PM localization was not affected by wortmannin treatment (Fig. 2A).
FIG 2.

PI(3,4,5)P3-independent membrane targeting of the isolated PH domain of SKAP55. (A) Jurkat T cells were transfected with plasmids encoding GFP, GFP-PH-PLCδ, GFP-PH-AKT, or PH-SKAP55-GFP. Twenty-four hours after transfection, cells were treated with dimethyl sulfoxide (DMSO) or wortmannin for 1 h at 37°C. Cells were fixed, permeabilized, stained with TRITC-phalloidin to visualize F-actin, and then imaged by confocal laser scanning microscopy. A histogram tool was used to determine the fluorescence intensity at the PM of individual cells (see Fig. S1 in the supplemental material) to calculate the ratios of fluorescence intensity after the subtraction of background GFP levels (n = 3 or 4). In the right panel, lysates were prepared from these transfectants to assess the inhibition of PI3K, as monitored by Western blotting for pAKT S473 (n = 3). (B) Primary human T cells were transfected with plasmids as described above for panel A. Twenty-four hours after transfection, cells were pretreated with DMSO or wortmannin for 1 h at 37°C and subsequently stimulated with anti-CD3/CD28 antibodies. Cells were analyzed as described above for panel A. In the bottom panel, the CD3/CD28-induced activation of PI3K in the presence or absence of wortmannin was monitored by Western blotting for pAKT S473 (n = 3) (means ± standard deviations) (**, P ≤ 0.01; ***, P ≤ 0.001).
We next investigated the subcellular localization of PH-SKAP55-GFP in resting and CD3/CD28-stimulated primary human T cells. PI(3,4,5)P3 levels are low in resting primary T cells, and consequently, GFP-PH-AKT localizes mostly in the cytosol (Fig. 2B). CD3/CD28 stimulation enhances PI3K activity, thereby inducing the membrane recruitment of GFP-PH-AKT, a process that is counteracted by wortmannin treatment (Fig. 2B). In marked contrast to GFP-PH-AKT, the PH domains of both PLCδ and SKAP55 constitutively localize at the PM in nonactivated primary human T cells. Moreover, neither CD3/CD28 stimulation nor wortmannin treatment affected the ratios of the fluorescence intensities of both PH domains at the PM (Fig. 2B). These data collectively suggest that in contrast to GFP-PH-AKT, the PM localization of PH-SKAP55-GFP does not depend on PI(3,4,5)P3 binding.
Lysine 152 is essential for PM targeting through actin binding of the isolated PH domain of SKAP55.
Besides PIP binding, particular PH domains are able to mediate protein-protein interactions with actin through positively charged amino acids such as lysine and arginine (25, 26). We therefore sought to test whether the PH domain of SKAP55 might interact with actin. To assess this point, we performed coimmunoprecipitation studies in Jurkat T cells expressing GFP, PH-SKAP55-GFP, or GFP-PH-PLCδ. Here we observed that PH-SKAP55-GFP coprecipitated actin, while (as previously reported [25, 26]) GFP-PH-PLCδ did not (Fig. 3A). This result suggests that the PH domain of SKAP55 interacts with actin.
FIG 3.

PM targeting of the PH domain of SKAP55 is mediated by interaction with actin and depends on lysine 152. (A) Jurkat T cells were transfected with plasmids encoding GFP, GFP-PH-PLCδ, GFP-PH-AKT, or PH-SKAP55-GFP. (Left) Twenty-four hours after transfection, the expression of GFP and GFP fusion proteins was assessed by Western blotting with the indicated antibodies. Lysates from these transfectants were used for anti-GFP immunoprecipitation. (Right) Precipitates were analyzed by Western blotting for panactin and GFP. The lysate of Jurkat T cells (JE6) served as a positive control for the detection of panactin (n = 3). (B) Jurkat T cells were transfected with plasmids encoding GFP, wild-type PH-SKAP55-GFP, or mutants (K116, R131M, K152E, and K152M). (Left) Twenty-four hours after transfection, the expression of GFP and GFP fusion proteins was assessed by Western blotting with the indicated antibodies. Lysates of these transfectants were used for anti-GFP immunoprecipitation (IP). (Right) Precipitates were analyzed by Western blotting for panactin and GFP. The lysate of Jurkat T cells (JE6) served as a positive control for the detection of panactin (n = 2). (C and D) Jurkat T cells (C) or primary human T cells (D) were transfected with plasmids encoding GFP, wild-type PH-SKAP55-GFP, or mutants (K116M, R131M, K152E, and K152M). Twenty-four hours after transfection, cells were fixed, permeabilized, stained with TRITC-phalloidin, and then imaged by confocal laser scanning microscopy. The ratios of fluorescence intensity after the subtraction of background GFP levels were calculated as described in the legend to Fig. 2A (n = 3 or 4) (means ± standard deviations) (*, P ≤ 0.05; **, P ≤ 0.01).
Given that actin is a highly acidic protein itself, we asked whether the residues important for the interaction with the negatively charged phosphoinositides would also affect actin binding. As shown in Fig. 3B, the K152E (or K152M) mutant completely abolished actin binding of the SKAP55 PH domain, whereas the K116M and R131M mutants retained their actin binding capability.
We performed cosedimentation and coprecipitation assays employing purified G- and F-actin to assess whether the PH domain of SKAP55 would directly interact with actin. The data shown in Fig. S2 in the supplemental material demonstrate that this is not the case. Indeed, neither the wild-type PH domain nor the K152E mutant cosedimented with F-actin (Fig. S2A). Moreover, neither the wild-type PH domain of SKAP55 nor the K152E mutant sequestered G-actin for F-actin polymerization (Fig. S2B). Finally, anti-His precipitates of the wild-type PH domain did not coprecipitate either F- or G-actin (Fig. S2C). Hence, the data shown in Fig. 3 and in Fig. S2 show that (i) the isolated PH domain of SKAP55 interacts with actin, (ii) this interaction is mediated via K152, and (iii) the interaction between the two molecules is indirect.
We next asked whether the K152E (or K152M) mutant would interfere with PM targeting of PH-SKAP55-GFP. As shown in Fig. 3C and D, Jurkat T cells and primary human T cells expressing PH-SKAP55(K152E/M)-GFP displayed strongly attenuated ratios of fluorescence intensity at the PM. In contrast to PH-SKAP55(K152E/M)-GFP, mutations K116M and R131M showed only a slight reduction in PM localization. These data strongly suggest that K152 is a critical residue for membrane targeting of the isolated PH domain of SKAP55, probably due to its ability to indirectly interact with actin rather than impacting PIP binding.
K152 of SKAP55 is crucial for TCR-mediated adhesion.
In order to determine the contribution of the K116, R131, and K152 residues of the PH domain of SKAP55 to regulate TCR-mediated T cell adhesion, we generated “suppression/reexpression” plasmids that simultaneously encode both a specific short hairpin RNA (shRNA) to knock down endogenous SKAP55 and a cDNA that allows the reexpression of an shRNA-insensitive FLAG-tagged SKAP55 molecule (Fig. 4A). As shown in Fig. 4B, the suppression/reexpression plasmids are capable of suppressing endogenous SKAP55 and allow comparable levels of reexpression of either wild-type SKAP55 or variants of SKAP55 carrying the K116M, R131M, or K152E point mutation. Figure 4C further shows that Jurkat T cells in which SKAP55 was downregulated by the shRNA approach are inhibited in their ability to adhere to ICAM-1. Reexpression of shRNA-resistant wild-type SKAP55 rescued TCR-mediated adherence to ICAM-1. In contrast to wild-type SKAP55 (and in line with previously reported data [21]), the reexpression of the SKAP55-R131M mutant moderately impaired adhesion, while the reexpression of the SKAP55-K116M mutant showed a behavior similar to that for the reexpression of wild-type SKAP55 (Fig. 4C). However, differently from the R131M and K116M mutants, cells expressing the SKAP55-K152E mutant showed strongly impaired adhesion similar to that of SKAP55 knockdown cells (Fig. 4C). Notably, that functional effects exerted by individual SKAP55 mutants on adhesion were not due to an altered expression of the TCR or LFA-1 (see Fig. S3A in the supplemental material).
FIG 4.

The K152 mutant of SKAP55 impairs TCR-mediated adhesion. (A) Schematic representation of the suppression/reexpression vector used in this study. (B) Jurkat T cells were transfected with suppression/reexpression constructs that do not suppress endogenous SKAP55 (shC), reduce the endogenous protein level of SKAP55 (shSKAP55), or reexpress FLAG-tagged shRNA-resistant wild-type SKAP55 (WT) or its mutants (K116M, R131M, and K152E). Forty-eight hours after transfection, lysates were analyzed by Western blotting with the indicated antibodies. (C) Transfected Jurkat T cells as described above for panel B were analyzed for their ability to adhere to ICAM-1-coated wells in a resting state or upon stimulation with two different concentrations of CD3 antibodies (1 μg/ml and 5 μg/ml). Adherent cells were counted and calculated as a percentage of the input (n = 3) (means ± standard deviations) (*, P ≤ 0.05; **, P ≤ 0.01).
PM fractions from nonactivated or TCR-stimulated Jurkat T cells reexpressing wild-type SKAP55 or the above-described mutants were prepared to biochemically investigate the PM recruitment of the individual SKAP55 mutants. Figure 5A shows that wild-type SKAP55 as well as all SKAP55 mutants tested (K116M, R131M, and K152E) were properly recruited to the PM upon TCR triggering. The same was true for the constitutive interaction partners of SKAP55 (RAPL, RIAM, and ADAP) or the inducible binding partner Rap1 (inducibly binding to RAPL and RIAM), SLP-76 (inducibly binding to ADAP), or talin (inducibly binding to RIAM and LFA-1) (Fig. 5A). Thus, none of the mutations within the PH domain, including K152E, has an impact on the TCR-mediated recruitment of SKAP55 and its associated signaling complex to the PM.
FIG 5.

The K152E mutation abolishes the TCR-mediated interaction of LFA-1 with actin and talin. (A) Jurkat T cells were transfected with suppression/reexpression constructs that reexpress FLAG-tagged shRNA-resistant wild-type (WT) SKAP55 or its mutants (K116M, R131M, and K152E). After 48 h, cells were left untreated or were stimulated with CD3 antibodies (CD3). PM fractions were isolated and analyzed by Western blotting with the indicated antibodies. Detection of CD11a served as a control for the fractionation of PM (n = 2). (B and C) Jurkat T cells transfected as described above for panel A were left untreated or stimulated with CD3 antibodies (CD3). Lysates were used for the immunoprecipitation of either SKAP55 using anti-FLAG antibodies or LFA-1 using anti-CD11a antibodies. Precipitates were analyzed by Western blotting using the indicated antibodies (n = 2).
We next asked whether any of the mutations would affect the TCR-mediated interaction between the ADAP/SKAP55 module and actin, talin, and/or LFA-1. To test this, FLAG (SKAP55) immunoprecipitates were obtained from postnuclear lysates of nonstimulated or TCR-activated Jurkat T cells reexpressing wild-type FLAG-SKAP55 or the corresponding K116M, R131M, or K152E mutant. Figure 5B shows that, as reported previously by us (19) and others (18, 20, 21), ADAP, RIAM, and RAPL constitutively interact with wild-type SKAP55 and with all three SKAP55 mutants, whereas Rap1 and SLP-76 associate with the whole complex only after TCR stimulation. However, in marked contrast to wild-type SKAP55 and the K116M and R131M mutants of SKAP55, the TCR-mediated association between the ADAP/SKAP55 complex and either LFA-1, talin, or actin was strongly reduced in cells reexpressing the K152E mutant of SKAP55 (Fig. 5B). Reciprocal immunoprecipitation studies for LFA-1 showed that the TCR-mediated association of LFA-1 with the ADAP/SKAP55 module, actin, and talin was strongly attenuated in cells reexpressing the K152E mutant of SKAP55 (Fig. 5C). Taken together, these data indicate that the expression of the K152E mutant affects the inducible association between the ADAP/SKAP55 complex and LFA-1 via the actin/talin axis. The failure of LFA-1 to interact with its targets abrogates the ability of the K152E mutant to mediate adhesion upon T cell activation.
D120 of SKAP55 is essential for TCR-independent PM targeting, adhesion, and LFA-1 activation.
In contrast to the isolated PH domain of SKAP55 (Fig. 2A), full-length SKAP55 localizes primarily in the cytosol in nonstimulated Jurkat T cells and relocalizes to the PM only after CD3 stimulation (Fig. 6A). A previous study showed that in nonactivated macrophages, the N-terminal DM domain of the SKAP55 homologue SKAP-HOM prevents membrane targeting of SKAP-HOM by blocking the PH domain via an autoinhibitory mechanism: the SKAP-HOM PH domain β1-β2-loop forms a small interface with the DM domain and is predicted to thereby hinder PI(3,4,5)P3 binding by the formation of a short helix (27). Assuming that the structure of the tandem DM-PH domain in SKAP55 is similar to that of SKAP-HOM (Protein Data Bank [PDB] accession number 2OTX), one would expect chemical shift changes in the IP4 binding pocket when the isolated PH domain of SKAP55 is compared with the PH domain as part of the tandem DM-PH construct of SKAP55. This was indeed observed by NMR (see Fig. S4B in the supplemental material) and shows that the interface between the PH and the DM domains is localized in the vicinity of the critical residues K152 (see the model in Fig. S4C) and aspartate 120 (D120), homologous to D129 in SKAP-HOM (Fig. S4A), where it regulates the intramolecular switch by charge inversion in the critical interface.
FIG 6.

The D120K mutant of SKAP55 induces TCR-independent adhesion and LFA-1 activation. (A) Jurkat T cells were transfected with plasmids encoding GFP, full-length wild-type (WT) SKAP55-GFP, or mutants (D120K and D120K/K152E). Twenty-four hours after transfection, cells were left untreated or were stimulated with CD3 antibodies. Subsequently, cells were fixed, permeabilized, probed with TRITC-phalloidin, and imaged by confocal laser scanning microscopy. The ratios of fluorescence intensity after the subtraction of background GFP levels were calculated as described in the legend to Fig. 2A (n = 3). TCR stimulation was assessed by analyzing pERK1/2 using Western blotting (right). (B) Jurkat T cells were transfected with suppression/reexpression constructs that do not suppress endogenous SKAP55 (shC) or reduce the endogenous protein level of SKAP55 (shSKAP55) and reexpress FLAG-tagged shRNA-resistant wild-type SKAP55 or its mutants (D120K, K152E, or D120K/K152E). Forty-eight hours after transfection, lysates were analyzed by Western blotting using the indicated antibodies. (C) Jurkat T cells were transfected as described above for panel B. Cells were analyzed for their ability to adhere to ICAM-1-coated wells in a resting state or upon stimulation with two different concentrations of CD3 antibodies (1 μg/ml and 5 μg/ml), respectively. Adherent cells were counted and calculated as a percentage of the input (n = 3). (D) Jurkat T cells transfected as described above for panel B were left untreated (ICAM-1) or were stimulated with CD3 antibodies (ICAM-1/CD3), followed by staining with the anti-LFA-1 antibody mAb24. mAb24 epitope expression was assessed by flow cytometry within the GFP gate, and the mean fluorescence intensity of untreated shC-transfected Jurkat T cells was set to 1 to calculate fold induction (n = 3) (means ± standard deviations) (*, P ≤ 0.05; **, P ≤ 0.01).
We generated a plasmid encoding the D120K mutant of SKAP55, which we expressed in Jurkat T cells to study the localization of this mutant. As shown in Fig. 6A, the D120K mutant displayed constitutive PM localization at a level that was comparable to that found in TCR-stimulated cells expressing wild-type SKAP55 or the D120K/K152E double mutant. Of note, TCR stimulation did not further increase the ratios of fluorescence intensity of the D120K mutant at the PM (Fig. 6A). Thus, similarly to D129 in SKAP-HOM, D120 in the PH domain is a critical residue that regulates the PM recruitment of SKAP55.
Previous results from us and others showed that constitutive PM targeting of SKAP55 (either by using a LAT/SKAP55 chimera protein or by introducing an N-terminal myristoylation tag) induces T cell adhesion even in the absence of TCR-mediated stimuli (16, 21). Given the fact that the D120K mutant of SKAP55 displayed a constitutive PM association, we hypothesized that this mutant would also induce the TCR-independent activation of LFA-1. Indeed, the reexpression of SKAP55-D120K (Fig. 6B) induced basal adhesion to ICAM-1 (Fig. 6C) and LFA-1 affinity modulation (Fig. 6D). Note that functional effects exerted by the D120K mutant of SKAP55 on adhesion were not due to an increased expression level of the TCR or LFA-1 (see Fig. S3B in the supplemental material). In summary, D120K-mediated PM targeting of SKAP55 induces the spontaneous and constitutive activation of LFA-1.
To provide a biochemical basis for the functional behavior of the D120K mutant, we prepared PM fractions (Fig. 7A), anti-FLAG (SKAP55) immunoprecipitates (Fig. 7B), or anti-LFA-1 immunoprecipitates (Fig. 7C) from nonstimulated or TCR-stimulated Jurkat cell transfectants expressing the SKAP55-D120K mutant. In line with the functional data, the D120K mutant as well as the known SKAP55 interaction partners ADAP, RIAM, and RAPL localized to the PM even in the absence of TCR stimulation (Fig. 7A). Similarly, anti-FLAG (SKAP55) immunoprecipitates obtained from lysates of nonstimulated D120K transfectants showed a constitutive association of the SKAP55-D120K mutant with actin, talin, and LFA-1 (Fig. 7B). LFA-1 immunoprecipitates prepared from D120K-expressing cells showed TCR-independent associations between LFA-1, actin, RIAM, RAPL, ADAP, SKAP55, and talin. Hence, mutation D120K fully activates the ADAP/SKAP55 module at the PM.
FIG 7.

The K152E mutant of SKAP55 overwrites LFA-1 activation by the D120K mutant. (A) Jurkat T cells were transfected with suppression/reexpression constructs that reexpress FLAG-tagged shRNA-resistant wild-type (WT) SKAP55 or its mutants (D120K, K152E, or D120K/K152E). Forty-eight hours after transfection, cells were left untreated or were stimulated with CD3 antibodies. PM fractions were isolated and analyzed by Western blotting using the indicated antibodies. Detection of CD11a served as control for the fractionation of PMs (n = 2). (B and C) Jurkat T cells transfected as described above for panel A were left untreated or stimulated with CD3 antibodies (CD3). Lysates were used for the immunoprecipitation of SKAP55 (using anti-FLAG antibodies) or LFA-1 (using anti-CD11a antibodies). Precipitates were analyzed by Western blotting using the indicated antibodies (n = 2).
Next, we were interested in determining whether the D120K mutant would dominate/override the negative regulatory effect of the K152E mutant (or vice versa). To assess this question, we generated a SKAP55 mutant carrying the D120K/K152E double mutation within the PH domain. Figure 7A shows that the D120K/K152E double mutant no longer localized to the PM in nonstimulated cells and that cells expressing this mutant had lost their ability to adhere and to activate LFA-1 (Fig. 6C and D). Hence, the constitutive PM targeting of SKAP55 as well as the spontaneous integrin activation induced by the D120K mutant depend on K152.
In addition to abrogating the constitutive PM association of SKAP55, the D120K/K152E double mutant completely abrogated the TCR-induced binding of T cells to ICAM-1 and LFA-1 affinity modulation (Fig. 6C and D). Moreover, the TCR-mediated association of the ADAP/SKAP55 module with LFA-1, talin, and actin (Fig. 7B) as well as the TCR-mediated interaction of LFA-1 with the ADAP/SKAP55 module, talin, RIAM, and RAPL were severely attenuated (Fig. 7C). Hence, all functional effects exerted by the D120K single mutant were counteracted by the additional mutation K152E. Thus, K152E is dominant over D120K.
Constitutive adhesion of the D120K mutant of SKAP55 requires talin but not Rap1.
Unexpectedly, the experiments shown in Fig. 7A and B revealed that despite constitutive PM targeting of the ADAP/SKAP55 module and despite the constitutive activation of LFA-1, Rap1 was not constitutively targeted to the PM in cells expressing the D120K mutant (Fig. 7A) and did not coprecipitate with SKAP55 in nonstimulated transfectants expressing the D120K mutant (Fig. 7B). Rather, even in D120K mutant-expressing cells, membrane targeting of Rap1 as well as its interaction with the ADAP/SKAP55 module required the stimulation of the TCR (Fig. 7A and B). This finding suggested that the constitutive activation of LFA-1 in cells expressing the D120K mutant occurs independently of Rap1.
To address the role of Rap1 in more detail, we reexpressed the D120K mutant in Jurkat T cells in which Rap1 had been downregulated by small interfering RNA (siRNA). Figure 8A shows that in cells reexpressing either wild-type SKAP55 or the D120K mutant, the knockdown of Rap1 did not affect basal adhesion, whereas TCR-mediated adhesion was almost completely blocked. This finding confirms that Rap1 is dispensable for steady-state adhesion, while it is necessary for TCR-mediated LFA-1 activation even in cells reexpressing the D120K mutant. In marked contrast to Rap1, the knockdown of talin reverted the spontaneous and TCR-mediated integrin activation of cells expressing the D120K mutant (Fig. 8B). Hence, spontaneous adhesion induced by the D120K mutant depends on talin.
FIG 8.
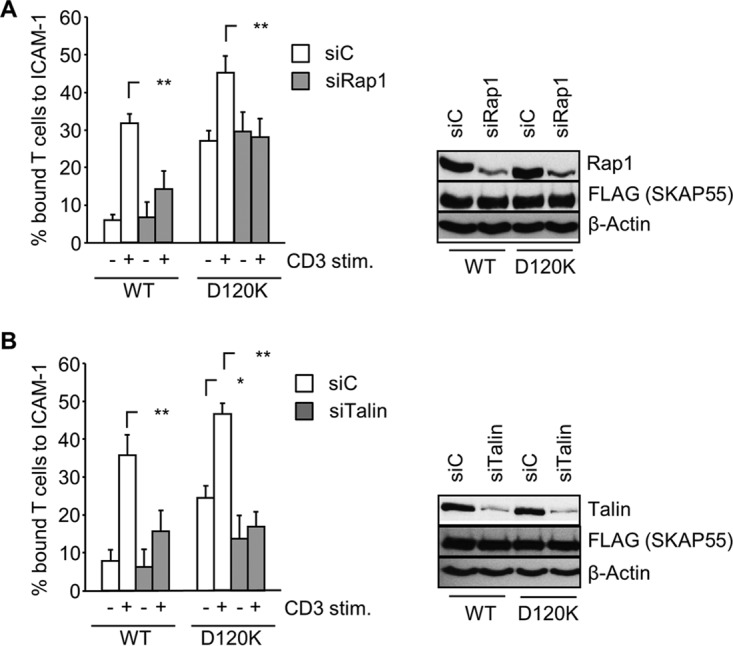
TCR-independent adhesion induced by the D120K mutant of SKAP55 is dependent on talin but not on Rap1. (A) Jurkat T cells were cotransfected with suppression/reexpression constructs that reexpress FLAG-tagged shRNA-resistant wild-type (WT) SKAP55 or the D120K mutant in combination with either control siRNA (siC) or siRNA against Rap1 (siRap1). Forty-eight hours after transfection, cells were left untreated or were stimulated with CD3 antibodies (1 μg/ml). Cells were analyzed for their ability to adhere to ICAM-1-coated wells in a resting state or upon stimulation with CD3 antibodies. Adherent cells were counted and calculated as a percentage of the input. Lysates were prepared from transfectants and analyzed by Western blotting for FLAG, Rap1, and β-actin (right) (n = 3). (B) Jurkat T cells were cotransfected with suppression/reexpression constructs as described above for panel A in combination with either control siRNA (siC) or siRNA against talin (siTalin). Forty-eight hours after transfection, cells were left untreated or were stimulated CD3 antibodies (1 μg/ml). Cells were analyzed for their ability to adhere to ICAM-1-coated wells in a resting state or upon stimulation with CD3 antibodies. Adherent cells were counted and calculated as a percentage of the input. Lysates were prepared from transfected cells and analyzed by Western blotting using the indicated antibodies (right) (n = 3) (means ± standard deviations) (*, P ≤ 0.05; **, P ≤ 0.01).
DISCUSSION
PH domains are generally known to bind lipids and to facilitate the translocation of proteins to the PM (28). In this study, we characterized the lipid binding properties as well as the functional properties of the PH domain of SKAP55 with a focus on the TCR-mediated activation of the β2-integrin LFA-1. We identified two critical amino acids within the PH domain of SKAP55, D120 and K152, that appear to regulate the membrane recruitment of the ADAP/SKAP55 module and T cell adhesion.
Structural inspection of the isolated PH domain of SKAP55 revealed that this domain contains a classical lipid-binding pocket that is present in many PH domains. It is built up by the variable β1-β2-, β3-β4-, and β6-β7-loops (28). The PH domain of SKAP55 contains all amino acids (except serine 118 in the β1 strand) at homologous positions that have been predicted for the lipid binding of its homologue SKAP-HOM (27). By using NMR spectroscopy, we showed that the PH domain of SKAP55 has a preference for PI(3,4,5)P3 over PI(4,5)P2 binding. However, in comparison to its homologue SKAP-HOM, the isolated PH domain of SKAP55 shows a much lower affinity for PI(3,4,5)P3 binding (74 ± 12 μM for SKAP55 versus 8 μM for SKAP-HOM) (27). In part, this difference could result from the different methods used in these two studies, namely, NMR (this study) and fluorescence polarization (27). However, SKAP-HOM contains an arginine instead of serine (SKAP55) at position 118, a residue that is localized not far from the lipid binding site. Thus, SKAP55 likely harbors a nonoptimal PIP binding pocket.
Studies employing a PI3K inhibitor showed that in both Jurkat T cells as well as primary human T lymphocytes, the constitutive PM recruitment of the isolated PH domain of SKAP55 likely does not require PI(3,4,5)P3 binding. In line with this finding, the individual mutation of the putative PI(3,4,5)P3-interacting residues R131 and K116 only moderately affected PM targeting of the isolated PH domain of SKAP55 in either Jurkat or human primary T cells. Functionally, using a genetic suppression/reexpression system, we showed that the K116M mutant of SKAP55 behaved almost like wild-type SKAP55, while cells reexpressing the R131M mutant showed only slightly impaired adhesion to ICAM-1, a finding that is in line with previously reported data (21). Hence, in our experimental system using suppression/reexpression plasmids for SKAP55, residue R131 appears to be of moderate importance for SKAP55 function.
In marked contrast to R131 and K116, the K152E mutation almost completely abolished the PM localization of the PH domain of SKAP55. Previous studies showed that positively charged amino acids (e.g., lysine residues such as K152) within particular PH domains allow these domains to interact with actin (25, 26). Immunoprecipitation experiments revealed that this also appears to be true for the PH domain of SKAP55. Indeed, while the wild-type SKAP55 PH domain coprecipitated actin, the corresponding K152E(M) mutants completely failed to do so. These findings strongly suggest that the isolated PH domain of SKAP55 is primarily targeted to the PM via a K152-mediated interaction with actin. The identification of the PM-cytoskeleton linker molecules (e.g., ezrin/radixin/moesin [ERM] proteins) that mediate the link between the isolated PH domain of SKAP55 and actin requires further investigations.
The suppression of endogenous SKAP55 and reexpression of the SKAP55-K152E mutant in Jurkat T cells strongly impaired the TCR-induced adhesion and activation of LFA-1. Surprisingly, however, biochemical analysis of PM fractions revealed that in contrast to the isolated PH domain, the K152 mutation did not alter the TCR-induced recruitment of SKAP55 and its association partners to the PM in the context of the full-length SKAP55 molecule. Indeed, all components known to interact with SKAP55 in T cells were either constitutively (ADAP, RIAM, and RAPL) or inducibly (SLP-76 and Rap1) found in membrane preparations of TCR-stimulated cells expressing the K152E mutant. This finding indicates that redundant pathways for membrane targeting of the whole complex exist, which rescues the K152E mutant of full-length SKAP55. However, further analysis of anti-FLAG/SKAP55 and anti-LFA-1 immunoprecipitates showed that despite normal PM targeting, the K152E mutation completely abrogated the ability of SKAP55 and its associated signaling complex to interact with actin, talin, and LFA-1. Hence, K152 within the PH domain of SKAP55 is mandatory for the activation of LFA-1 by generating a molecular bridge between the ADAP/SKAP55 module, actin, and talin.
As noted above, an important difference between the isolated PH domain of SKAP55 and the full-length SKAP55 molecule is that mutation of K152 in the isolated PH domain displaces the latter from the PM, whereas mutation of K152 in the context of the full-length SKAP55 molecule does not. One possibility to explain this difference would be that in contrast to the isolated PH domain, full-length SKAP55 could use two independent (or complementary) routes for PM targeting. One route would involve actin/talin binding via the PH domain, whereas the second route would encompass an inducible association of the ADAP/SKAP55 complex with SLP-76 via ADAP (17). SLP-76 binds via Gads to LAT following the phosphorylation of LAT by TCR/Lck-activated ZAP70. It is important to note that in T cells, the chemokine-mediated membrane association of the ADAP/SKAP55 complex as well as the chemokine-induced activation of LFA-1 occur in the absence of detectable LAT phosphorylation and do not depend on an inducible association between SLP-76 and ADAP (29). Thus, it appears that SKAP55-mediated integrin activation does not necessarily depend on the ADAP/SLP76/Gads/pLAT signaling pathway.
Still, the fact that SKAP55 can activate integrins independently of membrane recruitment via the ADAP/SLP76/Gads/pLAT route raises the important question of an inducible (TCR or chemokine) membrane recruitment mechanism of SKAP55 that involves the PH domain. Our approach to elucidating this question was based on a previous report by Swanson et al. in which the authors investigated the functional properties of the PH domain of the SKAP55 homologue SKAP-HOM (27). In that publication, those authors proposed a model in which an intramolecular switch mechanism occurring between the N-terminal DM domain and the PH domain of SKAP-HOM dynamically regulates the PM targeting of the protein (27). In the closed conformation (i.e., in the absence of external stimuli), an interface between the DM domain and the PH domain supports a short helical segment in the β1-β2-loop incompatible with lipid binding. Those authors identified D129 within the PH domain of SKAP-HOM as being critically involved in the DM-PH switch mechanism. Indeed, the D129K mutation strongly increased the affinity of the PH domain of SKAP-HOM for PI(3,4,5)P3. Consequently, the expression of the D129K mutant of SKAP-HOM in macrophages induced the spontaneous localization of the mutated molecule to actin membrane ruffles (27). We identified D120 within the PH domain of SKAP55, representing the equivalent of D129 in SKAP-HOM. The reexpression of the SKAP55-D120K mutant in Jurkat T cells induced the constitutive PM targeting of this mutant (as well as the constitutive binding partners of SKAP55, ADAP, RIAM, and RAPL). Moreover, Jurkat T cells reexpressing the SKAP55 D120K mutant adhered to ICAM-1 in the absence of TCR-mediated stimuli (Fig. 6). These findings corroborate data from previous reports that showed that the forced PM recruitment of SKAP55 (by fusion with a myristoylation or a LAT tag) bypasses the need for TCR stimulation to induce adhesion (16, 21).
Using the D120K mutant, we made three important observations. First, we noted that the constitutive activation of LFA-1 induced by D120K did not require the membrane recruitment of Rap1 or the binding of Rap1 to either RIAM or RAPL. Indeed, the knockdown of Rap1 in D120K mutant-expressing cells did not affect the spontaneous adhesion that was induced by the D120K mutant. Hence, it appears as if Rap1 is not required for this basally induced adhesion. Still, the TCR-mediated augmentation of adhesion was found to be dependent on Rap1 in both cells expressing wild-type SKAP55 as well as cells expressing the D120K mutant, which is in line with previously reported data showing that Rap1 deficiency reduces TCR-mediated adhesion but not adhesion in nonstimulated T cells (5).
Second, in contrast to the knockdown of Rap1, the downregulation of talin partly blocked the basal adhesion of cells expressing the D120K mutant, and enhanced TCR-induced adhesion by the D120K mutant was also attenuated. We previously identified two independent ADAP/SKAP55 modules interacting with either RIAM or RAPL (30). RIAM has been reported to be inducibly associated with talin. This interaction promotes the PM targeting of talin, releases talin from its autoinhibition, and allows binding to the cytoplasmic tail of the β-chain of LFA-1 (7, 31). Beside RIAM and talin, RAPL interacts with the cytoplasmic tail of the α-chain of LFA-1. To our knowledge, it is not known whether a RAPL deficiency would affect the inside-out signaling of LFA-1 upon TCR stimulation. Independent of the exact mechanism, our experiments clearly show that actin binding by SKAP55 crucially connects the cytoskeleton to LFA-1.
Third, we found that the additional mutation of K152 (SKAP55-D120K/K152E double mutant) completely abrogated both the constitutive membrane localization of the D120K mutant as well as the constitutive activation of LFA-1 induced by the D120K mutant. Biochemical analysis of the SKAP55- and LFA-1-associated signaling complexes revealed that this was due to a failure of the double mutant to interact with actin and talin and, hence, a failure to interact with and to activate LFA-1. In summary, the D120K mutant can activate integrins only in the presence of K152, a residue that is crucial for both lipid and actin binding.
Our data suggest a model where in nonstimulated T cells, the spontaneous membrane recruitment of SKAP55 and its associated signaling complex is prevented by an intramolecular switch mechanism. Here the N-terminal DM domain of SKAP55 folds back on the PH domain, thereby blocking the interaction of the SKAP55-associated signaling complex with actin and talin. Upon TCR stimulation, the DM-PH domain switch undergoes a conformational change that releases the PH domain from autoinhibition and allows the signaling complex to interact with actin/talin, thereby activating LFA-1. In parallel (or subsequently), the PM localization of the ADAP/SKAP55 module is stabilized by the SLP-76/Gads signaling complex that binds to phosphorylated LAT. Together, the two membrane-targeting mechanisms facilitate stable T cell adhesion (see Fig. S5 in the supplemental material).
The obvious question emerging from the proposed model is how the DM-PH domain switch is released from autoinhibition following TCR stimulation. Dimerization domains are often involved in intra- or intermolecular protein-protein interactions, which can be used for the autoinhibition of functional domains (32). Moreover, Ophir and colleagues recently showed that SKAP55 forms homodimers (and heterodimers with SKAP-HOM) and that this dimerization is mediated by the DM domain of SKAP55 (18). The TCR-induced dimerization (or dedimerization) of SKAP55 upon T cell stimulation could therefore induce conformational changes within the DM domain that could release the PH domain from autoinhibition and thus allow PM targeting of SKAP55. An alternative scenario would be that the local production of PI(3,4,5)P3 upon T cell stimulation shifts the equilibrium toward the open, actin binding conformation of SKAP55.
Another puzzling question that needs clarification is why single point mutations within the PH domain of SKAP55 (or SKAP-HOM) produce dramatic functional effects in terms of integrin activation (16, 27), whereas a deletion of the whole PH domain of SKAP55 (or of SKAP-HOM) does not alter integrin functions (see Fig. S6 in the supplemental material) or subcellular localization, as shown in various model systems (16, 18, 27). In striking contrast to cells expressing the D120K/K152E double mutant of SKAP55 (in which the binding of the ADAP/SKAP55 module to LFA-1, talin, and actin was reduced) (Fig. 6), we did not find a significant alteration of the LFA-1-associated signaling complex in cells expressing a SKAP55 variant where the whole PH domain was deleted (Fig. S6C). Therefore, we currently cannot provide a model that would allow us to explain the contradicting and previously reported results that have been obtained by using deletion mutants versus single point mutations. As indicated by the structure (27), the interactions of the PH and the DM domains are intertwined, and a released DM domain will certainly be able to exert certain functions of the molecule. Furthermore, within the context of the larger complex of the SKAP55/ADAP module, redundant actin and lipid binding properties might render the PH domain function seemingly dispensable. However, the tuning of the integrin response, as shown here to be strongly modulated by the PH domain of SKAP55, might become visible only at certain relative concentrations of SKAP55 and its direct binding partners, including PIPs and actin.
Here we have identified two critical residues within the PH domain of SKAP55, D120 and K152, that regulate SKAP55 functions with regard to PM targeting. D120 appears to mediate the intramolecular release of the DM-PH switch. However, K152 overrides this feature and shows that the release of autoinhibition has to be followed by productive actin binding in order to allow SKAP55 and its associated signaling complex to promote the functional activation of LFA-1.
MATERIALS AND METHODS
Cell culture and transfection.
The Jurkat T cell line (JE6.1; ATCC) was cultured in RPMI 1640 medium (Biochrom AG) supplemented with 10% fetal calf serum (FCS; PAN-Biotech) and stable l-glutamine at 37°C with 5% CO2. Transient transfections of Jurkat T cells were performed as previously described (16, 29), and cells were cultured for 24 h (for cDNA constructs encoding GFP-tagged PH domains or wild-type SKAP55 [including its mutants]) or for 48 h (upon transfection with suppression/reexpression plasmids or along with siRNAs) before use. siRNAs siC (control siRNA), siRap1, and siTalin were purchased from Santa Cruz. Primary human T cells were isolated from healthy donors by standard separation methods using the AutoMACS system (Miltenyi Biotech) and maintained in RPMI 1640 medium containing 10% FCS, stable l-glutamine, and 2 μg/ml ciprofloxacin (Roche). Approval for these studies was obtained from the Ethics Committee of the Medical Faculty (89/13) at the Otto von Guericke University, Magdeburg, Germany. Informed consent was obtained in accordance with the Declaration of Helsinki. T cells (5 × 106 to 8 × 106) were transfected with 5 μg DNA by using the Amaxa human T cell Nucleofector kit (Lonza) according to the manufacturer's instructions. Transfected human primary T cells were cultured for 24 h before use. The anti-CD3 MAb alone or in combination with the anti-CD28 MAb (clones OKT3 and CD28.2; both from eBioscience) was used for the stimulation of T cells. The viability of Jurkat T cells and human primary T cells upon treatment with wortmannin (Calbiochem) was assessed by trypan blue exclusion (Sigma-Aldrich).
Flow cytometry and mAb24 binding assay.
For flow cytometry analysis, LFA-1 (clone MEM48; provided by V. Horejsi [Institute of Molecular Genetics, Academy of Sciences, Prague, Czech Republic]) or CD3 (clone OKT3) was used in combination with allophycocyanin (APC)-conjugated anti-mouse IgG (Dianova), and samples were analyzed by using a FACSCalibur flow cytometer and CellquestPro software (BD Bioscience). mAb24 binding was assessed as previously reported (33, 34). Briefly, cells were left untreated or stimulated for 30 min with plate-immobilized anti-CD3 antibody (clone OKT3) in the presence of plate-bound human Fc-tagged ICAM-1 (10 μg/ml; R&D Systems). Cells were harvested and incubated with mAb24 antibody (10 μg/ml; provided by N. Hogg, Cancer Research UK London Research Institute, London, UK). Bound mAb24 antibody was detected by using APC-conjugated anti-mouse IgG1 (Dianova) and analyzed by flow cytometry.
cDNA constructs and generation of plasmids.
The isolated PH domain of SKAP55 cloned into the pEGFP-N1 vector (Clontech) was described previously (16). The pEF-BOS expression vectors encoding N-terminally FLAG-tagged SKAP55 or a deletion of the PH domain within SKAP55 were described previously (35). The isolated PH domain of PLCδ was cloned into the pEGFP-C1 vector (a gift from Jacob Rullo, Toronto General Research Institute, University Health Network, Toronto, Canada). The isolated PH domain of AKT cloned into the pEGFP-C1 vector was provided by D. Cantrell (Department of Cell Signaling and Immunology, College of Life Sciences, University of Dundee, Dundee, UK). The isolated PH domain (residues 97 to 216) or the DM-PH domain (residues 7 to 222) of SKAP55 was subcloned into pET28a to generate a recombinant N-terminally His-tagged (MGSSHHHHHHSSGLVPRGSHMASMTGGQQMGRGSEF) fusion protein. Full-length human SKAP55 cDNA was cloned into the pEGFP-N1 vector. QuikChange site-directed mutagenesis kit II from Agilent Technologies was used to create point mutations within full-length SKAP55 or the isolated PH domain of SKAP55 by using the indicated primers for the K116M (K116M SKAP55-for/K116M SKAP55-rev), R131M (R131M SKAP55-for/R131M SKAP55-rev), K152E (K152E SKAP55-for/K152E SKAP55-rev), K152M (K152M SKAP55-for/K152M SKAP55-rev), and D120K (D120K SKAP55-for/D120K SKAP55-rev) mutants. The gene coding for the N-terminally His-tagged PH domain containing the mutation K152E, which was used for NMR measurements, was obtained by gene synthesis (Invitrogen) and additionally contained the S161G polymorphism. For the construction of the suppression/reexpression vector of SKAP55 and its mutants, the 19-nucleotide sequence GAAAGAATCCTGCTTTGAA was used to target SKAP55 and cloned into the pCMS4 vector (a gift from D. Billadeau, Department of Biochemistry and Molecular Biology, Division of Oncology Research, Mayo Clinic, Rochester, MN) by using the primer pair SK55-shRNA-for/SK55-shRNA-rev. cDNA sequences of full-length SKAP55 and mutants were amplified by PCR to introduce two restriction sites, MluI and NotI sites, which were needed for cloning into the pCMS4 vector (MluISKAP55-for/Not1SKAP55-rev). An shRNA-resistant form of SKAP55 was created by mutagenesis in which the targeting sequence within the DNA was changed to AAgGAgTCgTGtTTcGAg (lowercase letters indicate changed nucleotides) by using SKAP55 sh-res-for and SKAP55 sh-res-rev. All amplified PCR products were cloned into the pJET1.2 vector (Thermo Fischer) and sequenced prior to subcloning in the designated vectors or after mutagenesis. See Table S1 in the supplemental material for sequences of primers used for cloning, PCR, and mutagenesis.
Adhesion assay.
Adhesion assays were performed by using a 96-well plate precoated with 0.5 μg ICAM-1/well (R&D Systems). Transfected Jurkat T cells were left untreated or stimulated with anti-CD3 MAb (clone OKT3) (1 or 5 μg/ml) for 30 min at 37°C prior to the adhesion assay. Cells were then allowed to adhere for 30 min at 37°C, and unbound cells were carefully washed off with Hanks' buffered saline (Biochrom AG). Bound cells were counted and calculated as a percentage of the input (2 × 105 cells) in triplicates. Conjugate formation was performed as previously described (19, 29).
Western blotting, immunoprecipitation, and plasma membrane fractions.
Cell lysis and immunoprecipitations were performed as previously described (35, 36). Equivalent amounts of protein were used for precipitation experiments and Western blot analyses. Cell lysates or immune complexes were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred onto nitrocellulose. Western blot analyses were conducted with the indicated antibodies (see below), and blots were developed with the appropriate horseradish peroxidase-conjugated secondary antibodies (Dianova) and the Luminol detection system (Carl Roth). The isolation of plasma membrane fractions was described previously (16). Anti-FLAG antibodies (mouse MAb clone M2 or rabbit; both from Sigma-Aldrich), anti-CD11a MAb (clone 38; Calbiochem), anti-GFP MAb (or conjugated to agarose [both from Santa Cruz]), anti-SKAP55 rat MAb (clone 13B6F2) (16), anti-RAPL rat MAb (clone 104B4G12) (30), anti-RIAM rat MAb (clone 15B7E8) (29), phospho-AKT-S473 rabbit MAb, anti-phospho-extracellular signal-regulated kinase 1/2 (anti-phospho-ERK1/2) rabbit serum (Cell Signaling), antipanactin MAb AC40 (antikoerper-online), antitalin MAb (clone 8D4, Sigma-Aldrich), anti-ADAP mouse MAb, anti-human ADAP sheep serum (37), anti-mouse MAb Rap1 (both from BD Bioscience), and anti-mouse MAb for His (Santa Cruz) were used for Western blotting and/or immunoprecipitation.
Immunofluorescence and confocal microscopy.
Transfected Jurkat T cells or primary human T cells (rested or stimulated with 10 μg/ml of anti-CD3/CD28 MAbs for 15 min at 37°C) were plated onto 12-well slides coated with poly-l-lysine (Marienfeld KG) for 15 min at 4°C and fixed with 3.5% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 10 min. Cells were permeabilized with 0.1% Triton X-100 in PBS, blocked with 5% horse serum (Biochrom AG) in PBS, and incubated with tetramethyl rhodamine isothiocyanate (TRITC)-labeled phalloidin (Sigma-Aldrich). Coverslips were mounted in Mowiol 488 and imaged with a Leica TCS SP2 laser scanning confocal system (Leica Microsystems) using an apochromatic 63× oil immersion objective (numerical aperture [NA], 1.4).
Analysis of images.
Image analysis for the cellular localization of GFP-tagged proteins was done by using Adobe Photoshop CS3. The fluorescence signals near the plasma membrane were calculated, and a curve (fluorescence intensity versus length of the measured area [micrometers]) was generated by using a self-made MATLAB (Mathworks Inc.) tool. The fluorescence signals were measured at 4 different positions (at 3, 6, 9, and 12 o'clock) in each cell. The generated curves were analyzed by determining the middle of the F-actin curve (red) and dropping a perpendicular that divides the green or yellow curve (GFP-tagged proteins) into two halves (see Fig. S1 in the supplemental material). Area A (close to the plasma membrane) and area B (cytoplasm) were determined by using Adobe Photoshop CS3. The obtained values for areas A and B were used to calculate the ratios of fluorescence intensity at the plasma membrane. The calculation was done as follows: (i) calculation of the ratio of (A)/(A+B) for each curve, (ii) averaging of all four positions within one cell, and (iii) averaging of the calculated values for 10 to 25 cells. The mean average number of all cells expressing GFP alone for each experiment was subtracted from the average number of cells expressing individual GFP-tagged fusion proteins.
Protein purification, NMR spectroscopy, and titration experiments.
N-terminally His-tagged PH and DM-PH domain constructs as well as mutant variants were expressed in BL21(DE3) cells and purified via Ni2+-nitrilotriacetic acid (NTA) affinity chromatography followed by size exclusion chromatography. NMR spectra were acquired on a Bruker AV 700-MHz spectrometer equipped with a 5-mm triple-resonance cryoprobe, processed with Topspin (Bruker), and analyzed with CcpNMR Analysis (38). Backbone assignments of ∼83% of residues 97 to 216 were obtained by standard three-dimensional (3D) experiments in combination with 15N amino acid selective labeling. Subsequently, assignments were transferred to the nearest-neighbor peak in DM-PH or mutant spectra. For ligand titration experiments, HSQC spectra of 150 μM the isolated SKAP55 PH domain were recorded in the presence of ligand concentrations of 0, 50, 150, 300, 500, 750, 1,050, 1,500, 2,000, and 2,670 μM IP4, IP3, PIP2-C4, and PIP3-C4 (all purchased from Echelon Bioscience). For weakly binding IP3, two additional concentrations, 3,200 und 3,740 μM, were considered. The HSQC titration with IP4 was repeated with 270 μM the PH domain of SKAP55 (0, 50, 150, 300, 600, 1,130, and 2,466 μM IP4) in order to improve the spectral quality and compared to spectra of 270 μM R131M-PH and K152E-PH in the presence of no or 2,466 μM IP4, respectively. For KD determinations, the titration curves of the most strongly shifting resonances were individually fit to a two-state binding model, and the KD was determined by linear regression analysis. Structural representations were generated with PyMOL (PyMOL molecular graphics system, version 1.8; Schrödinger LLC).
Actin binding assays.
The nonmuscle actin binding protein spin-down assay Biochem kit (Cytoskeleton Inc.) was used to assess polymer F-actin binding and to sequester monomeric actin (G-actin) for polymerization to F-actin by ultracentrifugation to separate F-actin from G-actin according to the manufacturer's instructions. Samples were analyzed by SDS-PAGE and Coomassie blue staining (Roth).
Statistical analysis.
Statistical differences were analyzed by using Student's t test. A P value of ≤0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anke Ramonat for excellent technical assistance and Nirdosh Dadwal for purification of proteins.
This work was supported by the medical faculty of the Otto von Guericke University (stipend for A.W.) and DFG grants CRC 854 (B10, B12, B19, and Z01 for C.F., S.K., B.S., A.J.M., and/or L.P.) and CRC 958/765 (A7 or C4 for C.F.).
We declare no competing financial interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00509-16.
REFERENCES
- 1.Abram CL, Lowell CA. 2009. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol 27:339–362. doi: 10.1146/annurev.immunol.021908.132554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hogg N, Patzak I, Willenbrock F. 2011. The insider's guide to leukocyte integrin signalling and function. Nat Rev Immunol 11:416–426. doi: 10.1038/nri2986. [DOI] [PubMed] [Google Scholar]
- 3.Kinashi T. 2005. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol 5:546–559. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- 4.Moser M, Legate KR, Zent R, Fassler R. 2009. The tail of integrins, talin, and kindlins. Science 324:895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- 5.Su W, Wynne J, Pinheiro EM, Strazza M, Mor A, Montenont E, Berger J, Paul DS, Bergmeier W, Gertler FB, Philips MR. 2015. Rap1 and its effector RIAM are required for lymphocyte trafficking. Blood 126:2695–2703. doi: 10.1182/blood-2015-05-644104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klapproth S, Sperandio M, Pinheiro EM, Prunster M, Soehnlein O, Gertler FB, Fassler R, Moser M. 2015. Loss of the Rap1 effector RIAM results in leukocyte adhesion deficiency due to impaired beta2 integrin function in mice. Blood 126:2704–2712. doi: 10.1182/blood-2015-05-647453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calderwood DA. 2015. The Rap1-RIAM pathway prefers beta2 integrins. Blood 126:2658–2659. doi: 10.1182/blood-2015-09-668962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witte A, Degen J, Baumgart K, Waldt N, Kuropka B, Freund C, Schraven B, Kliche S. 2012. Emerging roles of ADAP, SKAP55, and SKAP-HOM for integrin and NF-kB signaling in T cells. J Clin Cell Immunol S12:002. [Google Scholar]
- 9.Griffiths EK, Krawczyk C, Kong YY, Raab M, Hyduk SJ, Bouchard D, Chan VS, Kozieradzki I, Oliveira-Dos-Santos AJ, Wakeham A, Ohashi PS, Cybulsky MI, Rudd CE, Penninger JM. 2001. Positive regulation of T cell activation and integrin adhesion by the adapter Fyb/Slap. Science 293:2260–2263. doi: 10.1126/science.1063397. [DOI] [PubMed] [Google Scholar]
- 10.Peterson EJ, Woods ML, Dmowski SA, Derimanov G, Jordan MS, Wu JN, Myung PS, Liu QH, Pribila JT, Freedman BD, Shimizu Y, Koretzky GA. 2001. Coupling of the TCR to integrin activation by Slap-130/Fyb. Science 293:2263–2265. doi: 10.1126/science.1063486. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Liu H, Lu Y, Lovatt M, Wei B, Rudd CE. 2007. Functional defects of SKAP-55-deficient T cells identify a regulatory role for the adaptor in LFA-1 adhesion. Mol Cell Biol 27:6863–6875. doi: 10.1128/MCB.00556-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burbach BJ, Srivastava R, Ingram MA, Mitchell JS, Shimizu Y. 2011. The pleckstrin homology domain in the SKAP55 adapter protein defines the ability of the adapter protein ADAP to regulate integrin function and NF-kappaB activation. J Immunol 186:6227–6237. doi: 10.4049/jimmunol.1002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell JS, Burbach BJ, Srivastava R, Fife BT, Shimizu Y. 2013. Multistage T cell-dendritic cell interactions control optimal CD4 T cell activation through the ADAP-SKAP55-signaling module. J Immunol 191:2372–2383. doi: 10.4049/jimmunol.1300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marie-Cardine A, Bruyns E, Eckerskorn C, Kirchgessner H, Meuer SC, Schraven B. 1997. Molecular cloning of SKAP55, a novel protein that associates with the protein tyrosine kinase p59fyn in human T-lymphocytes. J Biol Chem 272:16077–16080. doi: 10.1074/jbc.272.26.16077. [DOI] [PubMed] [Google Scholar]
- 15.Huang Y, Norton DD, Precht P, Martindale JL, Burkhardt JK, Wange RL. 2005. Deficiency of ADAP/Fyb/SLAP-130 destabilizes SKAP55 in Jurkat T cells. J Biol Chem 280:23576–23583. doi: 10.1074/jbc.M413201200. [DOI] [PubMed] [Google Scholar]
- 16.Kliche S, Breitling D, Togni M, Pusch R, Heuer K, Wang X, Freund C, Kasirer-Friede A, Menasche G, Koretzky GA, Schraven B. 2006. The ADAP/SKAP55 signaling module regulates T-cell receptor-mediated integrin activation through plasma membrane targeting of Rap1. Mol Cell Biol 26:7130–7144. doi: 10.1128/MCB.00331-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, McCann FE, Gordan JD, Wu X, Raab M, Malik TH, Davis DM, Rudd CE. 2004. ADAP-SLP-76 binding differentially regulates supramolecular activation cluster (SMAC) formation relative to T cell-APC conjugation. J Exp Med 200:1063–1074. doi: 10.1084/jem.20040780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ophir MJ, Liu BC, Bunnell SC. 2013. The N terminus of SKAP55 enables T cell adhesion to TCR and integrin ligands via distinct mechanisms. J Cell Biol 203:1021–1041. doi: 10.1083/jcb.201305088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menasche G, Kliche S, Chen EJ, Stradal TE, Schraven B, Koretzky G. 2007. RIAM links the ADAP/SKAP-55 signaling module to Rap1, facilitating T-cell-receptor-mediated integrin activation. Mol Cell Biol 27:4070–4081. doi: 10.1128/MCB.02011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raab M, Wang H, Lu Y, Smith X, Wu Z, Strebhardt K, Ladbury JE, Rudd CE. 2010. T cell receptor “inside-out” pathway via signaling module SKAP1-RapL regulates T cell motility and interactions in lymph nodes. Immunity 32:541–556. doi: 10.1016/j.immuni.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raab M, Smith X, Matthess Y, Strebhardt K, Rudd CE. 2011. SKAP1 protein PH domain determines RapL membrane localization and Rap1 protein complex formation for T cell receptor (TCR) activation of LFA-1. J Biol Chem 286:29663–29670. doi: 10.1074/jbc.M111.222661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim D, Lu Y, Rudd CE. 2016. Non-cleavable talin rescues defect in the T-cell conjugation of T-cells deficient in the immune adaptor SKAP1. Immunol Lett 172:40–46. doi: 10.1016/j.imlet.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB. 1995. Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83:1037–1046. doi: 10.1016/0092-8674(95)90219-8. [DOI] [PubMed] [Google Scholar]
- 24.Costello PS, Gallagher M, Cantrell DA. 2002. Sustained and dynamic inositol lipid metabolism inside and outside the immunological synapse. Nat Immunol 3:1082–1089. doi: 10.1038/ni848. [DOI] [PubMed] [Google Scholar]
- 25.Yao L, Janmey P, Frigeri LG, Han W, Fujita J, Kawakami Y, Apgar JR, Kawakami T. 1999. Pleckstrin homology domains interact with filamentous actin. J Biol Chem 274:19752–19761. doi: 10.1074/jbc.274.28.19752. [DOI] [PubMed] [Google Scholar]
- 26.Macia E, Partisani M, Favard C, Mortier E, Zimmermann P, Carlier MF, Gounon P, Luton F, Franco M. 2008. The pleckstrin homology domain of the Arf6-specific exchange factor EFA6 localizes to the plasma membrane by interacting with phosphatidylinositol 4,5-bisphosphate and F-actin. J Biol Chem 283:19836–19844. doi: 10.1074/jbc.M800781200. [DOI] [PubMed] [Google Scholar]
- 27.Swanson KD, Tang Y, Ceccarelli DF, Poy F, Sliwa JP, Neel BG, Eck MJ. 2008. The Skap-hom dimerization and PH domains comprise a 3′-phosphoinositide-gated molecular switch. Mol Cell 32:564–575. doi: 10.1016/j.molcel.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemmon MA. 2007. Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp 74:81–93. doi: 10.1042/BSS0740081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horn J, Wang X, Reichardt P, Stradal TE, Warnecke N, Simeoni L, Gunzer M, Yablonski D, Schraven B, Kliche S. 2009. Src homology 2-domain containing leukocyte-specific phosphoprotein of 76 kDa is mandatory for TCR-mediated inside-out signaling, but dispensable for CXCR4-mediated LFA-1 activation, adhesion, and migration of T cells. J Immunol 183:5756–5767. doi: 10.4049/jimmunol.0900649. [DOI] [PubMed] [Google Scholar]
- 30.Kliche S, Worbs T, Wang X, Degen J, Patzak I, Meineke B, Togni M, Moser M, Reinhold A, Kiefer F, Freund C, Forster R, Schraven B. 2012. CCR7-mediated LFA-1 functions in T cells are regulated by 2 independent ADAP/SKAP55 modules. Blood 119:777–785. doi: 10.1182/blood-2011-06-362269. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Zhu L, Zhang H, Hirbawi J, Fukuda K, Dwivedi P, Liu J, Byzova T, Plow EF, Wu J, Qin J. 2014. Conformational activation of talin by RIAM triggers integrin-mediated cell adhesion. Nat Commun 5:5880. doi: 10.1038/ncomms6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mason JM, Arndt KM. 2004. Coiled coil domains: stability, specificity, and biological implications. Chembiochem 5:170–176. doi: 10.1002/cbic.200300781. [DOI] [PubMed] [Google Scholar]
- 33.Cabanas C, Hogg N. 1993. Ligand intercellular adhesion molecule 1 has a necessary role in activation of integrin lymphocyte function-associated molecule 1. Proc Natl Acad Sci U S A 90:5838–5842. doi: 10.1073/pnas.90.12.5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reedquist KA, Ross E, Koop EA, Wolthuis RM, Zwartkruis FJ, van Kooyk Y, Salmon M, Buckley CD, Bos JL. 2000. The small GTPase, Rap1, mediates CD31-induced integrin adhesion. J Cell Biol 148:1151–1158. doi: 10.1083/jcb.148.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marie-Cardine A, Hendricks-Taylor LR, Boerth NJ, Zhao H, Schraven B, Koretzky GA. 1998. Molecular interaction between the Fyn-associated protein SKAP55 and the SLP-76-associated phosphoprotein SLAP-130. J Biol Chem 273:25789–25795. doi: 10.1074/jbc.273.40.25789. [DOI] [PubMed] [Google Scholar]
- 36.Togni M, Swanson KD, Reimann S, Kliche S, Pearce AC, Simeoni L, Reinhold D, Wienands J, Neel BG, Schraven B, Gerber A. 2005. Regulation of in vitro and in vivo immune functions by the cytosolic adaptor protein SKAP-HOM. Mol Cell Biol 25:8052–8063. doi: 10.1128/MCB.25.18.8052-8063.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Musci MA, Hendricks-Taylor LR, Motto DG, Paskind M, Kamens J, Turck CW, Koretzky GA. 1997. Molecular cloning of SLAP-130, an SLP-76-associated substrate of the T cell antigen receptor-stimulated protein tyrosine kinases. J Biol Chem 272:11674–11677. doi: 10.1074/jbc.272.18.11674. [DOI] [PubMed] [Google Scholar]
- 38.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. 2005. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.