

ABSTRACT
The mammalian target of rapamycin complex 1 (mTORC1) is activated by extracellular factors that control bone accrual. However, the direct role of this complex in osteoblast biology remains to be determined. To investigate this question, we disrupted mTORC1 function in preosteoblasts by targeted deletion of Raptor (Rptor) in Osterix-expressing cells. Deletion of Rptor resulted in reduced limb length that was associated with smaller epiphyseal growth plates in the postnatal skeleton. Rptor deletion caused a marked reduction in pre- and postnatal bone accrual, which was evident in skeletal elements derived from both intramembranous and endochondrial ossification. The decrease in bone accrual, as well as the associated increase in skeletal fragility, was due to a reduction in osteoblast function. In vitro, osteoblasts derived from knockout mice display a reduced osteogenic potential, and an assessment of bone-developmental markers in Rptor knockout osteoblasts revealed a transcriptional profile consistent with an immature osteoblast phenotype suggesting that osteoblast differentiation was stalled early in osteogenesis. Metabolic labeling and an assessment of cell size of Rptor knockout osteoblasts revealed a significant decrease in protein synthesis, a major driver of cell growth. These findings demonstrate that mTORC1 plays an important role in skeletal development by regulating mRNA translation during preosteoblast differentiation.
KEYWORDS: Raptor, mTORC1, osteoblast, osteogenesis
INTRODUCTION
Intramembranous ossification (most of the craniofacial skeleton) and endochondrial ossification (the vertebrate appendicular and axial skeleton) are the two cellular processes required to form the skeleton (reviewed in reference 1). In both processes, osteoblasts (OBs), the bone-forming cells of the skeleton, play an essential role. OBs differentiate from mesenchymal progenitors through distinct developmental stages marked by expression of key transcription factors, including runt-related transcription factor 2 (Runx2 product), osterix (Sp7 and Osx product), and β-catenin (Ctnnb1 product). Runx2 is the master regulator of OB lineage commitment and directly regulates Osx expression as evidenced by a lack of osterix transcript in Runx2 knockout animals (2). The homozygous deletion of Runx2 or Osx results in the formation of a complete cartilaginous skeleton, but no bone formation takes place in either the endochondral or the membranous skeleton due to the absence of OBs (2, 3).
Runx2, osterix, and β-catenin are regulated by extracellular factors to control embryonic and postnatal skeletal development. Bone morphogenetic proteins (BMPs), including BMP2 and BMP4, are potent bone anabolic factors that promote osteogenesis via the activation of SMAD signaling molecules, in turn regulating Runx2 and osterix expression and activity (4–6). Wnt ligands bind to a receptor complex comprising Frizzled and LRP5/6, inducing a signaling cascade that directs OB differentiation at the pre-OB stage, enhancing Runx2 expression (7, 8). Insulin and insulin-like growth factor (IGF-1) indirectly regulate Runx2 to promote postnatal bone accrual. Insulin downregulates Twist1 and Twist2 expression (9), negative regulators of Runx2 that bind and inhibit Runx2 activity (10). IGF-1 and insulin also inhibit Foxo1-mediated suppression of Runx2 activity by promoting Foxo1 phosphorylation and nuclear exclusion (11).
Bone anabolic signals require activation of cellular processes that govern growth. The phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway is activated by a variety of extracellular factors and promotes cell cycle progression, cell survival and growth signals. In mice, the OB-specific deletion of Pten, a negative regulator of PI3K, results in a progressive increase in bone mineral density throughout life (12), whereas Akt1−/−/Akt2−/− mice have shorter bones (13) and delayed bone ossification (14, 15), demonstrating an important role for this pathway in skeletal development. Activation of this pathway in OBs through the deletion of Pten or expression of myristoylated-Akt (15) leads to the constitutive activation of mTOR, the nutrient-sensing pathway in mammalian cells that controls cell growth (reviewed in reference 16). mTOR forms two functionally distinct multiprotein complexes, termed mTORC1 and mTORC2. Each complex is defined by the unique adaptor proteins raptor (regulatory-associated protein of mTOR) and rictor (rapamycin-insensitive companion of mTOR), respectively (17, 18). mTORC2 regulates cell survival, metabolism, and cytoskeletal organization through the phosphorylation of several members of the AGC kinase subfamily, including Akt (reviewed in reference 19). mTORC1 plays a central role in the regulation of anabolic processes, including mRNA translation and ribosome biogenesis (reviewed in reference 20). mTORC1 regulates translation through the inhibition of 4E-BP1, a negative regulator of translation that interferes with translation initiation (21), and via activation of p70 S6K (S6K) and its downstream effectors, including ribosomal protein S6 (rS6) and eukaryotic elongation factor 2 kinase (22).
Extracellular factors that control skeletal development and bone accrual activate mTORC1. Most notably, insulin strongly activates the PI3K/Akt/mTORC1 pathway to promote anabolic cell processes, including translation, lipid biogenesis, and nutrient storage (23, 24). Insulin activates mTORC1 via inhibition of the tuberous sclerosis complex, which is a GTPase-activating protein for Rheb, a small GTPase. In its GTP-bound state, Rheb binds to and activates mTORC1 (25). Inhibition of mTORC1, using rapamycin, blocks insulin and IGF-1 stimulated osteogenesis in vitro (26, 27). Furthermore, deletion of the insulin receptor or insulin-like growth factor receptor 1 (Igflr) in OBs causes a postnatal low bone phenotype (9, 27), suggesting that mTORC1 plays an important role in transducing insulin and IGF-1 bone anabolic signals. Consistent with this view, inhibition of mTORC1 by administration of rapamycin to C57BL/6 mice resulted in a significant reduction in mineral apposition and a reduction in trabecular bone volume and thickness (27).
Taken together, these findings suggest that mTORC1 plays an important role in bone accrual. To specifically investigate this question, we deleted Rptor, the unique component of mTORC1, in pre-OBs and assessed the effects on skeletal development. Deletion of Rptor in pre-OBs blocked OB differentiation, leading to defective intramembranous and endochondrial ossification, low bone mass, and skeletal fragility.
RESULTS
OB-specific deletion of raptor in Rptorob−/− mice.
Recent evidence suggests extracellular factors that regulate osteogenesis and bone accrual utilize the mTORC1 pathway to transduce anabolic signals important for pre- and postnatal skeletal development (9, 27, 28). To investigate this question, we generated mice in which Rptor, a unique and essential component of mTORC1, was disrupted in preosteoblastic cells. Male progeny from the mating of Osx1-GFP::cre-raptorfl/wt and raptorfl/fl mice with the genotypes Osx1-GFP::cre-raptorfl/wt and Osx1-GFP::cre-raptorfl/fl (here referred to as Rptorob−/+ [het] and Rptorob−/− [hom], respectively) were analyzed. In light of the skeletal phenotype of Osx1-GFP::cre mice (here referred to as Osx:cre) (29), age-matched male Osx:cre mice were used as controls.
To confirm bone-specific deletion of Rptor, genomic DNA was isolated from multiple tissues of Osx:cre and knockout animals, and PCR was used to amplify a product generated by cre-mediated recombination of the Rptor gene (30). A deletion-specific product was amplified from DNA isolated from the long bones of Rptorob−/+ and Rptorob−/− mice, confirming tissue-specific Rptor deletion (Fig. 1A). To confirm deletion at the transcriptional level, we bred Raptorfl/fl mice with the R26-stop-EYFP reporter line and then mated the progeny with Osx:cre to generate eYFP-Rptorob−/+ and eYFP-Rptorob−/− mice in which cells that have expressed Osterix are labeled eYFP+ cells for their life span (31). RT-PCR was performed on RNA isolated from eYFP+ cells recovered (using fluorescence-activated cell sorting [FACS]) from the long bones of control (eYFP-Osx:cre), eYFP-Rptorob−/+, and eYFP-Rptorob−/− mice using Rptor-specific primers. A significant reduction in Rptor transcript was detected in eYFP+ cells isolated from het and hom mice relative to control, confirming gene inactivation in the majority of cells isolated by FACS (purity, ∼95% eYFP+) (Fig. 1B).
FIG 1.
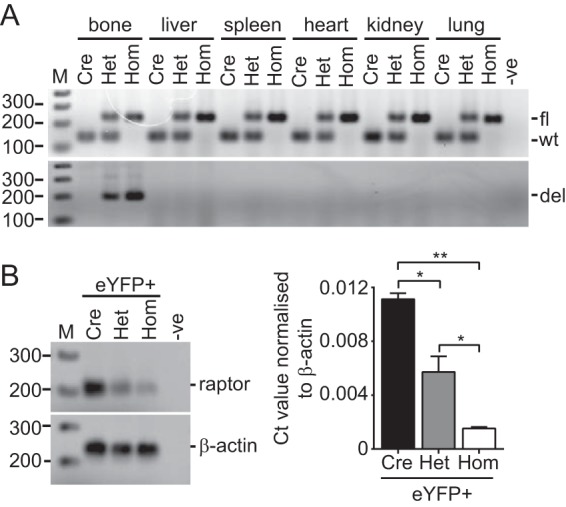
OB-specific deletion of Rptor. (A) Genomic DNA was isolated from the indicated tissues of 4-week-old Osx:cre (cre), Rptorob−/+ (het), and Rptorob−/− (hom) mice, and the genotypes were confirmed (upper panel). An Osx:cre-mediated gene deletion product was amplified in genomic DNA isolated from the long bones of het and hom mice (lower panel). (B) Quantitative PCR was performed on RNA extracted from eYFP+ cells isolated from eYFP-Osx:cre mice (control), eYFP-Rptorob−/+ mice, and eYFP-Rptorob−/− mice using Rptor exon 6-specific primers. Amplified product (left panel) and relative transcript levels normalized to β-actin (right panel) are shown. fl, floxed; wt, wild type; del, deletion. *, P < 0.05; **, P < 0.01.
Prenatal skeletal phenotype in Rptorob−/− mice.
Rptorob−/+ and Rptorob−/− mice were born at the expected Mendelian frequencies. No gross phenotypic differences or difference in weight or length were evident in Rptorob−/− animals compared to either wild-type (WT) littermates (data not shown) or age-matched Osx:cre mice at birth (Fig. 2A). To investigate the effect of Rptor deletion on prenatal skeletal development, we stained the skeletons of newborn mice to assess the level of mineralized bone (alizarin red) and cartilage (alcian blue). Vertebral spinous and transverse processes and bones of the pelvic cage (illium and ischium) were less mineralized in Rptorob−/− mice than in Osx:cre mice (Fig. 2B). The skulls of newborn Rptorob−/− mice were narrower and less mineralized (Fig. 2B). Furthermore, the level of alizarin red labeling of the jawbones of Rptorob−/− mice was reduced relative to Osx:cre mice (Fig. 2B). The long bones of the forelimbs and hind limbs (the humerus and the femur, respectively) were significantly shorter in Rptorob−/− mice (mean decrease of 8.5% ± 1% for both bones) (Fig. 2C and D), and the percentage of bone that was mineralized was also significantly less in new born Rptorob−/− mice relative to Osx:cre mice (17.4% ± 2.2% for both bones) (Fig. 2C and D). In summary, the deletion of Rptor in osterix-expressing cells (from embryonic day 13.5 [E13.5]) (8) caused a significant reduction in the lengths of long bones and in the level of mineralization of the long bones in the skeletons of newborn mice, demonstrating an important role for mTORC1 in prenatal skeletal development.
FIG 2.

Prenatal skeletal phenotype in Rptorob−/− mice. (A) Images of newborn (P0) Osx:cre (cre) and Rptorob−/− (hom) mice. Scale bar, 10 mm. (B) Newborn mice were stained with alizarin red and alcian blue to visualize mineralized bone (red) and cartilage (blue). (Upper panel) Images of the pelvic cage. Scale bar, 5 mm. (Middle panels) Superior view of the skull. (Lower panels) Lateral view of the skull. Scale bar, 1 mm. (C) Forelimbs of Osx:cre and Rptorob−/− mice. The length of the humerus and the percentage of the humerus that is mineralized were measured. Scale bar, 5 mm. (D) Hind limbs of Osx:cre and Rptorob−/− mice. The length of the femur and the percentage of the femur that is mineralized were measured. Scale bar, 5 mm. Data are presented as means ± the SD (n = 6 per group). **, P < 0.01; ***, P < 0.001.
Reduced weight and bone length in Rptorob−/− mice.
To assess the effects of OB-specific Rptor deletion on postnatal skeletal development, we analyzed animals at 4 and 12 weeks of age since these time points represent the early and later phases of long-bone growth in mice (32). At 4 weeks of age, Rptorob−/− mice were smaller (Fig. 3A) and weighed significantly less (mean decrease, 22.9% ± 7.8%) than Osx:cre mice (Fig. 3B). The reduced weight of Rptorob−/− mice was still evident at 12 weeks of age relative to Osx:cre and Rptorob−/+ mice (15.2% ± 3.5% and 15.4% ± 3.5% reductions, respectively).
FIG 3.
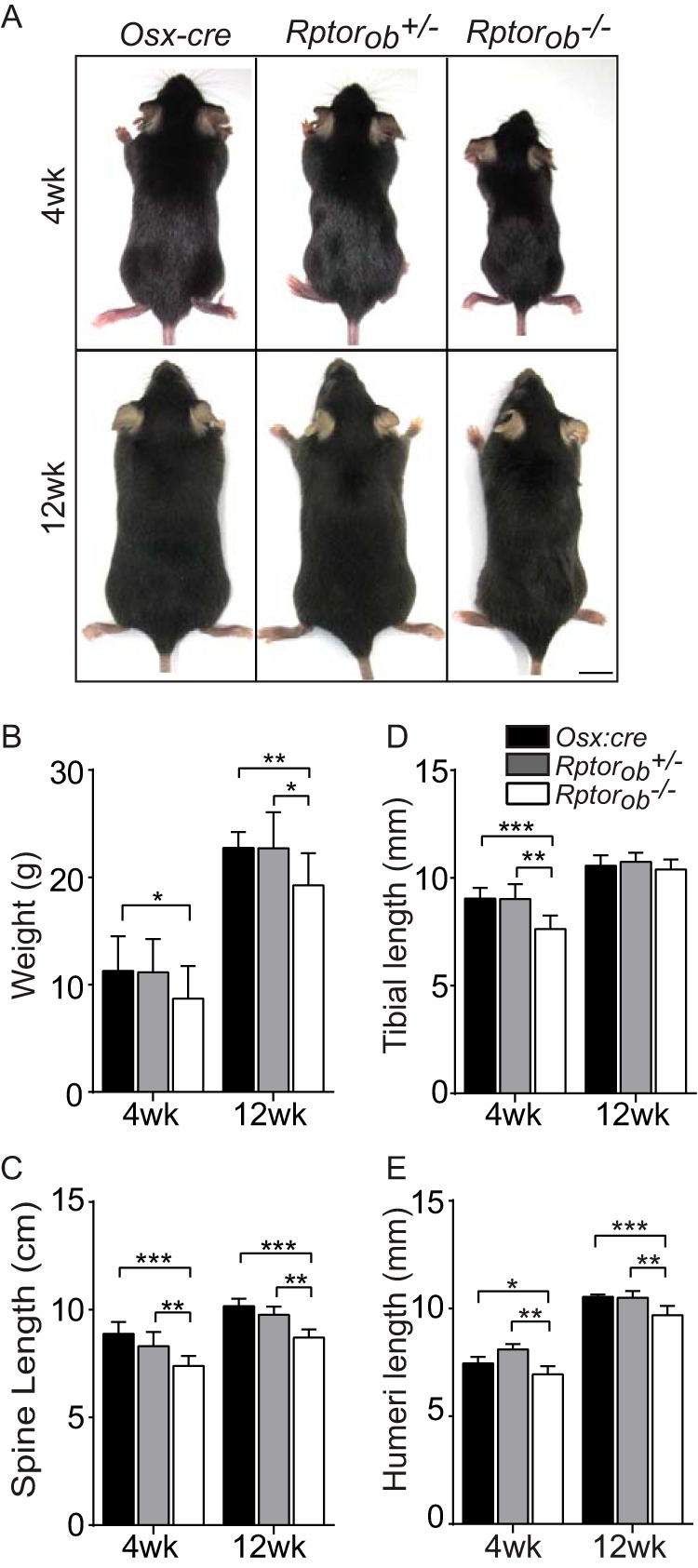
Phenotypic assessment of Rptorob−/− mice. (A) Images of 4- and 12-week-old mice. Scale bar, 1 cm. (B) Weights of mice. (C) Tibial lengths (distance from the proximal tibial head to the fibular notch). (D) Spinal lengths (the distance from C1 to L5 vertebrae). (E) Humeral lengths (distance from the greater tubercle to the epicondyle). Measurements are presented as means ± the SD (n ≥ 6 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To evaluate the macroscopic effects of OB-specific Rptor deletion on skeletal development, we measured the lengths of the spines, tibiae, and humeri at 4 and 12 weeks of age. At 4 weeks, the spines of Rptorob−/− mice were significantly shorter relative to het and Osx:cre mice (10.9% ± 1.8% and 16.8% ± 2%, respectively) (Fig. 3C). Similarly, the lengths of the tibiae were significantly shorter in Raptorob−/− mice (Fig. 3D), as were the lengths of the humeri (Fig. 3E). At 12 weeks, the spine length and humeral length remained reduced in Rptorob−/− mice relative to Osx:cre mice (Fig. 3C and E), but tibial length had normalized to control lengths (Fig. 3D).
Low endochondral bone mass in Rptorob−/− mice.
To assess the effects of OB-specific Rptor deletion on the microarchitecture of the skeleton, cortical and trabecular bone parameters were assessed using micro-computed tomography (μCT) (Fig. 4A). For the assessment of cortical bone, the tibial diaphysis was analyzed. At 4 and 12 weeks of age, cortical thickness (Ct.Th) was significantly reduced in Raptorob−/− mice relative to both Raptorob−/+ mice and Osx:cre mice (Fig. 4A and B). Consistent with a reduction in Ct.Th, an increase in intrameduallary diameter was observed in het and hom mice relative to Osx:cre mice at 12 weeks of age (Fig. 4C).
FIG 4.

Low postnatal bone masses in Rptorob−/− mice. (A) Cross-sectional μCT images of cortical and trabecular bone (tibia) of Osx:cre, Rptorob+/−, and Rptorob−/− mice. (B and C) Cortical thickness (Ct.Th) (B) and intramedullary diameter (C) of the cortical bone. (D to G) Quantitative assessment of the trabecular bone of the tibia. BV, bone volume; TV, total volume; Tb.Th, trabecular thickness; Tb.N, trabecular number; Tb.Sp, trabecular seperation. (H and I) Quantitative assessment of structural indices of the trabecular bone architecture of the tibia. Tb.Pf, trabecular pattern factor; SMI, structural model index. (J and K) Femurs from control and knockout animals were subjected to 3-point load testing. (L) μCT images of the skulls of 4-week-old Osx:cre and Rptorob−/− mice. (Left panels) Superior view of skulls; (right panels) lateral view of skulls. Measurements are presented as means ± the SD (n ≥ 6 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To analyze changes in trabecular bone parameters, a region in the secondary spongiosa proximal to the tibial growth plate was analyzed. At 4 weeks of age, bone volume normalized to the total tissue volume (BV/TV) was profoundly reduced in Rptorob−/− mice (mean decrease, 29% ± 3.5%). This reduction was increased at 12 weeks, at which point the bone volume was significantly reduced in both Rptorob−/+ mice (31% ± 5.2%) and Rptorob−/− mice (45% ± 7%) relative to Osx:cre mice (Fig. 4D). The trabecular number (Tb.N) and thickness (Tb.Th) were reduced in Rptorob−/− mice relative to the Rptorob−/+ and Osx:cre mice at 4 weeks (29.4% ± 4.2% and 15.7% ± 1.2%, respectively). Notably, at 12 weeks, Tb.N was significantly lower for both Rptorob−/+ mice (24.8% ± 5.9%) and Rptorob−/− mice (35.5% ± 5.1%) relative to Osx:cre mice (Fig. 4E and F). Consistent with a reduction in Tb.N and Tb.Th, trabecular separation (Tb.Sp) was increased in both Rptorob−/+ mice (mean increase, 44.3% ± 7.5%) and Rptorob−/− mice (45.7% ± 9.4%) relative to Osx:cre mice at 4 weeks of age (Fig. 4G).
An assessment of trabecular structural indices revealed an increase in the trabecular bone pattern factor (Tb.Pf), a measure of connectivity that increases as the level of connections between trabecular structures decreases, in Rptorob−/− mice relative to Osx:cre mice at 12 weeks of age (Fig. 4H). This increase in Tb.Pf was associated with an increase in the structural model index (SMI), a measure of the shape of the trabeculae that increases as the trabeculae transition from a plate-like to rod-like architecture, for Rptorob−/− mice relative to Osx:cre mice at 12 weeks of age (Fig. 4I).
We next evaluated the biomechanical properties of OB-specific Rptor-deficient bone by subjecting isolated bones to load testing. The femurs from Rptorob−/− mice displayed a reduced bending stiffness (flexural rigidity; mean decrease, 71.9% ± 0.2% and 74.8% ± 10.1%, respectively) relative to Rptorob−/+ and Osx:cre controls (Fig. 4J). The femurs from Rptorob−/− mice were also found to have a lower strength (ultimate load; mean decrease, 56.3% ± 10%) relative to Osx:cre controls (Fig. 4K).
We next assessed the effects of Rptor gene deletion on intramembranous ossification, the developmental process that accounts for the majority of bones of the skull. Three-dimensional reconstructions of μCT scans of 4-week-old mice revealed that the skulls of Rptorob−/− mice appeared to be smaller and malformed compared to Osx:cre mice (Fig. 4L). Furthermore, regions of the skulls of Rptorob−/− mice appeared to be incompletely mineralized, especially in the frontal region adjacent to the coronal suture line and also in the nasal region. The lambdoidal and sagittal sutures in the skulls of Rptorob−/− mice were also incompletely mineralized, suggesting that the deletion of Rptor in calvarial preosteoblasts disrupts intramembranous ossification. In summary, the deletion of Rptor in preosteoblasts causes a significant reduction in both cortical and trabecular bone parameters that culminates in more brittle bones, highlighting an important role for mTORC1 in postnatal skeletal development.
Increased intramedullary adipose in Rptorob−/− mice.
In addition to the low bone mass, high levels of intramedullary adipocytes were observed in the tibiae of Rptorob−/− mice compared to Osx:cre mice (Fig. 5A). At 4 and 12 weeks of age, the numbers of adipocytes per intramedullary area (N.Adc/M.Ar) were significantly higher in Rptorob−/− mice relative to Osx:cre and Rptorob−/+ mice (Fig. 5B). An assessment of the area occupied by adipocytes per number of adipocytes (AdC.Ar/N.AdC) suggested that there was no difference in adipocyte size in knockout mice relative to Osx:cre mice (Fig. 5C).
FIG 5.
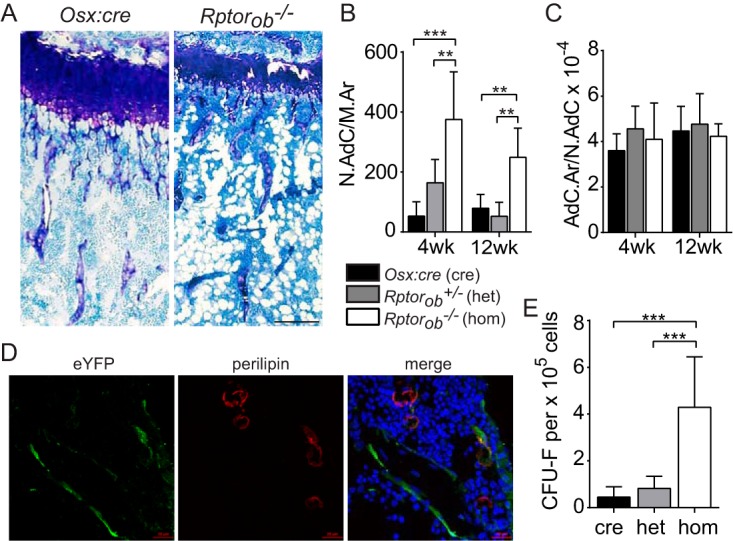
Elevated levels of intramedullary adipose in Rptorob−/− mice. (A) Toluidine blue-stained tibial sections of Osx:cre (cre) and Rptorob−/− (hom) mice. Adipocytes appear as cleared circles. Scale bar, 200 μm. (B and C) The numbers of adipocytes per area of the intramedullary space (N.Adc/M.Ar) (B) and the areas occupied by adipocyes per number of adipocytes (AdC.Ar/N.AdC) (C) were quantified using histomorphometry. (D) Two-color confocal microscopy was used to detect overlapping eYFP and perilipin staining in frozen tibial sections isolated from 4-week-old eYFP-Osx:cre and eYFP-Rptorob−/− mice. Nuclei are stained with DAPI. Scale bar, 20 μm. (E) Compact bone cells were isolated from the long bones of Osx:cre (cre), Rptorob−/+ (het), and Rptorob−/− (hom) mice, and the numbers of CFU-F per 105 cells plated were enumerated. Measurements are presented as means ± the SD. **, P < 0.01; ***, P < 0.001.
The increase in intramedullary adiposity in Rptorob−/− mice suggests that the deletion of Rptor in preosteoblasts affected the lineage commitment of the osteoadipogenic precursor pool toward the adipogenic lineage or that adipocytes arose from the trans- or dedifferentiation of Rptor knockout osteoblastic cells. To investigate the latter, we examined the origin of the adipocytes using mice bred with the eYFP reporter. Tibial sections from eYFP-Osx:cre control and eYFP-Rptorob−/− mice were stained for eYFP and perilipin, an adipocyte integral membrane protein, and overlapping signals assessed using two-color confocal microscopy. Only a small proportion of adipocytes in the bone marrow of eYFP-Rptorob−/− mice were found to be eYFP+ perilipin+, suggesting that most of the adipocytes did not arise from the trans- or dedifferentiation of Rptor-null cells (Fig. 5D).
We next assessed whether the deletion of Rptor in preosteoblastic cells affected the mesenchymal stem cell (MSC) pool by measuring the number of CFU/fibroblast (CFU-F) in cell populations isolated from the long bones of Rptorob−/− and control mice. Enumeration of morphologically discrete fibroblastic colonies revealed a significant increase in CFU-F in Rptorob−/− mice relative to Osx-cre and het mice (fold increase, 9.6 ± 4.8 and 5.2 ± 0.9, respectively) (Fig. 5E). These data suggest that deletion of Rptor in preosteoblasts stimulates progenitor cell proliferation as a mechanism to compensate for impaired bone formation in Rptorob−/− mice.
Narrow and disorganized growth plate in postnatal Rptorob−/− mice.
Longitudinal bone growth is primarily driven by the coordinated proliferation and differentiation of chondrocytes. At the growth plate, chondrogenic cells are arranged into vertical columns that can be stratified into horizontal layers (zones) corresponding to different phases of chondrocyte differentiation (1, 33). Osterix expression has previously been observed in the prehypertrophic chondrocytes of the developing skeleton (2, 34) and the postnatal skeleton (35). Consistent with these findings, chondrocytes within the growth plates of eYFP-Osx:cre and eYFP-Rptorob−/− mice were found to be eYFP+ (Fig. 6A), suggesting that the deletion of Rptor in chondrogenic cells could contribute to the reduction in limb length observed in Rptorob−/− mice.
FIG 6.

Narrow and disorganized growth plate in Rptorob−/− mice. (A) eYFP-labeled cells within the hypertrophic chondrocyte zones of epiphyseal growth plates of 4-week-old eYFP-Osx:cre and eYFP-Rptorob−/− mice. Nuclei were stained with DAPI. Scale bar, 50 μm. (B) Safranin O staining of acidic proteoglycan in the epiphyseal growth plate of the tibia. Scale bar, 200 μm. (C) Growth plate widths based on Safranin O staining. (D to F) Width of chondrocyte zones within the growth plate based on chondrocyte morphology. (G) Immunohistochemical staining of PCNA in the growth plates of Osx:cre and Rptorob−/− mice. Scale bar, 50 μm. (H) Quantitative PCR was performed on RNA extracted from eYFP+ cells isolated from eYFP-Osx:cre and eYFP-Rptorob−/− mice using primers specific to the indicated genes. The relative transcript levels were normalized to β-actin. PZ, proliferating zone; HZ, hypertrophic zone; PS, primary spongiosa. Measurements are presented as means ± the SD (n ≥ 6 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To investigate this question, we analyzed the morphology of the tibial growth plate using sections in which acidic proteoglycan, present in cartilage tissues, was stained red (Fig. 6B). In Rptorob−/− mice, chondrocytes within the proliferating zone were disorganized, with fewer cells arranged into vertical columns relative to Osx:cre mice (Fig. 6B). Furthermore, at 4 weeks of age, the width of the growth plates in Rptorob−/+ and Rptorob−/− mice was significantly reduced relative to Osx:cre mice (Fig. 6C). We next assessed the width of the chondrocyte zones within the growth plates of knockout and Osx:cre mice. At 4 and 12 weeks of age there was no difference in the width of the zone of resting chondrocytes (Fig. 6D), precursor cells which, when activated, proliferate and synthesize a cartilagenous matrix. In contrast, at 4 weeks of age the width of the proliferating zone (where chondrocytes undergo rapid division by mitosis) in Rptorob−/+ and Rptorob−/− mice and the width of the hypertrophic zone (terminally differentiated chondrocytes) were significantly reduced relative to Osx:cre mice (Fig. 6E and F). No differences in the width of the growth plate or the width of chondrocyte zones were evident in 12-week-old control and knockout mice.
We next assessed the proliferative state of chondrocytes within the growth plate of 4-week-old Rptorob−/− mice as a possible mechanism to account for the marked reduction in the size of the proliferating and hypertrophic zones. Using immunohistochemistry (IHC), proliferating cell nuclear antigen (PCNA) expression, a nuclear marker of proliferating cells (36), was clearly evident in chondrocytes within the growth plate of Osx:cre mice but was otherwise absent in the growth plate in chondrocytes from Rptorob−/− mice (Fig. 6G).
We next assessed whether Rptor deletion in osterix-expressing chondrocytes affects the stages of chondrocyte differentiation. Chondrocyte maturation stimulates mineralizeation of the cartilage and ingress of the vasculature, allowing OB recruitment and differentiation (37). To achieve this, we assessed the transcript levels of immature and mature chondrocyte markers in eYFP+ cells recovered from the long bones of eYFP-Osx:cre and eYFP-Rptorob−/− mice. There was no difference in the expression of early chondrocyte markers Sox9 and Col2a1 (isoform 2) in Rptorob−/− mice relative to Osx:cre mice (Fig. 6H). However, the transcript levels of Col10a1 and Mmp13, markers of mature and terminally differentiated chondrocytes, were significantly reduced in Rptorob−/− mice relative to Osx:cre mice (Fig. 6H). In summary, these findings suggest that mTORC1 plays an important role in long bone growth by regulating chondrocyte proliferation and differentiation.
Reduced OB function in Rptorob−/− mice.
Bone mass is maintained through the coordinated action of bone-resorbing osteoclasts (OCs) and bone-forming OBs. To determine whether the low bone mass phenotype of Rptorob−/− mice was due to a reduction in OB numbers and/or an increase is OC numbers, OB and OC cells were enumerated in toluidene blue-stained tibial sections. No significant differences in the numbers of OBs or OCs per bone perimeter (N.Ob/B.Pm and N.Oc/B.Pm, respectively) were found in knockout animals relative to Osx:cre mice at 4 and 12 weeks of age (Fig. 7A and B). Similarly, there were no differences in the bone surfaces occupied by either OBs or OCs (Fig. 7C and D). No change in OB and OC numbers or in the surface areas occupied by OBs suggests that the phenotype primarily results from defective OB function. In support of this hypothesis, the mineral apposition rate (MAR) was significantly reduced in Rptorob−/− animals relative to Osx:cre and het mice (Fig. 7E and F). The low MAR in Rptorob−/− mice was associated with a significant reduction in the bone formation rate (BFR) relative to both Osx:cre and Rptorob−/+ mice (Fig. 7G). Furthermore, the serum osteocalcin levels, a marker of OB function, were significantly reduced in Rptorob−/− mice at 4 weeks of age relative to age-matched Osx:cre mice (Fig. 7H). These data suggest that the deletion of Rptor in preosteoblastic cells has a significant effect on OB function that could be associated with either a functional defect in mineralizeation and/or a defect in osteogenesis.
FIG 7.

Reduced OB function in Rptorob−/− mice. (A and B) OBs lining the bone perimeter (N.OB/B.Pm) (A) and OB perimeter per bone perimeter (OB.Pm/B.Pm) (B) were measured in tibial sections using histomorphometry. (C and D) OCs lining the per bone perimeter (N.Oc/B.Pm) (C) and the OC perimeter per bone perimeter (Oc.Pm/B.Pm) (D) were measured in tibial sections using histomorphometry. (E) Dually labeled osteoid after consecutive intraperitoneal injections (3 days apart) of calcein. Arrowheads indicate the interlabel distance. (F) The mineral apposition rate (MAR) was calculated by dividing the interlabel distance by the time interval. (G) The bone formation rate (BFR) was calculated as an expression of the amount of newly formed mineralized bone. (H) Serum osteocalcin levels at 4 weeks of age. Measurements are presented as means ± the SD (n ≥ 7 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
OBs from Rptorob−/− mice have an immature transcription profile and reduced osteogenic potential.
To determine whether the reduction in OB function in Rptorob−/− mice was due to a block in OB differentiation, we assessed the expression of OB differentiation markers in tibial sections from knockout and control mice. Osterix expression was clearly evident in cells of the primary spongiosa immediately adjacent to the zone of hypertrophic chondrocytes in the growth plate of Osx:cre controls but was otherwise absent in sections derived from Rptorob−/− mice (Fig. 8A). Similarly, osteocalcin expression, a marker of mature OBs, was evident in tibial sections from Osx:cre controls but was absent from Rptorob−/− mice (Fig. 8A).
FIG 8.
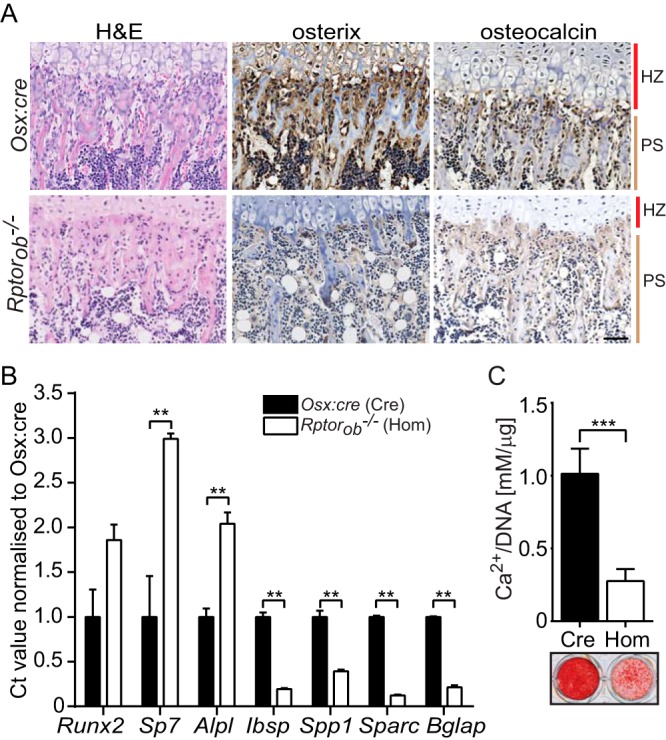
Stalled osteogenic differentiation in Rptorob−/− OBs. (A) Hematoxylin and eosin staining (H&E) and IHC staining of osterix and osteocalcin in the primary spongiosa of tibial sections from 4-week-old Osx:cre (cre) and Rptorob−/− (hom) mice. Scale bar, 50 μm. (B) Quantitative PCR was performed on RNA extracted from eYFP+ cells isolated from eYFP-Osx:cre and eYFP-Rptorob−/− mice. The expression levels of the indicated genes relative to β-actin are shown. (C) Calvarial cells isolated from 4-week-old Osx:cre and Rptorob−/− mice were cultured under osteogenic conditions, and the levels of acid-soluble mineral were quantified and expressed relative to the total DNA. Mineralized matrix was stained using alizarin red. HZ, hypertrophic zone; PS, primary spongiosa. Measurements are presented as means ± the SD from three independent experiments.*, P < 0.05; **, P < 0.01; ***, P < 0.001.
To gain further insight into the osteoblastic phenotype of the Rptor knockout cells, we analyzed the transcript level of genes involved at different stages of osteogenesis in eYFP+ cells isolated from the long bones of eYFP-Osx:cre and eYFP-Rptorob−/− mice using reverse transcription-PCR. The expression of Runx2 and Sp7 (osterix), transcription factors essential for the initial stages of osteogenesis, and Alpl, a marker of early osteogenic differentiation, was significantly elevated in eYFP+ cells isolated from Rptorob−/− mice relative to eYFP-Osx:cre mice (Fig. 8B). In contrast, the expression of late OB markers, including integrin-binding saloprotein (Ibsp), osteopontin (Spp1), osteonectin (Sparc), and osteocalcin (Bglap), was significantly downregulated in Rptorob−/− mice relative to Osx:cre mice (Fig. 8B). These data suggest that the deletion of Rptor in preosteoblasts, using the Osx-cre driver, stalls OB differentiation, resulting in osteogenic cells with an immature transcript profile.
We next investigated how the immature OB phenotype of Rptor knockout cells affects OB function. Calvarial cells, isolated from knockout and control mice, were cultured under osteogenic conditions and the ability to form a mineralized matrix was assessed. Deletion of Rptor caused a significant reduction in mineralized matrix production as evidenced by a decrease in the level of acid-solubilized calcium and calcium-dependent alizarin red staining relative to Osx:cre mice (Fig. 8C). These data suggest that mTORC1 function in preosteoblastic cells is essential for osteogenic differentiation.
Mechansisms leading to low bone mass in Rptorob−/− mice.
To investigate the mechanism leading to the stall in osteogenesis, we isolated calvarial cells from newborn raptorfl/fl mice and used a tamoxifen-inducible Cre lentivirus to generate Rptor knockout OBs (Rap−/−). The deletion of Rptor caused a reduction in basal, insulin-stimulated and IGF-1-stimulated phosphorylation of the key mTORC1 substrates p70 S6K and 4E-BP1 and ribosomal protein S6, a p70 S6K substrate, findings consistent with reduced mTORC1 activity (Fig. 9A). When cultured under osteoinductive conditions, the loss of mTORC1 function led to a significant decrease in osteogenic potential, as evidenced by the reduced expression of osteoblast-related genes in Rap−/− osteoblasts relative to control (flox) cells (Fig. 9B). Despite this reduced osteogenic potential, no differences in Runx2 RNA or protein levels were detected in Rap−/− OBs relative to control cells (Fig. 9B and C). In contrast, the levels of osterix (Sp7), a Runx2-regulated transcription factor essential for osteogenesis, were significantly reduced in Rap−/− osteoblasts (Fig. 9B to D). These findings suggested that Runx2 transcriptional activity is reduced in Rap−/− cells, and this may account for the reduction in osteogenic potential of these cells.
FIG 9.

Mechanisms leading to low bone mass in Rptorob−/− mice. Calvarial cells isolated from Rptorfl/fl mice (P4) were infected with a lentivirus harboring a tamoxifen-inducible cre recombinase. Infected cells were treated with tamoxifen (Rap−/−) to stimulate Rptor gene deletion. Control cells (flox) were treated with vehicle. (A) The effect of Rptor gene deletion on mTORC1 substrate activation at steady-state and following stimulation with either insulin or IGF-1 was assessed by Western blotting. (B) Assessment of osteogenic gene expression in vehicle (flox)- or tamoxifen (Rap−/−)-treated cells after 21 days of osteogenic induction. (C) Western blots showing Runx2 and osterix protein levels in vehicle (flox)- or tamoxifen (Rap−/−)-treated cells after 7, 14, and 21 days of osteogenic induction. (D) Densitometry showing osterix protein levels normalized to β-actin in vehicle (flox)- or tamoxifen (Rap−/−)-treated cells after 7, 14, and 21 days of osteogenic induction. (E) Western blots showing the activation status of STAT1 and STAT3 in vehicle (flox)- or tamoxifen (Rap−/−)-treated cells. (F) Western blots showing the subcellular localization of the indicated proteins in vehicle (flox)- or tamoxifen (Rap−/−)-treated cells. (G) Metabolic labeling in vehicle (flox), tamoxifen (Rap−/−)- or vehicle plus AZD8055 (flox+AZD)-treated cells. (H) Cell size analysis in vehicle (flox)-, tamoxifen (Rap−/−)-, or vehicle plus AZD8055 (flox+AZD)-treated cells. Measurements are presented as means ± the SD from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To investigate how mTORC1 regulates Runx2 activity, we looked at known mTORC1 substrates and their relationship with Runx2 in osteoblasts. From this investigation, we identified STAT1 and STAT3 as potential candidates. STAT1 is a known substrate of mTORC1 (38, 39) and unphosphorylated STAT1 has been shown to attenuate Runx2 activity by retaining Runx2 in the cytoplasm (40). STAT3 is essential for normal skeletal development (41, 42), and STAT3 activation (serine 727 phosphorylation) is dependent on mTORC1 activity (43). Based on these findings, we investigated the phosphorylation status of STAT1 and STAT3 in control and Rptor-null osteoblasts, and this revealed no significant change in the phosphorylation status of Ser727 of either protein in control versus Rap−/− osteoblasts (Fig. 9E). Furthermore, an assessment of the subcellular localization of Runx2, STAT1, and STAT3 failed to reveal any significant changes in Rap−/− OBs relative to control cells (Fig. 9F).
In light of the significant effect of Rptor gene deletion on the phosphorylation status of S6K and 4E-BP1 (Fig. 9A), we looked at the protein synthetic capacity of these cells as a possible mechanism to explain the reduction in bone mass in Rptorob−/− mice. Protein synthesis was significantly reduced in Rap−/− OBs relative to control OBs (Fig. 9G). Furthermore, this reduction was similar to the decrease observed in cells treated with AZD8055, a potent inhibitor of both mTORC1 and mTORC2 (44), suggesting that the mTOR signaling plays a substantial role in the control of ribosome biogenesis and protein synthesis in OBs. We next examined the effect of Rptor gene deletion on cell size since mTORC1 and its substrate S6K are key determinants of this process (45, 46). Deletion of Rptor or treatment of control cells with AZD8055 significantly reduced the size of calvarial OBs (Fig. 9H). Taken together, these data suggest that deletion of Rptor in preosteoblasts reduces the protein synthetic capacity and sizes of these cells, which is likely to inhibit their ability to differentiate into mature OBs.
DISCUSSION
In this study, we investigated the role of mTORC1 in pre- and postnatal skeletal development by deleting Rptor, an essential component of mTORC1, in preosteoblasts. The deletion of Rptor using Osx:cre had a profound effect on postnatal bone accrual. By 4 weeks of age, cortical bone thickness (Ct.Th) was reduced by 32.3%, and the trabecular bone volume (TB/BV) was reduced by 29%, decreasing to 45% by 12 weeks. Notably, this reduction occurred relative to Osx:cre mice, which already display reduced cortical thickness and trabecular bone volume compared to wild-type mice (29). These data demonstrate that mTORC1 plays an important role in postnatal bone accrual.
The low-bone-mass phenotype of Rptorob−/− mice was associated with a significant decrease in the mineral apposition rate and the bone formation rate. This was not due to a reduction in OB numbers but to a decrease in OB function. In vitro, calvarial OBs isolated from Rptorob−/− mice and Rptor knockout OBs, generated using lentiviral Cre, were shown to have a reduced mineralizing capacity and reduced osteogenic potential. To investigate the mechanism, we bred our mice lines with an eYFP reporter strain that allowed us to purify the osteoblastic cells in which Rptor was deleted. Using this approach, we found that OB differentiation is stalled at an early phase in the osteogenic program, as evidenced by a decrease in the expression of late OB markers and an increase in the expression of Runx2, Sp7, and ALPL. Runx2 controls OB differentiation by regulating the expression of key osteoblastic genes, including Osx (2) and the genes for integrin-binding saloprotein (Ibsp), osteopontin (Spp1), osteonectin (Sparc), and osteocalcin (Bglap) (47, 48). Mechanistically, mTORC1 controls mRNA translation and ribosome biogenesis (21), suggesting that the suppression of translation in Rptorob−/− OBs reduced the ability of preosteoblasts to proceed with the osteogenic program once Osx expression was initiated by Runx2. Consistent with this view, we showed that Rap−/− OBs have a reduced osteogenic potential that was associated with a significant reduction in the synthetic properties of these cells, as measured by the level of protein synthesis and assessment of cell size (Fig. 9). These findings are iconsistent with an important role for mTORC1 function in the control of mRNA translation during the early phase of osteogenic differentiation.
Suppression of insulin and IGF-1 signaling is likely to play an important role in the skeletal phenotype of Rptorob−/− mice. In vitro, both insulin and IGF-1 regulate primary mTORC1 substrates in OBs, and this was significantly reduced upon knockout of Rptor (Fig. 9). Mice in which the insulin receptor (Ob-ΔIR) or insulin-like growth factor receptor (Igflr−/−) is deleted in OBs have a low-bone-mass phenotype (9, 27). In Ob-ΔIR mice, the failure to accumulate trabecular bone is due, in part, to a significant reduction in OB numbers, whereas the low bone mass in Igflr−/− mice appears to be due to a failure of Igflr-null OBs to fully differentiate into mature osteocalcin-positive cells. IGF-1 activates the PI3K/Akt/mTOR pathway, and IGF-1-induced osteogenic differentiation of Sca-1+ MSC progenitor cells is blocked by rapamycin (27), suggesting that the low-bone-mass phenotype in Rptorob−/− mice could be due, in part, to impaired IGF-1 signaling. mTORC1 has also been shown to play a role in the anabolic effects of Wnt7b using mice in which the Wnt7b cDNA, flanked by a floxed transcriptional stop, was knocked-in to the Rosa26 locus (R26-Wnt7b) (28). Osx:cre-mediated activation of Wnt7b expression, initiated postnatally, promoted bone formation by stimulating OB number and function. In this postnatal context, deletion of Rptor in compound mice overexpressing Wnt-7b (Osx:Cre; R26-Wnt7b; Raptorfl/fl) reduced Wnt-7b-induced bone mass by inhibiting OB function (as evidenced by decreased mineral apposition rate and bone formation rate) but not OB numbers (28). These results are consistent with our findings that demonstrate an important role for mTORC1 in the control of OB function.
In this study, we found that Rptorob−/− mice undergo a significant reduction in limb length and mineralizeation at birth and postnatally. Osx expression has previously been reported in the growth plate of prenatal (2, 34, 49) and postnatal mice (35) and, consistent with these findings, chondrocytes were labeled eYFP+ in eYFP-Rptorob−/− and control eYFP-Osx:cre mice. Labeling of chondrocytes has also been observed in several lineage tracing studies using reporter mice and Osx:cre mice (8, 50–52). In the developing skeleton, Osx transcripts are found transiently in differentiating chondrocytes at E13.5 and weakly in the prehypertrophic chondrocytes of E15.5 embyros (2). At E18.5, osterix protein is evident in prehypertrophic chondrocytes and is abundant in preosteoblasts in the primary spongiosa but absent from the more mature hypertrophic cells (53). This pattern of Osx expression overlaps with mTORC1 activity in the developing skeleton. Activated prS6, a major substrate of mTORC1, is evident in the proliferating columnar chondrocytes and prehypertrophic chondrocytes of embyros from E16.5 to E18.5. The activation status of prS6 is markedly reduced in the hypertrophic zone but is reactivated in chondrocytes at the final stage of hypertrophy (53, 54). Activated prS6 is also abundantly evident in OB precursor cells within the primary spongiosa at E18.5 (53). Recent evidence suggests that correct regulation of mTORC1 activity during chondrogeneis is essential for the normal progression of prehypertrophic chondrocytes to a terminal hypertrophic fate. Deletion of Lkb1, a negative regulator of mTORC1 (55) in immature chondrocytes using Col2α1-Cre causes a rapamycin-sensitive expansion of immature chondrocytes and the formation of enchondroma-like tumors (53). Deletion of mTOR or raptor in the developing head and limb mesenchyme at E9.0, prior to skeletogenesis, using Prx-1-cre mice has also been shown to profoundly affect chondrogenesis (54). Most notably, mTORC1 is important for the transition of chondrocytes to the hypertrophic program and may be involved in the phases of hypertrophy that drive the increase in dry cell mass (54, 56). Given the importance of mTORC1 activity in chondrogenesis, deletion of Rptor in osterix-expressing prehypertrophic chondrocytes may have affected chondrocyte differentiation by prematurely terminating mTORC1 activity. We did note that Col10a1 and Mmp13 expression was reduced in eYFP+ chondrocytes isolated from the long bones of eYFP-Rptorob−/− mice, suggesting that fewer terminally differentiated chondrocytes were present in Rptorob−/− mice. It was also evident that deletion of Rptor in osterix-expressing hypertrophic chondrocytes caused an overall reduction in chondrocyte proliferation, as measured by a decrease in the expression of PCNA (Fig. 6G), suggesting that decreased chondrocyte proliferation may also account for the decrease in expression of mature chondrocyte markers.
Recently, gain-of-function mouse models have been used to investigate the role of mTORC1 in osteogenesis. Deletion of either Tsc1 in preosteoblasts using Osx:cre (ΔTsc1 mice [57]) or Tsc2 in mature osteoblasts using Ocn:cre (ΔTsc2 mice [58]), results in constitutive activation of mTORC1. In both models, hyperactivation of mTORC1 leads to an increase in postnatal bone mass that was evident in cortical and trabecular regions of the long bones and calvarial bones of the skull. The increase in bone mass is associated with an increase in OB number; however, in both models, OB function is impaired. In vitro, the addition of low-dose rapamycin (10 pM to 100 pM) to ΔTsc1 or ΔTsc2 OBs is able to rescue the impaired osteoblast function, implicating mTORC1 as a negative regulator of osteogenesis. However, in ΔTsc2 mice (58) the impaired OB function can be attributed, at least in part, to a reduction in insulin signaling as mTORC1 promotes negative-feedback inhibition of insulin signaling via mTORC1-p70 S6K-mediated phosphorylation and degradation of IRS-1 (59). Hyperactivation of this feedback loop in ΔTsc2 OBs may contribute to the reduction in OB activity, as seen in mice in which the insulin receptor (Ob-ΔIR) is deleted in OBs (9). Constitutive activation of mTORC1 in preosteoblasts (ΔTsc1 mice) causes more severe skeletal abnormalities than those observed in ΔTsc2 mice, possibly reflecting the earlier embryonic time point of Cre activation (Osx promoter E13.5 days postcoitus [dpc] versus Ocn promoter E17 dpc [2, 26]). The addition of rapamycin to ΔTsc1 mice, from E13.5 dpc, rescues the bone defects, suggesting that mTORC1 function must be switched off to allow osteogenesis to proceed correctly. In contrast to gain-of-function mouse models, the evidence from loss-of-function studies suggests that mTORC1 may play a role in promoting osteoblast differentiation. Mice with the hom deletion of mTOR in mature osteoblasts (Ocn:cre ΔmTOR) demonstrate reduced tibial trabecular bone volume and trabecular number (58). This phenotype can be attributed to impaired mTORC1 function since mice with homozygous deletion of Rictor, an essential component of mTORC2, in mature osteoblasts have no tibial bone phenotype (60). As reported here, Rptorob−/− mice have significantly reduced mineral apposition and bone formation rates and no change in OB number per bone perimeter (N.OB/B.Pm, Fig. 7), suggesting impaired OB function. Analysis of the transcriptional profile of OBs isolated from Rptorob−/− mice revealed an immature bone phenotype, suggesting that mTORC1 signaling promotes osteoblast differentiation from preosteoblasts, a finding supported by the recent work of others (61). Taken together, the findings from loss- and gain-of-function studies indicate that temporal regulation of mTORC1 plays an essential role in the osteogenic program.
Clinically, rapamycin (Sirolimus) and the related analogue (Everolimus) are used as immunosuppressants to prevent organ transplant rejection and to treat a variety of cancers (62). Given the role of mTORC1 in osteogenesis, it is conceivable that the use of inhibitors in the pediatric context could lead to adverse skeletal outcomes. To date, despite extended treatment periods (2 to 5 years), these concerns have not been borne out with respect to rates of linear growth in young children (age < 5 years) (63, 64). Nonetheless, continued assessment of bone mineral density is warranted in these children. Notably, in adults, adjuvant treatment of estrogen receptor-positive (ER+) breast cancer with Everolimus appears to have bone-protective effects (65), which have been attributed to its potent antiosteoclastic properties rather than to a bone anabolic effect.
In summary, our studies have revealed a novel role for mTORC1 in osteogenesis. Notably, mTORC1 is required for the progression of the osteogenic program from the preosteoblast stage to the mature osteoblast stage. Mechanistically, mTORC1-mediated control of mRNA translation is likely to be a key determinant governing this progression. Finally, our studies further highlight the importance of pathways that coordinate cellular growth and energy metabolism in the accrual of bone mass.
MATERIALS AND METHODS
Transgenic mice.
All mice were bred and housed at the SA Pathology Animal Care Facility, and studies were performed with Institutional Ethics approval (SA Pathology Animal Ethics Committee approval Project 63/10). Raptorfl/fl mice (30), Osx1-GFP::cre mice (8), and R26-stop-EYFP mice (31) were used in this study. The following forward and reverse primer pairs were used for genotyping: Raptorfl/fl, 5′-ATGGTAGCAGGCACACTCTTCATG-3′ and 5′-GCTAAACATTCAGTCCCTAATC-3′; Osx1-GFP::cre, 5′-CGCGGTCTGGCAGTAAAAACTATC-3′ and 5′-CCCACCGTCAGTACGTGAGATATC-3′; and R26-stop-EYFP, 5′-AAAGTCGCTCTGAGTTGTTAT-3′ and 5′-GCGAAGAGTTTGTCCTCAACC-3′. Detection of Rptor gene deletion was performed using 5′-ATGGTAGCAGGCACACTCTTCATG-3′ and 5′-CTCAGAGAACTGCAGTGCTGAAGG-3′, as previously described (30). Genomic DNA was isolated from tissues using a DNeasy blood and tissue kit (Qiagen, Melbourne, Australia).
Skeletal staining.
Alizarin red and alcian blue staining of new born mice was performed as previously described (66). The percentage of the bone (humerus and tibia) that was mineralized was calculated by dividing the length of the mineralized (alizarin red) region of the bone by the total length of the bone using ImageJ (67). Images were aquired using bright field optics.
High-resolution μCT scanning.
X-ray microtomography was performed using a SkyScan 1076 (Bruker, Kontich, Belgium). Calvariae and tibiae were scanned at 74 kV/100 mA using a 0.5-mm aluminum filter, a 0.8 rotation step, and two-frame averaging with an isometric resolution of 8.65 μm/pixel. Three-dimensional reconstruction of the scan data (smoothing, 1; beam hardening, 30%) was performed using NRecon (SkyScan). Analysis of the bone microarchitecture was performed using CTAn (SkyScan). Cortical bone measurements were calculated as the average of measurements in the lateral, medial, caudal, and cranial orientation from three slices of cortical bone. Slices were selected in relation to the primary spongiosa. For analysis of the proximal tibia, a trabecular region of interest was manually defined to exclude the cortex. Transverse micro-computed tomography (μCT) slices of the tibia were acquired using a 6.5-μm slice increment. A total of 150 slices (0.97 mm) were analyzed for each tibia, commencing 20 slices distal to the primary spongiosa. Images were reconstructed and thresholded using a specimen-specific threshold. For trabecular bone regions, the percent bone volume (BV/TV), trabecular thickness (Tb.Th.), trabecular number (Tb.N), trabecular separation (Tb.Sp), structure model index (SMI), and trabecular pattern factor (Tb.Pf) were calculated using CTAn. To ensure that the analysis of trabecular bone was conducted in corresponding regions of the secondary spongiosa proximal to the growth plate, the distance from the primary spongiosa and the area analyzed was normalized to the length of the tibia. Calvarial images were acquired using Avizo Fire (FEI Visualization Sciences Group, Burlington, MA). Tibial length measurements were performed using CTAn software (SkyScan) and taken from the proximal tibial head to the fibrual notch. Humeral lengths were measured using CTAn, with measurements taken from the greater tubercle to the epicondyle. For spine length, the distance from cervical 1 (C1) to lumbar 5 (L5) was measured in whole-body X-ray images (LX-60; Faxitron, Lincolnshire, IL) and quantitated using ImageJ.
Histology.
Methyl methacrylate processing and embedding of nondecalcified bone and staining of sections were conducted as previously described (68, 69). Briefly, sections (5 μm) were deplasticized with acetone for 15 min and then rinsed in water. Toluidine blue sections were stained in 2% toluidine blue solution for 3 min and washed twice with sodium phosphate-citrate buffer. Safranin O sections were stained in 0.2% Fast Green (Sigma) for 5 min and then stained in 0.1% Safranin O stain for 5 min. Quantitation of images was conducted using OsteoMeasure XP (OsteoMetrics, Inc.). Histomorphometric analysis of OB numbers (N.Ob/B.Pm), OB-occupied bone surface (Ob.S/BS), osteoclast numbers (N.Oc/B.Pm), osteoclast-occupied bone surface (Oc.S/BS), the number of adipocytes per intramedullary area (total area − bone area) (N.AdC/M.Ar), and the percentage adipose area per intramedullary area (AdC.Ar/M.Ar) was conducted on toluidine blue-stained sections. The region of interest for the proximal mouse tibiae was defined as a region of secondary spongiosa distal from the primary spongiosa to a distance of 1.8 mm.
Double fluorochrome labeling and mineral apposition rate.
To measure bone formation in vivo, mice were subcutaneously injected with calcein at 20 μg per g of body weight and then injected again after a 3-day interval. At 24 h after the second injection, the tibias were collected, fixed in 10% formalin, and then embedded in methyl methacrylate as previously described (68). Dynamic measurements of bone formation were conducted on calcein-labeled sections of the tibiae in the secondary spongiosa using the Osteomeasure XP system (Osteometrics, Inc., Atlanta, GA), and the distance between the two calcein-labeled mineralization fronts was quantitated. The MAR was calculated by dividing the interlabel distance by the time interval.
Immunohistochemistry.
Bones were fixed in formalin for 1 week, demineralized in EDTA (14%, wt/vol), and embedded in paraffin. Sections (5 μm) were subjected to antigen retrieval (10 mM citrate buffer [pH 6.0], 95°C) for 10 min and then cooled to room temperature. Endogenous peroxidase was quenched with 0.5% H2O2 (vol/vol) in methanol for 30 min, and sections were blocked in 3% normal horse serum in phosphate-buffered saline (pH 7.4; PBS) and then incubated with the primary antibody at 4°C overnight. Sections were washed in PBS and then incubated with species-specific biotinylated secondary antibodies (anti-goat [BA-5000] and anti-rabbit [BA-1000] antibodies; Vector Laboratories, Burlingame, CA) for 1 h at room temperature, followed by 30 min of incubation with streptavidin-HRP (Vector Laboratories). Antibody binding was visualized using diaminobenzidine (DAB) according to the manufacturer's instructions (Vector Laboratories). The antibodies used for IHC were as follows: osterix (ab22552; Abcam, Cambridge, United Kingdom), eGFP (A600-101-215; Rockland, Limerick, PA), eYFP (A600-401-215; Rockland), osteocalcin (sc-18319; Santa Cruz Biotechnology, Santa Cruz, CA), and PCNA (sc-7907; Santa Cruz Biotechnology).
Confocal microscopy.
For immunofluorescence staining, paraffin-embedded sections were antigen retrieved as described above and blocked in 1% bovine serum albumin, 2.5% goat serum, and 2.5% horse serum in PBS (blocking buffer), followed by incubation with primary antibodies overnight at 4°C. Sections were washed with ice-cold PBS and then incubated with fluorescently tagged species-specific secondary antibodies (anti-chicken [Alexa Fluor 488 conjugated, ab150173; Abcam] and anti-rabbit [Alexa Fluor 647 conjugated, ab150067; Abcam] antibodies) at room temperature for 1 h. The sections were washed and then briefly fixed for 10 min (0.1% formalin in PBS), and the nuclei were labeled with DAPI (4′,6′-diamidino-2-phenylindole; 300 nM in PBS) for 5 min. Sections were mounted with ProLong Gold antifade reagent (Invitrogen).
For frozen sections, the bones were fixed overnight in formalin at 4°C and then demineralized in EDTA (14% [wt/vol] in PBS) and 10% sucrose at 4°C until no calcified tissue was detectable by X-ray examination. Tissues were frozen in optimum-cutting-temperature resin (OCT; Sakura, Alphen aan den Rijn, Netherlands) using liquid nitrogen chilled isopentane then sectioned using a cryostat (CM 3050s; Leica, Sydney, Australia). Sections (8 μm) were air dried and then stained and mounted as described above. Fluorescent labeling was detected using a LSM400 confocal microscope (Zeiss, Oberkochen, Germany). Antibodies to eYFP (ab13970; Abcam) and perilipin (catalog no. 9349; Cell Signaling, Danvers, MA) were used for immunofluorescence analyses.
Biomechanical testing.
Three-point bending analyses to failure were conducted using femurs isolated from 4-week-old Osx:cre, Rptorob−/+, and Rptorob−/− mice as previously described (70).
Isolation of eYFP+ cells and real-time PCR.
Compact bone cells (CB-MSCs) were obtained from the long bones of 4-week-old eYFP-Osx:cre, eYFP-Raptorob−/+, and eYFP-Raptorob−/− mice in accordance to published methods (71). In brief, the tibias and femurs from 8 to 12 mice were excised, cleaned thoroughly, and then crushed in ice-cold PBS supplemented with 2% fetal calf serum (FCS) and 2 mM EDTA, washed several times, chopped into small fragments, and digested into type 1 collagenase (Worthington Biochemical Corporation, Lakewood, NJ) at 3 mg/ml at 37°C for 45 min with shaking. The resulting cell suspension was filtered through a 70-μm-pore size strainer, and then eYFP+ cells were isolated by FACS using a MoFlo Astrios (Beckman Coulter, Brea, CA). Total RNA was isolated from eYFP+ cells by using a RNAqueous micro kit (Life Technologies, Melbourne, Australia), and cDNA was prepared from 70 ng of total RNA using Sensiscript reverse transcriptase according to the manufacturer's instructions (Qiagen). Real-time PCRs were performed using RT2 SYBR green ROX reagent (Qiagen) in a Rotor-Gene 3000 thermal cycler (Corbett Research/Qiagen). Forward and reverse primer pairs, designed to amplify across intron-exon boundries, are listed in Table 1. Changes in gene expression were calculated relative to β-actin.
TABLE 1.
Oligonucleotides used for real-time PCR
| Gene | Accession no. | Sequence (5′–3′) |
Size (bp) | |
|---|---|---|---|---|
| Forward | Reverse | |||
| Rptor | NM_028898.2 | TGGGTCTTCAACAAGAACTACACT | TCTGGGCAAGTGGATGGTTT | 199 |
| Runx2 | NM_001146038.2 | ATGATGACACTGCCACCTCTG | ATGAAATGCTTGGGAACTGC | 120 |
| Sp7 | NM_130458.3 | ATGGCGTCCTCTCTGCTTG | GTCCATTGGTGCTTGAGAAGG | 211 |
| Bglap | NM_007541.3 | AAGCAGGAGGGCAATAAGGT | TCAAGCCATACTGGTCTGATAGC | 142 |
| Spp1 | NM_001204201.1 | AGCAAGAAACTCTTCCAAGCAA | GATTCGTCAGATTCATCCGAGT | 130 |
| Sparc | NM_009242.5 | ACTACATCGGACCATGCAAATAC | CGTACAAGGTGACCAGGACAT | 108 |
| Ibsp | NM_008318.3 | CAGTCCAGGGAGGCAGTG | GGAAAGTGTGGAGTTCTCTG | 158 |
| Col10a1 | NM_009925.4 | CATCTCCCAGCACCAGAATC | GCTAGCAAGTGGGCCCTTTA | 142 |
| Sox9 | NM_011448.4 | CACAAGAAAGACCACCCCGA | GGACCCTGAGATTGCCCAGA | 209 |
| Mmp13 | NM_008607.2 | AGCAGTTCCAAAGGCTACAACT | CCTCGGAGACTGGTAATGGC | 269 |
| Col2a1(II) | NM_001113515.2 | GCGACCGGGAGCATATAACT | GCCCTAATTTTCGGGCATCC | 290 |
| Actb | NM_007393.4 | TTGCTGACAGGATGCAGAAG | AAGGGTGTAAAACGCAGCTC | 236 |
| Alpl | NM_007431.3 | GCCTTACCAACTCTTTTGTGC | GGCTACATTGGTGTTGAGCTT | 116 |
| Gli1 | NM_010296.2 | TACCATGAGCCCTTCTTTAGGA | GCATCATTGAACCCCGAGTAG | 116 |
Primary cell cultures and lentiviral transduction.
Primary calvarial OB cultures were established from 4-week-old Osx:cre, Rptorob−/+, and Rptorob−/− mice and Rptorfl/fl mice (P4) by using serial collagenase digestion as previously described (26). Cells were cultured in α-modified Eagle medium (Sigma-Aldrich, St. Louis, MO) supplemented with 20% FCS, 2 mM l-glutamine, 1 mM sodium pyruvate, 15 mM HEPES buffer, penicillin (50 U/ml), and streptomycin (50 μg/ml) as the growth medium. OB cultures generated from raptorfl/fl mice (passages 2 and 3) were infected with a lentivirus harboring a tamoxifen-inducible self-deleting Cre recombinase (LEGO-CreERT2-iG2) in the presence of 4 μg of Polybrene/ml as previously described (72). Calvarial cells with stable lentiviral integration were purified using FACS on the basis of eGFP expression. After in vitro expansion, the cells were treated with 0.5 mM 4-hydroxytamoxifen (4-OHT; Sigma-Aldrich) or vehicle (0.05% ethanol) for 8 days. The 4-OHT-treated cultures are labeled “Rap−/−” to signify Rptor gene deletion, and the vehicle-treated cultures are labeled “flox” to signify intact floxed Rptor allelles. No 4-OHT or vehicle was included in cultures beyond the 8-day Cre induction period.
CFU-F assays.
For CFU-F assays, compact bone MSCs were isolated from the long bones of 4-week-old mice as previously described (73) and plated at concentrations of 1 × 105, 3 × 105, and 6 × 105 cells/well in duplicate in six-well plates. Plated cells were incubated for 9 days in a hypoxic in vitro cabinet (Coy Laboratory Products) at 37°C before staining for CFU-F colonies with 0.1% (wt/vol) toluidine blue in 1% paraformaldehyde as previously described (74). Aggregates of >50 cells were scored as CFU-F-derived colonies, and aggregates of >10 and <50 cells were scored as clusters.
Osteogenesis assays.
Primary calvarial osteoblast cultures were seeded into 24- or 6-well plates (9 × 103/cm2) in growth medium supplemented with β-glycerol phosphate (10 mM) and l-ascorbic acid 2-phosphate (100 μg/ml) (osteogenic induction medium). The medium was changed twice weekly for 21 days, and the mineral content was quantified by measuring the concentration of Ca2+ in an acid-solubilized matrix using an Arsenazo kit (Thermo Fisher Scientific, Inc., Middletown, VA), normalized to the number of cells per well using a Quant-iT PicoGreen double-stranded DNA (dsDNA) assay (Life Technologies). The mineralized matrix was stained with alizarin red S (2% [wt/vol]) for 1 h, rinsed with water, and allowed to air dry. For lentivirus-infected OB cultures, RNA was harvested using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA), and real-time PCR was performed as described above.
Western blotting and subcellular fractionation.
Protein lysates were prepared from vehicle- and tamoxifen-treated lentivirus-transduced cultures, and proteins (50 μg) were resolved by SDS-PAGE, transferred to a polyvinylidene difluoride nylon membrane, and Western blotted as previously described (69). For insulin and IGF-1 pulse experiments, the cells were incubated under serum-free conditions for 3 h and then pulsed with 100 nM insulin or IGF-1 (ProSpec-Tany TechnoGene, Ltd., Ness-Ziona, Israel) at 100 μg/ml for 10 min. Nuclear and cytoplasmic extracts were prepared using NE-PER extraction reagents according to the manufacturer's instructions (Thermo Fisher Scientific). Antibodies to the following (catalog numbers are in parentheses) were used for Western blotting: Raptor (2280), P-p70 S6K T389 (9206), p70 S6K (9202), P-S6 S240/S244 (2215), S6 (2317), P-STAT1 S727 (8826), STAT1 (14994), P-STAT3 S727 (9134), STAT3 (9139), Runx2 (8486), and histone H3 (4499) (Cell Signaling Technologies, Beverly, MA); osterix (ab22552; Abcam); HSP90α/β (sc-7947; Santa Cruz Biotechnology); and β-actin (A5441; Sigma-Aldrich). Serum osteocalcin levels were measured by using a commercial enzyme-linked immunosorbent assay (Alfa Aesar, Ward Hill, MA) according to the manufacturer's instructions.
Metabolic labeling and cell size assessment.
Metabolic labeling experiments were performed as previously described (75). In brief, vehicle- and tamoxifen-treated lentivirus-infected calvarial cultures were seeded into 6-well plates (2 × 105 cells/plate) and cultured for 15 h, a time frame in which there is no significant difference in the proliferative capacity of control versus Rap−/− cells (data not shown). Growth medium was replaced with Cys-Met-free Dulbecco modified Eagle medium (Thermo Fisher) supplemented with 10% dialyzed FCS for 1 h. AZD8055 (1 μM; Santa Cruz Biotechnology) was added after 30 min. [35S]methionine-cysteine (Perkin-Elmer, Waltham, MA) was then added (to a final concentration of 10 μCi/ml), and the cells were cultured for 1 h. [35S]methionine-cysteine incorporation was determined using scintillation counting and normalized to the total cell number. Analysis of the cell size was performed as described previously (76).
Statistical analysis.
All data are graphed as means ± the standard deviations (SD). Statistical analyses were performed using a one-way or two-way analysis of variance with Dunnett's post hoc test or an unpaired Student t test using Prism (GraphPad Software, Inc., La Jolla, CA). In all cases, a P value <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We gratefully acknowledge Vicki Wilczek for monitoring mouse breeding and genotyping and the assistance of Kate Pilkington and Rebecca Salmon with cell sorting (Detmold Family Imaging Facility, SA Pathology). We also thank T. Jack Martin (St Vincent's Institute) for reviewing the manuscript, Kristoffer Riecken (University Medical Centre, Hamburg, Germany) for the LeGO-iG2 vector, and Pierre Chambon (IGBMC, France) for the Cre-ERT2 expression vector.
This study was supported by grants awarded by the National Health and Medical Research Council of Australia (grant APP1030528 awarded to A.C.W.Z., S.G., and S.F.), a Mary Overton Early Career Fellowship (S.K.M.), and an Australian Postgraduate Award (M.P.M.).
REFERENCES
- 1.Kronenberg HM. 2003. Developmental regulation of the growth plate. Nature 423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 2.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. 2002. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108:17–29. doi: 10.1016/S0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 3.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. 1997. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89:755–764. doi: 10.1016/S0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 4.Lee KS, Kim HJ, Li QL, Chi XZ, Ueta C, Komori T, Wozney JM, Kim EG, Choi JY, Ryoo HM, Bae SC. 2000. Runx2 is a common target of transforming growth factor β1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol Cell Biol 20:8783–8792. doi: 10.1128/MCB.20.23.8783-8792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsubara T, Kida K, Yamaguchi A, Hata K, Ichida F, Meguro H, Aburatani H, Nishimura R, Yoneda T. 2008. BMP2 regulates Osterix through Msx2 and Runx2 during osteoblast differentiation. J Biol Chem 283:29119–29125. doi: 10.1074/jbc.M801774200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L, Einhorn T, Tabin CJ, Rosen V. 2006. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet 38:1424–1429. doi: 10.1038/ng1916. [DOI] [PubMed] [Google Scholar]
- 7.Day TF, Guo X, Garrett-Beal L, Yang Y. 2005. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell 8:739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 8.Rodda SJ, McMahon AP. 2006. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation, and maintenance of osteoblast progenitors. Development 133:3231–3244. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 9.Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, Chen D, Faugere MC, Aja S, Hussain MA, Bruning JC, Clemens TL. 2010. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 142:309–319. doi: 10.1016/j.cell.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bialek P, Kern B, Yang X, Schrock M, Sosic D, Hong N, Wu H, Yu K, Ornitz DM, Olson EN, Justice MJ, Karsenty G. 2004. A twist code determines the onset of osteoblast differentiation. Dev Cell 6:423–435. doi: 10.1016/S1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- 11.Yang S, Xu H, Yu S, Cao H, Fan J, Ge C, Fransceschi RT, Dong HH, Xiao G. 2011. Foxo1 mediates insulin-like growth factor 1 (IGF1)/insulin regulation of osteocalcin expression by antagonizing Runx2 in osteoblasts. J Biol Chem 286:19149–19158. doi: 10.1074/jbc.M110.197905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, Bruxvoort KJ, Zylstra CR, Liu J, Cichowski R, Faugere MC, Bouxsein ML, Wan C, Williams BO, Clemens TL. 2007. Lifelong accumulation of bone in mice lacking Pten in osteoblasts. Proc Natl Acad Sci U S A 104:2259–2264. doi: 10.1073/pnas.0604153104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, Sundararajan D, Chen WS, Crawford SE, Coleman KG, Hay N. 2003. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev 17:1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ulici V, Hoenselaar KD, Agoston H, McErlain DD, Umoh J, Chakrabarti S, Holdsworth DW, Beier F. 2009. The role of Akt1 in terminal stages of endochondral bone formation: angiogenesis and ossification. Bone 45:1133–1145. doi: 10.1016/j.bone.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Rokutanda S, Fujita T, Kanatani N, Yoshida CA, Komori H, Liu W, Mizuno A, Komori T. 2009. Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Dev Biol 328:78–93. doi: 10.1016/j.ydbio.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Laplante M, Sabatini DM. 2013. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci 126:1713–1719. doi: 10.1242/jcs.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. 2002. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110:163–175. doi: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 18.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. 2004. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 19.Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sengupta S, Peterson TR, Sabatini DM. 2010. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell 40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. 2012. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kenney JW, Moore CE, Wang X, Proud CG. 2014. Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv Biol Regul 55:15–27. doi: 10.1016/j.jbior.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 23.Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, Blenis J. 2011. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332:1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. 2011. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao X, Pan D. 2001. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev 15:1383–1392. doi: 10.1101/gad.901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fulzele K, DiGirolamo DJ, Liu Z, Xu J, Messina JL, Clemens TL. 2007. Disruption of the insulin-like growth factor type 1 receptor in osteoblasts enhances insulin signaling and action. J Biol Chem 282:25649–25658. doi: 10.1074/jbc.M700651200. [DOI] [PubMed] [Google Scholar]
- 27.Xian L, Wu X, Pang L, Lou M, Rosen CJ, Qiu T, Crane J, Frassica F, Zhang L, Rodriguez JP, Xiaofeng J, Shoshana Y, Shouhong X, Argiris E, Mei W, Xu C. 2012. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat Med 18:1095–1101. doi: 10.1038/nm.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Tu X, Esen E, Joeng KS, Lin C, Arbeit JM, Ruegg MA, Hall MN, Ma L, Long F. 2014. WNT7B promotes bone formation in part through mTORC1. PLoS Genet 10:e1004145. doi: 10.1371/journal.pgen.1004145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davey RA, Clarke MV, Sastra S, Skinner JP, Chiang C, Anderson PH, Zajac JD. 2012. Decreased body weight in young Osterix-Cre transgenic mice results in delayed cortical bone expansion and accrual. Transgenic Res 21:885–893. doi: 10.1007/s11248-011-9581-z. [DOI] [PubMed] [Google Scholar]
- 30.Bentzinger CF, Romanino K, Cloetta D, Lin S, Mascarenhas JB, Oliveri F, Xia J, Casanova E, Costa CF, Brink M, Zorzato F, Hall MN, Ruegg MA. 2008. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab 8:411–424. doi: 10.1016/j.cmet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. 2001. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silbermann M, Kadar T. 1977. Age-related changes in the cellular population of the growth plate of normal mouse. Acta Anat 97:459–468. doi: 10.1159/000144767. [DOI] [PubMed] [Google Scholar]
- 33.Long F, Ornitz DM. 2013. Development of the endochondral skeleton. Cold Spring Harb Perspect Biol 5:a008334. doi: 10.1101/cshperspect.a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koyama E, Young B, Nagayama M, Shibukawa Y, Enomoto-Iwamoto M, Iwamoto M, Maeda Y, Lanske B, Song B, Serra R, Pacifici M. 2007. Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development 134:2159–2169. doi: 10.1242/dev.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou X, Zhang Z, Feng JQ, Dusevich VM, Sinha K, Zhang H, Darnay BG, de Crombrugghe B. 2010. Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proc Natl Acad Sci U S A 107:12919–12924. doi: 10.1073/pnas.0912855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurki P, Ogata K, Tan EM. 1988. Monoclonal antibodies to proliferating cell nuclear antigen (PCNA)/cyclin as probes for proliferating cells by immunofluorescence microscopy and flow cytometry. J Immunol Methods 109:49–59. doi: 10.1016/0022-1759(88)90441-3. [DOI] [PubMed] [Google Scholar]
- 37.Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, Carmeliet G, Kronenberg HM. 2010. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell 19:329–344. doi: 10.1016/j.devcel.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kristof AS, Marks-Konczalik J, Billings E, Moss J. 2003. Stimulation of signal transducer and activator of transcription 1 (STAT1)-dependent gene transcription by lipopolysaccharide and interferon-gamma is regulated by mammalian target of rapamycin. J Biol Chem 278:33637–33644. doi: 10.1074/jbc.M301053200. [DOI] [PubMed] [Google Scholar]
- 39.Fielhaber JA, Han YS, Tan J, Xing S, Biggs CM, Joung KB, Kristof AS. 2009. Inactivation of mammalian target of rapamycin increases STAT1 nuclear content and transcriptional activity in α4- and protein phosphatase 2A-dependent fashion. J Biol Chem 284:24341–24353. doi: 10.1074/jbc.M109.033530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S, Koga T, Isobe M, Kern BE, Yokochi T, Chin YE, Karsenty G, Taniguchi T, Takayanagi H. 2003. Stat1 functions as a cytoplasmic attenuator of Runx2 in the transcriptional program of osteoblast differentiation. Genes Dev 17:1979–1991. doi: 10.1101/gad.1119303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Itoh S, Udagawa N, Takahashi N, Yoshitake F, Narita H, Ebisu S, Ishihara K. 2006. A critical role for interleukin-6 family-mediated Stat3 activation in osteoblast differentiation and bone formation. Bone 39:505–512. doi: 10.1016/j.bone.2006.02.074. [DOI] [PubMed] [Google Scholar]
- 42.Zhou H, Newnum AB, Martin JR, Li P, Nelson MT, Moh A, Fu XY, Yokota H, Li J. 2011. Osteoblast/osteocyte-specific inactivation of Stat3 decreases load-driven bone formation and accumulates reactive oxygen species. Bone 49:404–411. doi: 10.1016/j.bone.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 43.Yokogami K, Wakisaka S, Avruch J, Reeves SA. 2000. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol 10:47–50. doi: 10.1016/S0960-9822(99)00268-7. [DOI] [PubMed] [Google Scholar]
- 44.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, James D, Howard Z, Dudley P, Hughes G, Smith L, Maguire S, Hummersone M, Malagu K, Menear K, Jenkins R, Jacobsen M, Smith GC, Guichard S, Pass M. 2010. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 70:288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 45.Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. 1999. Drosophila S6 kinase: a regulator of cell size. Science 285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- 46.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. 2002. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev 16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. 1997. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89:747–754. doi: 10.1016/S0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 48.Karsenty G, Ducy P, Starbuck M, Priemel M, Shen J, Geoffroy V, Amling M. 1999. Cbfa1 as a regulator of osteoblast differentiation and function. Bone 25:107–108. doi: 10.1016/S8756-3282(99)00111-8. [DOI] [PubMed] [Google Scholar]
- 49.Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F. 2005. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development 132:49–60. [DOI] [PubMed] [Google Scholar]
- 50.Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC, Fonhoue BD, Caron A, Bronson R, Bouxsein ML, Mukherjee S, Lees JA. 2008. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc Natl Acad Sci U S A 105:11851–11856. doi: 10.1073/pnas.0805462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y, Strecker S, Wang L, Kronenberg MS, Wang W, Rowe DW, Maye P. 2013. Osterix-cre labeled progenitor cells contribute to the formation and maintenance of the bone marrow stroma. PLoS One 8:e71318. doi: 10.1371/journal.pone.0071318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen J, Shi Y, Regan J, Karuppaiah K, Ornitz DM, Long F. 2014. Osx-Cre targets multiple cell types besides osteoblast lineage in postnatal mice. PLoS One 9:e85161. doi: 10.1371/journal.pone.0085161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lai LP, Lilley BN, Sanes JR, McMahon AP. 2013. Lkb1/Stk11 regulation of mTOR signaling controls the transition of chondrocyte fates and suppresses skeletal tumor formation. Proc Natl Acad Sci U S A 110:19450–19455. doi: 10.1073/pnas.1309001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen J, Long F. 2014. mTORC1 signaling controls mammalian skeletal growth through stimulation of protein synthesis. Development (Cambridge, England) 141:2848–2854. doi: 10.1242/dev.108811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. 2004. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev 18:1533–1538. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cooper KL, Oh S, Sung Y, Dasari RR, Kirschner MW, Tabin CJ. 2013. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 495:375–378. doi: 10.1038/nature11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang B, Wang Y, Wang W, Chen J, Lai P, Liu Z, Yan B, Xu S, Zhang Z, Zeng C, Rong L, Liu B, Cai D, Jin D, Bai X. 2015. mTORC1 prevents preosteoblast differentiation through the Notch signaling pathway. PLoS Genet 11:e1005426. doi: 10.1371/journal.pgen.1005426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Riddle RC, Frey JL, Tomlinson RE, Ferron M, Li Y, Digirolamo DJ, Faugere MC, Hussain MA, Karsenty G, Clemens TL. 2014. Tsc2 is a molecular checkpoint controlling osteoblast development and glucose homeostasis. Mol Cell Biol 34:1850–1862. doi: 10.1128/MCB.00075-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhande R, Mitchell JJ, Wu J, Sun XJ. 2002. Molecular mechanism of insulin-induced degradation of insulin receptor substrate 1. Mol Cell Biol 22:1016–1026. doi: 10.1128/MCB.22.4.1016-1026.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu DM, Zhao L, Liu TT, Jiao PL, Zhao DD, Shih MS, Tao B, Sun LH, Zhao HY, Liu JM. 2016. Rictor/mTORC2 loss in osteoblasts impairs bone mass and strength. Bone 90:50–58. doi: 10.1016/j.bone.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 61.Chen J, Long F. 2015. mTORC1 signaling promotes osteoblast differentiation from preosteoblasts. PLoS One 10:e0130627. doi: 10.1371/journal.pone.0130627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saran U, Foti M, Dufour JF. 2015. Cellular and molecular effects of the mTOR inhibitor everolimus. Clin Sci 129:895–914. doi: 10.1042/CS20150149. [DOI] [PubMed] [Google Scholar]
- 63.Hymes LC, Warshaw BL. 2011. Five-year experience using sirolimus-based, calcineurin inhibitor-free immunosuppression in pediatric renal transplantation. Pediatr Transplant 15:437–441. doi: 10.1111/j.1399-3046.2011.01477.x. [DOI] [PubMed] [Google Scholar]
- 64.Kotulska K, Chmielewski D, Borkowska J, Jurkiewicz E, Kuczynski D, Kmiec T, Lojszczyk B, Dunin-Wasowicz D, Jozwiak S. 2013. Long-term effect of everolimus on epilepsy and growth in children under 3 years of age treated for subependymal giant cell astrocytoma associated with tuberous sclerosis complex. Eur J Paediatr Neurol 17:479–485. doi: 10.1016/j.ejpn.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 65.Hadji P, Coleman R, Gnant M. 2013. Bone effects of mammalian target of rapamycin (mTOR) inhibition with everolimus. Crit Rev Oncol Hematol 87:101–111. doi: 10.1016/j.critrevonc.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 66.Ovchinnikov D. 2009. Alcian blue/alizarin red staining of cartilage and bone in mouse. Cold Spring Harb Protoc 2009:pdb.prot5170. doi: 10.1101/pdb.prot5170. [DOI] [PubMed] [Google Scholar]
- 67.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vandyke K, Fitter S, Dewar AL, Hughes TP, Zannettino AC. 2010. Dysregulation of bone remodeling by imatinib mesylate. Blood 115:766–774. doi: 10.1182/blood-2009-08-237404. [DOI] [PubMed] [Google Scholar]
- 69.Fitter S, Dewar AL, Kostakis P, To LB, Hughes TP, Roberts MM, Lynch K, Vernon-Roberts B, Zannettino AC. 2008. Long-term imatinib therapy promotes bone formation in CML patients. Blood 111:2538–2547. doi: 10.1182/blood-2007-07-104281. [DOI] [PubMed] [Google Scholar]
- 70.Arthur A, Panagopoulos RA, Cooper L, Menicanin D, Parkinson IH, Codrington JD, Vandyke K, Zannettino AC, Koblar SA, Sims NA, Matsuo K, Gronthos S. 2013. EphB4 enhances the process of endochondral ossification and inhibits remodeling during bone fracture repair. J Bone Miner Res 28:926–935. doi: 10.1002/jbmr.1821. [DOI] [PubMed] [Google Scholar]
- 71.Short BJ, Brouard N, Simmons PJ. 2009. Prospective isolation of mesenchymal stem cells from mouse compact bone. Methods Mol Biol 482:259–268. doi: 10.1007/978-1-59745-060-7_16. [DOI] [PubMed] [Google Scholar]
- 72.Isenmann S, Arthur A, Zannettino AC, Turner JL, Shi S, Glackin CA, Gronthos S. 2009. TWIST family of basic helix-loop-helix transcription factors mediate human mesenchymal stem cell growth and commitment. Stem Cells 27:2457–2468. doi: 10.1002/stem.181. [DOI] [PubMed] [Google Scholar]
- 73.Martin SK, Fitter S, Dutta AK, Matthews MP, Walkley CR, Hall MN, Ruegg MA, Gronthos S, Zannettino AC. 2015. The differential roles of mTORC1 and mTORC2 in mesenchymal stem cell differentiation. Stem Cells 33:1359–1365. doi: 10.1002/stem.1931. [DOI] [PubMed] [Google Scholar]
- 74.Gronthos S, Zannettino AC, Hay SJ, Shi S, Graves SE, Kortesidis A, Simmons PJ. 2003. Molecular and cellular characterisation of highly purified stromal stem cells derived from human bone marrow. J Cell Sci 116:1827–1835. doi: 10.1242/jcs.00369. [DOI] [PubMed] [Google Scholar]
- 75.Huo Y, Iadevaia V, Yao Z, Kelly I, Cosulich S, Guichard S, Foster LJ, Proud CG. 2012. Stable isotope-labeling analysis of the impact of inhibition of the mammalian target of rapamycin on protein synthesis. Biochem J 444:141–151. doi: 10.1042/BJ20112107. [DOI] [PubMed] [Google Scholar]
- 76.Xie J, Mikolajek H, Pigott CR, Hooper KJ, Mellows T, Moore CE, Mohammed H, Werner JM, Thomas GJ, Proud CG. 2015. Molecular mechanism for the control of eukaryotic elongation factor 2 kinase by pH: role in cancer cell survival. Mol Cell Biol 35:1805–1824. doi: 10.1128/MCB.00012-15. [DOI] [PMC free article] [PubMed] [Google Scholar]