

ABSTRACT
The promising results seen in studies of secondary bile acids in experimental colitis suggest that they may represent an attractive and safe class of drugs for the treatment of inflammatory bowel diseases (IBD). However, the exact mechanism by which bile acid therapy confers protection from colitogenesis is currently unknown. Since the gut microbiota plays a crucial role in the pathogenesis of IBD, and exogenous bile acid administration may affect the community structure of the microbiota, we examined the impact of the secondary bile acid ursodeoxycholic acid (UDCA) and its taurine or glycine conjugates on the fecal microbial community structure during experimental colitis. Daily oral administration of UDCA, tauroursodeoxycholic acid (TUDCA), or glycoursodeoxycholic acid (GUDCA) equally lowered the severity of dextran sodium sulfate-induced colitis in mice, as evidenced by reduced body weight loss, colonic shortening, and expression of inflammatory cytokines. Illumina sequencing demonstrated that bile acid therapy during colitis did not restore fecal bacterial richness and diversity. However, bile acid therapy normalized the colitis-associated increased ratio of Firmicutes to Bacteroidetes. Interestingly, administration of bile acids prevented the loss of Clostridium cluster XIVa and increased the abundance of Akkermansia muciniphila, bacterial species known to be particularly decreased in IBD patients. We conclude that UDCA, which is an FDA-approved drug for cholestatic liver disorders, could be an attractive treatment option to reduce dysbiosis and ameliorate inflammation in human IBD.
IMPORTANCE Secondary bile acids are emerging as attractive candidates for the treatment of inflammatory bowel disease. Although bile acids may affect the intestinal microbial community structure, which significantly contributes to the course of these inflammatory disorders, the impact of bile acid therapy on the fecal microbiota during colitis has not yet been considered. Here, we studied the alterations in the fecal microbial abundance in colitic mice following the administration of secondary bile acids. Our results show that secondary bile acids reduce the severity of colitis and ameliorate colitis-associated fecal dysbiosis at the phylum level. This study indicates that secondary bile acids might act as a safe and effective drug for inflammatory bowel disease.
KEYWORDS: bile acids, colitis, dysbiosis
INTRODUCTION
Inflammatory bowel diseases (IBD) are chronic inflammatory disorders of the gastrointestinal tract characterized by intestinal dysbiosis. Restricted bacterial diversity and underrepresentation of anti-inflammatory microorganisms, such as Clostridium cluster XIVa and Akkermansia muciniphila, represent typical dysbiotic features in IBD (1–4). Since the intestinal microbial community performs a wide range of bile acid modifications, including deconjugation, dehydroxylation, oxidation, and epimerization (5), shifts in the composition of the gut microbiota are associated with perturbations of the fecal bile acid profile (6, 7). Of particular interest, Duboc and colleagues demonstrated that the conversion of primary bile acids (synthesized in the liver from cholesterol) to secondary bile acids (generated by bacterial modifications) is impaired in IBD patients (7). Because secondary bile acids exhibit immunomodulatory functions (7–10), increasing secondary bile acid levels in the intestinal lumen could be an efficient therapeutic approach for IBD. In line with this hypothesis, the administration of the secondary hydrophilic bile acid ursodeoxycholic acid (UDCA) ameliorates experimental colitis, but the exact mechanism protecting from colitogenesis is not fully understood (11).
When administered orally, unconjugated UDCA is rapidly conjugated with glycine in humans, and to a lesser extent with taurine, on its first pass through the liver (12, 13). Based on the observation that fecal bile acid hydrophobicity correlates with the severity of colitis (14), it is reasonable to assume that conjugates of UDCA, which are more hydrophilic than unconjugated UDCA, might be more favorable therapeutic agents for intestinal inflammation. In this regard, we and others have shown that tauroursodeoxycholic acid (TUDCA) alleviates dextran sodium sulfate (DSS)-induced colitis in mice (15, 16). The potential beneficial effect of glycoursodeoxycholic acid (GUDCA) in colitis, however, has not been addressed so far, and studies comparing the therapeutic effectiveness of these different bile acid species are lacking.
While the composition of the luminal bile acid pool is controlled by intestinal bacteria, it is well established that bile acids, in turn, also shape the gut microbiota. Bile acids restrict bacterial proliferation and overgrowth directly by causing membrane damage, which is positively correlated with bile acid hydrophobicity (17–19). Thus, the bactericidal activity of bile acids decreases with increasing numbers of hydroxyl groups and by conjugation of the bile acid side chain with taurine or glycine (19). In addition to their role as antimicrobial agents, bile acids also stimulate the growth of selected bacterial species (5). Similarly, these properties are determined both by the hydroxylation pattern and the conjugation status of the bile acid steroid nucleus. For example, increased intestinal levels of bile acids carrying a hydroxyl group at position C-7 of the steroid core favor the growth of 7α-dehydroxylating bacteria, such as Clostridium cluster XIVa members (20, 21). Furthermore, the amino acids in conjugated bile acids act as microbial substrates for distinct bacterial groups; glycine is metabolized by Clostridium species (22, 23), while taurine is a source of sulfite from which Bilophila wadsworthia derives energy for its growth (24, 25). Interestingly, a diet high in saturated fat promotes taurine-associated conjugation of hepatic bile acids, resulting in the outgrowth of B. wadsworthia and exacerbation of colitis (26).
Considering that the gut microbial architecture and metabolism contribute to the course of IBD (27, 28), we compared the therapeutic effectiveness of UDCA and its taurine or glycine conjugates in DSS-induced colitis in mice and investigated their impact on the fecal microbial community.
RESULTS
Oral administration of UDCA or its taurine- or glycine-conjugated species is equally protective in acute DSS-induced colitis.
To compare the therapeutic effects of UDCA and its taurine or glycine derivatives on DSS-induced colitis, C57BL6/J mice were challenged with 4% DSS for 7 days and treated daily with UDCA, TUDCA, or GUDCA by oral gavage. Bile acid therapy reduced the rate of body weight loss, with no differences in efficacy between the three bile acid treatments (Fig. 1A). At day 10 after the initiation of colitis, body weight loss was significantly higher in placebo-treated mice than in bile acid-treated mice (P = 0.027, 0.021, and 0.021 for treatment with UDCA, TUDCA, and GUDCA, respectively). Accordingly, the clinical disease activity score, colonic shortening, and colonic concentrations of chemokine (C-X-C motif) ligand 1 (CXCL1), granulocyte colony-stimulating factor (G-CSF), and interleukin-6 (IL-6), which have been reported to show enhanced expression in the acute phase of DSS-induced colitis (29, 30), were all attenuated following bile acid treatment (Fig. 1B to D). At the systemic level, lower levels of CXCL1 and G-CSF, but not IL-6, were detected in the serum of bile acid-treated mice than in that of the placebo-treated group (Fig. 1E). Together, these data demonstrate that UDCA and its taurine or glycine conjugates decrease the severity of DSS-induced colitis with similar effectiveness.
FIG 1.

Oral administration of UDCA, TUDCA, and GUDCA improves clinical parameters and inflammatory markers in acute DSS-induced colitis. C57BL/6J mice received 4% DSS in the drinking water for 7 days, followed by normal water for 3 days. Control mice received water alone. From the start of DSS administration, mice were treated with UDCA, TUDCA, or GUDCA (500 mg/kg/day) by oral gavage. (A) Body weight changes during acute DSS-induced colitis. Body weights are represented as a percentage of their initial body weight at day 0. Results of a Mann-Whitney U test on day 10 are shown. (B) Clinical disease activity score. Results of a Mann-Whitney U test on day 9 are shown. (C) Colon lengths were assessed upon euthanasia on day 10. (D and E) Cytokine levels of CXCL1, G-CSF, and IL-6 in colonic tissue (D) and serum (E) collected on day 10. Data are represented as the mean ± SEM (n = 8 in each group). *, P < 0.05; **, P < 0.01. C, control; P, placebo; T, TUDCA; U, UDCA; G, GUDCA.
Bile acid supplementation to mice challenged with DSS prevents colitis-associated dysbiosis at the phylum level.
Because bile acids have been recognized as modulators of the intestinal microbiota (21, 26), which is believed to play a critical role in colitis (1, 28), we questioned whether the administration of UDCA or its conjugated derivatives prevented dysbiosis during experimental colitis. Fecal samples were collected 2 days after removing DSS from the drinking water (day 9), when colitis was fully established, and microbiota profiles were determined by 16S rRNA Illumina MiSeq sequencing. Four out of 40 mice were excluded from microbiota analysis because of insufficient amounts of feces available for DNA extraction. As expected, administration of DSS resulted in a significant reduction in species richness (measured by the number of operational taxonomic units; P = 0.002; Fig. 2A) and microbial diversity (Shannon index, which takes into account both species abundance and evenness; P = 0.002; Fig. 2B). In addition, the total fecal bacterial load had dropped significantly to 22% of the values seen in non-DSS control mice (P = 0.002; Fig. 2C). Compared to the placebo-treated group, microbial richness and diversity were slightly declined when colitic mice were treated with UDCA (P = 0.086 and 0.015, respectively), but no changes could be observed after the administration of TUDCA or GUDCA. Furthermore, daily administration of UDCA or its taurine or glycine conjugates during colitis did not affect the decrease in fecal bacterial load (Fig. 2A to C).
FIG 2.

Oral administration of UDCA, TUDCA, and GUDCA during DSS-induced colitis prevents colitis-associated dysbiosis at the phylum level. Fecal samples were collected on day 9 of colitis, and microbiota profiles were characterized by 16S rRNA Illumina MiSeq sequencing. (A to C) Estimation of species richness (i.e., total number of operational taxonomic units) (A), species diversity (i.e., Shannon index) (B), and bacterial load in the fecal microbiota (C). Bacterial load was calculated as 2ΔCT/[total DNA concentration]. (D) Ratio of the percentage of 16S rRNA gene sequences belonging to Firmicutes and Bacteroidetes. (E) Composition of the fecal microbial community at the phylum level. Data are represented as the mean ± SEM (n ≥ 6 in each group). *, P < 0.05; **, P < 0.01. C, control; P, placebo; T, TUDCA; U, UDCA; G, GUDCA.
We next analyzed the fecal microbial composition at the major taxonomic hierarchy levels to determine if particular bacterial phyla were altered following bile acid therapy during active disease. Taxonomic assignment of the sequence reads revealed two dominant phyla, Bacteroidetes and Firmicutes, accounting for 58.31% and 31.05% of the fecal communities of non-DSS control mice, respectively. Other phyla present with an average relative abundance ranging from 4.50% to 0.09% were Proteobacteria, Deferribacteria, “Candidatus Saccharibacteria,” and Actinobacteria. Administration of DSS significantly altered the structure of the fecal microbiome. At day 9 of colitis, the Firmicutes/Bacteroidetes ratio increased in mice that were challenged with DSS (Fig. 2D). More specifically, bacteria of the Bacteroidetes phylum were underrepresented in fecal samples of placebo-treated mice, while the relative abundance of Firmicutes bacteria tended to increase (Fig. 2E). Interestingly, oral administration of TUDCA or UDCA normalized the Firmicutes/Bacteroidetes ratio, while GUDCA treatment showed a slight but nonsignificant tendency to reduce this ratio (P = 0.12, Fig. 2D). Compared to placebo-treated mice, the fecal microbial community of bile acid-treated mice with colitis showed a lower abundance of Firmicutes and an increased abundance of Bacteroidetes (Fig. 2E). These results show that bile acid therapy in DSS-induced colitis neither mitigates nor aggravates altered microbial richness and population diversity but corrects fecal microbiota dysbiosis at the phylum level.
Bile acid supplementation to mice challenged with DSS alters the fecal microbiota at lower taxonomic levels.
To examine which bacterial populations accounted for the changes at the phylum level, we investigated the relative abundance of bacteria at lower taxonomic levels. The abundance of unclassified Bacteroidetes was reduced upon DSS administration, whereas no changes were noted for Bacteroidia levels (Fig. 3A). Interestingly, bile acid supplementation did not elicit an effect on unclassified Bacteroidetes but was associated with a significant increase in the abundance of Bacteroidia compared with placebo-treated mice (Fig. 3A). Examination at the family level indicated that upon DSS challenge, the relative proportions of Bacteroidaceae and Porphyromonadaceae increased (Fig. 3B and C), while Prevotellaceae family members nearly completely disappeared in fecal samples of placebo-treated mice (0.03% versus 6.64% in non-DSS control mice; Fig. 3D). Interestingly, the relative abundance of Prevotellaceae increased to 2.24% and 2.61% when mice were treated with UDCA and TUDCA, respectively (Fig. 3D). Moreover, Bacteroidaceae tended to further increase upon bile acid supplementation, especially when mice were treated with UDCA (Fig. 3B). Within Firmicutes, Erysipelotrichia expanded in the fecal samples of mice that were challenged with DSS, although bile acid therapy did not abrogate nor amplify this increase (Fig. 3A). However, the relative abundance of Clostridia remained unchanged in the placebo-treated group but declined in mice that were administered UDCA or derivatives (Fig. 3A). Comparisons at the genus level further demonstrated that DSS-induced colitis was associated with the outgrowth of Clostridium clusters XI and XIVb and with a depletion of Clostridium cluster XIVa (Fig. 3E; data not shown). Compared to the placebo-treated group, colitic mice that were treated with UDCA or conjugates exhibited increased numbers of Clostridium cluster XIVa bacteria (Fig. 3E). Clostridium species belonging to the clusters XI and XIVb, however, were not altered upon bile acid treatment (data not shown). Interestingly, sequences that were aligned to the Verrucomicrobia phylum were completely absent in the fecal communities of non-DSS control mice. Within this phylum, however, one genus (Akkermansia) expanded in the fecal samples of mice that were challenged with DSS. Akkermansia accounted for 0.047% of the detectable bacteria in the fecal samples of placebo-treated mice, and the relative abundance further increased upon treatment with UDCA, TUDCA, and GUDCA (0.145%, 0.114%, and 0.159%, respectively; Fig. 3F). Given the importance of these species in IBD (3, 4), the bile acid-induced enrichment of A. muciniphila was confirmed by quantitative real-time PCR (qRT-PCR) analysis (Fig. 3G).
FIG 3.

Oral administration of UDCA, TUDCA, and GUDCA during DSS-induced colitis alters the fecal microbiota at lower taxonomic levels. Fecal samples were collected on day 9 of colitis and microbiota profiles were characterized by 16S rRNA Illumina MiSeq sequencing. (A) Composition of the fecal microbial community at the class level. (B to F) Percentage of 16S rRNA gene sequences belonging to Bacteroidaceae (B), Porphyromonadaceae (C), Prevotellaceae (D), Clostridium cluster XIVa (E), and Akkermansia (F). (G) qRT-PCR results for A. muciniphila. Copy numbers were normalized to the 16S rRNA gene copy number in each sample. Data are represented as the mean ± SEM (n ≥ 6 in each group). *, P < 0.05; **, P < 0.01; ***, P < 0.001. C, control; P, placebo; T, TUDCA; U, UDCA; G, GUDCA.
Oral administration of UDCA, TUDCA, and GUDCA results in a similar fecal bile acid pool.
To compare the extent of biotransformation of orally administered UDCA, TUDCA, and GUDCA, we performed ultrahigh-performance liquid chromatography with high-resolution mass spectrometry (UHPLC-HRMS) analysis on fecal samples collected at day 4 of colitis. This time point was chosen because changes in the bile acid composition have been suggested to reach steady state already within 4 days of bile acid administration (31). Due to insufficient quantities of feces, two mice were excluded from UHPLC-HRMS analysis. UDCA concentrations were significantly higher in fecal samples of mice administered UDCA or its taurine or glycine conjugates than in those of placebo-treated mice (Fig. 4A). Concomitant with these changes, we observed increased fecal levels of TUDCA and lithocholic acid (LCA) in bile acid-treated mice (Fig. 4B and C). Of note, these changes occurred irrespective of whether animals were treated with UDCA, TUDCA, or GUDCA and without significant differences between the three bile acid treatments. In contrast, administration of GUDCA caused a substantial increase in fecal GUDCA levels, while no elevation was observed in animals that were administered UDCA or TUDCA (Fig. 4D). This finding is consistent with the fact that bile acids are predominantly conjugated with taurine (>97%), instead of glycine (<0.1%), in mice (32). Together, these data indicate that orally administered UDCA, TUDCA, and GUDCA are extensively metabolized in vivo, resulting in a similar fecal bile acid composition.
FIG 4.
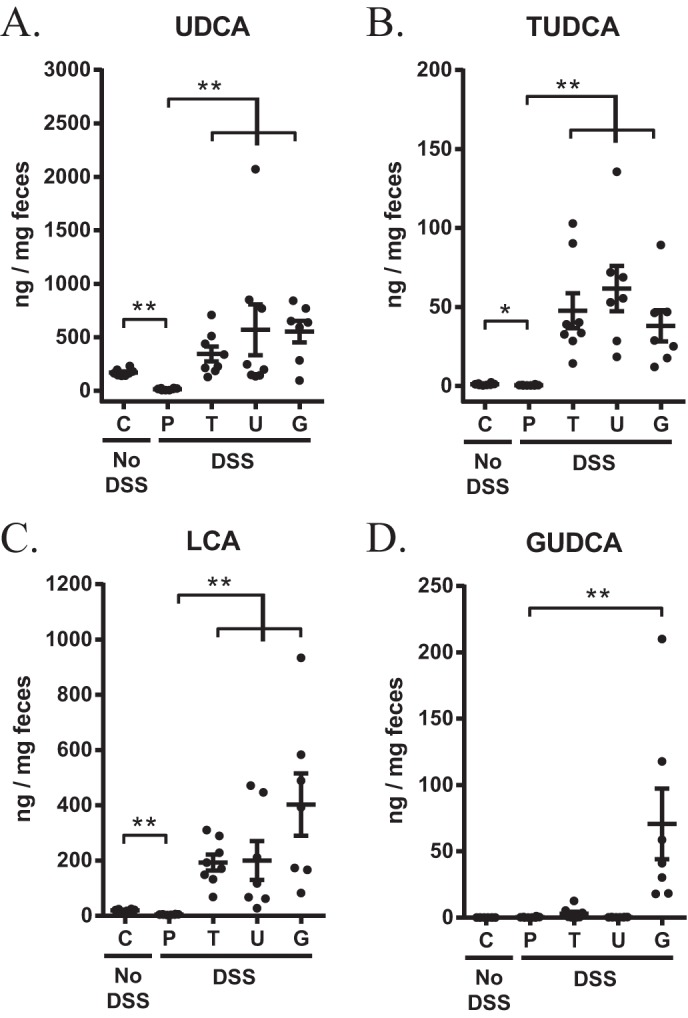
Orally administered UDCA, TUDCA, and GUDCA undergo extensive biotransformation. Fecal samples were collected at day 4 of colitis, and bile acids were quantified using UHPLC-HRMS. (A) UDCA, (B) TUDCA, (C) LCA, and (D) GUDCA. Data are represented as the mean ± SEM (n ≥ 7 in each group). *, P < 0.05; **, P < 0.01. C, control; P, placebo; T, TUDCA; U, UDCA; G, GUDCA.
DISCUSSION
Previous studies have reported the therapeutic potential of the hydrophilic bile acids UDCA and TUDCA in experimental colitis (11, 15, 16), albeit without comparing their respective effectiveness. In the present study, we showed that daily administration of UDCA and its taurine- and glycine-coupled conjugates equally attenuated body weight loss, disease activity, and colonic shortening caused by DSS. Moreover, oral bile acid therapy reduced proinflammatory cytokine concentrations in the colon and serum to a similar extent.
Bile acids are important regulators of the intestinal microbiota (21), which plays a crucial role in the pathogenesis of IBD (27). In order to examine the role of UDCA and its taurine- or glycine-conjugated species in the regulation of the intestinal microbiota during colitis, we orally administered UDCA, TUDCA, or GUDCA to mice that were challenged with DSS and determined fecal microbiota profiles by 16S rRNA Illumina MiSeq sequencing. Consistent with previous studies, DSS-induced colitis was associated with distinct alterations in the population structure of the gut microbiota (33, 34). These changes were generally characterized by a reduced overall microbial diversity and a decrease in species richness and bacterial load, which are also main hallmarks of dysbiosis in IBD patients (35, 36). We showed that oral bile acid administration did not prevent the DSS-induced changes in microbial richness, diversity, and bacterial load. Interestingly, species diversity decreased even more when colitic mice were treated with UDCA. However, although reduced richness and diversity of the gut microbiota have been associated with human disease (37–39), it seems unlikely that this effect is also clinically relevant; the clinical outcome in UDCA-treated mice was similar to that of mice that were treated with TUDCA or GUDCA.
At the phylum level, TUDCA, UDCA, and to a lesser extent, GUDCA, normalized the Firmicutes/Bacteroidetes ratio that was increased in placebo-treated mice with colitis. This ratio is often used as a proxy for microbial health status (40–45) and, more specifically, to describe the degree of dysbiosis in IBD (43–45). Of note, the phylum-level population shifts from Bacteroidetes to Firmicutes induced by DSS in our study resemble those observed in obese individuals and in animals on a high-fat diet and have been associated with low-grade intestinal and systemic inflammation in obesity (46, 47). In this context, fecal calprotectin and plasma C-reactive protein levels showed a positive correlation with bacteria belonging to the Firmicutes, whereas a negative correlation was found between C-reactive protein levels and specific groups within the Bacteroidetes (46). It is therefore likely that bile acid therapy counteracts the development of proinflammatory microbiota during colitis. This is speculative since it remains unknown if the changes seen in the Firmicutes/Bacteroidetes ratio following bile acid treatment are a cause, rather than a consequence, of the bile acid anti-inflammatory effect. However, bile acid therapy did not prevent the DSS-induced decrease in unclassified members of the phylum Bacteroidetes but increased the relative abundance of Bacteroidia, which were not affected by DSS. Thus, we can speculate that bile acid therapy directly interferes with an imbalanced microbial environment.
We demonstrated that Clostridium cluster XIVa species were significantly underrepresented upon DSS challenge, confirming previous observations in both human and experimental IBD (2, 48–50). However, oral administration of UDCA or its taurine or glycine conjugates was able to provoke an enrichment of these species compared with placebo-treated mice. It has been shown that selected members within the clostridial cluster XIVa possess 7α-dehydroxylation activity (20), which is involved in a multistep biochemical pathway that converts UDCA to LCA (51, 52). In our experiment, orally administered TUDCA and GUDCA were rapidly deconjugated to UDCA, so either bile acid treatment created a substrate-rich environment for these species. This may explain the bloom of Clostridium cluster XIVa that was observed in colitic mice that were treated with bile acids. Clostridium spp. belonging to cluster XIVa are important inducers of regulatory T cells in the colon (53). In addition, 80% of the butyrate-producing strains isolated from human fecal samples belong to the clostridial cluster XIVa (54). Butyrate is a short-chain fatty acid with distinctive anti-inflammatory properties that has already proven its efficacy in Crohn's disease (55). However, butyrate-producing bacteria are depleted in the fecal microbiota of IBD patients (49, 50, 56). Thus, our observation that UDCA or its taurine or glycine conjugates increased the abundance of Clostridium cluster XIVa during colonic inflammation is of particular interest and may suggest an immunomodulatory role of these bile acids.
Another finding of this study was the overrepresentation of Bacteroidaceae, Prevotellaceae, and Akkermansia in fecal samples of bile acid-treated mice following DSS exposure. These results might be related to the stimulatory effect of bile acids on mucin secretion as a defense mechanism to protect the gastrointestinal epithelium against potential bile acid toxicity (57–59). Bacterial species belonging to the genera Bacteroides, Prevotella, and Akkermansia produce one or more enzymes required for mucin degradation (60), which is enhanced during the acute phase of DSS-induced colitis (34). Therefore, it is reasonable to assume that these bacteria can grow better in a mucin-rich environment. A. muciniphila is a commensal bacterium residing in the mucus layer of the intestinal tract and has been shown to be reduced in IBD patients (3, 4). Although conflicting results were obtained in studies assessing the role of Akkermansia in colonic inflammation, these species are thought to play a key role in the regulation of gut barrier function and mucosal immune responses toward the commensal microbiota (61, 62).
The molecular structure of a bile acid determines its metabolism, physicochemical properties, and biological effects (63). In the present study, we used three bile acids sharing the same steroidal hydroxylation pattern but differing in their amino acid conjugation pattern. Neither bile acid species tested proved to be more or less efficacious than the other in reducing colonic inflammation. Likewise, administration of UDCA induced changes in the bacterial community similar to those with its taurine- or glycine-conjugated species. These observations can be explained by the rapid in vivo biotransformation of orally administered bile acids by the liver and by the intestinal microbiota. With the exception of fecal GUDCA concentrations, which were only increased following GUDCA therapy, there were no differences in fecal concentrations of UDCA, TUDCA, or LCA between mice that were administered UDCA or its conjugates. This is in contrast to data from previous studies in patients with primary biliary cirrhosis (12) and rats (31) showing that, compared with UDCA, orally administered TUDCA undergoes reduced 7-dehydroxylation to LCA. It is conceivable that interspecies differences in intestinal microbiota account for these discrepancies. For example, deconjugation of TUDCA or GUDCA is a prerequisite for further 7-dehydroxylation and is catalyzed by bile salt hydrolases (31). Because lactobacilli, which express bile salt hydrolases, are more abundant in the mouse gut microbiota than in the human gut microbiota (64), it is likely that biotransformation of these conjugated bile acids occurs to a larger extent in mice.
In summary, we report that UDCA and its taurine- or glycine-conjugated species ameliorate colonic inflammation in mice without differing in therapeutic effectiveness and reduce DSS-induced fecal dysbiosis at the phylum level, irrespective of the bile acid conjugation status. As we demonstrated no advantage of using either the taurine or glycine conjugate of UDCA, we suggest that UDCA could be a safe and readily available treatment option for IBD. This conclusion is further supported by the current therapeutic use of UDCA in cholestatic patients (65) and by its preventive effects on IBD-associated colorectal carcinogenesis (66–68).
MATERIALS AND METHODS
Animals.
Male 8-week-old C57Bl/6J mice were obtained from Harlan Laboratories (Horst, The Netherlands) and maintained under standard laboratory conditions with ad libitum access to food (mice maintenance chow, Carfil Labofood; Pavan Service, Belgium) and water. Prior to the experiment, mice were cohoused to homogenize gut microbiota between experimental groups. After a 1-week acclimatization, mice were assigned to the treatment groups based on body weights. In order to avoid bacterial cross-contamination between groups, mice of different treatment groups were housed in separate cages. The study was approved by the institutional review board of the Faculty of Medicine and Health Science of Ghent University (ECD 2014-25).
Bile acid treatment.
Mice were divided into five groups (n = 8 in each group). Three of them received bile acid treatment: UDCA (Tokyo Chemical Industry Co. Ltd., Toshima-Ku, Tokyo, Japan), TUDCA (Calbiochem, Darmstadt, Germany), or GUDCA (Sigma-Aldrich, Diegem, Belgium). Bile acids were dissolved in phosphate-buffered saline (PBS) or Labrafil M1944 (Gattefossé, Saint-Priest, France) and administered daily by oral gavage (500 mg/kg of body weight/day). Treatments started at day 0 of DSS exposure. A non-DSS control group and DSS control group (referred to as placebo-treated group) received the vehicle (PBS or Labrafil) alone.
Induction and assessment of colitis.
Acute colitis was established by adding 4% (wt/vol) DSS (molecular weight, 36,000 to 50,000; MP Biomedicals, Illkirch, France) to the drinking water for 7 days, followed by normal water for 3 days. A non-DSS control group received normal drinking water throughout the experiment. Body weight and disease activity were recorded daily. A disease activity index (DAI) was calculated as the combined score of body weight loss (0, none; 1, 0 to 10%; 2, 10 to 20%; 3, >20%), stool consistency (0, normal droppings; 1, loose droppings; 2, diarrhea), and fecal blood loss (0, none; 1, hemoccult positive; 2, gross bleeding). Occult blood was detected using the ColoScreen hemoccult kit (Helena Laboratories, Inc., Beaumont, TX, USA). Ten days after initiation of the experiment, mice were anesthetized and blood was collected from the retro-orbital sinus. The mice were then sacrificed by cervical dislocation, and the colons were removed and their lengths were measured. Segments of distal colon were cut, rinsed with PBS, and frozen in liquid nitrogen. The blood was centrifuged (10,000 rpm for 10 min at 4°C) and serum was collected. All samples were stored at −80°C until further processing.
Luminex.
Colonic tissues were homogenized in PBS containing protease and phosphatase inhibitors, and total protein concentration was measured using the Bradford method (Bio-Rad, Nazareth, Belgium). Protein levels of chemokine (C-X-C motif) ligand 1 (CXCL1), granulocyte colony-stimulating factor (G-CSF), and interleukin-6 (IL-6) were determined in colon homogenates and serum using the Bio-Plex Pro mouse cytokine group I multiplex kit (Bio-Rad), according to the manufacturer's instructions. Measurements were performed with the Bio-Plex MagPix multiplex reader, and data were analyzed using the Bio-Plex Manager 6.1 software (Bio-Rad).
DNA extraction from fecal samples.
Fresh fecal pellets were collected at day 9 of colitis and immediately stored at −80°C. Total DNA was extracted from the fecal samples using the QIAamp DNA stool minikit (Qiagen Benelux, Venlo, The Netherlands). First, 180 to 220 mg of stool was resuspended in 1.4 ml of buffer ASL. Then, 0.5 g of 0.1-mm-diameter Zirconia beads (BioSpec Products, Bartlesville, OK) and 4 glass beads (BioSpec Products) were added, and samples were homogenized by vortexing. The suspension was then heated at 95°C for 15 min, according to the manufacturer's instructions.
Illumina sequencing.
The V1 to V2 region of the 16S rRNA gene was amplified as previously described (69). Briefly, in a first 20-cycle PCR, the 16S rRNA gene target was enriched using the well-documented 27F and 338R primers (70, 71), as previously specified (72). This reaction mixture was used as the template in a second 15-cycle PCR with primers comprising sequences complementary to the Illumina-specific adaptors to the 5′ ends (69). The second reaction mixture was then used as the template in a third 10-cycle PCR with primers designed to integrate both the sequence of the Illumina-specific multiplexing sequencing primers and the index primers. Libraries prepared by pooling equimolar ratios of amplicons were finally sequenced on a MiSeq (Illumina, Hayward, CA, USA). Afterwards, reads were annotated as described by Verstraelen et al. (73).
Illumina data analysis.
Data analysis was performed as previously described (73). After resampling to the minimum sequencing depth using the phyloseq package (74) from the R program (75), a total of 8,911 reads were obtained. Rarefaction curves were generated using the vegan package from R (76). All phylotypes were assigned a taxonomic affiliation based on the naive Bayesian classification (RDP Classifier) (77) with a threshold of 80%. The relative abundances of all phylotypes were then compared between different experimental groups.
qRT-PCR.
Total fecal DNA was diluted 1:2 in water, and 3 μl was used in qRT-PCR with SYBR green (SensiMix SYBR no-ROX kit; Bioline Reagents, United Kingdom) and 250 nM each primer (BioLegio, Nijmegen, The Netherlands). Primer sequences used for amplification of A. muciniphila were 5′-CAGCACGTGAAGGTGGGGAC-3′ and 5′-CCTTGCGGTTGGCTTCAGAT-3′ (78). A two-step program was performed on the LightCycler 480 (Roche). The cycling conditions were 95°C for 10 min, 45 cycles of 95°C for 10 s, and 60°C for 1 min. The amount of A. muciniphila 16S rRNA gene in each sample was normalized to the total amount of bacterial 16S rRNA gene. For the quantification of total 16S rRNA gene copies, fecal DNA was diluted 1:10 in water, and the universal bacterial 16S rRNA gene primers PRBA338f (5′-ACTCCTACGGGAGGCAGCAG-3′) and PRUN518r (5′-ATTACCGCGGCTGCTGG-3′) were used (79). Total bacterial load was calculated using the formula 2ΔCT/[total DNA concentration].
Bile acid quantification. (i) Sample preparation.
Fresh fecal pellets were collected at day 4 of colitis and immediately stored at −80°C. Before bile acids were extracted from fecal samples, 20 μl of an internal standard solution (TUDCA-d5 [Santa-Cruz Biotechnology, Heidelberg, Germany] at 25 ng/μl in methanol [VWR International, Merck Millipore, Darmstadt, Germany]) was added to 25 mg of feces. The extraction protocol started with the addition of 5 ml of ice-cold acetonitrile (VWR International) containing 5% ammonium hydroxide (Merck Millipore). The solution was homogenized with an Ultra-Turrax homogenizer, thoroughly mixed by vortexing for 1 min, and then placed in an ultrasonic bath for 30 min. The resultant mixture was centrifuged at 9,000 × g for 10 min, and the supernatant was collected. The extraction procedure was repeated once more, and the combined supernatants were subsequently evaporated under nitrogen at 40°C. Each dry extract was then resuspended in 200 μl of a 40:60 mixture of solvent A (7.5 mM ammonium acetate [Merck Millipore] in ultrapure water [pH 4.0]) and solvent B (5% acetonitrile in methanol), centrifuged at 9,000 × g for 10 min, and the supernatant was collected. To compensate for matrix effects, the standard addition method was applied for bile acid quantification (80). Briefly, the supernatant was divided into two equal aliquots and transferred to liquid chromatography-mass spectrometry vials. One aliquot was spiked with 20 μl of the 40:60 mixture of solvents A and B. The other aliquot was spiked with 20 μl of a bile acid solution (a 40:60 mixture of solvents A and B, supplemented with 16.5 ng/μl lithocholic acid [LCA; Sigma-Aldrich], 89.4 ng/μl UDCA [Sigma-Aldrich], 0.75 ng/μl TUDCA [Calbiochem], and 0.75 ng/μl GUDCA [Sigma-Aldrich]). A 10-μl aliquot of each sample was injected into the ultrahigh-performance liquid chromatography with high-resolution mass spectrometry (UHPLC-HRMS) system.
(ii) UHPLC-HRMS analysis.
Chromatographic separation of bile acids was carried out on an Accela UHPLC system (Thermo Fisher Scientific, San Jose, CA, USA), with an Acquity UPLC HSS C18 column (1.8 μm, 50 mm by 2.1 mm; Waters). The binary solvent system consisting of two solvents, A and B, was set at a constant flow rate of 300 μl/min at 35°C. For elution, a gradient profile was applied with the following proportions (vol/vol) of solvent A: 0 to 1.0 min at 40%, 1.0 to 6.0 min from 40% to 1%, 6.0 to 8.0 min at 1%, 8.0 to 8.1 min from 1% to 40%, followed by 3.9 min of reequilibration.
HRMS analysis was performed on an Exactive standalone benchtop mass spectrometer (Thermo Fisher Scientific), equipped with a heated electrospray ionization source (HESI-II), operating in the negative ionization mode. Ionization source working parameters were optimized and were set to a sheath, auxiliary, and sweep gas of 40, 5, and 1 arbitrary units (au), respectively; heater and capillary temperature of 120°C and 375°C; and tube lens, skimmer, capillary, and spray voltages of 123 V, 22 V, 43.5 V, and 4 kV (+/−), respectively. A scan range of m/z 300 to 550 was selected, and the resolution was set at 100,000 full width at half maximum (FWHM) at 1 Hz (1 scan per s). The automatic gain control (AGC) target was set at high dynamic range (3 × E6 ions), and the maximum injection time was 100 ms.
(iii) Data processing.
HRMS data processing was performed with Xcalibur 3.0 (Thermo Fisher Scientific). The concentration of a selected bile acid was calculated using the following formula (80):
with Cunk being the unknown concentration of the bile acid in the original fecal sample, CSA being the spiked concentration of the bile acid in the fecal sample after standard addition, ARunk being the area ratio of the bile acid in the original sample, and ARSA being the area ratio of the bile acid in the fecal sample after standard addition.
Statistical analysis.
Statistical analysis was performed using SPSS Statistics version 22.0 (IBM SPSS, Chicago, IL, USA) and GraphPad Prism version 4 (GraphPad, CA, USA). All data are expressed as mean ± standard error of the mean (SEM). Data were tested for normality using the Kolmogorov-Smirnov test. Statistical significant differences between groups were assessed using the unpaired Student's t test for normally distributed data, applying the Welch's correction in case of unequal variances, or the Mann-Whitney U test for nonnormally distributed data. Two-tailed probabilities were calculated, and P values of less than 0.05 were considered statistically significant.
Accession number(s).
The sequence data were submitted to the European Nucleotide Archive (ENA) with accession numbers LT700235 to LT702885 and can be accessed at http://www.ebi.ac.uk/ena/data/view/LT700235-LT702885.
ACKNOWLEDGMENTS
We are grateful to Falk Pharmaceuticals, who kindly provided TUDCA for research purposes. We also thank Hilde Devlies, Griet Driesschaert, and Petra Van Wassenhove for providing technical assistance.
We declare no conflicts of interest.
This work was supported by research grants from the Research Foundation Flanders (FWO; grants 11J9915N and 1298213N), a concerted action grant (GOA) from the Special Research Fund (BOF; grant GOA 2012/01G00812) of Ghent University, and a grant from the Belgian foundation for Crohn's disease and ulcerative colitis patients (CCV vzw, research grant 2014).
REFERENCES
- 1.Nagao-Kitamoto H, Kitamoto S, Kuffa P, Kamada N. 2016. Pathogenic role of the gut microbiota in gastrointestinal diseases. Intest Res 14:127–138. doi: 10.5217/ir.2016.14.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andoh A, Imaeda H, Aomatsu T, Inatomi O, Bamba S, Sasaki M, Saito Y, Tsujikawa T, Fujiyama Y. 2011. Comparison of the fecal microbiota profiles between ulcerative colitis and Crohn's disease using terminal restriction fragment length polymorphism analysis. J Gastroenterol 46:479–486. doi: 10.1007/s00535-010-0368-4. [DOI] [PubMed] [Google Scholar]
- 3.Png CW, Lindén SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin THJ. 2010. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol 105:2420–2428. doi: 10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- 4.Vigsnæs LK, Brynskov J, Steenholdt C, Wilcks A, Licht TR. 2012. Gram-negative bacteria account for main differences between faecal microbiota from patients with ulcerative colitis and healthy controls. Benef Microbes 3:287–297. doi: 10.3920/BM2012.0018. [DOI] [PubMed] [Google Scholar]
- 5.Wahlström A, Sayin SI, Marschall H-U, Bäckhed F. 2016. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 24:41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, Takei H, Muto A, Nittono H, Ridlon JM, White MB, Noble NA, Monteith P, Fuchs M, Thacker LR, Sikaroodi M, Bajaj JS. 2013. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol 58:949–955. doi: 10.1016/j.jhep.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert M-A, Quervain E, Thomas G, Barbu V, Humbert L, Despras G, Bridonneau C, Dumetz F, Grill J-P, Masliah J, Beaugerie L, Cosnes J, Chazouillères O, Poupon R, Wolf C, Mallet J-M, Langella P, Trugnan G, Sokol H, Seksik P. 2013. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62:531–539. doi: 10.1136/gutjnl-2012-302578. [DOI] [PubMed] [Google Scholar]
- 8.Calmus Y, Weill B, Ozier Y, Chéreau C, Houssin D, Poupon R. 1992. Immunosuppressive properties of chenodeoxycholic and ursodeoxycholic acids in the mouse. Gastroenterology 103:617–621. doi: 10.1016/0016-5085(92)90855-S. [DOI] [PubMed] [Google Scholar]
- 9.Greve JW, Gouma DJ, Buurman WA. 1989. Bile acids inhibit endotoxin-induced release of tumor necrosis factor by monocytes: an in vitro study. Hepatology 10:454–458. doi: 10.1002/hep.1840100409. [DOI] [PubMed] [Google Scholar]
- 10.Yoneno K, Hisamatsu T, Shimamura K, Kamada N, Ichikawa R, Kitazume MT, Mori M, Uo M, Namikawa Y, Matsuoka K, Sato T, Koganei K, Sugita A, Kanai T, Hibi T. 2013. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn's disease. Immunology 139:19–29. doi: 10.1111/imm.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martínez-Moya P, Romero-Calvo I, Requena P, Hernández-Chirlaque C, Aranda CJ, González R, Zarzuelo A, Suárez MD, Martínez-Augustin O, Marín JJG, de Medina FS. 2013. Dose-dependent antiinflammatory effect of ursodeoxycholic acid in experimental colitis. Int Immunopharmacol 15:372–380. doi: 10.1016/j.intimp.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 12.Invernizzi P, Setchell KD, Crosignani A, Battezzati PM, Larghi A, O'Connell NC, Podda M. 1999. Differences in the metabolism and disposition of ursodeoxycholic acid and of its taurine-conjugated species in patients with primary biliary cirrhosis. Hepatology 29:320–327. doi: 10.1002/hep.510290220. [DOI] [PubMed] [Google Scholar]
- 13.Rudolph G, Kloeters-Plachky P, Sauer P, Stiehl A. 2002. Intestinal absorption and biliary secretion of ursodeoxycholic acid and its taurine conjugate. Eur J Clin Invest 32:575–580. doi: 10.1046/j.1365-2362.2002.01030.x. [DOI] [PubMed] [Google Scholar]
- 14.Stenman LK, Holma R, Forsgård R, Gylling H, Korpela R. 2013. Higher fecal bile acid hydrophobicity is associated with exacerbation of dextran sodium sulfate colitis in mice. J Nutr 143:1691–1697. doi: 10.3945/jn.113.180810. [DOI] [PubMed] [Google Scholar]
- 15.Cao SS, Zimmermann EM, Chuang B-M, Song B, Nwokoye A, Wilkinson JE, Eaton KA, Kaufman RJ. 2013. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 144:989–1000.e6. doi: 10.1053/j.gastro.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laukens D, Devisscher L, Van den Bossche L, Hindryckx P, Vandenbroucke RE, Vandewynckel Y-P, Cuvelier C, Brinkman BM, Libert C, Vandenabeele P, De Vos M. 2014. Tauroursodeoxycholic acid inhibits experimental colitis by preventing early intestinal epithelial cell death. Lab Invest 94:1419–1430. doi: 10.1038/labinvest.2014.117. [DOI] [PubMed] [Google Scholar]
- 17.Kurdi P, Kawanishi K, Mizutani K, Yokota A. 2006. Mechanism of growth of inhibition by free bile acids in lactobacilli and bifidobacteria. J Bacteriol 188:1979–1986. doi: 10.1128/JB.188.5.1979-1986.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taranto MP, Perez-Martinez G, Font de Valdez G. 2006. Effect of bile acid on the cell membrane functionality of lactic acid bacteria for oral administration. Res Microbiol 157:720–725. doi: 10.1016/j.resmic.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Sonomoto K, Yokota A. 2011. Lactic acid bacteria and bifidobacteria: current progress in advanced research. Caister Academic Press, Haverhill, United Kingdom. [Google Scholar]
- 20.Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. 2013. Cirrhosis, bile acids and gut microbiota. Gut Microbes 4:382–387. doi: 10.4161/gmic.25723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Islam KBMS, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. 2011. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 141:1773–1781. doi: 10.1053/j.gastro.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 22.Dürre P, Andreesen JR. 1983. Purine and glycine metabolism by purinolytic clostridia. J Bacteriol 154:192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lebertz H, Andreesen JR. 1988. Glycine fermentation by Clostridium histolyticum. Arch Microbiol 150:11–14. doi: 10.1007/BF00409710. [DOI] [Google Scholar]
- 24.Laue H, Denger K, Cook AM. 1997. Taurine reduction in anaerobic respiration of Bilophila wadsworthia RZATAU. Appl Environ Microbiol 63:2016–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laue H, Smits THM, Schumacher UK, Claros MC, Hartemink R, Cook AM. 2006. Identification of Bilophila wadsworthia by specific PCR which targets the taurine:pyruvate aminotransferase gene. FEMS Microbiol Lett 261:74–79. doi: 10.1111/j.1574-6968.2006.00335.x. [DOI] [PubMed] [Google Scholar]
- 26.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. 2012. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487:104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wlodarska M, Kostic AD, Xavier RJ. 2015. An integrative view of microbiome-host interactions in inflammatory bowel diseases. Cell Host Microbe 17:577–591. doi: 10.1016/j.chom.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brinkman BM, Becker A, Ayiseh RB, Hildebrand F, Raes J, Huys G, Vandenabeele P. 2013. Gut microbiota affects sensitivity to acute DSS-induced colitis independently of host genotype. Inflamm Bowel Dis 19:2560–2567. doi: 10.1097/MIB.0b013e3182a8759a. [DOI] [PubMed] [Google Scholar]
- 29.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen T-E, Conklin LS, Centola M, Li X. 2009. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis 15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peter MR, Jerkic M, Sotov V, Douda DN, Ardelean DS, Ghamami N, Lakschevitz F, Khan MA, Robertson SJ, Glogauer M, Philpott DJ, Palaniyar N, Letarte M. 2014. Impaired resolution of inflammation in the endoglin heterozygous mouse model of chronic colitis. Mediators Inflamm 2014:767185. doi: 10.1155/2014/767185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodrigues CM, Kren BT, Steer CJ, Setchell KD. 1995. Tauroursodeoxycholate increases rat liver ursodeoxycholate levels and limits lithocholate formation better than ursodeoxycholate. Gastroenterology 109:564–572. doi: 10.1016/0016-5085(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 32.Alnouti Y, Csanaky IL, Klaassen CD. 2008. Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 873:209–217. doi: 10.1016/j.jchromb.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagalingam NA, Kao JY, Young VB. 2011. Microbial ecology of the murine gut associated with the development of dextran sodium sulfate-induced colitis. Inflamm Bowel Dis 17:917–926. doi: 10.1002/ibd.21462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwab C, Berry D, Rauch I, Rennisch I, Ramesmayer J, Hainzl E, Heider S, Decker T, Kenner L, Müller M, Strobl B, Wagner M, Schleper C, Loy A, Urich T. 2014. Longitudinal study of murine microbiota activity and interactions with the host during acute inflammation and recovery. ISME J 8:1101–1114. doi: 10.1038/ismej.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, Roca J, Dore J. 2006. Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut 55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wills ES, Jonkers DMAE, Savelkoul PH, Masclee AA, Pierik MJ, Penders J. 2014. Fecal microbial composition of ulcerative colitis and Crohn's disease patients in remission and subsequent exacerbation. PLoS One 9:e90981. doi: 10.1371/journal.pone.0090981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerritsen J, Smidt H, Rijkers GT, de Vos WM. 2011. Intestinal microbiota in human health and disease: the impact of probiotics. Genes Nutr 6:209–240. doi: 10.1007/s12263-011-0229-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto J-M, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jørgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clément K, Doré J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, de Vos WM, Zucker J-D, Raes J, Hansen T, MetaHIT Consortium, Bork P, Wang J, Ehrlich SD, Pedersen O. 2013. Richness of human gut microbiome correlates with metabolic markers. Nature 500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 39.Vandeputte D, Falony G, Vieira-Silva S, Tito RY, Joossens M, Raes J. 2016. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 65:57–62. doi: 10.1136/gutjnl-2015-309618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. 2006. Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 41.Hong SN, Rhee P-L. 2014. Unraveling the ties between irritable bowel syndrome and intestinal microbiota. World J Gastroenterol 20:2470–2481. doi: 10.3748/wjg.v20.i10.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wen L, Ley RE, Volchkov PV, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV. 2008. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brinkman BM, Hildebrand F, Kubica M, Goosens D, Del Favero J, Declercq W, Raes J, Vandenabeele P. 2011. Caspase deficiency alters the murine gut microbiome. Cell Death Dis 2:e220. doi: 10.1038/cddis.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ritchie LE, Sturino JM, Carroll RJ, Rooney LW, Azcarate-Peril MA, Turner ND. 2015. Polyphenol-rich sorghum brans alter colon microbiota and impact species diversity and species richness after multiple bouts of dextran sodium sulfate-induced colitis. FEMS Microbiol Ecol 91:fiv008. doi: 10.1093/femsec/fiv008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabeerdoss J, Jayakanthan P, Pugazhendhi S, Ramakrishna BS. 2015. Alterations of mucosal microbiota in the colon of patients with inflammatory bowel disease revealed by real time polymerase chain reaction amplification of 16S ribosomal ribonucleic acid. Indian J Med Res 142:23–32. doi: 10.4103/0971-5916.162091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verdam FJ, Fuentes S, de Jonge C, Zoetendal EG, Erbil R, Greve JW, Buurman WA, de Vos WM, Rensen SS. 2013. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity (Silver Spring) 21:E607–E615. doi: 10.1002/oby.20466. [DOI] [PubMed] [Google Scholar]
- 47.Lau E, Marques C, Pestana D, Santoalha M, Carvalho D, Freitas P, Calhau C. 2016. The role of I-FABP as a biomarker of intestinal barrier dysfunction driven by gut microbiota changes in obesity. Nutr Metab (Lond) 13:31. doi: 10.1186/s12986-016-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Fazio L, Cavazza E, Spisni E, Strillacci A, Centanni M, Candela M, Praticò C, Campieri M, Ricci C, Valerii MC. 2014. Longitudinal analysis of inflammation and microbiota dynamics in a model of mild chronic dextran sulfate sodium-induced colitis in mice. World J Gastroenterol 20:2051–2061. doi: 10.3748/wjg.v20.i8.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang W, Chen L, Zhou R, Wang X, Song L, Huang S, Wang G, Xia B. 2014. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J Clin Microbiol 52:398–406. doi: 10.1128/JCM.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, Ballet V, Claes K, Van Immerseel F, Verbeke K, Ferrante M, Verhaegen J, Rutgeerts P, Vermeire S. 2014. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63:1275–1283. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 51.Hirano S, Masuda N. 1982. Enhancement of the 7 alpha-dehydroxylase activity of a Gram-positive intestinal anaerobe by Bacteroides and its significance in the 7-dehydroxylation of ursodeoxycholic acid. J Lipid Res 23:1152–1158. [PubMed] [Google Scholar]
- 52.Lepercq P, Gérard P, Béguet F, Grill J-P, Relano P, Cayuela C, Juste C. 2004. Isolates from normal human intestinal flora but not lactic acid bacteria exhibit 7a- and 7b-hydroxysteroid dehydrogenase activities. Microb Ecol Health Dis 16:195–201. doi: 10.1080/08910600410033393. [DOI] [Google Scholar]
- 53.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K. 2011. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, Henderson C, Flint HJ. 2000. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl Environ Microbiol 66:1654–1661. doi: 10.1128/AEM.66.4.1654-1661.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Di Sabatino A, Morera R, Ciccocioppo R, Cazzola P, Gotti S, Tinozzi FP, Tinozzi S, Corazza GR. 2005. Oral butyrate for mildly to moderately active Crohn's disease. Aliment Pharmacol Ther 22:789–794. doi: 10.1111/j.1365-2036.2005.02639.x. [DOI] [PubMed] [Google Scholar]
- 56.Kumari R, Ahuja V, Paul J. 2013. Fluctuations in butyrate-producing bacteria in ulcerative colitis patients of North India. World J Gastroenterol 19:3404–3414. doi: 10.3748/wjg.v19.i22.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klinkspoor JH, Tytgat GN, Lee SP, Groen AK. 1996. Mechanism of bile salt-induced mucin secretion by cultured dog gallbladder epithelial cells. Biochem J 316:873–877. doi: 10.1042/bj3160873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shekels LL, Lyftogt CT, Ho SB. 1996. Bile acid-induced alterations of mucin production in differentiated human colon cancer cell lines. Int J Biochem Cell Biol 28:193–201. doi: 10.1016/1357-2725(95)00125-5. [DOI] [PubMed] [Google Scholar]
- 59.Klinkspoor JH, Mok KS, Van Klinken BJ, Tytgat GN, Lee SP, Groen AK. 1999. Mucin secretion by the human colon cell line LS174T is regulated by bile salts. Glycobiology 9:13–19. doi: 10.1093/glycob/9.1.13. [DOI] [PubMed] [Google Scholar]
- 60.Derrien M, van Passel MW, van de Bovenkamp JH, Schipper RG, de Vos WM, Dekker J. 2010. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 1:254–268. doi: 10.4161/gmic.1.4.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Derrien M, Van Baarlen P, Hooiveld G, Norin E, Müller M, de Vos WM. 2011. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin degrader Akkermansia muciniphila. Front Microbiol 2:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD. 2013. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A 110:9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ridgway N, McLeod R. 2015. Biochemistry of lipids, lipoproteins and membranes. Elsevier, Amsterdam, The Netherlands. [Google Scholar]
- 64.Nguyen TLA, Vieira-Silva S, Liston A, Raes J. 2015. How informative is the mouse for human gut microbiota research? Dis Model Mech 8:1–16. doi: 10.1242/dmm.017400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roma MG, Toledo FD, Boaglio AC, Basiglio CL, Crocenzi FA, Sánchez Pozzi EJ. 2011. Ursodeoxycholic acid in cholestasis: linking action mechanisms to therapeutic applications. Clin Sci (Lond) 121:523–544. doi: 10.1042/CS20110184. [DOI] [PubMed] [Google Scholar]
- 66.Pardi DS, Loftus EV Jr, Kremers WK, Keach J, Lindor KD. 2003. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology 124:889–893. doi: 10.1053/gast.2003.50156. [DOI] [PubMed] [Google Scholar]
- 67.Kohno H, Suzuki R, Yasui Y, Miyamoto S, Wakabayashi K, Tanaka T. 2007. Ursodeoxycholic acid versus sulfasalazine in colitis-related colon carcinogenesis. Clin Cancer Res 13:2519–2525. [DOI] [PubMed] [Google Scholar]
- 68.Huang W-K, Hsu H-C, Liu J-R, Yang T-S, Chen J-S, Chang JW-C, Lin Y-C, Yu K-H, Kuo C-F, See L-C. 2016. The association of ursodeoxycholic acid use with colorectal cancer risk. Medicine (Baltimore) 95:e2980. doi: 10.1097/MD.0000000000002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Camarinha-Silva A, Jáuregui R, Chaves-Moreno D, Oxley APA, Schaumburg F, Becker K, Wos-Oxley ML, Pieper DH. 2014. Comparing the anterior nare bacterial community of two discrete human populations using Illumina amplicon sequencing. Environ Microbiol 16:2939–2952. doi: 10.1111/1462-2920.12362. [DOI] [PubMed] [Google Scholar]
- 70.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175. In Stackebrandt E, Goodfellow M (ed), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, New York, NY. [Google Scholar]
- 71.Etchebehere C, Tiedje J. 2005. Presence of two different active nirS nitrite reductase genes in a denitrifying Thauera sp. from a high-nitrate-removal-rate reactor. Appl Environ Microbiol 71:5642–5645. doi: 10.1128/AEM.71.9.5642-5645.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chaves-Moreno D, Plumeier I, Kahl S, Krismer B, Peschel A, Oxley APA, Jauregui R, Pieper DH. 2015. The microbial community structure of the cotton rat nose. Environ Microbiol Rep 7:929–935. doi: 10.1111/1758-2229.12334. [DOI] [PubMed] [Google Scholar]
- 73.Verstraelen H, Vilchez-Vargas R, Desimpel F, Jauregui R, Vankeirsbilck N, Weyers S, Verhelst R, De Sutter P, Pieper DH, Van De Wiele T. 2016. Characterisation of the human uterine microbiome in non-pregnant women through deep sequencing of the V1–2 region of the 16S rRNA gene. PeerJ 4:e1602. doi: 10.7717/peerj.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.R Core Team. 2012. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 76.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H. 2016. vegan: community ecology package. https://cran.r-project.org/web/packages/vegan/index.html.
- 77.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schneeberger M, Everard A, Gómez-Valadés AG, Matamoros S, Ramírez S, Delzenne NM, Gomis R, Claret M, Cani PD. 2015. Akkermansia muciniphila inversely correlates with the onset of inflammation, altered adipose tissue metabolism and metabolic disorders during obesity in mice. Sci Rep 5:16643. doi: 10.1038/srep16643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ovreås L, Forney L, Daae FL, Torsvik V. 1997. Distribution of bacterioplankton in meromictic Lake Sælenvannet, as determined by denaturing gradient gel electrophoresis of PCR-amplified gene fragments coding for 16S rRNA. Appl Environ Microbiol 63:3367–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cai X, Liu Y, Zhou X, Navaneethan U, Shen B, Guo B. 2012. An LC-ESI-MS method for the quantitative analysis of bile acids composition in fecal materials. Biomed Chromatogr 26:101–108. doi: 10.1002/bmc.1633. [DOI] [PubMed] [Google Scholar]