

Abstract
Synthetic biology and metabolic engineering experiments frequently require the fine-tuning of gene expression to balance and optimize protein levels of regulators or metabolic enzymes. A key concept of synthetic biology is the development of modular parts that can be used in different contexts. Here, we have applied a computational multifactor design approach to generate de novo synthetic core promoters and 5′ untranslated regions (UTRs) for yeast cells. In contrast to upstream cis-regulatory modules (CRMs), core promoters are typically not subject to specific regulation, making them ideal engineering targets for gene expression fine-tuning. 112 synthetic core promoter sequences were designed on the basis of the sequence/function relationship of natural core promoters, nucleosome occupancy and the presence of short motifs. The synthetic core promoters were fused to the Pichia pastoris AOX1 CRM, and the resulting activity spanned more than a 200-fold range (0.3% to 70.6% of the wild type AOX1 level). The top-ten synthetic core promoters with highest activity were fused to six additional CRMs (three in P. pastoris and three in Saccharomyces cerevisiae). Inducible CRM constructs showed significantly higher activity than constitutive CRMs, reaching up to 176% of natural core promoters. Comparing the activity of the same synthetic core promoters fused to different CRMs revealed high correlations only for CRMs within the same organism. These data suggest that modularity is maintained to some extent but only within the same organism. Due to the conserved role of eukaryotic core promoters, this rational design concept may be transferred to other organisms as a generic engineering tool.
Keywords: core promoter, promoter library, transcriptional fine-tuning, synthetic biology, yeast, computational design
Introduction
Metabolic pathways and genetic circuits are commonly introduced into microbes such as Saccharomyces cerevisiae or Escherichia coli to produce chemicals or to implement novel functions.1,2 Such experiments typically require the fine-tuning of gene expression to balance and optimize protein levels of metabolic enzymes or regulators. In prokaryotes, protein production can be controlled relatively easily using synthetic ribosomal binding sites.3 However, to fine-tune gene expression and protein levels in unicellular eukaryotes, transcription is the most targeted step4−7 and, to this end, various engineering tools have been developed.4,8−10 Most promoter engineering efforts in eukaryotes were focused on yeasts, since they are the most commonly used eukaryotic expression systems for complex multigene pathways.11−13S. cerevisiae has most commonly been used for metabolic engineering endeavors, but recently also alternative yeasts such as Pichia pastoris have been increasingly used.14,15 Yeast promoter libraries were designed either by random sequence modifications9,16 or by rational approaches8,17−19 with a focus on cis-regulatory modules (CRMs).20 CRM is a general term referring to regulatory DNA sequences, also named enhancers in higher eukaryotes, while in yeasts rather the terms upstream activating/repressing sequences (UAS/URS) are used.20,21 CRMs interact with particular transcription factors conferring specific activation/repression regulatory mechanisms.
CRMs alone are however nonfunctional, requiring a core (minimal) promoter sequence to recruit general transcription factors and RNA polymerase II for transcription initiation.4,22,23 Similarly, the core promoter alone results in basal to no expression at all, and requires a CRM for strong expression and specific regulation. Engineering the core promoter and 5′ untranslated region (UTR) has mainly an impact on transcription strength, translation initiation and most probably mRNA stability. In contrast, engineering CRMs affects transcription strength but also impacts regulation (i.e., constitutive or inducible). For instance, studies on the methanol inducible AOX1 (alcohol oxidase 1) promoter (PAOX1) in P. pastoris(8,24,25) showed that deletions or insertions of CRMs (more specifically in predicted transcription factor binding sites, TFBSs) resulted in promoter activity variations and also in regulatory differences. One example for altered regulation were derepressed PAOX1 variants.8 The wild type PAOX1 is tightly repressed on glucose, remaining repressed even when glucose is depleted and strictly requires methanol for induction. Depressed variants however start expression once glucose is depleted not requiring methanol induction.
In contrast to such mutations in CRMs, modifications of the core promoter sequence impacted only promoter strength, leaving induction/repression profiles unchanged.25 Additionally, studies on CRMs are typically limited to one promoter, i.e., its conclusions cannot be easily transferred to other promoters, even in the same organism. For instance, information gained from deletion studies of PAOX1(8,24,26) cannot be transferred to other methanol inducible promoters in P. pastoris due to the low sequence similarly between these coregulated promoters.5 In contrast, core promoter function is conserved even between related species.8,27
Hence, we hypothesized that de novo designed synthetic core promoters could be used as interchangeable parts between related organisms. Such universal “tuning knobs” could be used for regulating the strength of gene expression without interfering with specific regulation in a given organism for different promoters, or in different species. Since the designed promoters are artificial, they have lower probability of recombining with natural sequences in the genome which favors strain stability and also facilitates the expression cassette assembly. To design such promoters, we used a genome scale data set available for S. cerevisiae.28,29
S. cerevisiae is the most commonly used yeast for basic research on transcription regulation and synthetic promoter design.4,30,31 Recently, comprehensive studies have also addressed the sequence/function relationship of natural core promoters27,28,32−34 and 5′UTRs.34 Two genome-scale studies were performed in this yeast by measuring the expression of 859 natural promoters under different conditions29 and using this data set to deduce core promoter properties affecting expression.28 Also, nucleosome affinity in the core promoter was shown to be an effective modification target for designing core promoters.18 For the interspecies comparisons we selected S. cerevisiae and P. pastoris.
P. pastoris is, after E. coli, the most commonly used expression system for single proteins.35 The exceptionally strong and tightly methanol regulated PAOX1, has motivated several research studies on transcriptional regulation mechanisms (reviewed by36 and summarized in Supporting Information S1 alongside S. cerevisiae studies7−9,16,18,19,24−27,32−34,37−44). Recently, it has been reported that at least 15 promoters of genes involved in methanol utilization (MUT) are coregulated with PAOX1, some of which show higher expression.5 Hence, P. pastoris offers one of the largest sets of promoters that are coregulated and easily applicable strategies for regulating their strengths would be desirable.
In the present study, we designed generic synthetic core promoters for protein production fine-tuning in yeasts. Acknowledging the fact that manifold structural features contribute to the promoter strength, we have incorporated in our design several factors, which were derived from a S. cerevisiae core promoter data set (e.g., TATA box position and nucleosome affinity).28,29 Using this design approach, we have created a library of 112 synthetic core promoters and 5′UTRs that were validated with the P. pastoris PAOX1 CRM (PAOX1-R). Additionally, we tested the best performing synthetic core promoters with alternative CRMs of P. pastoris and S. cerevisiae promoters, demonstrating their applicability in different contexts.
Results
Computational Design of Synthetic Core Promoters
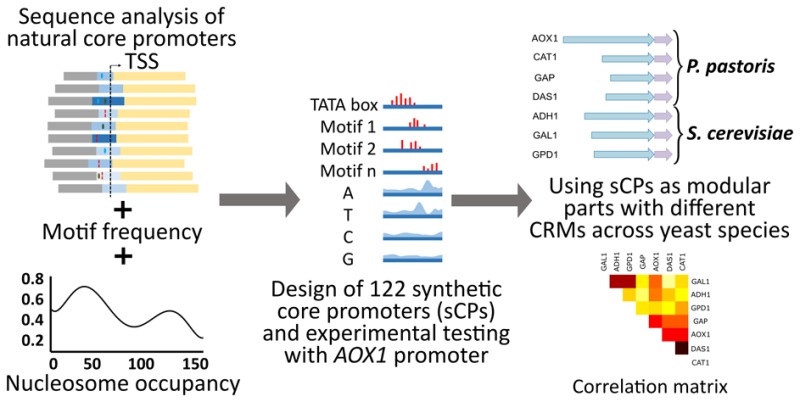
Several factors were simultaneously incorporated in the synthetic core promoter design: (i) nucleotide occurrence along the sequence of 140 strong natural S. cerevisiae core promoters (as reported by28), (ii) the presence and position of the TATA box, (iii) the position and number other motifs (other than TATA box, as defined by28) and (iv) nucleosome occupancy profiles.28,45 Using this approach, we have created a library of synthetic core promoters and 5′UTRs for generic yeast cells. The method adopted in this study is represented schematically in Figure 1 and described in detail in Supporting Information S2.
Figure 1.

Design strategy for synthetic core promoters. Three steps were followed: (A) Computation of (i) nucleotide probability distribution, (ii) TATA box position, (iii) position and frequency distribution of motifs and (iv) average nucleosome occupancy along the sequence of 140 S. cerevisiae natural strong core promoters28 aligned by the transcription start site. (B) Generation of 400 random sequences using information on nucleotide probability distribution. (C) Partitioning of sequences in four groups (dubbed P, T, M and A), to which TATA boxes and motifs were added, according to the group they belonged to (group P: without TATA box nor motifs; group T: with TATA box and without motifs; group M: with motifs and without TATA box; group A: with TATA box and motifs). Subsequently, the nucleosome occupancy profile of each of the generated sequences was compared to the average profile for the natural strong promoters. The generated sequences with higher similarity to the average natural nucleosome occupancy were selected to be tested in vivo.
The input sequences were taken from a library of S. cerevisiae natural core promoter sequences.28 We used the genome wide S. cerevisiae core promoter sequences data published by Lubliner et al.,28 in which 729 native S. cerevisiae promoters were segmented into four groups (low, medium, high and very high maximal expression). Subsequently, different structural features were examined such as nucleotide frequency, nucleosome occupancy and presence/number of short motifs (up to four nucleotides). Lubliner et al.(28) showed that some of these features are highly predictive of maximal promoter activity, namely the high A and T content and TATA-box like elements around the TSS. Also, it was demonstrated that there is a correlation between promoter strength and low nucleosome affinity.18
We reasoned that this data set (input sequences) could also be used in a reverse way to generate a model and create synthetic core promoters de novo. We started from the subset of 140 strong core promoters and the respective 5′UTRs. First, we have selected sequences of 150 bp (50 bp downstream and 100 bp upstream of the transcriptional start site (TSS)) for analysis. Then, to extract important sequence features, we have applied the following computational procedure (Figure 1 A):
-
(i)
Computation of the nucleotide probability distribution along the sequence, calculated with a 20 bp windows size and 10 bp windows step;
-
(ii)
Computation of the TATA box position distribution along the sequence;
-
(iii)
Computation of the position and frequency distribution of motifs along the sequence. Only the subset of motifs with highest effect (positive or negative) on the promoter strength were considered (defined by Lubliner et al.(28));
-
(iv)
Computation of the average nucleosome occupancy along the promoter sequence (using the software package described in ref (45)).
Using this information, we have designed 4 groups (named P, T, M, and A) of 28 sequences each for experimental screening (Figure 1, Figure 2 B and Supplementary Tables S2–S5). They differ in the presence or absence of a TATA box and/or selected motifs (group P: without TATA box nor motifs; group T: with TATA box and without motifs; group M: with motifs and without TATA box; group A: with TATA box and motifs). In this way, the synthetic core promoters were termed according to their group and to the respective measured activity, i.e., the 4 groups with 28 sequences each were termed “P#”, “T#”, “M#”, “A#”, where the letters stand for P: (nucleotide) probability, T: TATA box, M: motifs and A: all, respectively. They were ordered in increasing expression strength. The general properties of the designed sequences are available in Supplementary Tables S7–S10.
Figure 2.

Establishing the PAOX1-R screening system (A) and testing the 112 synthetic core promoters (B–F). Promoter activity was measured by fluorescence intensity of the reporter protein after cultivation in 96-well deep-well plates and under methanol induction for 48h. (A) Promoter activity mean and respective standard deviation of control constructs: (i) wild type PAOX1 (green), (ii) PAOX1-R fused to HHF2 core promoter, (iii) PAOX1-R without core promoter, (iv) AOX1 core promoter without CRM and (v) seven completely random sequences fused to PAOX1-R. (B) Overview of the groups of synthetic core promoters tested. Box plot of the minimum, first quartile, average, third quartile and maximum promoter activities for each of the four groups of synthetic core promoters (Groups P, M, T and A). (C–F) Landscape of mean promoter activity, and respective standard deviation, for each of the four groups of synthetic core promoters (Group P, T, M and A, respectively). The individual synthetic core promoter activity is presented in increasing activity order. The legend of panel C applies as well to panels D−F. Mean values and standard deviations shown in this figure were calculated from at least three independent cultivations in separate deep-well plates.
The sequences were computed in a 4-step procedure as follows:
Step 1: Generation of 400 random sequences using information on nucleotide probability distribution only (Figure 1 B). TATA boxes or any of the selected motifs were searched and replaced by a newly generated synthetic sequence. This procedure was repeated until no motif or TATA-box were found in the generated sequences. Start codons upstream of the protein codon region were also removed to avoid frame shift mutations or different N-termini of the reporter protein. Lastly, due to the known relevance of the nucleotides adjacent to the start codon,34 this region was replaced by the PAOX1 Kozak sequence (CGAAACG) in the generated sequences. These 400 sequences were partitioned in four groups of 100 sequences each.
Step 2: Addition of a TATA box to groups T and A. The TATA box positioning followed a Gaussian distribution model with mean and standard deviation computed on the natural strong core promoter sequences. One TATA box was inserted per core promoter sequence (Figure 1 C).
Step 3: Addition of motifs to groups M and A. The frequency and position of each motif in each sequence also followed a Gaussian distribution model inferred from the natural sequences, meaning that some motifs might be present more than once while others might be absent in a given sequence (Figure 1 C).
Step 4: Design space reduction. Twenty-eight synthetic sequences out of the 100 sequences of each group were selected for experimental screening based on the nucleosome occupancy.45 The 28 synthetic sequences with higher similarity to natural promoters concerning the predicted nucleosome average occupancy were selected for screening.
Before fusing these final 112 synthetic core promoters to the PAOX1-R (AOX1 promoter CRM), we aimed to validate the core promoter structure of this promoter.
Assessing Core Promoter-CRM Structure in the P. pastoris PAOX1 System
The natural (wild type) P. pastoris PAOX1 fused to an eGFP (enhanced green fluorescent protein) reporter gene was used as positive control in this study. eGFP has widely been used as reporter for promoter characterization studies in P. pastoris.5,8,25,26 All reporter protein fluorescence measurements of promoter variants were performed with a 96-well plate reader and are given relative to the wild type level normalized to 100% (shown in green the bar plots in Figure 2 A and C–F). The plate reader based fluorescence measurements were also validated by flow cytometry measurements yielding excellent reproducibility (r2 = 0.96, see Supplementary Figure S6).
A negative control variant was generated by deleting the P. pastoris PAOX1-R (−769 to −172 bp from start codon) to probe for its function. In a second negative control, the core promoter was deleted (−171 to −1 bp from start codon). In both control variants there was no detectable fluorescence thus the expression was completely disrupted (Figure 2 A). This confirms that the core promoter sequence with high affinity to RNA polymerase II was completely removed in the variant without core promoter. Likewise, the variant in which the CRM was removed showed no fluorescence, confirming that all the relevant regulatory protein binding sites were removed resulting in complete functionality loss.
To ascertain the principle of modularity in this system, we characterized a variant in which the AOX1 core promoter was replaced by another strong core promoter, of the HHF2 gene.46,47 The promoter activity level was identical to the natural PAOX1, showing that different core promoters can be used interchangeably (Figure 2 A).
Given the complete loss of functionality when the core promoter or CRM are removed, as well as the modularity verified in this system, the determined core promoter-CRM boundary was maintained in all subsequent core promoter replacements. Namely, the core promoter boundary was set to 10 bp upstream of the TATA box.
Establishing a Baseline Expression Level
Seven control variants were generated in which the P. pastoris AOX1 core promoter was replaced by completely random sequences (Figure 2 A R1–R7). The resulting expression levels measure the basal expression of the PAOX1-R given that there is enough spacing between the CRM and the protein coding sequence for RNA polymerase II to bind. We performed this experiment to test basic background transcription in our system. The average relative promoter activity of the seven control variants was 5.9% of the wild type promoter fluorescence (Figure 2 A). We have used this value as threshold to evaluate whether the synthetic core promoters are significantly different from random sequences. In this way, synthetic core promoters with an expression value significantly lower than 5.9% were considered nonfunctional. For this purpose, we have adopted the one-way analysis of variance (ANOVA) statistical test.
Synthetic Core Promoters under the Control of the P. pastoris PAOX1-R
The aforementioned 112 synthetic constructs were assessed by replacing the native P. pastoris AOX1 core promoter by each of the 112 synthetic sequences and measuring eGFP reporter gene fluorescence. The overall promoter activity landscape is shown for each group (P, T, M and A) in Figure 2 C–F, respectively. Seventy-eight percent of sequences showed a statistically significant (p-value of 0.05) higher activity than baseline expression and are thus considered as functional. Within the functional subset, reporter protein fluorescence levels ranged between 6.5% to 70.6% with mean 17.0% and standard deviation 11.5%. Additionally, it was observed that the mean activity levels in groups T and A, 18.7% and 19.3%, respectively, are roughly 2-fold higher than groups P and M, 9.2% and 9.1%, respectively. Furthermore, 16 out of the 25 nonfunctional core promoters do not have a TATA box. This is a strong indication that the TATA box is a key sequence element in the PAOX1 system (Figure 2 B).
Regarding the presence of motifs (group M and A), our data suggest that their presence does not significantly affect the expression level, given that the mean activity level is similar in groups with or without motifs (group P and T, respectively). However, we might speculate that the presence of motifs in association with other factors may explain the higher expression levels observed for promoters A28 and A27, given that both have motifs (Figure 2 F).
Focusing the analysis on the ten promoters with highest activity (orange in Figure 2 C–F) it is striking that the presence of a TATA box is a common feature, whereas the presence of motifs is not. The only exception might be the M28 promoter, which belongs to a TATA less group. M28 has, however, a TATA box like sequence in position −115 from the start codon.
Analysis of the Top-Ten Synthetic Core Promoter Sequences
The top ten synthetic core promoter sequences obtained in the screening with the PAOX1-R (T22, T23, T24, T25, T26, T27, T28, M28, A27 and A28) were scrutinized in detail. They were examined by (i) BLAST analysis against the P. pastoris genome to search for similarities to naturally occurring sequences, (ii) multiple sequence alignment to assess the presence of common motifs and (iii) nucleosome occupancy analysis to evaluate its importance and common patterns.
To search for fragments of natural sequences, a standard nucleotide BLAST searching procedure against the whole P. pastoris CBS 7435 genome was adopted and no significant matches were found. The detailed results are provided as Supplementary Table S11. The highest e-value (0.083) was obtained for A28, T27, T26 and M28 sequences BLAST. The A28 and M28 matches were in protein coding regions and in an inter gene sequence in the case of T26, thus making it unlikely to be characteristic regulatory sequences. In the case of T27, the match was in a possible promoter region in the P. pastoris genome (10 bp upstream of nucleolar protein coding sequence). The match position in the synthetic core promoter sequence was however further upstream, close to the PAOX1-R (−147 to −130 bp).
To perform the multiple sequence alignments we used the EMBL-EBI Clustal Omega tool.48 The resulting alignment (Figure 3 A) shows the conserved positions in seven or more sequences (shaded in blue in Figure 3 A). Some of the marked positions are isolated, possibly caused by the higher adenine and thymine content, characteristic of strong core promoter sequences.28 In addition, three different common motifs (with more than one consecutive position conserved) were identified. The first one is located close to the TATA box region (position 40 in Figure 3 A). However, two sequences had the respective TATA boxes positioned downstream from this region (T28 and T23), around position 70. This may influence the subsequent AT rich motif (position 74). The last conserved region is a thymine rich sequence (position 146), followed by an adenine rich sequence (not marked), which may be related to the TSS as suggested by Lubliner et al.(28)
Figure 3.

Analysis of the top ten synthetic core promoter sequences obtained from screenings with the PAOX1-R. (A) Multiple sequence alignment, using Clustal Omega,48 (top 10 synthetic promoters used as input, ranked in increasing order of promoter activity). The positions conserved in seven or more of sequences are marked with a blue shade. The AOX1 core promoter sequence is shown as an example of a strong natural core promoter to highlight the synthetic core promoter sequence diversity. The synthetic core promoters and AOX1 core promoter alignment was performed separately to highlight sequence features of the synthetic core promoters. (B) Nucleosome occupancy profile heatmap of the top ten synthetic core promoters when fused to PAOX1-R. The nucleosome occupancy was calculated with Kaplan et al. prediction package.45 The core promoter is limited to 150 bp form the protein start codon. The TATA box location is marked in blue. (C) 112 synthetic core promoter activity mean and standard as a function of the respective cumulative nucleosome affinity scores calculated using the Xi et al. software package.65
Lastly, we calculated the nucleosome occupancy for the 10 best synthetic core promoters (Figure 3 B) using the model developed by Kaplan et al.(45)Figure 3 C shows the sum of nucleosome affinity for all the synthetic promoters. The data in Figure 3 B unveil relatively low nucleosome occupancy in several synthetic core promoters (e.g., T28, A27 and T26) but without a clear pattern. There are however a few exceptions (T27 and T25) with relatively high nucleosome occupancy. To ascertain a possible correlation between promoter expression and nucleosome affinity, we calculated nucleosome affinity for all the synthetic promoters and compared it with the respective expression levels. It revealed no statistically significant correlation, with a correlation coefficient of 0.07 (Figure 3 C). This somewhat unexpected result might be explained by the diversity of synthetic sequences (discussed later).
The average position of the TATA box in the ten best promoters is position −120 (Figure 3 B) with variations of 20 base pair around the mean. There are some promoters with lower activity with TATA boxes considerably downstream of this interval. Yet it is not possible to draw a direct causal relationship between TATA box position and promoter strength since many other features differ between them.
Second Round Screening: Top-Ten Synthetic Core Promoters in Different Yeasts and CRMs
In the previous section, we validated the designed method and its capacity to create completely novel core promoters, demonstrating its functionality with the PAOX1-R. Yet, we aimed to use synthetic core promoters as general tools for fine-tuning expression. Thus, they should be functional when fused to CRMs of any promoters. Hence, the top ten synthetic core promoters obtained from fusion with the PAOX1-R (Figure 2 and summarized in Figure 4 B) were fused to six different CRMs (Figure 4 A), three from P. pastoris (PDAS1-R, PCAT1-R and PGAP-R: Figure 4 C to E, respectively), and the other three from S. cerevisiae (PScGAL1-R, PScGPD1-R and PScADH1-R: Figure 4 F to H, respectively). These additional CRMs were chosen so that we could benchmark the synthetic core promoters in different conditions, i.e., under the control of inducible (PDAS1-R, PCAT1-R and PScGAL1-R) and constitutive (PGAP-R, PScGPD1-R andPScADH1-R) CRMs. In all constructs, the synthetic core promoter was delimited to 10 bp upstream of the TATA box. Therefore, the core promoters have a different length depending on the location of the TATA box and on the CRM length.
Figure 4.

Testing modularity of the synthetic core promoters by fusing them to CRMs of different promoters in P. pastoris and S. cerevisiae. (A) Relative size and CRM-core promoter fusion location of the different CRMs (PAOX1-R, PCAT1-R, PGAP-R, PDAS1-R, PScADH1-R, PScGAL1-R and PScGPD1-R, respectively) tested. (B–H) Mean promoter activity (normalized reporter protein fluorescence), and respective standard deviation, of fusions of the ten synthetic core promoters with highest activity (with PAOX1-R, Figure 2) to different CRMs (blue arrows in panel A). CRMs of the following promoters were tested in two different yeasts: The PAOX1-R, PDAS1-R, PCAT1-R and PGAP-R were tested in P. pastoris (B–E), while PScGAL1-R, PScGPD1-R and PScADH1-R were tested in S. cerevisiae (F–H). Fluorescence measurements under optimal induction conditions are shown for the respective promoters: P. pastoris PAOX1-R, PCAT1-R and PDAS1-R were induced with methanol, S. cerevisiae PScGAL1-R was induced with Galactose. Constitutive P. pastoris and S. cerevisiae promoters PGAP-R, PScGPD1-R and PScADH1-R were cultivated on glucose containing media. All values represent single measurements of at least three independent cultivations in separate 96-well deep-well plates. In each case, the corresponding wild type promoter activity is represented in green. The order of the synthetic core promoters is kept the same (increasing promoter activity when fused to PAOX1-R), to facilitate interpretation. The data of the core promoter fusions to PAOX1-R is also shown dispersed in Figure 2 and summarized here in panel B.
A key result of these experiments is that the top-ten synthetic promoters show significantly higher expression when fused to CRMs of inducible promoters, irrespectively of the yeast and inducible mechanism, i.e., the tested CRMs are inducible by methanol (PCAT1-R, PDAS1-R, and PAOX1-R in P. pastoris) and galactose (PScGAL1-R in S. cerevisiae). The minimum relative promoter activity was 38% for PCAT1-R and PDAS1-R, 27% and 53% in PAOX1-R and PScGAL1-R, respectively. With all these CRMs, the strongest synthetic core promoter gave a higher relative expression than the PAOX1-R, namely 82%, 122% and 176% for PCAT1-R, PDAS1-R and PScGAL1-R, respectively, compared to 70% for the PAOX1-R. Notably, PDAS1-R and PScGAL1-R gave even a higher expression value than the respective natural wild type core promoters, 122% and 176%, respectively. It should be stressed that these synthetic core promoters seem to be independent of the regulatory mechanism, since they are functional under the control of CRMs that respond to different stimuli (methanol and galactose) and in different yeasts.
Fusions of the core promoters to CRMs of constitutive promoters show a limited functionality with the maximum relative promoter activity around 20% in PGAP-R, PScADH1-R and PScGPD1-R. All these CRMs have a TATA box in their natural sequence. In yeast there are mainly two types of promoters, TATA-positive and TATA-less promoters.31 Most of the available promoter studies were developed for the former group of promoters,31 thus we lack detailed understanding of critical sequence elements for transcription initiation in the TATA-less promoters. Hence, we have hypothesized that, although these promoters have a TATA box in their sequence, the transcription initiation might be TATA box independent. This would explain the apparent failure of the constitutive CRMs since the presence of a TATA box and adjacent nucleotides in the synthetic core promoters favor a TATA box dependent transcription initiation mechanism. To test this hypothesis, we have disrupted the TATA box in the natural promoter sequence by mutating it. We have replaced three nucleotides of this motif by cytosine in the PAOX1 (control), PGAP, PScADH1 and PScGPD1. The resulting activity data showed that the expression is disrupted after the TATA box mutation in all promoters (18%, 20%, 8% and 2% of the wild type promoters for PAOX1, PGAP, PScGPD1 and PScADH1, Supplementary Figure S4). Expression is therefore depending on the TATA box element in all cases. This finding does not confirm our hypothesis and suggests that other so far unknown elements seem to be essential for strong transcription from constitutive TATA box dependent yeast promoters.
Correlation between the Activities of Synthetic Core Promoters Fused to Different CRMs
We have evaluated context dependency and modularity of the top ten synthetic promoters by correlating the activity data of each synthetic core promoter under the control of different CRMs in different yeasts. This resulted in the correlation matrix depicted in Figure 5 A (heatmap showing all possible combinations of CRMs and yeasts, Supplementary Figure S5). It was observed that the highest correlation coefficients are obtained within the subset of inducible CRMs in P. pastoris, PCAT1-R, PDAS1-R and PAOX1-R (e.g., Figure 5 B), with correlation coefficients ranging between 0.40 and 0.63. Also relatively high correlation coefficients (around 0.5) were found when comparing the PScGAL1-R CRM and the constitutive CRMs in S. cerevisiae (e.g., Figure 5 C). On the other hand, relatively low correlations are observed when comparing CRMs of P. pastoris against CRMs of S. cerevisiae (e.g., Figure 5 D). The apparent low correlation observed between the synthetic promoters controlled by the PScADH1-R and PScGPD1-R might be explained by the much lower expression levels in these particular experiments. Finally, it should be noted that even when correlation is high, the relative expression levels of the same synthetic core promoter under the control of two different CRMs varies significantly, which means that although functional and correlated, the synthetic core promoters are not completely independent of the CRM to which they are fused.
Figure 5.

Correlation analysis of the top ten synthetic core promoter activities fused to seven different CRMs. (A) Heatmap of the correlation coefficients of the top-ten synthetic core promoter activities fused to different CRMs. All the possible combinations of CRMs are shown. The correlation coefficients were calculated based on the average of single measurements of at least three independent cultivations in separate 96-well deep-well plates. The samples were taken 48 h after induction (PAOX1-R, PDAS1-R, PCAT1-R cases) or inoculation (PGAP-R, PScGPD1-R, PScADH1-R and PScGAL1-R CRMs cases). PAOX1-R, PDAS1-R, PCAT1-R and PGAP1-R were tested in P. pastoris, while the remaining CRMs were tested in S. cerevisiae. Panels B–D show representative data on the correlation coefficients: (B) Synthetic core promoter activities when fused to PDAS1-R as a function of its respective activities when fused to PCAT1-R. (C) Synthetic core promoter activities when fused to PGPD1-R as a function of its respective activities when fused to PScGAL1-R. (D) Synthetic core promoter activities when fused to PDAS1-R as a function of its respective activities when fused to PScGAL1-R. The correlation diagrams between other promoters are shown in Supplementary Figure S5.
Discussion
Functionality of Synthetic Core Promoters
In this study we have followed a de novo design approach to generate synthetic core promoter sequences for yeast cells. The design was based on natural S. cerevisiae core promoters resulting in synthetic core promoters that were at first experimentally tested in P. pastoris. We have chosen this approach, because we were primarily interested in developing regulatory elements for P. pastoris, where generally applicable promoter engineering strategies are scarce.36 In contrast to S. cerevisiae, where large sets of experimental data on core promoters from large scale high throughput studies are available, no such studies have been performed in the widely used protein production host P. pastoris. Hence we used the data set from S. cerevisiae due to the reported conservation of core promoters27 and previous studies which demonstrated functionality of S. cerevisiae core promoters in P. pastoris.8
This design method delivered 77.6% of functional core promoter sequences with the P. pastoris PAOX1-R (Figure 2). These sequences are markedly different from naturally occurring sequences (no clear matches to natural promoters were found by BLAST search), between each other and substantially more diverse than variants typically obtained by local random mutations of a natural core promoter.7,16,43 This lack of resemblance to natural sequences is an important feature of this set of promoters. It may increase the genetic stability in the genomic context, as these sequences have low probability of recombining with any natural sequence in the genome. This feature will be valuable for future in vivo and in vitro pathway assembly,49 when assembling a multigene pathway using a different promoters for each enzyme, with the objective of fine-tuning the production of each one while employing a single inductor.
In a recent study, 11 artificial core promoter sequences were assessed in P. pastoris.25 Of these, only two were generated de novo by consensus sequence analysis of natural core promoters of PAOX1, PGAP, PHIS4 and PADH2. The other nine sequences were generated by replacements of short stretches in the natural PAOX1 core promoter. For the two consensus derived sequences, the activity levels were within the range of the basal activity level obtained from randomized sequences in this study (Figure 2 A), suggesting that the previous design considerations had a nonsignificant effect over using random sequences. The replacement method was more successful, with activity levels as high as 117% of the natural PAOX1. However, with the replacement method the resulting sequences share a high degree of similarity with the natural sequence, thus questioning the ability of the method to generate truly synthetic sequences. As discussed by Dehli et al., a diversity inherent component design approach as the one adopted here, is advantageous for synthetic biology problems, as it facilitates orthogonality, modularity and standardization of new components.50
We have obtained an average activity level of 17% with a dispersion of 11.5% and a maximum activity of 70% of the wild type PAOX1. Overall, this reflects the ability of the design method to span a wide spectrum of highly diverse synthetic sequences. However, the relatively low average activity might be in part explained by the way the experimental input data from S. cerevisiae was obtained.28 Lubliner et al. deduced core promoter functionality from reporter protein fluorescence measurements of the entire promoter (including the CRM), whereas we fused all core promoters to the same CRM. Hence expression strength of the S. cerevisiae measurements may also be influenced by the CRM and not solely the core promoter. Additionally, the phylogenetic distance between S. cerevisiae and P. pastoris may have complicated our efforts. It has been shown that core promoters in distant related yeasts maintain their functionality but with lower expression.27 To further support this statement, it should be underlined that the highest relative expression levels (176%) were obtained for PScGAL1-R in S. cerevisiae.
Another characteristic that could compromise the synthetic core promoters’ strength is the boundary between the core promoter and the CRM. Here we maintained the same boundary condition in all experiments (−10 bp from the TATA box), however, it might have some influence in promoters’ strength and might be targeted for optimization in future studies.
No Motifs Except the TATA Box Clearly Affect Expression
Figure 2 B shows an expression box plot for the four groups of sequences. The comparison between groups P and M and groups T and A show that the introduction of motifs does not affect the mean expression level, but might have an effect in specific cases (e.g., A28 and A27). Indeed, the effect of motifs in core promoter strength is not consensual in the literature. Recently, Seizl et al.(51) suggested that the GAAAA 5-mer is a conserved yeast promoter element, functioning as a TATA binding protein binding site in promoters lacking a consensus TATA box element. However, Lubliner et al.(52) studied knockout mutations of 122 GAAAA 5-mers that showed little to no effect on protein expression. Other studies have concluded that, with the possible exception of the TATA box (when present), motifs are not determinant for S. cerevisiae core promoter functionality.32,53
The comparison of groups P and M with groups T and A reveals that the presence of a TATA box motif is a key effector of high expression levels, which corroborates the data presented in previous studies.32,53 Indeed, within the top ten promoters only one sequence (M28) does not have a TATA box. This apparent exception is however discarded after a careful sequence analysis revealing that M28 has a TATA box like sequence, namely TATTTAATA at position −115. Several previous studies have shown that mutations in the TATA box region greatly affect promoter strength.54,55 In another study, Mogno et al.(56) analyzed libraries of TATA-positive and TATA-less promoters in S. cerevisiae showing that the TATA box mainly affects the transcription rate by enhancing it. It was also shown that the location, orientation and flanking bases critically affect TATA box function and core promoter activity.52 However, given the size of our data set (56 synthetic core promoters with TATA box), we cannot draw solid conclusion regarding these aspects.
The Role of Nucleosome Occupancy
Nucleosome occupancy has been reported as having a fundamental role in transcription initiation.18,57 Variations in nucleosome occupancy alone may cause large differences in promoter strength. Raveh-Sadka et al.(57) showed that AT rich sequences are associated with low nucleosome affinity and high promoter activity. Curran et al.(18) have redesigned nucleosome architecture in natural S. cerevisiae promoters with a 1.5- to 6-fold expression increase of a reporter protein (β-GAL). They have hypothesized that nucleosome occupancy is an important causative factor limiting the strength of native promoters and is likely an evolutionary mechanism for controlling transcriptional strength.18 In our study, we observe no statistically meaningful correlation between promoter strength and nucleosome occupancy (Figure 3 C). This suggests that other factors might have an even higher effect than nucleosome occupancy, which was the main design factor studied by Curran et al.(18) Similar results were obtained by Lam et al.,58 who have shown that the interplay of nucleosomes and motifs is important to explain promoter activity variations in S. cerevisiae. Experimental data for P. pastoris nucleosome occupancy are still not available and might help to explain our observations in the future.
Effects of Core Promoter and 5′UTR
Our design approach of synthetic sequences implicitly included the 5′UTR, as this region is interwoven with the core promoter (the beginning of the 5′UTR, downstream of the transcription start site, was found to be important for transcription initiation28). Therefore, the variation in reporter fluorescence measurements from our library of synthetic core promoters may be influenced by transcriptional or translational effects. The mRNA levels may be affected by the rate of transcription initiation as well as by the transcript stability. In our setting, translation initiation at the start codon was designed to be identical between all synthetic sequences, as we have used the same Kozak sequence in every design. Namely, the Kozak sequences of the AOX1 5′UTR was chosen, as the respective protein is translated at exceptionally high levels.36 Hence, the Kozak sequence in our synthetic core promoters should provide a best case scenario and translation initiation should not be limiting. Ribosome scanning for the start codon may be influenced by different secondary structures of the 5′UTRs. However, as the 5′UTRs of the top ten synthetic core promoters are AT-rich (and hence do not favor the formation of strong secondary structures) little influence is expected in our setting.
We also tested fusions of the synthetic core promoters/5′UTRs to the CRMs of different promoters (Figure 4). The transition/spacing of the synthetic core promoter to the CRMs may influence expression whereas the function of the 5′UTR is expected to be independent of the upstream CRM it is fused to as the 5′UTR of fusions of the same synthetic core promoter to different CRMs is identical. If there was a strong effect from the 5′UTR, it should influence reporter protein fluorescence independently of the fusions to the CRM. A strongly positive/negative effect of the 5′UTR of a synthetic promoter would increase/limit expression in every context.
However, the measurements shown in Figure 4 demonstrate, that the core promoter fusions showed in part varying responses when fused to different CRMs. Most notably, synthetic core promoters fused to CRMs of constitutive promoters showed considerably lower reporter fluorescence levels than when fused to inducible promoters. It appears that the nature of the CRM/core promoter transitions, influencing transcription, show a considerably stronger effect, than 5′UTR function.
Gaining deeper mechanistic insights on transcriptional/translation effects requires further studies. Reverse transcription quantitative real-time PCR (RT-qPCR) experiments would therefore be ideal to discriminate between transcriptional/translational effects. RT-qPCR using specific primers for the eGFP reporter gene would allow to compare transcript levels with the eGFP reporter protein fluorescence.
Such experiments appeared too extensive for the initial library of 112 synthetic core promoters, as for each promoter/strain RNA needs to be isolated separately (in case of biological replicates, the number would further multiply). However, RT-qPCRs may be performed to mechanistically characterize a subset of particularly interesting constructs (e.g., core promoters showing exceptionally high reporter protein fluorescence or surprising results depending on the design group [for example promoters A27 and A28 in Figure 2 F]). We validated the functionality and general applicability of the best 10 core promoters by fusing them to CRMs from different promoters demonstrating that they are not strictly context dependent (i.e., only functional if fused to the PAOX1 CRM, see Figures 4, 5 and section below).
Nonetheless, RT-qPCR experiments would be paramount to gain mechanistic insights and may be run as concluding experiment in a similar setting to quantify expression differences.
Independently of the underlying mechanisms governing reporter fluorescence output, the applicability of the synthetic core promoters generated in this study for modular expression fine-tuning was validated by fusions to the CRMs of different promoters.
Modularity of Synthetic Core Promoters
To assess modularity, we have inserted the top ten synthetic core promoters in P. pastoris and in S. cerevisiae under the control of seven different CRMs, four of which are inducible (PCAT1-R, PDAS1-R, PScGAL1-R and PAOX1-R) and three constitutive (PGAP-R, PScADH1-R and PScGPD1-R) (Figure 4 and 5). The fusions of synthetic core promoters to the different inducible CRMs controlled expression strength under different conditions, while leaving the regulatory mode unaffected: Fusions of the synthetic core promoters to the repressible AOX1 and DAS1 CRMs remained tightly repressed whereas fusions to the derepressed CAT1 promoter showed an expected increase in reporter protein fluorescence (Supplementary Figure S7).
The expression levels of the constitutive promoters are consistently lower than the inducible promoters (Figure 4). Although the compatibility between CRMs and core promoters has previously been proven even between different organisms,8,27 it appears, according to our data, it is not universal. For instance, in S. cerevisiae the CRM of the RPS5 gene is compatible with ADH1 and CUP1 core promoters, thus being able to initiate transcription. This is however not reciprocal, i.e., the ADH1 and CUP1 CRMs cannot initiate transcription when coupled with the RPS5 core promoter.59 We have hypothesized that the tested constitutive promoters have a TATA box independent transcription initiation, hence being incompatible with this set of TATA box containing synthetic core promoters. We tested this hypothesis by mutating the TATA box in the respective natural promoter sequences. The results show that the expression is disrupted, indicating that the transcription initiation of all constitutive promoters in this study is TATA box dependent. Hence, the lower expression of synthetic promoters fused to constitutive CRMs must rather be attributed to unknown regulatory mechanisms specific for constitutive promoters.
Within the group of inducible promoters, expression levels are high, irrespective of the yeast and CRM specific regulatory mechanism. Although the different CRMs in different yeasts respond to different stimuli (namely, methanol and galactose) it had no effect on its functionality. Some CRMs outperform the activity levels of the wild type promoter, namely PDAS1-R and PScGAL1-R in P. pastoris and S. cerevisiae, respectively. S. cerevisiae PScGAL1-R showed the highest relative activity level (176% of the wild type PScGAL1). This may reflect the fact that our design was based on S. cerevisiae core promoters.
The correlation analysis of synthetic core promoters’ expression levels under the control of different CRMs (Figure 5) shows that the correlations are higher when comparing CRMs in the same organism. This is the case of PCAT1-R against PDAS1-R in P. pastoris and PScGAL1-R against PGPD1-R in S. cerevisiae. Correlations are in general very low (r2 lower than 0.2) when comparing CRMs of different organisms. For instance, in the case of PDAS1-R and PScGAL1-R in S. cerevisiae and P. pastoris, respectively. These data suggest that comparable expression strength irrespective of the context, i.e., modularity is maintained only within the same organism, although the core promoters are also functional in other organisms. Zeevi et al. described the conservation of orthologous ribosomal promoter activity within closely related genus of yeasts.27 For instance, S. paradoxus, showed high correlation with S. cerevisiae while Kluyveromyces lactis diverged considerably. Likewise, we can anticipate that the low correlation observed in our study is due to the phylogenetic distance between P. pastoris and S. cerevisiae genus.27
All in all, our work demonstrated the feasibility of a multi factor rational synthetic core promoter design and its applicability as general engineering tool for gene expression fine-tuning. Due to their sequence diversity and independence of natural sequences, similarly designed synthetic core promoters may become valuable tools for synthetic biology and metabolic engineering applications in other eukaryotic organisms.
Materials and Methods
Strains
The P. pastoris CBS7435 (Komagataella phaffii, NRLLY-1143060) wild type strain and the S. cerevisiae FY 1679–01B strain (isogenic to S. cerevisiae S288c with an uracil auxotrophy61) were used as host organisms to screen the synthetic promoter activity, while E. coli TOP10 F′ was used to perform the cloning work.
Vectors and Cloning: Controls and Synthetic Core Promoters Fused to the PAOX1-R
Ten different controls were created using the genomic wild type PAOX1 sequence as template: deletion of the entire upstream regulatory region (CRM) upstream of the core promoter, deletion of the core promoter, replacement of the natural AOX1 core promoter with the core promoter of the HHF2 gene46,47 and seven completely random sequences. For the first control (deletion of CRM) primers C-WO-CRM1 and eGFP-pAOX1–3prime were used. For the remaining controls, pAOX1_Syn_dBamHI_SwaI-forward was used as forward primer, while as reverse primers were C-WO-Core1, C–W–HHF2+10 and R1 to R7, respectively. The primers sequences are provided in Supplementary Table S1.
The synthetic core promoters were ordered as long primers (Ultramer DNA Plate Oligo by Integrated DNA Technologies (Leuven, Belgium) in 96-well microtiter plates), attached by PCR to the PAOX1-R and cloned into the P. pastoris/E. coli shuttle vector pPpT4_SB-truncatedAOX1-eGFP, reported by Vogl et al.(25) The plasmid genbank file and respective map are available in the Supporting Information and Supplementary Figure S1. The synthetic promoters were amplified using forward primer pAOX1_Syn_dBamHI_SwaI-forward and the reverse primers listed in Supplementary Tables S2–S5.
The final PCR product was gel purified and cloned by assembly cloning into the SwaI and NheI digested vector backbone. All constructs were verified by Sanger sequencing.
Controls and Entry Vectors to Assess Synthetic Core Promoters with Different CRMs in P. pastoris and S. cerevisiae
The best synthetic core promoters were tested when fused to the CRMs of six additional promoters (CAT1, DAS1, GAP, ADH1, GAL1 and GPD1, named PCAT1-R, PDAS1-R, PGAP-R, PScADH-R, PScGAL1-R and PScGPD1-R, respectively). Three CRMs were tested in P. pastoris (PCAT1-R, PDAS1-R and PGAP-R), while the remaining three were tested in S. cerevisiae (PScADH-R, PScGAL1-R and PScGPD1-R). At first, the positive controls were created. To do so, the genomic wild type sequences of the P. pastoris promoters were amplified using the following three primers groups: CAT-core and CAT-CRM-forw, DAS-core and DAS-CRM-forw and GAP-core and GAP-CRM-forw (Supplementary Table S6), resulting in promoter fragments of 500, 552, and 486 bp, respectively. In each of the three PCR reactions, the respective wild type whole promoter sequence was used as template. It was then cloned into the P. pastoris/E. coli shuttle vector used in the previous screening, where the AOX1 truncated sequence had been removed (digestion with SwaI and NheI). For the S. cerevisiae whole promoter plasmids (used as positive controls), the promoter sequences were amplified from S. cerevisiae genomic DNA and cloned into a reporter vector (named Sc_eGFP_RFP_ARS) comprised by pUC origin of replication for E. coli, the ARS/CEN sequence for low-copy replication in S. cerevisiae, URA3–3′ and URA3–5′ integration sequences, a stuffer sequence flanked by eGFP and RFP and the two transcriptional terminators PRM9 and SPG5 as well as a Kanamycin resistance cassette, consisting of TEF1 and EM72 promoters for expression in yeast and E. coli, respectively, the KanMX6 resistance gene and terminator TIF51A (plasmids kindly provided by Pitzer, J., unpublished results). The plasmid genbank file is available in the Supporting Information and the respective map is shown in Supplementary Figure S2.
For each CRM, an entry vector was created to facilitate cloning of the synthetic core promoter fusions. Such entry vectors had a CRM sequence (without core promoter), a placeholder fragment and the eGFP coding sequence. The primers used to amplify the CRMs sequences for P. pastoris were the following three groups: CAT-CRM-rev and CAT-CRM-forw, DAS-CRM-rev and DAS-CRM-forw and GAP-CRM-rev and GAP-CRM-forw (Supplementary Table S6). While for S. cerevisiae CRMs sequences amplification the reverse primer used were: ADH-CRM-rev, GAL-CRM-rev and GPD-CRM-rev (Supplementary Table S6). The forward primer was, in these three cases, seqTomato19–41rev. The backbones used were Sc_eGFP_RFP_ARS for S. cerevisiae and pPpT4-bidi-sTomato-eGFP (Vogl, T., unpublished results) for P. pastoris (both genbank files are available in Supporting Information and respective maps in Supplementary Figure S2–S3). The S. cerevisiae vector was digested with AscI while the P. pastoris vector was linearized with AscI and SwaI. The digestion was gel purified and an assembly cloning was performed for each of the PCR results, yielding a six entry vectors (one for each CRM) and three plasmids containing a wild type promoter of interest each (PCAT1, PDAS1, PGAP: to be tested in P. pastoris), which were verified by Sanger sequencing.
Cloning a Subset of Synthetic Core Promoters with Different CRMs in P. pastoris and S. cerevisiae
Each of the ten best synthetic core promoters identified with the PAOX1-R (T22, T23, T24, T25, T26, T27, T28, M28, A27 and A28) was amplified by PCR six times to include the different CRMs overhangs, to be used for assembly cloning. The reverse primers used for each of the 10 best core promoters were T22-GFP-rev, T23-GFP-rev, T24-GFP-rev, T25-GFP-rev, T26-GFP-rev, T27-GFP-rev, T28-GFP-rev, M28-GFP-rev, A27-GFP-rev and A28-GFP-rev. Different forward primers were used depending on the CRM to be fused. For instance, to amplify the 10 synthetic promoters to be cloned in the PCAT1-R plasmid, the following forward primers were used: T22-CAT-rev, T23-CAT-rev, T24-CAT-rev, T25-CAT-rev, T26-CAT-rev, T27-CAT-rev, T28-CAT-rev, M28-CAT-rev, A27-CAT-rev and A28-CAT-rev. The three different entry vectors for P. pastoris containing the PGAP-R, PDAS1-R and PCAT1-R were digested by AscI and NheI to remove the placeholder fragment. The digestion products were gel purified. The linearized plasmids were used for assembly cloning with each of the respective 10 PCR core promoter fragments.
A similar approach was performed to screen the top 10 synthetic promoters in S. cerevisiae. The synthetic core promoters used the same reverse primers, while the forward primers vary according to the CRM sequence, as explained above. The entry vectors containing the PScADH-R, PScGAL1-R and PScGPD1-R were digested by AscI and NheI to remove the placeholder fragment. They were gel purified. The linearized plasmids were used for assembly cloning with each of the respective 10 PCR core promoter fragments.
All the primers used to clone the ten synthetic promoters with highest activity with different CRMs in P. pastoris and S. cerevisiae and the respective entry vectors are listed in Supplementary Table S6.
Transformation of P. pastoris and Cultivations
The aforementioned plasmids were digested with SwaI for linearization. P. pastoris was transformed with low amounts of linearized plasmid (approximately 1 μg of DNA) using the condensed protocol reported by Lin-Cereghino et al.(62) This low amount of expression cassette was used to reduce multi copy integration and variability between transformants.25 Then, from the resulting transformants, 28 were screened using a previously reported high throughput method.25,63 Briefly, cells were grown for 60h on 250 μL BMD1 and subsequently induced with methanol (250 μL BMM2 [1% methanol] at 60h and 50 μL BMM10 [5% methanol] at 72h). The transformants were screened for uniformity and three representative transformants from the linear range of the landscape were selected for rescreening, using the same protocol. Lastly, one transformant per construct was used for comparison of the variants under the same growth conditions. Fluorescence measurements were performed using a 96-well microtiter plate reader (Synergy MX, Biotek, Winooski, VT, USA) as described previously.5,25 Biological replicates from at least 3-fold cultivations of the same transformant were used to calculate the mean and standard deviations values, which are shown in Figures 2–5. These values represent the eGFP fluorescence values normalized per OD600, where the background measurements of diluted medium were subtracted. eGFP fluorescence (excitation at 488 nm and emission at 507 nm) and absorption at 600 nm (OD600, optical density 600) were measured in micro titer plates, 48 h after the first induction for the methanol inducible promoters (derived of PAOX1-R, PCAT1-Rand PDAS1-R), while the fluorescence values of the constitutive PGAP1-R variants were taken 48h after the inoculation. A subset of strains was also measured by flow cytometry using a BD LSRFortessa cell analyzer (results shown in Supplementary Figure S6). Cells were grown identically to plate reader measurements in deep well plates in biological 8-fold replicates and diluted 1:20 in PBS buffer and 30.000 events measured for each replicate (doublets were consistently <5% in all samples).
Transformation of S. cerevisiae and Cultivations
S. cerevisiae was transformed with circular plasmids (0.5 μg of DNA) using chemically competent cells.64 Then, from the resulting transformants, 28 were screened using a similar protocol to the one used for P. pastoris. Briefly, cells were grown for 24h on 250 μL YPD. The PScGAL1-R variants were additionally screened using YPGal medium instead of YPD. The transformants were screened for uniformity and three representative transformants from the linear range of the landscape were selected for rescreening, using the previous protocol. Lastly, one transformant per construct was used for comparison of the variants under the same growth conditions. Measurements were made in an identical way as to the P. pastoris protocol.
Acknowledgments
We gratefully acknowledge Julia Pitzer for kindly providing the S. cerevisiae wild type strain and initial S. cerevisiae plasmids used in this work. Financial support for this work was provided by the Portuguese Fundação para a Ciência e Tecnologia [PhD grant SFRH/BD/51577/2011 to R.M.C.P.]; and we gratefully acknowledge the Austrian Science Fund (FWF) project number W901 (DK “Molecular Enzymology” Graz) for funding and support from NAWI Graz. We would like to thank Astrid Weninger and Anna-Maria Hatzl for organizational support. We would like to thank Tobias Eisenberg (University of Graz) and the NAWI Graz Central Lab Gracia for support and excellent assistance in performing the FACS measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.6b00178.
S1 (Summary of literature references on S. cerevisiae and P. pastoris core promoters), S2 (Detailed computational design of synthetic core promoters), Supplementary Tables S1–S6 (Lists of primers), Supplementary Tables S7–S10 (properties of synthetic core promoters), Supplementary Table S11 (BLAST results), Supplementary Figures S1–S7 (PDF)
Plasmid maps in GenBank format (ZIP)
Author Present Address
§ Sandoz GmbH, Biochemiestrasse 1, A-6336 Langkampfen/Tirol, Austria.
Author Contributions
R.M.C.P. implemented all in silico designs, did the molecular cloning, performed all experiments and analyzed the data. T.V. provided input for the design process, experimental execution and cross-species applicability. R.M.C.P. and T.V. interpreted the results and wrote the manuscript. J.E.F. and C.K. performed the flow cytometry measurements. T.V., A.G., R.M.C.P and R.O. conceived of the study. A.G. and R.O. supervised the research. All authors read and approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Stephanopoulos G. (2012) Synthetic biology and metabolic engineering. ACS Synth. Biol. 1, 514–25. 10.1021/sb300094q. [DOI] [PubMed] [Google Scholar]
- Tabor J. J.; Salis H. M.; Simpson Z. B.; Chevalier A. A.; Levskaya A.; Marcotte E. M.; Voigt C. A.; Ellington A. D. (2009) A synthetic genetic edge detection program. Cell 137, 1272–81. 10.1016/j.cell.2009.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis H. M.; Mirsky E. A.; Voigt C. A. (2010) Automated Design of Synthetic Ribosome Binding Sites to Precisely Control Protein Expression. Nat. Biotechnol. 27, 946–950. 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazeck J.; Alper H. S. (2013) Promoter engineering: recent advances in controlling transcription at the most fundamental level. Biotechnol. J. 8, 46–58. 10.1002/biot.201200120. [DOI] [PubMed] [Google Scholar]
- Vogl T.; Sturmberger L.; Kickenweiz T.; Wasmayer R.; Schmid C.; Hatzl A.-M.; Gerstmann M. A.; Pitzer J.; Wagner M.; Thallinger G. G.; Geier M.; Glieder A. (2016) A Toolbox of Diverse Promoters Related to Methanol Utilization: Functionally Verified Parts for Heterologous Pathway Expression in Pichia pastoris. ACS Synth. Biol. 5, 172–86. 10.1021/acssynbio.5b00199. [DOI] [PubMed] [Google Scholar]
- Ellis T.; Wang X.; Collins J. J. (2009) Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nat. Biotechnol. 27, 465–71. 10.1038/nbt.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevoigt E.; Kohnke J.; Fischer C. R.; Alper H.; Stahl U.; Stephanopoulos G. (2006) Engineering of promoter replacement cassettes for fine-tuning of gene expression in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 72, 5266–73. 10.1128/AEM.00530-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner F. S.; Ruth C.; Langenegger D.; Johnson S. N.; Hyka P.; Lin-Cereghino G. P.; Lin-Cereghino J.; Kovar K.; Cregg J. M.; Glieder A. (2008) Promoter library designed for fine-tuned gene expression in Pichia pastoris. Nucleic Acids Res. 36, e76. 10.1093/nar/gkn369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper H.; Fischer C.; Nevoigt E.; Stephanopoulos G. (2005) Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. U. S. A. 102, 12678–83. 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. J.; Sweeney B.; Mainwaring D. O.; James D. C. (2014) Synthetic promoters for CHO cell engineering. Biotechnol. Bioeng. 111, 1638–47. 10.1002/bit.25227. [DOI] [PubMed] [Google Scholar]
- Nielsen J.; Larsson C.; van Maris A.; Pronk J. (2013) Metabolic engineering of yeast for production of fuels and chemicals. Curr. Opin. Biotechnol. 24, 398–404. 10.1016/j.copbio.2013.03.023. [DOI] [PubMed] [Google Scholar]
- Paddon C. J.; Westfall P. J.; Pitera D. J.; Benjamin K.; Fisher K.; McPhee D.; Leavell M. D.; Tai A.; Main A.; Eng D.; Polichuk D. R.; Teoh K. H.; Reed D. W.; Treynor T.; Lenihan J.; Fleck M.; Bajad S.; Dang G.; Dengrove D.; Diola D.; Dorin G.; Ellens K. W.; Fickes S.; Galazzo J.; Gaucher S. P.; Geistlinger T.; Henry R.; Hepp M.; Horning T.; Iqbal T.; Jiang H.; Kizer L.; Lieu B.; Melis D.; Moss N.; Regentin R.; Secrest S.; Tsuruta H.; Vazquez R.; Westblade L. F.; Xu L.; Yu M.; Zhang Y.; Zhao L.; Lievense J.; Covello P. S.; Keasling J. D.; Reiling K. K.; Renninger N. S.; Newman J. D. (2013) High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496, 528–32. 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- Hong K.-K.; Nielsen J. (2012) Metabolic engineering of Saccharomyces cerevisiae: a key cell factory platform for future biorefineries. Cell. Mol. Life Sci. 69, 2671–90. 10.1007/s00018-012-0945-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Redden H.; Alper H. S. (2013) Frontiers of yeast metabolic engineering: diversifying beyond ethanol and Saccharomyces. Curr. Opin. Biotechnol. 24, 1–8. 10.1016/j.copbio.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Wagner J. M.; Alper H. S. (2015) Synthetic biology and molecular genetics in non-conventional yeasts: Current tools and future advances. Fungal Genet. Biol. 89, 1–11. 10.1016/j.fgb.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Berg L.; Strand T. A.; Valla S.; Brautaset T. (2013) Combinatorial mutagenesis and selection to understand and improve yeast promoters. BioMed Res. Int. 2013, 926985. 10.1155/2013/926985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazeck J.; Garg R.; Reed B.; Alper H. S. (2012) Controlling promoter strength and regulation in Saccharomyces cerevisiae using synthetic hybrid promoters. Biotechnol. Bioeng. 109, 2884–95. 10.1002/bit.24552. [DOI] [PubMed] [Google Scholar]
- Curran K. A.; Crook N. C.; Karim A. S.; Gupta A.; Wagman A. M.; Alper H. S. (2014) Design of synthetic yeast promoters via tuning of nucleosome architecture. Nat. Commun. 5, 4002. 10.1038/ncomms5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redden H.; Alper H. S. (2015) The development and characterization of synthetic minimal yeast promoters. Nat. Commun. 6, 7810. 10.1038/ncomms8810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli K. M.; Slattery M.; Mann R. S. (2012) Disentangling the many layers of eukaryotic transcriptional regulation. Annu. Rev. Genet. 46, 43–68. 10.1146/annurev-genet-110711-155437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison L. A. (2007) Transcription in eukaryotes. Fundam. Mol. Biol. 312–391. [Google Scholar]
- Juven-Gershon T.; Kadonaga J. T. (2010) Regulation of gene expression via the core promoter and the basal transcriptional machinery. Dev. Biol. 339, 225–9. 10.1016/j.ydbio.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale S. T.; Kadonaga J. T. (2003) The RNA polymerase II core promoter. Annu. Rev. Biochem. 72, 449–79. 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- Ruth C.; Zuellig T.; Mellitzer A.; Weis R.; Looser V.; Kovar K.; Glieder A. (2010) Variable production windows for porcine trypsinogen employing synthetic inducible promoter variants in Pichia pastoris. Syst. Biol. Synth. Biol. 4, 181–91. 10.1007/s11693-010-9057-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl T.; Ruth C.; Pitzer J.; Kickenweiz T.; Glieder A. (2014) Synthetic Core Promoters for Pichia pastoris. ACS Synth. Biol. 3, 188–91. 10.1021/sb400091p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Y.; Zhou X.; Zhang W.; Zhang X.; Song Z.; Zhang Y. (2009) An upstream activation sequence controls the expression of AOX1 gene in Pichia pastoris. FEMS Yeast Res. 9, 1271–1282. 10.1111/j.1567-1364.2009.00571.x. [DOI] [PubMed] [Google Scholar]
- Zeevi D.; Lubliner S.; Lotan-Pompan M.; Hodis E.; Vesterman R.; Weinberger A.; Segal E. (2014) Molecular dissection of the genetic mechanisms that underlie expression conservation in orthologous yeast ribosomal promoters. Genome Res. 24, 1991–9. 10.1101/gr.179259.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubliner S.; Keren L.; Segal E. (2013) Sequence features of yeast and human core promoters that are predictive of maximal promoter activity. Nucleic Acids Res. 41, 5569–81. 10.1093/nar/gkt256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren L.; Zackay O.; Lotan-Pompan M.; Barenholz U.; Dekel E.; Sasson V.; Aidelberg G.; Bren A.; Zeevi D.; Weinberger A.; Alon U.; Milo R.; Segal E. (2013) Promoters maintain their relative activity levels under different growth conditions. Mol. Syst. Biol. 9, 1–17. 10.1038/msb.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon E.; Kalma Y.; Sharp A.; Raveh-Sadka T.; Levo M.; Zeevi D.; Keren L.; Yakhini Z.; Weinberger A.; Segal E. (2012) Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol. 30, 521–30. 10.1038/nbt.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn S.; Young E. T. (2011) Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 189, 705–36. 10.1534/genetics.111.127019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara F.; Kasahara K.; Kokubo T. (2011) Highly redundant function of multiple AT-rich sequences as core promoter elements in the TATA-less RPS5 promoter of Saccharomyces cerevisiae. Nucleic Acids Res. 39, 59–75. 10.1093/nar/gkq741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D.; Morris A. R.; Battenhouse A.; Iyer V. R. (2014) Simultaneous mapping of transcript ends at single-nucleotide resolution and identification of widespread promoter-associated non-coding RNA governed by TATA elements. Nucleic Acids Res. 42, 3736–49. 10.1093/nar/gkt1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir S.; Velten L.; Sharon E.; Zeevi D.; Carey L. B.; Weinberger A.; Segal E. (2013) Deciphering the rules by which 5′-UTR sequences affect protein expression in yeast. Proc. Natl. Acad. Sci. U. S. A. 110, E2792–801. 10.1073/pnas.1222534110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill R. M. (2014) Playing catch-up with Escherichia coli: using yeast to increase success rates in recombinant protein production experiments. Front. Microbiol. 5, 85. 10.3389/fmicb.2014.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl T.; Glieder A. (2013) Regulation of Pichia pastoris promoters and its consequences for protein production. New Biotechnol. 30, 385–404. 10.1016/j.nbt.2012.11.010. [DOI] [PubMed] [Google Scholar]
- Khalil A. S.; Lu T. K.; Bashor C. J.; Ramirez C. L.; Pyenson N. C.; Joung J. K.; Collins J. J. (2012) A synthetic biology framework for programming eukaryotic transcription functions. Cell 150, 647–58. 10.1016/j.cell.2012.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz J.; Siggia E. D.; Cohen B. A. (2009) Analysis of combinatorial cis-regulation in synthetic and genomic promoters. Nature 457, 215–8. 10.1038/nature07521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder M.; McMillen D. R. (2014) Design and characterization of a dual-mode promoter with activation and repression capability for tuning gene expression in yeast. Nucleic Acids Res. 42, 9514–22. 10.1093/nar/gku651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K. F.; Balázsi G.; Collins J. J. (2007) Combinatorial promoter design for engineering noisy gene expression. Proc. Natl. Acad. Sci. U. S. A. 104, 12726–31. 10.1073/pnas.0608451104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount B. A.; Weenink T.; Vasylechko S.; Ellis T. (2012) Rational diversification of a promoter providing fine-tuned expression and orthogonal regulation for synthetic biology. PLoS One 7, e33279. 10.1371/journal.pone.0033279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo W. S.; Chang M. W. (2014) Development and characterization of AND-gate dynamic controllers with a modular synthetic GAL1 core promoter in Saccharomyces cerevisiae. Biotechnol. Bioeng. 111, 144–51. 10.1002/bit.25001. [DOI] [PubMed] [Google Scholar]
- Qin X.; Qian J.; Yao G.; Zhuang Y.; Zhang S.; Chu J. (2011) GAP promoter library for fine-tuning of gene expression in Pichia pastoris. Appl. Environ. Microbiol. 77, 3600–8. 10.1128/AEM.02843-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley C. A.; Huang A.; Nattestad M.; Oshiro K. T.; Ray L. E.; Mulye T.; Li Z. H.; Le T.; Stephens J. J.; Gomez S. R.; Moy A. D.; Nguyen J. C.; Franz A. H.; Lin-Cereghino J.; Lin-Cereghino G. P. (2012) Analysis of the 5′ untranslated region (5′UTR) of the alcohol oxidase 1 (AOX1) gene in recombinant protein expression in Pichia pastoris. Gene 496, 118–27. 10.1016/j.gene.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N.; Moore I. K.; Fondufe-Mittendorf Y.; Gossett A. J.; Tillo D.; Field Y.; LeProust E. M.; Hughes T. R.; Lieb J. D.; Widom J.; Segal E. (2009) The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458, 362–366. 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geier M.; Fauland P.; Vogl T.; Glieder A. (2015) Compact multi-enzyme pathways in P. pastoris. Chem. Commun. 51, 1643–1646. 10.1039/C4CC08502G. [DOI] [PubMed] [Google Scholar]
- Weninger A.; Hatzl A.-M.; Schmid C.; Vogl T.; Glieder A. (2016) Combinatorial optimization of CRISPR/Cas9 expression enables precision genome engineering in the methylotrophic yeast Pichia pastoris. J. Biotechnol. 235, 139–49. 10.1016/j.jbiotec.2016.03.027. [DOI] [PubMed] [Google Scholar]
- McWilliam H.; Li W.; Uludag M.; Squizzato S.; Park Y. M.; Buso N.; Cowley A. P.; Lopez R. (2013) Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 41, 597–600. 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx H., Pflugl S., Mattanovich D., and Sauer M. (2016) Synthetic Biology Assisting Metabolic Pathway Engineering, In Synthetic Biology (Glieder A., Kubicek C. P., Mattanovich D., Wiltschi B., and Sauer M., Ed.), pp 255–280, Springer Press, London. [Google Scholar]
- Dehli T., Solem C., and Jensen P. R. (2012) Tunable promoters in synthetic and systems biology, In Reprogramming Microbial Metabolic Pathways, pp 181–201, Springer. [DOI] [PubMed] [Google Scholar]
- Seizl M.; Hartmann H.; Hoeg F.; Kurth F.; Martin D. E.; Söding J.; Cramer P. (2011) A conserved GA element in TATA-less RNA polymerase II promoters. PLoS One 6, e27595. 10.1371/journal.pone.0027595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubliner S.; Regev I.; Lotan-Pompan M.; Edelheit S.; Weinberger A.; Segal E. (2015) Core promoter sequence in yeast is a major determinant of expression level. Genome Res. 25, 1008–17. 10.1101/gr.188193.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basehoar A. D.; Zanton S. J.; Pugh B. F. (2004) Identification and distinct regulation of yeast TATA box-containing genes. Cell 116, 699–709. 10.1016/S0092-8674(04)00205-3. [DOI] [PubMed] [Google Scholar]
- Blake W. J.; Balázsi G.; Kohanski M. A.; Isaacs F. J.; Murphy K. F.; Kuang Y.; Cantor C. R.; Walt D. R.; Collins J. J. (2006) Phenotypic Consequences of Promoter-Mediated Transcriptional Noise. Mol. Cell 24, 853–865. [DOI] [PubMed] [Google Scholar]
- Raser J.; O’Shea E. (2013) Control of Stochasticity in Eukaryotic Gene Expression. Science 18, 1199–1216. 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogno I.; Vallania F.; Mitra R. D.; Cohen B. A. (2010) TATA is a modular component of synthetic promoters. Genome Res. 20, 1391–7. 10.1101/gr.106732.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh-Sadka T.; Levo M.; Shabi U.; Shany B.; Keren L.; Lotan-Pompan M.; Zeevi D.; Sharon E.; Weinberger A.; Segal E. (2012) Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat. Genet. 44, 743–50. 10.1038/ng.2305. [DOI] [PubMed] [Google Scholar]
- Lam F. H.; Steger D. J.; O’Shea E. K. (2008) Chromatin decouples promoter threshold from dynamic range. Nature 453, 246–250. 10.1038/nature06867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.-Y.; Bhaumik S. R.; Zhu X.; Li L.; Shen W.-C.; Dixit B. L.; Green M. R. (2002) Selective recruitment of TAFs by yeast upstream activating sequences. Implications for eukaryotic promoter structure. Curr. Biol. 12, 1240–4. 10.1016/S0960-9822(02)00932-6. [DOI] [PubMed] [Google Scholar]
- Sturmberger L.; Chappell T.; Geier M.; Krainer F.; Day K. J.; Vide U.; Trstenjak S.; Schiefer A.; Richardson T.; Soriaga L.; Darnhofer B.; Birner-Gruenberger R.; Glick B. S.; Tolstorukov I.; Cregg J.; Madden K.; Glieder A. (2016) Refined Pichia pastoris reference genome sequence. J. Biotechnol. 235, 121. 10.1016/j.jbiotec.2016.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston F.; Dollard C.; Ricupero-hovasse S. L. (1995) Construction of a Set of Convenient Saccharomyces cerevisiae Strains that are Isogenic to S288C. Yeast 11, 53–55. 10.1002/yea.320110107. [DOI] [PubMed] [Google Scholar]
- Lin-Cereghino J.; Wong W. W.; Xiong S.; Giang W.; Luong L. T.; Vu J.; Johnson S. D.; Lin-Cereghino G. P. (2005) Condensed protocol for competent cell preparation and transformation of the methylotrophic yeast Pichia pastoris. BioTechniques 38, 44–46. 10.2144/05381BM04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis R.; Luiten R.; Skranc W.; Schwab H.; Wubbolts M.; Glieder A. (2004) Reliable high-throughput screening with Pichia pastoris by limiting yeast cell death phenomena. FEMS Yeast Res. 5, 179–89. 10.1016/j.femsyr.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Amberg D., Burke D., and Strathern J. (2005) Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual, Cold Spring Harbor Laboratory Press. [Google Scholar]
- Xi L.; Fondufe-Mittendorf Y.; Xia L.; Flatow J.; Widom J.; Wang J.-P. (2010) Predicting nucleosome positioning using a duration Hidden Markov Model. BMC Bioinf. 11, 346. 10.1186/1471-2105-11-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.