

Summary
Cysteine residues on proteins serve diverse functional roles in catalysis and regulation and are susceptible to numerous posttranslational modifications. Methods to monitor the reactivity of cysteines within the context of a complex proteome have facilitated the identification and functional characterization of cysteine residues on disparate proteins. Here, we describe the use of a cysteine-reactive iodoacetamide probe coupled to isotopically labeled, cleavable linkers to identify and quantify cysteine-reactivity changes from two biological samples.
1. Introduction
Cysteine, one of the least abundant amino acids found in many organisms, is unique due to its intrinsically high nucleophilicity and sensitivity to oxidative modifications. These physicochemical properties enable cysteine to play critical roles in nucleophilic and redox catalysis, allosteric regulation, metal binding, and structural stabilization.1,2 Additionally, these functional cysteine residues are susceptible to a variety of posttranslational modifications (PTMs), including nitrosation, oxidation, palmitoylation, prenylation and Michael additions to oxidized lipids.3–7 The endogenous functions of a wide array of proteins (e.g. caspases, GAPDH, EGFR)8–10 are known to be modulated through these oxidative and lipid-derived cysteine PTMs. To better understand the ubiquity of cysteine PTMs and unearth novel regulatory functions for cysteines, proteomic methods to monitor changes in cysteine reactivity are essential.
Several proteomic methods exist for specifically monitoring each of the cysteine PTMs mentioned above, including selective probes for sulfenic acids,11 the biotin-switch technique12 and bioorthogonal lipid reporters.13 Other more general techniques that quantitatively reports on global changes of cysteine reactivity are able to report on any PTM that alters the nucleophilicity of the cysteine thiol. One such technology, termed isoTOP-ABPP, applies chemical probes and quantitative mass spectrometry (MS) to identify and quantify the relative abundance of hundreds of reactive cysteines within a proteome.14 An iodoacetamide-alkyne (IA) probe (Figure 1A) is used to covalently tag reactive cysteines within a proteome, and these probe-labeled proteins are then enriched and analyzed by quantitative MS with the aid of an isotopically labeled, cleavable linker that is incorporated through copper-catalyzed azide-alkyne cycloaddition (CuAAC).15 This method has been successfully applied to rank cysteine residues in the human proteome by reactivity with an iodoacetamide electrophile,14 to identify bacterial proteins sensitive to peroxide-mediated oxidation16 and to reveal cysteine residues susceptible to Michael addition reactions with lipid-derived electrophiles.17 We recently reported a variation of the isoTOP-ABPP platform that utilizes chemically cleavable azobenzene (Azo) linkers (Figure 1B) in place of the tobacco etch virus (TEV) protease-cleavable linkers in the original report.18 The Azo linkers incorporate either light (Azo-L) or heavy (Azo-H) stable-isotope signatures that enable comparative and quantitative profiling by mass spectrometry. We have successfully applied the Azo platform to monitor cysteine-reactivity changes within a complex proteome,18 and identify cysteine residues that are regulated through Zn2+ binding.19 Here, we provide the detailed protocol for applying this Azo platform to quantitatively monitor changes in cysteine reactivity in two biological samples.
Figure 1.
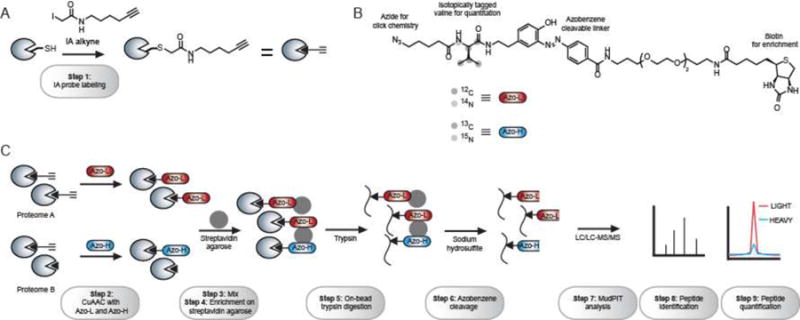
The reagents and experimental steps of the Azo platform to identify and quantify reactive cysteines from two biological samples. (A) Step 1 involves the addition of an iodoacetamide-alkyne (IA) probe to covalently modify reactive cysteines within a proteome. (B) The Azo tags contain an azide for click chemistry, an isotopically tagged valine for quantification, an azobenzene cleavable linker, and a biotin for enrichment. Azo-L and Azo-H have a mass difference of 6 Da. (C) The IA-labeled proteome is first subject to CuAAC to incorporate Azo-L and Azo-H (Step 2). The resulting samples are mixed together (Step 3) and subject to streptavidin enrichment (Step 4), on-bead trypsin digestion (Step 5), and sodium hydrosulfite treatment (Step 6). The resulting peptide mixture is analyzed by MudPIT (Step 7) and peptides are identified (Step 8) and quantified (Step 9).
The Azo platform (Figure 1C) begins with labeling of two proteome samples (e.g. healthy and diseased mouse liver proteomes) with the IA probe (Step 1). Probe-labeled proteins are then conjugated to either Azo-L or Azo-H linkers using CuAAC (Step 2). The resulting proteomes are then mixed together (Step 3) and subjected to enrichment on streptavidin agarose beads (Step 4), on-bead trypsin digestion (Step 5) and chemical cleavage of the azobenzene linker with sodium hydrosulfite (Step 6). The resulting isotopically labeled peptides are analyzed by multidimensional protein identification technology (MudPIT)20 on a high-resolution Orbitrap Mass Spectrometery (Thermo Scientific) (Step 7). Peptide identifications are obtained for every IA-labeled peptide using SEQUEST (Step 8),21 and quantification of the relative abundance of each peptide in the two biological samples is achieved using CIMAGE14 to generate a light:heavy ratio (R) (Step 9). In a typical experiment, approximately 1000 cysteines can be identified from a mammalian proteome and changes in the reactivity of these cysteines induced by a particular oxidative or electrophilic stress can be concurrently monitored.
2. Materials
2.1. Proteome preparation, IA labeling and CuAAC components
Phosphate-buffered saline (PBS) (Mediatech, Manassas, VA, USA)
Bio-Rad DC protein assay kit (Bio-Rad, Hercules, CA, USA)
IA probe (10 mM solution in DMSO) (see Note 1)
Tris 2-carboxyethyl phosphine (TCEP) (50 mM solution in water) (Sigma-Aldrich, St. Louis, MO, USA) (see Note 2)
Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (“ligand”) (1.7 mM solution in DMSO:t-butanol 1:4) (Sigma-Aldrich, St. Louis, MO, USA)
Azo-L/Azo-H (5 mM solutions in DMSO) (see Note 3)
Copper (II) sulfate (50 mM solution in water) (Sigma-Aldrich, St. Louis, MO, USA)
Sodium dodecyl sulfate (SDS) (0.2% and 1.2% solutions in PBS) (Bio-Rad, Hercules, CA, USA)
2.2. Streptavidin enrichment components
Streptavidin beads (Thermo, Rockford, IL, USA)
Urea (2 M and 6 M solution in PBS) (Sigma-Aldrich, St. Louis, MO, USA)
Dithiothreitol (DTT) (200 mM solution in water) (Invitrogen, Carlsbad, CA, USA)
Iodoacetamide (400 mM solution in water) (Acros, NJ, USA)
2.3. On-bead trypsin digestion and sodium hydrosulfite-cleavage components
Calcium chloride (100 mM solution in water) (Sigma-Aldrich, St. Louis, MO, USA)
Trypsin solution: 20 μg of sequencing-grade trypsin reconstituted in 40 μL of the trypsin buffer provided in package (Promega, Madison, WI, USA)
Bio-Spin columns (Bio-Rad, Hercules, CA, USA)
Formic acid (Fisher Scientific, Fair Lawn, NJ, USA)
Screw-top eppendorf (Bio-Rad, Hercules, CA, USA)
Sodium hydrosulfite (50 mM solution in PBS) (Sigma-Aldrich, St. Louis, MO, USA)
2.4. Mass-spectrometry analysis
Fused-silica capillary tubing (100 and 250 μm internal diameter) (Agilent, Santa Clara, CA, USA)
Aqua C18 reverse-phase resin (Phenomenex, Torrance, CA, USA)
Partisphere strong cation exchange (SCX) resin (Phenomenex, Torrance, CA, USA)
Buffer A: 95% water, 5% acetonitrile, 0.1% formic acid
Buffer B: 20% water, 80% acetonitrile, 0.1% formic acid
Buffer C: 95% water, 5% acetonitrile, 0.1% formic acid, 500 mM Ammonium acetate
3. Methods
3.1. Proteome preparation, IA labeling and CuAAC
Homogenize tissue or cell pellets (Samples A and B) in PBS, pH 7.4 and sonicate for 30 sec. Centrifuge at 200,000 × g for 45 min to obtain soluble proteomes (Proteomes A and B) and measure protein concentrations using the Bio-Rad DC protein assay kit according to manufacturer’s instructions.
Dilute Proteomes A and B to 2 mg/mL solutions in PBS (see Note 4). Add 500 uL of Proteomes A and B to each of two 1.5 mL eppendorf tubes (4 tubes total). Add 5 μL of 10 mM IA probe solution to each 500 μL sample to achieve a final concentration of 100 μM. After addition of the probe, vortex and incubate the reaction at room temperature (RT) for one hour.
Add 10 μL of 5 mM Azo-L (Proteome A) and Azo-H (Proteome B) stocks, 10 μL of fresh 50 mM TCEP solution (see Note 2), 30 μL of 1.7 mM “ligand” solution and 10 μL of 50 mM copper (II) sulfate to each tube and vortex. Incubate the reaction at RT for one hour with vortexing every 15 min. At this stage, the proteins will start to precipitate and the solution will turn cloudy.
After the one-hour incubation, combine the tubes pairwise (one Azo-L sample is combined with one Azo-H) sample, and centrifuge for 4 min at 6,500 × g at 4 °C. Remove the supernatant, add 500 μL cold methanol to each tube and sonicate for several seconds until the pellet is completely solubilized. Combine tubes pairwise again and centrifuge for 4 min at 6500 × g. Repeat methanol wash one more time (see Note 5).
3.2. Streptavidin enrichment
Add 1 mL 1.2% SDS/PBS to the final pellet, sonicate for several seconds until the solution turns clear and heat to 80–90 °C for 5 min. Transfer the 1 mL sample to a 15 mL conical tube containing 5 mL PBS. The final concentration of SDS in the sample is 0.2%.
Aliquot ~100 μL of the streptavidin agarose beads into an Eppendorf tube and wash 3 times with 1 mL PBS. Transfer the washed beads to the conical tube containing the proteome sample and incubate at 4°C overnight with constant rotation.
3.3. On-bead trypsin digestion
After the overnight 4°C incubation, warm the sample by rotating at RT over 1–2 hours. Centrifuge at 1400 × g for 3 min and remove the supernatant. Wash the beads with 5 mL of 0.2% SDS/PBS, rotate for 10 min, spin at 1400 × g for 3 min and remove the supernatant.
Wash the beads with 5 mL PBS, gently shaking for several seconds to resuspend the beads, centrifuge at 1400 × g for 3 min, and follow by removal of the supernatant. Repeat the PBS wash two more times.
Wash the beads with 5 mL water three times as described in step 2 (see Note 6).
Transfer the beads to a screw-capped Eppendorf tube using 500 μL of 6 M Urea/PBS. Add 25 μL freshly made 200 mM DTT solution and incubate at 65°C for 20 min with mixing every 10 min. Cool the tube for 1–2 min and add 25 μL of 400 mM iodoacetamide solution and incubate at 37°C for 30 min with rotation (see Note 7).
Dilute the reaction by adding 950 μL of PBS, centrifuge at 1400 × g for 2 min and remove the supernatant. Add a pre-mixed solution of 200 μL of 2 M Urea/PBS, 2 μL of 100 mM calcium chloride and 4 μL of Trypsin solution. Incubate the reaction at 37°C overnight with rotation (see Note 8).
3.4. Sodium-hydrosulfite cleavage
After overnight incubation, transfer the supernatant and beads to a Bio-Spin column and separate the beads by centrifugation at 1000 × g for 2 min. Wash the beads with 500 μL PBS 3 times and 500 μL water 3 times. Transfer the beads to a screw-capped Eppendorf tube using water, spin down the beads and remove the supernatant.
Add 75 μL 50 mM freshly made sodium hydrosulfite solution to the beads, rotate for 1 hour at RT, centrifuge at 1400 × g for 3 min and collect the supernatant (see Note 9).
Repeat step 2 two more times (see Note 10).
Wash beads with 100 μL of water and combine the wash with collected supernatant from step 2–3. The final sample volume should be around 325 μL.
Add 17.5 μL of formic acid and store the sample at −20°C until mass-spectrometry analysis.
3.5. Mass spectrometry analysis using MudPIT
Cut ~45 cm of the 100 μm fused-silica capillary tubing, burn off ~3 cm of the polyimide coating in the middle and wipe the burnt coating with methanol to expose the underlying silica. Generate two 5 μm tips using a laser puller to afford the columns for MS analysis. Pack each column with 10 cm of Aqua C18 reverse-phase resin and 3 cm of strong cation exchange (SCX) resin.
Cut 12 cm of the 250 μm fused-silica capillary tubing and generate a desalting column by connecting the tubing with an inline microfilter assembly. Pack the desalting column with 4 cm of Aqua C18 reverse phase resin.
Equilibrate the tip and the desalting column from step 1 and 2 on an Agilent 1100 series HPLC using a gradient of 40% Buffer A; 60% Buffer B to 100% Buffer A; 0% Buffer B over 30 min. Flow rate is set at 0.1 mL/min and a tee splitter is used to reduce the flow rate to 300–400 nL/min.
Thaw the MS sample and pressure load onto the equilibrated desalting column. Combine the loaded desalting column with the equilibrated tip and align onto a nanospray stage attached to an Orbitrap XL mass spectrometer (Thermo Scientific).
Elute the peptides using 5 steps (Table 1). The flow rate through the column is set to ~0.25 μL/min and the spray voltage is set to 2.75 kV. One full MS scan (FTMS) (400–1800 MW) was followed by 8 data dependent scans (ITMS) of the nth most intense ions with dynamic exclusion enabled (repeat count = 1; exclusion list size = 300; exclusion duration = 30s).
Table 1.
Solvent systems utilized for MudPIT analysis
| Step 1: 0% Buffer C
| |||
|---|---|---|---|
| Time [min] | % Buffer A | % Buffer B | % Buffer C |
| 0.00 | 100 | 0 | 0 |
| 5.00 | 100 | 0 | 0 |
| 60.00 | 55 | 45 | 0 |
| 70.00 | 0 | 100 | 0 |
| 100.00 | 0 | 100 | 0 |
| Step 2: 50% Buffer C
| |||
|---|---|---|---|
| Time [min] | % Buffer A | % Buffer B | % Buffer C |
| 0.00 | 100 | 0 | 0 |
| 6.00 | 100 | 0 | 0 |
| 6.10 | 45 | 5 | 50 |
| 8.00 | 45 | 5 | 50 |
| 8.10 | 95 | 5 | 0 |
| 15.00 | 85 | 15 | 0 |
| 35 00 | 75 | 25 | 0 |
| 75.00 | 45 | 55 | 0 |
| 80.00 | 45 | 55 | 0 |
| Step 3: 80% Buffer C
| |||
|---|---|---|---|
| Time [min] | % Buffer A | % Buffer B | % Buffer C |
| 0.00 | 100 | 0 | 0 |
| 5.00 | 100 | 0 | 0 |
| 5.10 | 15 | 5 | 80 |
| 8.00 | 15 | 5 | 80 |
| 8.10 | 95 | 5 | 0 |
| 18.00 | 85 | 15 | 0 |
| 63.00 | 75 | 25 | 0 |
| 115.00 | 45 | 55 | 0 |
| 120.00 | 45 | 55 | 0 |
| Step 4: 100% Buffer C
| |||
|---|---|---|---|
| Time [min] | % Buffer A | % Buffer B | % Buffer C |
| 0.00 | 100 | 0 | 0 |
| 4.00 | 100 | 0 | 0 |
| 4.10 | 0 | 0 | 100 |
| 20.00 | 0 | 0 | 100 |
| 20.10 | 93 | 7 | 0 |
| 25.00 | 85 | 15 | 0 |
| 100.00 | 70 | 30 | 0 |
| 184.00 | 0 | 100 | 0 |
| 194.00 | 0 | 100 | 0 |
| 195.00 | 100 | 0 | 0 |
| 200.00 | 100 | 0 | 0 |
| Step 5: 100% Buffer C
| |||
|---|---|---|---|
| Time [min] | % Buffer A | % Buffer B | % Buffer C |
| 0.00 | 100 | 0 | 0 |
| 4.00 | 100 | 0 | 0 |
| 4.10 | 0 | 0 | 100 |
| 14.00 | 0 | 0 | 100 |
| 14.10 | 93 | 7 | 0 |
| 30.00 | 70 | 30 | 0 |
| 50.00 | 0 | 100 | 0 |
| 55.00 | 0 | 100 | 0 |
| 56.00 | 100 | 0 | 0 |
| 60.00 | 100 | 0 | 0 |
3.6. Data analysis
The tandem MS data is searched against a protein sequence database using the SEQUEST algorithm for peptide identification.21
A static modification of +57.02146 on cysteine is specified to account for iodoacetamide alkylation and differential modifications of +456.2849 (Azo-L modification) and +462.2987 (Azo-H modification) are specified on cysteine to account for probe modifications.
SEQUEST output files are filtered using DTASelect 2.0., where reported peptides are required to be fully tryptic and contain the desired probe modification. Discriminant analyses are performed to achieve a peptide false-positive rate below 5%.
Quantification of light/heavy (Azo-L/Azo-H) ratios (R) is performed using the CIMAGE quantification package as previously described.14
5. Results
To demonstrate the quantitative accuracy of the Azo platform, Azo-L and Azo-H tagged mouse liver proteomes were mixed in pre-determined light:heavy ratios of 5:1, 2:1, 1:1, 1:2 and 1:5. These five samples were subject to enrichment and MS analysis according to the protocol described above. For each cysteine that is labeled by the IA probe, a light:heavy ratio was calculated in each of the five runs. Representative chromatography traces for one labeled peptide (K.AYDATC*LVK.A (where C* represents the cysteine modified by the IA probe) from S−formylglutathione hydrolase (ESD)) is shown in Figure 2. The co-elution of the light and heavy species, the high signal-to-noise, and the characteristic isotopic peak pattern demonstrate that our methods represent an accurate platform for relative quantification of cysteine reactivity changes from two or more proteomes.
Figure 2.
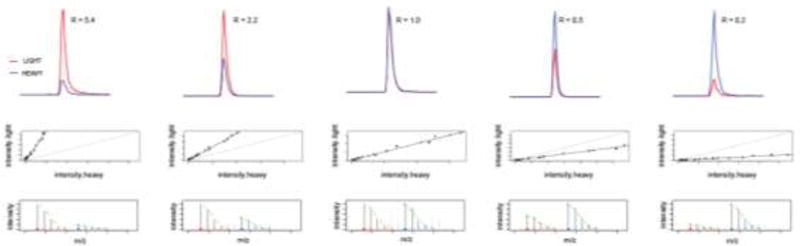
Representative data for one cysteine-containing peptide (K.AYDATC*LVK.A from S-formylglutathione hydrolase (ESD)) identified in an experiment in which Azo-L and Azo-H labeled proteomes were mixed together in 5:1, 2:1, 1:1, 1:2 and 1:5 ratios. The top panel shows the extracted ion chromatograms for the light and heavy species (in red and blue respectively), with the corresponding calculated light:heavy ratios (R). The middle panel shows plots of the intensity of the light species against the intensity of the heavy species, showing ideal co-elution of the two isotopically labeled peptides. The bottom panel shows the isotope envelope of the light and heavy species, demonstrating an ideal match to the expected isotope pattern (in green).
6. Discussion
The field of activity-based protein profiling applies active-site directed chemical probes to inform on the activity of a specific protein family. Here, we present a related technology in the area of ‘reactivity-based’ profiling, where instead of grouping proteins by functional class; we globally profile the reactivity of a particular amino acid. We focus on cysteine due to the abundance of this amino acid in functional loci; as sites of nucleophilic and redox catalysis, metal binding and regulation. The reactivity state of these functional cysteines directly informs on protein activity, and thereby enables the concurrent profiling of hundreds of proteins from diverse functional classes. Due to the susceptibility of cysteine to a variety of oxidative and lipid-based posttranslational modifications, analyzing the abundance of proteins containing functional cysteines does not often correlate with activity state. For this reason, we describe a chemical proteomic platform that is able to report on changes in cysteine reactivity as a means of identifying cysteine-mediated protein activity changes in two or more biological samples. This method couples the use of a general cysteine-reactive probe to a quantitative mass-spectrometry platform that utilizes an azobenzene cleavable linker for enrichment, selective release and quantification of probe-labeled peptides. Methods such as this, that globally quantify cysteine reactivity changes, can inform on targets of oxidative stress and the consequent protein-activity changes within a biological system.
Acknowledgments
This work was financially supported by the Smith Family Foundation, the Damon Runyon Cancer Research Foundation (DRR-18-12), NIH grants 1R01GM117004 and 1R01GM118431-01A1, and Boston College.
4. Notes
The IA probe is made in-house.14
TCEP should be stored in 4°C and the solution made fresh immediately prior to the CuAAC step.
Azo-L/Azo-H tags are made in-house.18
When comparing cysteine reactivity in two different proteomes, ensure that the two proteome samples are accurately normalized in terms of total protein concentrations.
This step should be performed at 4°C or on ice and the methanol should be pre-chilled at −20°C.
In the last water wash, carefully remove the supernatant using a pipette. The beads should appear faintly yellow tinted at this stage due to enrichment of the Azo-tagged proteins.
All reagents used in this step should be made fresh. Avoid vortexing and flip the tube gently instead to resuspend the beads.
Dilute the freshly made 6 M Urea/PBS to 2 M Urea/PBS using PBS. The trypsin solution should be made directly before addition to the sample and is good for reuse within a week if stored at 4°C.
50 mM sodium hydrosulfite solution should be made fresh with PBS. Using water instead of PBS slightly reduces the cleavage efficiency.
After incubating with sodium hydrosulfite solution, the beads should turn white as an indication of successful cleavage of the azobenzene linker.
References
- 1.Giles NM, Giles GI, Jacob C. Multiple roles of cysteine in biocatalysis. Biochem Biophys Res Commun. 2003;300:1–4. doi: 10.1016/s0006-291x(02)02770-5. [DOI] [PubMed] [Google Scholar]
- 2.Pace NJ, Weerapana E. Diverse functional roles of reactive cysteines. ACS Chem Biol. 2013;8:283–96. doi: 10.1021/cb3005269. [DOI] [PubMed] [Google Scholar]
- 3.Smith BC, Marletta MA. Mechanisms of S-nitrosothiol formation and selectivity in nitric oxide signaling. Curr Opin Chem Biol. 2012;16:498–506. doi: 10.1016/j.cbpa.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddie KG, Carroll KS. Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol. 2008;12:746–54. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 5.Tom CT, Martin BR. Fat chance! Getting a grip on a slippery modification. ACS Chem Biol. 2013;8:46–57. doi: 10.1021/cb300607e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–69. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 7.Fritz KS, Petersen DR. Exploring the biology of lipid peroxidation-derived protein carbonylation. Chem Res Toxicol. 2011;24:1411–9. doi: 10.1021/tx200169n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. S-Nitrosylation of mitochondrial caspases. J Cell Biol. 2001;154:1111–6. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–74. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 10.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2012;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leonard SE, Carroll KS. Chemical ‘omics’ approaches for understanding protein cysteine oxidation in biology. Curr Opin Chem Biol. 2010 doi: 10.1016/j.cbpa.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 12.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001 doi: 10.1126/stke.2001.86.pl1. pl1. [DOI] [PubMed] [Google Scholar]
- 13.Hang HC, Linder ME. Exploring protein lipidation with chemical biology. Chem Rev. 2011;111:6341–58. doi: 10.1021/cr2001977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Abstr Pap Am Chem S. 2011;241 doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weerapana E, Speers AE, Cravatt BF. Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)--a general method for mapping sites of probe modification in proteomes. Nat Protoc. 2007;2:1414–25. doi: 10.1038/nprot.2007.194. [DOI] [PubMed] [Google Scholar]
- 16.Deng X, Weerapana E, Ulanovskaya O, Sun F, Liang H, Ji Q, Ye Y, Fu Y, Zhou L, Li J, Zhang H, Wang C, Alvarez S, Hicks LM, Lan L, Wu M, Cravatt BF, He C. Proteome-wide quantification and characterization of oxidation-sensitive cysteines in pathogenic bacteria. Cell Host Microbe. 2013;13:358–70. doi: 10.1016/j.chom.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C, Weerapana E, Blewett MM, Cravatt BF. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods. 2013 doi: 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian Y, Martell J, Pace NJ, Ballard TE, Johnson DS, Weerapana E. An isotopically tagged azobenzene-based cleavable linker for quantitative proteomics. Chembiochem. 2013;14:1410–4. doi: 10.1002/cbic.201300396. [DOI] [PubMed] [Google Scholar]
- 19.Pace NJ, Weerapana E. A competitive chemical-proteomic platform to identify zinc-binding cysteines. ACS Chem Biol. 2014;9:258–65. doi: 10.1021/cb400622q. [DOI] [PubMed] [Google Scholar]
- 20.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 21.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–89. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]