

Abstract
Precolibactins and colibactins represent a family of natural products that are encoded by the clb gene cluster and are produced by certain commensal, extraintestinal, and probiotic E. coli. clb+ E. coli induce megalocytosis and DNA double-strand breaks in eukaryotic cells, but paradoxically, this gene cluster is found in the probiotic Nissle 1917. Evidence suggests precolibactins are converted to genotoxic colibactins by colibactin peptidase (ClbP)-mediated cleavage of an N-acyl- D-Asn side chain, and all isolation efforts have employed ΔclbP strains to facilitate accumulation of precolibactins. It was hypothesized that colibactins form unsaturated imines that alkylate DNA by cyclopropane ring opening (2 → 3). However, as no colibactins have been isolated, this hypothesis has not been tested experimentally. Additionally, precolibactins A–C (7–9) contain a pyridone that cannot generate the unsaturated imines that form the basis of this hypothesis. To resolve this, we prepared 13 synthetic colibactin derivatives and evaluated their DNA binding and alkylation activity. We show that unsaturated imines, but not the corresponding pyridone derivatives, potently alkylate DNA. The imine, unsaturated lactam, and cyclopropane are essential for efficient DNA alkylation. A cationic residue enhances activity. These studies suggest that precolibactins containing a pyridone are not responsible for the genotoxicity of the clb cluster. Instead, we propose that these are off-pathway fermentation products produced by a facile double cyclodehydration route that manifests in the absence of viable ClbP. The results presented herein provide a foundation to begin to connect metabolite structure with the disparate phenotypes associated with clb+ E. coli.
Graphical Abstract
INTRODUCTION
Precolibactins and colibactins are natural products produced by select commensal, extraintestinal, and probiotic E. coli. The metabolites are encoded by a hybrid polyketide synthase–nonribosomal peptide synthetase (PKS–NRPS) gene cluster termed clb or pks.1 clb+ E. coli strains induce DNA damage in eukaryotic cells and are thought to promote colorectal cancer formation,1c,2 but the gene cluster is also found in the probiotic strain Nissle 1917, which is used in Europe for the treatment of ulcerative colitis, diarrhea, and other gastrointestinal disorders.3 Mature precolibactins are substrates for the 12-transmembrane multidrug and toxic compound extrusion transporter ClbM, which mediates their transfer to the bacterial periplasm.4 There, the colibactin peptidase ClbP converts precolibactins to genotoxic colibactins via removal of an N-acyl-D-asparagine side chain.5 Mutation of ClbP abolishes cellular DNA damaging-activity,2a,5b and N-myristoyl-D-asparagine and closely related analogues have been identified in wild type clb+ E. coli cultures.5d,e Whether the differential production of biosynthetically related but distinct metabolites, or other factors (such as the requirement for cell-to-cell contact to observe cytopathic effects2a,6) underlie the seemingly contradictory phenotypes associated with the clb gene cluster, remains unresolved.
We have focused on understanding the molecular basis of colibactin-induced DNA damage. Advanced precolibactins arise from linear precursors of the generalized structure shown as 1 (Scheme 1). The linear precursors were suggested to transform to unsaturated lactams 2 that are processed by ClbP to generate unsaturated iminium ions 3 (colibactins), which alkylate DNA by cyclopropane ring-opening (gray pathway).7,8 However, this mechanistic hypothesis is ostensibly incompatible with subsequent isolation9 and synthesis10 efforts that lead to the identification and unequivocal structural assignment of precolibactins A (7),11 B (8), and C (9), which contain a pyridone residue (Figure 1). Precolibactins A–C (7–9) were obtained from clbP mutant strains; these deletion strains were employed to promote accumulation of the precolibactin metabolites. If 7–9 are the genotoxic precursors, the data outlined above5 suggests that amines such as 5 resulting from ClbP-mediated processing in the wild type strains are responsible for the cytopathicity of the clb cluster, as these cannot readily convert to unsaturated iminium ions such as 3. Precolibactin C (9) was demonstrated to be a substrate for ClbP.10
Scheme 1. Proposed Mechanisms of Action and Divergent Reactivity of Precolibactin Precursorsa.

aGray ball in 1–6 denotes the variable region of the structures.
Figure 1.

Structures of precolibactins A (7), B (8), and C (9).
In earlier synthetic work, we showed that the double dehydrative cyclization of the relatively stable N-acylated linear precursors (1) to pyridones such as 7–9 was facile under mildly acidic or basic conditions (cf., 1 → 2 → 4, Scheme 1).10 The unsaturated lactam intermediates (2) could be detected by LC/MS analysis, but they were not isolable, arguing against their interception by ClbP in the biosynthesis. We reasoned that the colibactins may instead form by ClbP processing of isolable linear precursors 1 directly. Sequential cyclodehydration reactions proceeding through the vinylogous ureas 6 would then provide 3 (red pathway). It follows from this analysis that precolibactins A–C (7–9) are non-natural cyclization products deriving from the absence of ClbP in the producing organisms, and are unlikely to be genotoxic. To test this hypothesis, we modified our synthetic strategy10 to allow access to 13 deacylated pyridone derivatives 5 and the analogous unsaturated iminium ions 3. We show that the iminium ions are potent DNA alkylation agents while the corresponding pyridone structures are not. In addition, we rigorously define the structure–function relationships of 3 that are required for or enhance DNA alkylation activity. Our synthetic studies support an alternative biosynthetic pathway involving the intermediacy of the vinylogous amide 6 en route to 3. Collectively, our data lend further support to the hypothesis that unsaturated iminium ions 3 are responsible for the genotoxic effects of the clb gene cluster and support the conclusion that precolibactins A–C (7–9) (and other pyridone-containing isolates) are off-pathway fermentation products derived from the absence of a functional clbP gene. This work constitutes the first structure–function studies of colibactin metabolites and provides a foundation to begin to connect the disparate phenotypic effects of the clb cluster with metabolite structure.
RESULTS
Comparison of DNA Binding and Alkylation Activities of Unsaturated Iminium Ion and Pyridone Structures
To assess the activities of candidate colibactin scaffolds, we required a route that would provide access to unsaturated imine and pyridone derivatives lacking the natural N-myristoyl-D-Asn prodrug fragment. Synthesis of the unsaturated imine derivatives was a significant challenge since we have previously shown that base- or acid-catalyzed cyclization of the linear precursors proceeded rapidly to the pyridone products.10 In addition, we targeted synthetic derivatives containing a cationic residue appended to a bithiazole fragment. The bithiazole of precolibactin C (9) is similar to that found in the DNA-cleavage agent bleomycin, and studies of that metabolite have established the importance of this substructure for DNA affinity.12 In addition, it has been shown that an α-aminomalonate building block is incorporated into the colibactin structure;9b,13 we speculated that this residue could enhance DNA affinity through an electrostatic interaction, similar to bleomycin12,14 and other known DNA-targeting agents.15 As the presumed natural C-terminal colibactin cationic substituent remains unknown, we initially targeted an N-(β-dimethylaminoethyl)amide derivative, as this is a common substructure in agents that target DNA. We later prepared a series of other derivatives to further explore this region and optimize activity (vide infra).
After some experimentation, we identified the linear colibactin derivative 12 as a common intermediate that could be selectively cyclized to provide either unsaturated imine or pyridone products (Scheme 2). Silver-mediated coupling of the β-ketothioester 10 (prepared in one step and 53% yield by the addition of lithium ethyl thioacetate to tert-butyl (S)-2-methyl- 5-oxopyrrolidine-1-carboxylate,16 see the Supporting Information) with the β-ketoamide 1110 provided the linear precursor 12 (1–3 g scale). The linear precursor 12 cyclized to a variable extent to the vinylogous imide 13 upon purification by anion exchange chromatography. To effect quantitative cyclization, the purified mixture of 12 and 13 was dissolved in 0.5% formic acid–5% methanol–acetonitrile and concentrated (3×). Following this protocol, the vinylogous imide 13 was obtained in 87% isolated yield (over two steps) and >95% purity. Coupling of the carboxylic acid function with N,N-dimethylethylenediamine mediated by propylphosphonic anhydride (T3P)17 provided the amide 14a (93%). Carbamate cleavage (trifluoroacetic acid) followed by neutralization with saturated aqueous sodium bicarbonate solution (to promote cyclodehydration) then provided the key unsaturated imine 15a (62%).
Scheme 2. Synthesis of the Unsaturated Imine 15a and the Pyridone 17a and Structures of the N-Methyl Amide Derivatives 15b and 17ba.

aStructures of the N-methylamides 15b and 17b. Reagents and conditions: (a) silver trifluoroacetate (AgOTFA), Et3N, DMF, 0 °C; (b) concentrate from 0.5% HCO2H–5% CH3OH–CH3CN, 23 °C, 87% (two steps); (c) propylphosphonic anhydride solution (T3P), N-methylmorpholine, N,Ndimethylethylenediamine, THF, 23 °C, 93%; (d) trifluoroacetic acid (TFA), CH2Cl2, 0 °C; aqueous NaHCO3, 23 °C, 62%; (e) K2CO3, CH3OH, 0 → 23 °C, 78% (two steps); (f) T3P, N-methylmorpholine, N,N-dimethylethylenediamine, THF, 23 °C, 46%; (g) TFA, CH2Cl2, 0 °C, 86%.
Alternatively, the direct addition of potassium carbonate (5 equiv) and methanol10 to the solution of the unpurified coupling product 12 induced double cyclodehydration to provide the pyridone 16 (78% from 10 and 11). T3P-mediated coupling with N,N-dimethylethylenediamine provided an amide (not shown) that was treated with trifluoroacetic acid to yield the amine 17a (40%, two steps). The N-methylamide derivatives 15b and 17b were targeted to directly evaluate the significance of a cationic terminal residue on DNA binding and damaging activity. These were prepared by strictly analogous sequences (see the Supporting Information).
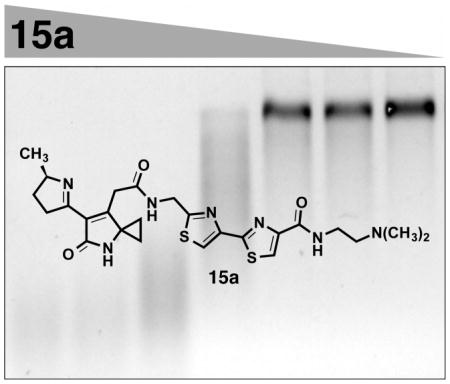
We evaluated the activity of 15a, 15b, 17a, and 17b in a DNA alkylation assay. Plasmid pBR322 DNA was treated with EcoR1-HF in Cutsmart buffer, and the resulting linearized DNA was incubated with 15a, 15b, 17a, or 17b for 15 h at 37 °C. The DNA was then denatured, and the sample was eluted on a 1% neutral agarose gel. DNA was visualized with SybrGold. Graphs of grayscale as a function of pixel location are presented in Figures S1–S5 of the Supporting Information. Cisplatin (CP, 100 μM) and methylmethanesulfonate (MMS, 500, 100, or 10 μM) were used as positive controls for DNA cross-linking and alkylation, respectively. As shown in Figure 2A, neither of the pyridone derivatives 17a or 17b displayed measurable activity in this assay at concentrations up to 500 μM. By comparison, both unsaturated imines 15a and 15b extensively alkylated DNA. The streaking of DNA induced by 15a and 15b parallels that observed with 500 μM of MMS and derives from the presence of shorter DNA strands that are formed by thermal or alkaline denaturation of extensively alkylated (or in the case of MMS, methylated) DNA.18 Further evidence for direct alkylation by 15a and 15b is presented in the cross-linking study that follows. The derivative 15a, which bears a tertiary amine substituent that can engage in an electrostatic interaction with the deoxyribose backbone, was significantly more active than the N-methylamide 15b. These data indicate that the pyridone residue is unlikely to constitute the genotoxic pharmacophore of the colibactins and establish the unsaturated lactam or imine, and cationic terminus as key functional groups in DNA alkylation activity.
Figure 2.

(A) DNA alkylation assay employing linearized pBR322 DNA and the pyridone derivatives 17a or 17b or the unsaturated imines 15a or 15b. Conditions: Linearized pBR322 DNA (20 μM in base pairs (bps)), 15a (100, 10, 1, 0.5, or 0.1 μM), 15b (500, 100, 10, or 1 μM), 17a (500 or 100 μM), or 17b (500 or 100 μM), 37 °C, 15 h. Cisplatin (CP; 100 μM) and methylmethanesulfonate (MMS; 500, 100, or 10 μM) were used as positive controls for DNA cross-linking and alkylation, respectively. DNA was visualized using SybrGold. B. DNA alkylation assay employing linearized pBR322 DNA and the unsaturated imine 15a. Conditions: Linearized pBR322 DNA (20 μM in base pairs), 15a (100, 10, 1, 0.1, 0.05, 0.01, or 0.005 μM), 37 °C, 15 h.
A full dose response of the most potent derivative 15a was conducted. As shown in Figure 2B, extensive DNA alkylation was observed at concentrations as low as 100 nM, and small amounts of DNA damage were detected using 50 or 10 nM of 15a. At 5 nM of 15a the extent of DNA damage was negligible.
We conducted a DNA melting temperature analysis to determine if the variance in DNA alkylation activity between the pyridones and the unsaturated imines was due to differences in DNA binding affinity. The hyperchromicity of calf thymus DNA (ctDNA) with increasing temperature was measured at 260 nm in the presence of varying concentrations of 15a or 17a. The Tm was defined as the temperature at which half of the duplex DNA was denatured and was determined by the maximum of the first derivative of the thermal denaturation profile. As shown in Figure 3A, treatment of ctDNA with 17a led to a dose-dependent increase in the melting temperature; an increase in Tm of 3.8 °C was observed at a ratio of 17a to base pairs (bps) of 4.0. By comparison, the unsaturated imine 15a exhibited time-dependent effects on duplex stability (Figure 3B). At times less than 1 h, stabilization of the duplex was observed, but upon longer incubations (3–15 h) duplex stability decreased. We hypothesized that this was due to an initial binding of 15a to the duplex followed by alkylation and slow degradation. To probe this, we repeated the plasmid alkylation assay using 15a and evaluated the extent of DNA degradation as a function of time. As shown in Figure 3C, degradation activity correlated approximately with the decrease in duplex stability observed in the melting point experiments (Figure 2B). These studies establish that the pyridone 17a can bind noncovalently to DNA. Therefore, the variance in DNA alkylation activity cannot be ascribed to lack of affinity of 17a with DNA.
Figure 3.

(A) Increase in the melting temperature of calf thymus DNA treated with increasing amounts of the pyridone 17a. Conditions: 2.09 mM NaH2PO4, 7.13 mM Na2HPO4, 928 μM Na2 EDTA, 1.01 mM DMSO, pH 7.18. The pyridone 17a was incubated with ctDNA for 3 h prior to UV thermal denaturation experiments (260 nm, heating rate: 0.5 °C/min). [DNA] = 32.0 mM bps. (B) Time-dependent modulation of the melting temperature of calf thymus DNA treated with 1 or 2 bp equiv of the imine 15a. Conditions: 2.09 mM NaH2PO4, 7.13 mM Na2HPO4, 928 μM Na2EDTA, 1.01 mM DMSO, pH 7.18. The imine 15a was incubated with ctDNA for 5 min, 1, 3, 6, or 15 h prior to UV thermal denaturation experiments (260 nm, heating rate: 0.5 °C/min). [DNA] = 32.0 mM bps. (C) Time-dependent DNA alkylation assay employing linearized pBR322 DNA and the unsaturated imine 15a. Conditions: Linearized pBR322 DNA (20 μM in base pairs), 15a (1 μM), 37 °C, 0.1–15 h.
Structure–Function Relationships of Unsaturated Imines
To gain further insights into the functional group requirements for DNA alkylation and to verify the observed activities, we prepared the dimer 15c and the gem-dimethyl derivative 15d (Figure 4A). As shown in Figure 4B, incubation of linearized pBR322 DNA with 10 μM of the dimer 15c and monitoring the mixture as a function of time revealed a clear cross-linked band at 3 h. Consistent with the time-dependent alkylation assay shown in Figure 3C, this cross-linked band diminished at later time points (6 or 15 h), suggesting extensive alkylation and degradation of the duplex. The gem-dimethyl derivative 15d did not alkylate DNA at concentrations up to 500 μM. These data further validate the nature of the DNA lesion as alkylation and indicate that the cyclopropane ring is essential for this activity.
Figure 4.

(A) Structures of the dimer 15c and the gem-dimethyl derivative 15d. (B) DNA alkylation assay employing linearized pBR322 DNA and the dimer 15c or the gem-dimethyl derivative 15d. Conditions: Linearized pBR322 DNA (20 μM in base pairs), 15c (10 μM) or 15d (500 or 100 μM), 37 °C, 0.1–15 h.
To gain insights into the molecular mechanism of alkylation, we studied the reactivity of 15b in vitro using propanethiol as a model nucleophile (Scheme 3). The N-methylamide 15b was chosen in preference to the β-(dimethylamino)ethyl amide 15a to simplify isolation. Treatment of the methylamide 15b with excess propanethiol and p-toluenesulfonic acid in 6:1 acetonitrile–N,N-dimethylformamide at 23 °C provided the ring-opened product 18 in 34% isolated yield. The adduct 18 was unstable but was characterized by NMR (1H, 13C, gCOSY, gHSQC, gHMBC) and LC/MS analyses. Cyclopropane ring-opening was evidenced by loss of the resonances at 1.37 and 1.68 ppm (positions a and b in 15b) and generation of a four-proton system centered at 2.57 ppm (positions a, b in 18; see Figure S11 and Table S1).
Scheme 3. Ring-Opening of the Unsaturated Imine 15b by Propanethiola.

aConditions: p-toluenesulfonic acid monohydrate, CH3CN–propanethiol–DMF (6:2:1), 23 °C, 34%.
A series of additional compounds were prepared to further interrogate structure–function relationships and optimize DNA alkylation activity (Figure 5A). The derivatives 15e–15i were synthesized to probe the influence of the nature of the cationic residue on DNA alkylation activity. All compounds were found to potently alkylate DNA although qualitative differences in activity were observed depending on the nature of the cationic residue (guanidine < primary amine < tertiary amine; see Figure S12 for full data). Finally, to rigorously determine if iminium ion formation is necessary for efficient alkylation, we prepared the unsaturated lactam 19 and the unsaturated imine 15j. Both compounds contain an N-methyl substituent at the central amide residue. This was installed to prevent competitive cyclization of 19 to the corresponding pyridone under the assay conditions; 15j was prepared to confirm that this modification does not abolish DNA alkylation activity (as compared to 15a). As shown in Figure 5B, the lactam 19 showed weak DNA alkylation activity at 500 μM, while the imine derivative 15j was significantly more potent, leading to extensive decomposition of the DNA at low (10 or 1 μM) concentrations, as expected. These final experiments show that the monocyclized derivatives 2 do display some (albeit weak) DNA alkylation activity at high concentrations, but that the unsaturated iminium ion 3 is a significantly more potent electrophile.
Figure 5.

(A) Structures of the analogues 15e–15i, 19, and 15j. (B) DNA alkylation assay employing linearized pBR322 DNA and the unsaturated lactam 19 or the unsaturated imine 15j. Conditions: Linearized pBR322 DNA (20 μM in base pairs), 19 (500, 100, 10, or 1 μM) or 15j (500, 100, 10, or 1 μM), 37 °C, 15 h.
DISCUSSION
The colibactins are PKS–NRPS-derived natural products and have been implicated in the genotoxic effects of commensal and extraintestinal E. coli.2a–c They are formed from the precolibactins upon removal of an N-acyl-D-Asn side chain by the colibactin peptidase ClbP.5 It was proposed that colibactins may generate unsaturated iminium ions that alkylate DNA (see 3, Scheme 1).7 However, this hypothesis has been impossible to evaluate because no colibactins have been obtained from the producing organisms, as researchers have employed clbP mutants. This modification is advantageous inasmuch as it allows for the accumulation of candidate precolibactions, determination of their structures, and elucidation of colibactin structures (by inference). However, direct isolation of colibactins is not possible using this strategy.
Many of the advanced isolates reported to date, such as precolibactins A–C (7–9), contain a pyridone nucleus, and the conversion of this aromatic substructure to an unsaturated iminium ion (5 → 3, Scheme 1) seems unlikely. We reasoned that removal of ClbP and persistence of an N-acyl-D-Asn side chain engenders unnatural cyclization events leading to the production of the pyridones. This hypothesis was based on our previous synthetic studies, which established a facile double dehydrative cyclization of N-acylated linear precursors 1 to pyridones under mildly acidic or basic conditions.10 It follows from this line of reasoning that pyridone-containing structures are unlikely to be genotoxic. To test this hypothesis, we prepared the linear precursor 12 (Scheme 2) and elaborated it to 13 pyridone and unsaturated imine derivatives, in which the substituents throughout the molecules were systematically modified. The successful synthesis of these unsaturated imines allowed us to evaluate their DNA alkylation activity for the first time.
The alkylation assay shown in Figure 2 demonstrates that the unsaturated imines, but not the corresponding pyridones, are potent DNA alkylation agents. The presence of a terminal cationic substituent enhances DNA alkylation activity (cf., 15a and 15b, Figure 2A), as was expected based on literature precedent.15 These results are consistent with earlier observations that an α-aminomalonate-derived residue is present in fully functionalized colibactins and is required for genotoxic effects.9b,13 Presumably this residue forms the basis for a cationic substituent that serves the same role as the non-natural cationic functional groups employed herein. It may also be essential for cellular efflux and trafficking to eukaryotic cells.
Our data support ClbP-mediated deacylation as a key step to prevent formation of pyridone products and facilitate cyclization to unsaturated imines such as 3 (Scheme 1). While these were originally proposed to form from unsaturated lactams 2, our synthetic studies (Scheme 2) suggest they may be generated from vinylogous ureas 6 instead. Regardless of the precise mechanism of formation, our structure–function studies show that the unsaturated lactam and cyclopropane are both necessary for DNA alkylation activity (Figure 6). Formation of an unsaturated imine significantly enhances DNA alkylation activity (c.f., 19 and 15j, Figure 5) and a cationic terminal residue further increases the extent of DNA alkylation. The observed ring-opening of 15b by propanethiol (Scheme 3) is consistent with DNA alkylation by cyclopropane ring-opening. In an earlier report, a shunt metabolite containing the unsaturated lactam substructure found in 19 was shown to weakly cross-link DNA.7a The activity of 19 is partially consistent with this result: this compound weakly alkylated DNA, although we did not obtain evidence of cross-linking.
Figure 6.

Structural elements of colibactins that are essential to (b, c) or significantly enhance (a, d) DNA alkylation activity.
These experiments constitute the first molecular-level analysis of candidate colibactin structure reactivity and provide support for the hypothesis previously proposed to involve DNA alkylation by an unsaturated iminium ion intermediate.7 They also clearly illustrate that the molecules are susceptible to several different modes of reactivity. These reactivities vary as a function of substituents, and metabolites produced by ΔclbP strains, such as precolibactins A–C (7–9), may arise from nonnatural reaction pathways. A macrocyclic precolibactin recently isolated from a ΔclbP strain may also similarly derive from an unnatural macrocyclization route.13b
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (R01GM110506 to S.B.H., 1DP2-CA186575 to J.M.C., and 5T32GM06754 3-12 to J.R.P.) and Yale University is gratefully acknowledged. We thank Professor Alison M. Sweeney (The University of Pennsylvania) for assistance with ImageJ.
Footnotes
ORCID
Jaymin R. Patel: 0000-0002-4380-4805
Seth B. Herzon: 0000-0001-5940-9853
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b10354.
Schemes, figures, and table; detailed experimental procedures; characterization data for all new compounds (PDF)
References
- 1.For reviews, see: Trautman EP, Crawford JM. Curr Top Med Chem. 2016;16:1705. doi: 10.2174/1568026616666151012111046.Balskus EP. Nat Prod Rep. 2015;32:1534. doi: 10.1039/c5np00091b.Taieb F, Petit C, Nougayrede JP, Oswald E. EcoSal Plus. 2016 doi: 10.1128/ecosalplus.ESP-0008-2016.
- 2.Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Science. 2006;313:848. doi: 10.1126/science.1127059.Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède JP. Proc Natl Acad Sci U S A. 2010;107:11537. doi: 10.1073/pnas.1001261107.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C. Science. 2012;338:120. doi: 10.1126/science.1224820.Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. Gut Microbes. 2014;5:675. doi: 10.4161/19490976.2014.969989.Grasso F, Frisan T. Biomolecules. 2015;5:1762. doi: 10.3390/biom5031762.Fais T, Delmas J, Cougnoux A, Dalmasso G, Bonnet R. Gut Microbes. 2016;7:329. doi: 10.1080/19490976.2016.1155020.For a review of relationships between gut microbiota composition and cancer, see: Brennan CA, Garrett WS. Annu Rev Microbiol. 2016;70:395. doi: 10.1146/annurev-micro-102215-095513.
- 3.(a) Olier M, Marcq I, Salvador-Cartier C, Secher T, Dobrindt U, Boury M, Bacquié V, Penary M, Gaultier E, Nougayrède JP, Fioramonti J, Oswald E. Gut Microbes. 2012;3:501. doi: 10.4161/gmic.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Scaldaferri F, Gerardi V, Mangiola F, Lopetuso LR, Pizzoferrato M, Petito V, Papa A, Stojanovic J, Poscia A, Cammarota G, Gasbarrini A. World J Gastroenterol. 2016;22:5505. doi: 10.3748/wjg.v22.i24.5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mousa JJ, Yang Y, Tomkovich S, Shima A, Newsome RC, Tripathi P, Oswald E, Bruner SD, Jobin C. Nat Microbiol. 2016;1:15009. doi: 10.1038/nmicrobiol.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Dubois D, Baron O, Cougnoux A, Delmas J, Pradel N, Boury M, Bouchon B, Bringer MA, Nougayrede JP, Oswald E, Bonnet R. J Biol Chem. 2011;286:35562. doi: 10.1074/jbc.M111.221960. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cougnoux A, Gibold L, Robin F, Dubois D, Pradel N, Darfeuille-Michaud A, Dalmasso G, Delmas J, Bonnet R. J Mol Biol. 2012;424:203. doi: 10.1016/j.jmb.2012.09.017. [DOI] [PubMed] [Google Scholar]; (c) Brotherton CA, Balskus EP. J Am Chem Soc. 2013;135:3359. doi: 10.1021/ja312154m. [DOI] [PubMed] [Google Scholar]; (d) Bian X, Fu J, Plaza A, Herrmann J, Pistorius D, Stewart AF, Zhang Y, Muller R. ChemBioChem. 2013;14:1194. doi: 10.1002/cbic.201300208. [DOI] [PubMed] [Google Scholar]; (e) Vizcaino MI, Engel P, Trautman E, Crawford JM. J Am Chem Soc. 2014;136:9244. doi: 10.1021/ja503450q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Outer membrane vesicles from Nissle 1917 undergo clathrin-mediated endocytotosis in several human epithelial cell lines, suggesting the probiotic effects of Nissle 1917 may not require cell-to- cell contact. See: Canas MA, Gimenez R, Fabrega MJ, Toloza L, Baldoma L, Badia J. PLoS One. 2016;11:e0160374. doi: 10.1371/journal.pone.0160374.
- 7.(a) Vizcaino MI, Crawford JM. Nat Chem. 2015;7:411. doi: 10.1038/nchem.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brotherton CA, Wilson M, Byrd G, Balskus EP. Org Lett. 2015;17:1545. doi: 10.1021/acs.orglett.5b00432. [DOI] [PubMed] [Google Scholar]
- 8.This mode of reactivity bears parallels to DNA alkylation by duocarmycin, CC-1065, and yatakemycin. For reviews, see: Boger DL, Garbaccio RM. Acc Chem Res. 1999;32:1043.Tichenor MS, Boger DL. Nat Prod Rep. 2008;25:220. doi: 10.1039/b705665f.
- 9.(a) Li ZR, Li Y, Lai JY, Tang J, Wang B, Lu L, Zhu G, Wu X, Xu Y, Qian PY. ChemBioChem. 2015;16:1715. doi: 10.1002/cbic.201500239. [DOI] [PubMed] [Google Scholar]; (b) Zha L, Wilson MR, Brotherton CA, Balskus EP. ACS Chem Biol. 2016;11:1287. doi: 10.1021/acschembio.6b00014. [DOI] [PubMed] [Google Scholar]
- 10.Healy AR, Vizcaino MI, Crawford JM, Herzon SB. J Am Chem Soc. 2016;138:5426. doi: 10.1021/jacs.6b02276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The structure of precolibactin A was originally predicted in ref 7a and was later revised to that shown as 7 in ref 10.
- 12.For reviews, see: Stubbe J, Kozarich JW, Wu W, Vanderwall DE. Acc Chem Res. 1996;29:322.Boger DL, Cai H. Angew Chem, Int Ed. 1999;38:448. doi: 10.1002/(SICI)1521-3773(19990215)38:4<448::AID-ANIE448>3.0.CO;2-W.
- 13.(a) Brachmann AO, Garcie C, Wu V, Martin P, Ueoka R, Oswald E, Piel J. Chem Commun. 2015;51:13138. doi: 10.1039/c5cc02718g. [DOI] [PubMed] [Google Scholar]; (b) Li ZR, Li J, Gu JP, Lai JY, Duggan BM, Zhang WP, Li ZL, Li YX, Tong RB, Xu Y, Lin DH, Moore BS, Qian PY. Nat Chem Biol. 2016;12:773. doi: 10.1038/nchembio.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Shipley JB, Hecht SM. Chem Res Toxicol. 1988;1:25. doi: 10.1021/tx00001a004. [DOI] [PubMed] [Google Scholar]; (b) Carter BJ, Reddy KS, Hecht SM. Tetrahedron. 1991;47:2463. [Google Scholar]
- 15.For reviews, see: Tse WC, Boger DL. Chem Biol. 2004;11:1607. doi: 10.1016/j.chembiol.2003.08.012.Paul A, Bhattacharya S. Curr Sci. 2012;102:212.Sirajuddin M, Ali S, Badshah A. J Photochem Photobiol, B. 2013;124:1. doi: 10.1016/j.jphotobiol.2013.03.013.
- 16.Tanaka A, Usuki T. Tetrahedron Lett. 2011;52:5036. [Google Scholar]
- 17.For a review, see: Basavaprabhu, Vishwanatha TM, Panguluri NR, Sureshbabu VV. Synthesis. 2013;45:1569.
- 18.Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman ASH, Helleday T. Nucleic Acids Res. 2005;33:3799. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.