

A comparative effectiveness trial indicates that dorsal root ganglion stimulation provided a higher rate of treatment success with less postural variation in paresthesia intensity compared to spinal cord stimulation.
Keywords: Chronic pain, Neurostimulation, Complex regional pain syndrome, Causalgia, Dorsal root ganglion stimulation
Abstract
Animal and human studies indicate that electrical stimulation of dorsal root ganglion (DRG) neurons may modulate neuropathic pain signals. ACCURATE, a pivotal, prospective, multicenter, randomized comparative effectiveness trial, was conducted in 152 subjects diagnosed with complex regional pain syndrome or causalgia in the lower extremities. Subjects received neurostimulation of the DRG or dorsal column (spinal cord stimulation, SCS). The primary end point was a composite of safety and efficacy at 3 months, and subjects were assessed through 12 months for long-term outcomes and adverse events. The predefined primary composite end point of treatment success was met for subjects with a permanent implant who reported 50% or greater decrease in visual analog scale score from preimplant baseline and who did not report any stimulation-related neurological deficits. No subjects reported stimulation-related neurological deficits. The percentage of subjects receiving ≥50% pain relief and treatment success was greater in the DRG arm (81.2%) than in the SCS arm (55.7%, P < 0.001) at 3 months. Device-related and serious adverse events were not different between the 2 groups. Dorsal root ganglion stimulation also demonstrated greater improvements in quality of life and psychological disposition. Finally, subjects using DRG stimulation reported less postural variation in paresthesia (P < 0.001) and reduced extraneous stimulation in nonpainful areas (P = 0.014), indicating DRG stimulation provided more targeted therapy to painful parts of the lower extremities. As the largest prospective, randomized comparative effectiveness trial to date, the results show that DRG stimulation provided a higher rate of treatment success with less postural variation in paresthesia intensity compared to SCS.
1. Introduction
The prevalence of neuropathic pain refractory to the current standard of care has been estimated to be 1.5% of the general population.26 Spinal cord stimulation (SCS), for which electrodes are placed into the dorsal epidural space, is an available treatment of a variety of chronic neuropathic pain conditions such as failed back surgery syndrome and complex regional pain syndrome (CRPS).8
Specific challenges for SCS remain, especially for pain conditions such as CRPS I and causalgia that differ by etiology and symptom profile from other chronic pain syndromes. An estimated 40% to 50% of CRPS subjects achieved clinically meaningful pain relief with SCS.11,14 Similar rates of successful pain relief are reported for heterogeneous populations that contain a significant CRPS population.23 Less than optimal results for some patients may be due to limitations of the selective targeting capabilities of SCS, unpleasant paresthesia, or from different mechanisms of action.
Lack of precision with SCS is attributed to shunting of energy by the cerebral spinal fluid, positional variations in stimulation, segmentation of spinal sensory input, and lead migrations postimplantation.18 In some cases, these challenges can be addressed with improved surgical techniques and device programming, but pain related to CRPS and causalgia remains difficult to treat; many SCS patients do not achieve high-level pain relief, despite efforts to improve techniques and programming.14
The dorsal root ganglion (DRG) plays a key role in the development and maintenance of neuropathic pain.13 The DRG, located between every spinal nerve and the spinal cord on the posterior root, houses the somas of the primary sensory neurons. These somas process and transmit sensory information from the periphery to the central nervous system. Animal models of chronic pain have shown that pathophysiologic changes occur in the DRG, including altered electrophysiological membrane properties, altered expression of integral membrane proteins, and altered expression of various genes that contribute to the hyperexcitability of neurons.15 The combination of the DRG's sensory function and accessibility through familiar epidural approaches make it an ideal target for neurostimulation. Pain therapies targeting the DRG included radiofrequency frequency ablation, steroid injections, and ganglionectomy.8
Initial evidence with 8 CRPS patients suggested that DRG stimulation may be successful in a larger proportion of subjects than SCS (71% vs 50%).28 Thus, the ACCURATE study, a randomized, controlled, multicenter trial, evaluated DRG stimulation compared to SCS stimulation for the treatment of chronic, intractable pain of the lower limbs attributed to CRPS or causalgia.
2. Methods
Under an Investigational Device Exemption, the ACCURATE study was designed as a prospective, randomized, controlled, multicenter study to evaluate the safety and efficacy of the DRG stimulation compared to traditional SCS for subjects with CRPS or causalgia (ClinicalTrials.gov: NCT01923285). The study was conducted in 22 US sites. Prior to any study initiation, all sites obtained approval from the institutional review board, and subjects were enrolled only after informed consent was obtained.
2.1. Patient selection
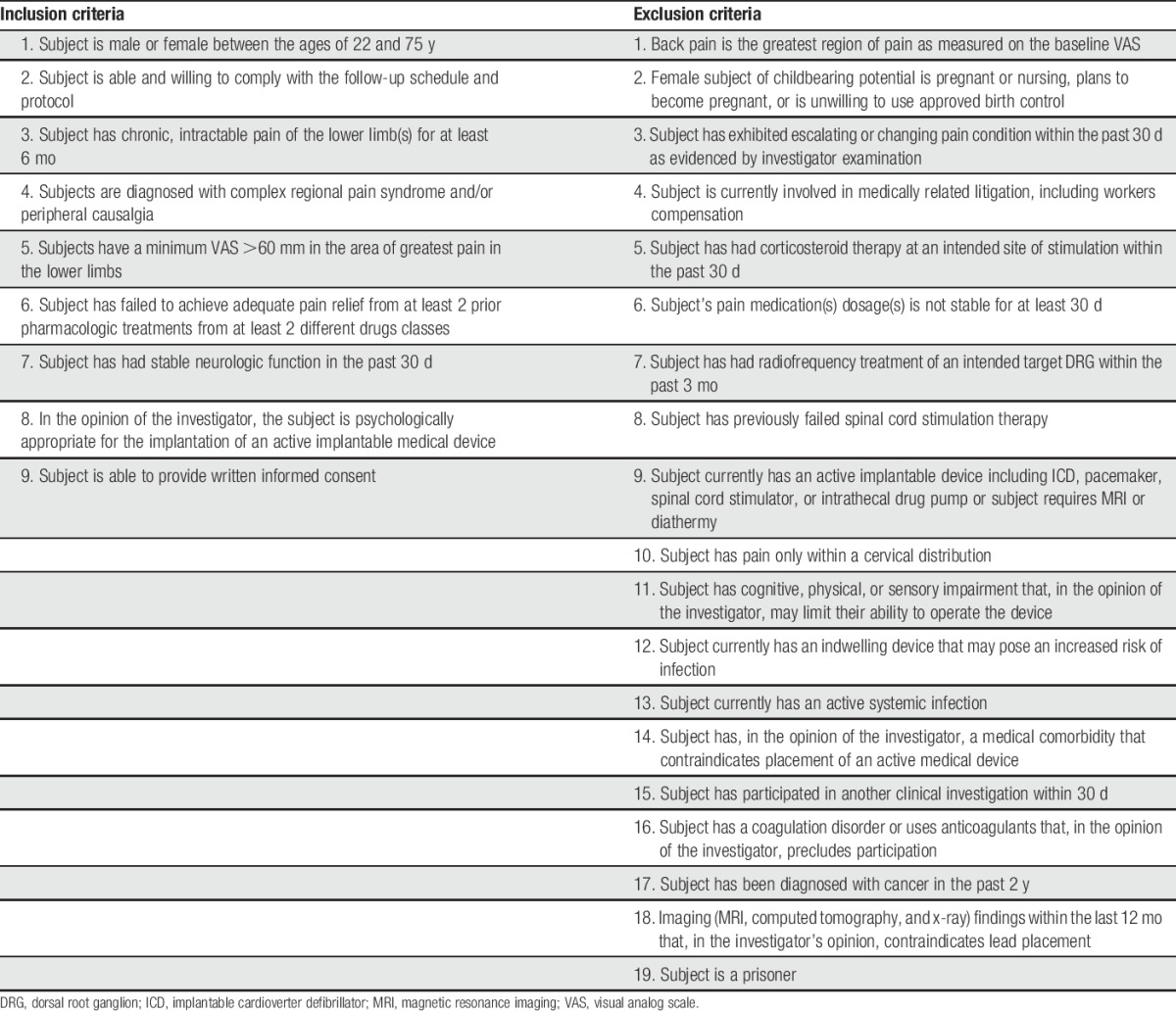
Subjects who had chronic, intractable neuropathic pain of the lower limbs associated with a diagnosis of CRPS or causalgia were screened and determined to be eligible according to the inclusion or exclusion criteria of the study (Table 1). Patients were diagnosed with CRPS type 1 based on the Budapest criteria.12 Causalgia was defined as a painful condition arising from damage to a nerve resulting in chronic pain, generally restricted to the innervation pattern of the damaged nerve or nerves, which may or may not have secondary symptoms.25 The diagnosis, in every case, was confirmed by an experienced medical monitor (N.M.) for strict adherence to these diagnostic criteria. Briefly, eligible subjects were naive to stimulation, had chronic, intractable pain for at least 6 months, tried and failed at least 2 prior pharmacologic treatments from 2 different drug classes, had stable neurologic function 30 days prior to screening, and were free from psychological pathology that contraindicated an implantable device. Subjects with changing or escalating pain condition or unstable use of pain medication 30 days prior to enrollment were not considered eligible to participate in the study. All subjects' medical, psychological, and imaging records were evaluated by an independent medical monitor to ensure appropriate patient selection.
Table 1.
Inclusion or exclusion criteria.
2.2. Study design
After signing informed consent, subjects underwent a baseline evaluation to determine enrollment eligibility. After enrollment, subjects were randomized to either DRG stimulation (DRG group) or traditional SCS (SCS group) in a 1:1 ratio. Randomization was based on random, permuted blocks and stratified by study center. The study's centralized electronic data collection system provided the subjects' randomized group assignments after subjects were enrolled. Subjects, investigators, and study site staff were not blinded to subjects' assigned therapy. Subjects proceeded to a temporary trial stimulation phase (ranging from 3 to 30 days based on each site's standard of care), using the device type stipulated by their randomization. The average trial stimulation phase in the DRG group was 5.8 (SD 2.8) days and 5.8 (SD 5.1) days for the SCS group (P = 0.206, Wilcoxon test).
Successful trial stimulation was determined by the subject achieving at least a 50% lower limb pain relief during the trial phase and expressing a desire to go on to a permanent implant. Subjects who were successful during the trial phase were eligible to continue on to permanent implantation. Subjects who failed the trial stimulation phase were exited from the study. However, data from the trial failures were included as treatment failures for the composite treatment success end point at 3 months and at subsequent time points through 12 months. Subjects in both arms, who achieved a successful outcome during the trial phase, were implanted with a permanent device and were followed for 12 months, with follow-ups at 3, 6, 9, and 12 months postimplant. Subjects were not allowed to change the maximum daily dose of their prescribed chronic lower limb pain medications from baseline to the 3-month follow-up visit at which time the primary and secondary end points were ascertained. Postoperative reprogramming to optimize therapy was allowed for both groups at any time during the study, per standard of care for neuromodulation devices. Programming occurred by respective companies (Medtronic and Spinal Modulation) under the guidance of appropriate clinical and technical industry personnel.
2.3. Description of devices and implant procedures
Dorsal root ganglion stimulation was delivered by the AXIUM Neurostimulator System (Spinal Modulation; LLC, Menlo Park, CA, a wholly owned subsidiary of St Jude Medical), which was recently approved by the US Food and Drug Administration for spinal column stimulation via epidural and intraspinal lead access to the DRG as an aid in the management of moderate to severe chronic intractable pain of the lower limbs in adult patients with CRPS type I and causalgia. The system is composed of percutaneous leads designed to stimulate the DRG, an external trial pulse generator, and an implantable pulse generator.
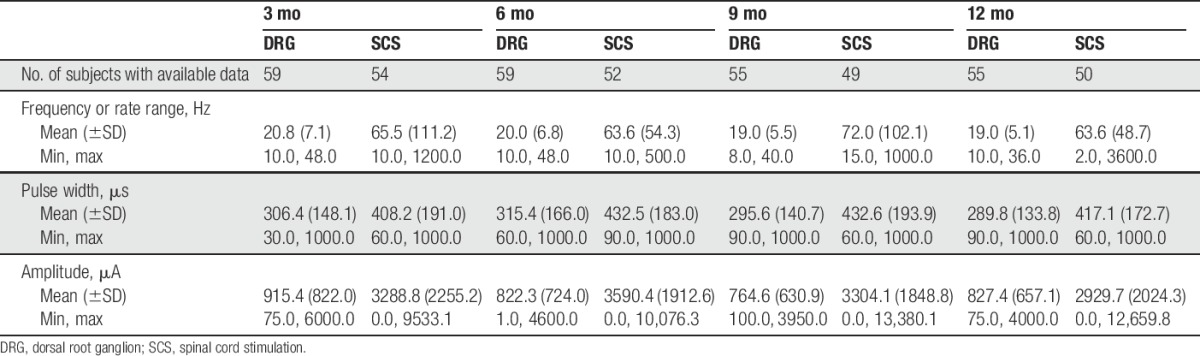
Traditional SCS was delivered with a commercially available system (RestoreUltra and RestoreSensor; Medtronic, Minneapolis, MN) indicated for a number of chronic pain conditions including CRPS I and causalgia. Both devices were programmed by separate technicians for each arm such that the programming was performed by experienced personnel for the specific device to achieve optimal analgesia. See Table 2 for a summary of programming parameters used during the study for both devices.
Table 2.
Programming settings.
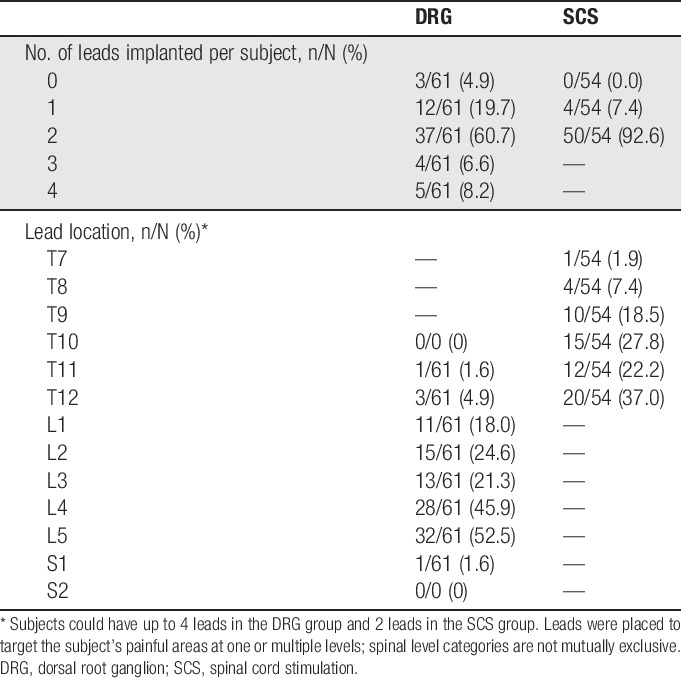
Standard procedures for trial and permanent implantations were used in the study. Dorsal root ganglion leads were placed in the lateral epidural space near the target DRG at levels from T10 to S2, depending on the dermatomal target corresponding to the subject's primary region of pain. Spinal cord stimulation leads were placed in the medial or paramedial epidural space such that the caudal-most electrical contact was not caudal to the top of the L1 vertebral body on an anterior–posterior fluoroscopic view. Depending on the anatomical target, up to 16 contacts were placed for both study arms. Intraoperative testing to determine stimulation overlap with subjects' painful areas was conducted during implantation. Figure 1 shows the lead placements for both groups. Table 3 summarizes the number and placement of leads for subjects in the study.
Figure 1.
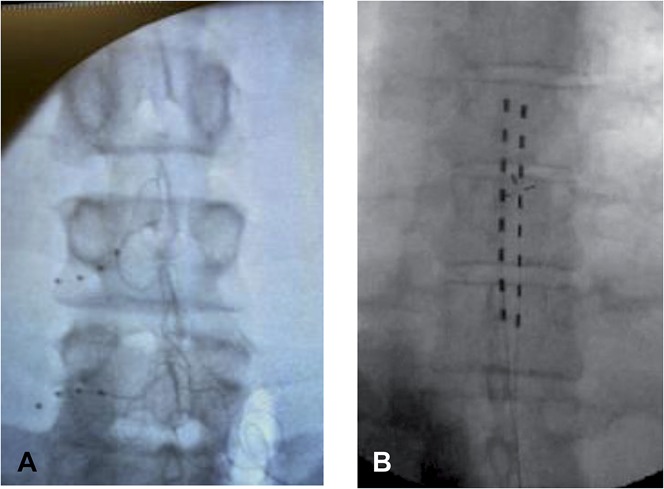
Lead placement. The lead for dorsal root ganglion (DRG) stimulation is specialized to provide percutaneous entry through the epidural space, exiting through the foramen, and resting around the DRG. As shown in panel A, DRG leads were placed in the lateral epidural space near the target DRG. For the SCS arm (panel B), leads were placed in the medial or paramedial epidural space such that the caudal-most electrical contact was not caudal to the top of the L1 vertebral body on an anterior–posterior fluoroscopic view. Depending on the anatomical target, up to 16 contacts were placed for both study arms. Intraoperative testing to determine paresthesia overlap over pain areas was conducted during trial evaluation period.
Table 3.
Summary of permanent leads implanted.
2.4. Sample size calculation and analysis populations
Sample size was determined based on the planned noninferiority test for the composite safety and effectiveness primary end point of treatment success. Treatment success was defined as ≥50% reduction in the visual analog scale (VAS) score in the primary area of pain during both trial and the 3-month visits with no incidence of stimulation-induced neurological deficits. Pilot data with 8 CRPS subjects and 22 causalgia subjects indicated that the success rate of DRG, defined as a 50% reduction in pain intensity, was 87% for CRPS subjects and 77% for causalgia subjects. Thus, an observed success rate at of least 15% above the 50% rate reported for SCS subjects was expected.14,28 Accounting for 15% attrition, an estimated 152 subjects (76 subjects in each arm) would provide greater than 85% power to test the primary end point hypothesis with a noninferiority margin of 10%.
The primary, secondary, and tertiary effectiveness analyses were based on the modified intention-to-treat (MITT) population including all randomized subjects who participated in the trial procedure (73 in each group). The MITT population was based on standard intention-to-treat principles, wherein subjects were analyzed based on their initial randomized treatments. The binary composite end points for success included subjects who failed the trial evaluation and exited the study as treatment failures. Safety data tabulations are based on the intention-to-treat analysis set including all randomized subjects (76 in each group).
2.5. Data collection and general statistical methods
Patient demographics and medical history were collected at baseline. At baseline and at each study visit, physical and neurological examinations, along with medication utilization, were recorded by study staff. Pain intensity was measured at baseline and at each study visit using the 100-mm visual analog scale (VAS), ranging from 0 (no pain) to 100 (worst imaginable pain) where higher scores represent greater pain severity. At baseline and each study visit, assessments of quality of life, psychological disposition, and experiential factors (measures described in detail below) were completed. All adverse events (AEs) through 12 months were reported and the occurrence of any stimulation-related neurological deficits was documented.
Descriptive statistics are presented as number of subjects, mean, SD, median, and range for all continuous variables and the number and percentage of subjects for categorical variables. As stipulated by the protocol and with the exception of the primary end point analysis, DRG stimulation and SCS were compared using a 2-sample t test (or Wilcoxon rank-sum test) for continuous outcomes and Pearson χ2 test (or Fisher exact test) for categorical outcomes. Choice of parametric or alternative tests was based on the data distributions for each measure, and the test used is reported in the results. Two-sided confidence intervals are also provided for certain outcome measures of interest to assess differences between the treatment arm and the control arm.
2.6. Primary composite end point
The predefined primary composite end point of the study was treatment success rates for the DRG subjects compared to the SCS subjects. To be considered a treatment success (1) a subject had a successful trial reporting ≥50% reduction in VAS score from baseline to the end of the trial phase, (2) reported a VAS score at 3 months that was reduced from preimplant baseline by ≥50%, and (3) did not experience a stimulation-related neurological deficit during either the trial phase or after permanent implant. A stimulation neurological deficit, different from AEs, was defined as a measurable 2-point worsening on the in-clinic sensory and motor neurological examination, within the appropriate concordant anatomy, that was induced by stimulation and subsided in the absence of stimulation for at least 24 hours. Sensory and motor examinations were conducted by the physician and rated as 2 (normal function), 1 (decreased function), or 0 (abnormal function); a score of 0 would indicate neurological deficit. No neurological deficits, as defined, were recorded for any subjects in either arm of the study. In addition, if a subject withdrew from the study due to a device-, procedure-, or stimulation-related AE, the subject was treated as a failure in the primary end point analysis.
As prespecified, the primary end point analyzed the success rate between the two treatment arms using Blackwelder methods for testing noninferiority between 2 proportions at a one-sided significance of 0.05.3 The noninferiority margin was set at 10%. If noninferiority of the primary end point was achieved, a superiority test was performed at a one-sided significance level of 0.025.
2.7. Secondary end point
2.7.1. Positional effects on paresthesia intensity
Paresthesia intensity, a prespecified secondary end point, was assessed at 3 months. Paresthesia intensity was rated by subjects using a previously published paresthesia intensity rating scale.16 Subjects rated the intensity of their perception of paresthesia, while upright and supine, on an 11-point numeric rating scale from 0 representing “No feeling” to 10 “Very intense.” Perceived paresthesia intensity difference between supine and upright positions was calculated and averaged across each group This end point was evaluated at a 2-sided significance level of 0.05.
2.8. Other end points
2.8.1. Short-Form-36
The Short-Form-36 (SF-36) is a self-reported health-related quality-of-life scale with 36 questions that yield scores on 8 dimensions of quality of life including physical functioning, role-physical, bodily pain, general health, vitality, social functioning, role-emotional, and mental health.27,29 These 8 dimensions also are combined to provide 2 summary scales for physical health (Physical Component Summary) and mental health (Mental Component Summary). Improvements on the SF-36 scale are represented by increased scores. Within- and between-group improvements were examined using the calculated change from baseline for each subscale or summary scales.
2.8.2. Profile of mood states
The profile of mood states (POMS) scale is a 65-item, 5-point Likert scale that measures mood states overall (total mood disturbance) as well as for 6 domains: tension, depression, anger, vigor, fatigue, and confusion. Higher scores indicate more negative mood states except for the vigor domain where higher scores indicate increased vigor.6 Within- and between-group improvements were examined using the calculated change from baseline for each domain and the total POMS score.
2.8.3. Brief pain inventory
The brief pain inventory (BPI) measures pain severity in the last 24 hours on a numeric pain rating scale from 0 “No pain” to 10 “Pain as bad as you can imagine,” and interference due to pain from 0 “Does not interfere” to 10 “Completely interferes.”5 The interference score was calculated as the mean of the interference items, and 2 subscales for the activity dimension and the affective dimensions of interference were tabulated. Within- and between-group improvements were examined using the calculated change from baseline for the pain and interference scales and for each interference subscale.
2.8.4. Subject satisfaction
Subjects completed a satisfaction scale at the end of trial phase and at 3, 6, and 12 months. Subjects rated satisfaction with pain relief and the therapy in general on an 11-point numeric rating scale with 0 indicating “Not Satisfied” and 10 indicating “Very Satisfied.” Subjects rated the likelihood of undergoing the therapy again on an 11-point numeric rating scale with 0 indicating “Not Likely” and 10 indicating “Very Likely.” Finally subjects rated the their subjective change in pain since baseline on a 7 point scale ranging from “Much Worse” to “Much Better.” Ratings were treated as interval data and summarized with descriptive statistics of central tendency.
2.8.5. Stimulation specificity
Stimulation specificity was evaluated to determine the extent to which paresthesia was felt by subjects in anatomical regions that were not painful at baseline. The pain and paresthesia diagram forms had identical diagrams of the human body on which subjects marked where they felt pain and paresthesia. The baseline pain diagrams completed by the subjects were compared to the subjects' paresthesia maps completed at the end of trial phase and at 3 months postimplant. Subjects were categorized based on the presence or absence of one or more paresthesia areas at follow-up that were not coincident with a pain area at baseline.
2.8.6. Percentage change in visual analog scale
The percentage of change in VAS score from baseline to each scheduled follow-up was computed for each subject and inspected using descriptive statistics and confidence intervals. Missing data were not imputed for this analysis; only subjects with VAS scores at baseline and follow-up were included in the analysis.
2.9. Safety analysis
Adverse events were collected and tabulated at all scheduled or unscheduled visits during the study. An AE was defined as any unfavorable and/or unintended sign, symptom or disease temporarily associated with the use of the implanted device, whether or not related to the device. A serious adverse event (SAE) was defined as any AE that is immediately life threatening; results in significant, persistent, or permanent disability; necessitates invasive intervention to prevent permanent impairment or death; results in the need for a 24-hour hospital stay or prolongation of a hospital stay; or results in death. Adverse event and SAE rates are expressed as the number of patients divided by the population at risk for each group (n = 76) through the 12-month study visit. All AEs reported were reviewed by an independent event committee that coded and adjudicated each event with regard to seriousness and relatedness to the implant procedure, device, and/or stimulation therapy.
3. Results
3.1. Patient accounting
See CONSORT diagram for full accounting (Fig. 2). Briefly, 320 subjects were consented and enrolled in the study from 22 investigational sites. Of these subjects, 168 were excluded for screen failures because they failed to meet the study's inclusion or exclusion criteria with the majority failing to meet the diagnostic criteria for inclusion. The remaining 152 subjects were enrolled and randomized to either the DRG or the SCS arm (76 in each arm). After randomization, 3 subjects from each group did not continue to the trial evaluation phase. Subjects who failed the success criterion at the end of the trial phase were exited from the study and considered treatment failures for composite end point analyses. A total of 61 DRG subjects and 54 SCS subjects met the success criteria at the end of their trial phase and continued to permanent implant. By the 12-month visit, 55 DRG subjects and 50 SCS subjects had evaluable data.
Figure 2.
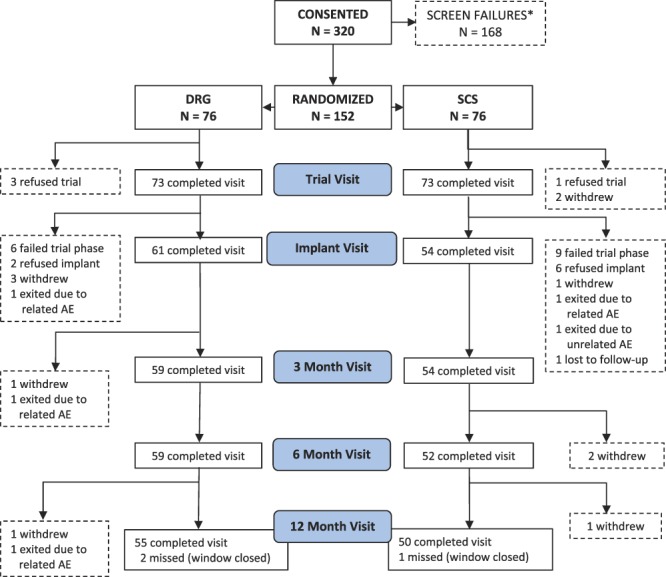
CONSORT diagram. *Subjects were enrolled if they met the inclusion criteria for the study. After consent, subjects were screened per exclusion criteria and exited if violations were revealed. AE, adverse event; DRG, dorsal root ganglion; SCS, spinal cord stimulation.
On average, each active study site randomized 3 subjects (range 0, 9) to each arm of the study. At any one site, the maximum number of randomized subjects was 11% (17/152) of the MITT population.
3.2. Baseline characteristics
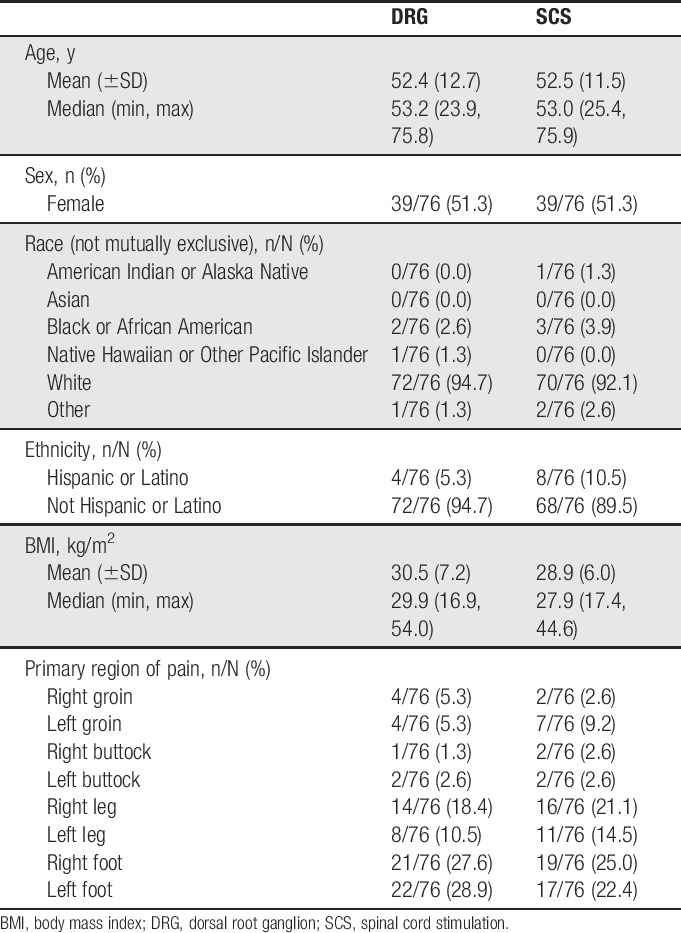
The average age of subjects was 52.4 years in the DRG stimulation arm and 52.5 years in the SCS arm. There were slightly more females than males in both arms (51.3% for both arms). Race was predominantly white (94.7% and 92.1% for DRG and SCS, respectively). Average body mass index was 30.5 for DRG and 28.9 for SCS. The average duration of chronic lower limb pain was 7.5 years for the DRG arm and 6.8 years for the SCS arm. Comorbidities and medications taken for subject conditions were similar in both arms. Overall, no statistically significant differences were found among the baseline characteristics between treatment arms. See Table 4 for a detailed summary of baseline characteristics.
Table 4.
Baseline demographics and characteristics.
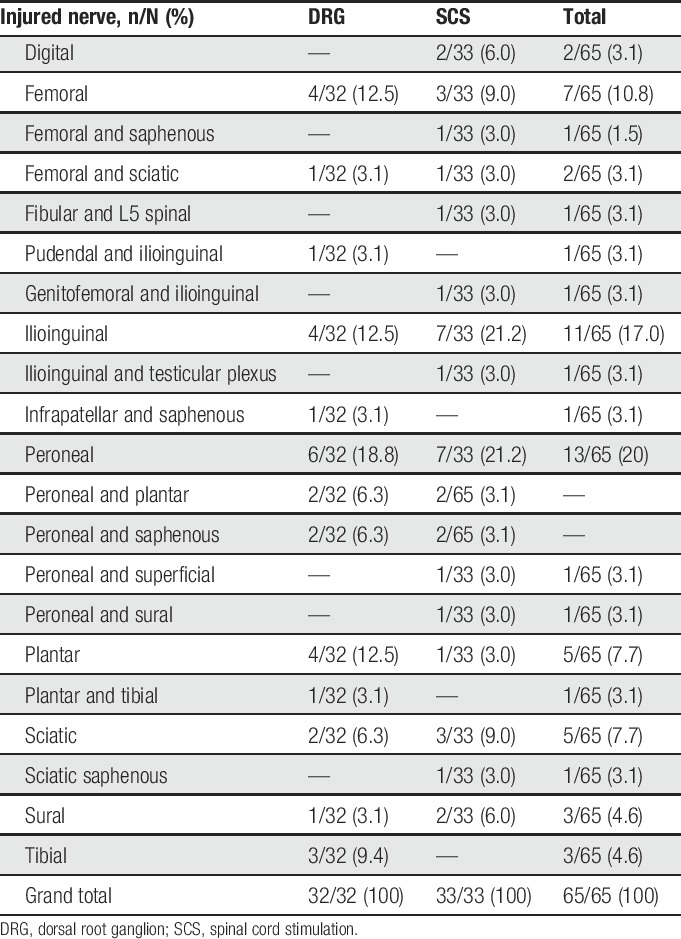
Similar distribution of CRPS (DRG: 44/76 [57.9%]; SCS: 43/76 [56.6%]) and causalgia (DRG: 32/76 [42.1%]; SCS: 33/76 [43.4%]) was reported between the arms. All CRPS subjects had sensory symptoms, 82/87 (94.3%) had motor trophic symptoms, 57/87 (65.5%) had vasomotor symptoms, and 58/87 (66.7%) had sudomotor or edema symptoms. A total of 79 of the 87 CRPS subjects had at least one symptom in each of 3 symptom categories documented at baseline; 8 CRPS subjects (3 in the DRG group and 5 in the SCS group) had one symptom in each of 2 symptom categories documented at the time of the baseline evaluation (sensory and motor). In the 8 subjects with only 2 secondary symptoms (sensory and motor) at enrollment, the medical monitor indicated that the reason that sudomotor or edema and vasomotor symptoms were not present at enrollment was a manifestation typically evident in the acute or early phase of the disease. The 8 patients who were enrolled in the study with only 2 symptoms documented had a range of 3 to 11 years of history of CRPS before enrollment. For subjects diagnosed with causalgia the injured nerves are documented in Table 5.
Table 5.
Injured nerves for causalgia subjects.
3.3. Primary composite end point
Figure 3 summarizes the primary composite end point results at 3 months, when the primary end point was ascertained, as well as over time through 12 months. No neurological deficits were reported during the study, so the rates of success at each time point include those subjects with a permanent implant who reported at least a 50% reduction in VAS from preimplant levels. Randomized subjects who did not proceed to permanent implant were considered treatment failures for this end point at each study visit. The proportion of subjects who achieved treatment success at 3 months in the DRG arm (81.2%; 56/69) was statistically greater than the SCS arm (55.7%; 39/70). The results demonstrated that DRG stimulation met not only noninferiority (P < 0.0001) but also statistical superiority (P < 0.0004). Long term, the proportion of subjects who achieved treatment success at 12 months in the DRG arm (74.2%; 49/66) also was greater than that in the SCS arm (53.0%; 35/66); these results demonstrated both noninferiority (P < 0.0001) and superiority (P < 0.0004) at the long-term follow-up.
Figure 3.
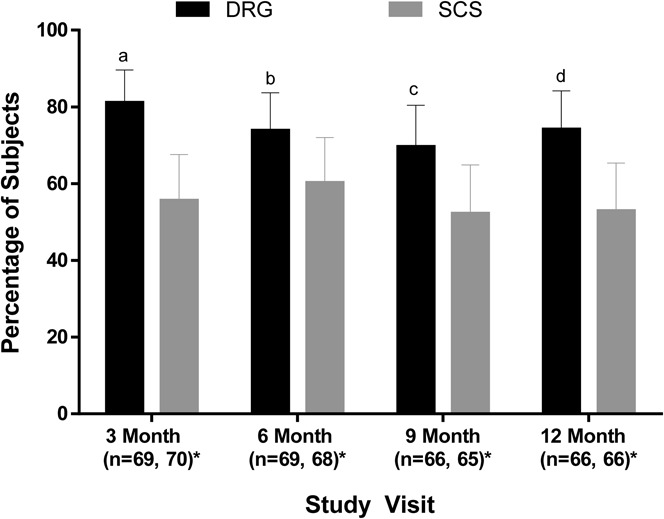
Proportion of subjects in each group who met the primary end point. The proportion of subjects who met the composite end point of success defined as 50% or greater pain reduction at both the trial phase and the indicated follow-up visit without a stimulation-related neurological deficit in the modified intent-to-treat population is shown. Subjects who exited the study after randomization were considered treatment failures. At all study visits, the proportion of subjects in the DRG stimulation group with successful therapy was noninferior to SCS (Blackwelder test of 2 proportions, all P < 0.01). Superiority was also established at each time point. aP < 0.001, bP = 0.04, cP = 0.02, and dP = 0.005. Error bars represent 95% confidence interval. *n for the DRG and SCS groups, respectively. DRG, dorsal root ganglion; SCS, spinal cord stimulation.
Similar results were observed at 3 months when the primary end point was stratified by primary diagnoses. For CRPS, a greater proportion of DRG subjects (82.5%) met the primary end point at 3 months than SCS subjects (57.5%) (noninferiority, P < 0.001; superiority, P = 0.006). For causalgia, the proportion of subjects who met the primary end point was higher for DRG (79.3%) than for SCS (53.3%) (noninferiority, P = 0.001; superiority, P = 0.014).
3.4. Secondary end point
On average, DRG subjects experienced significantly less postural variation in perceived paresthesia intensity than the SCS subjects (P < 0.001) at 3 months. Dorsal root ganglion subjects reported a mean difference between supine and upright paresthesia intensity rating of −0.1 ± 1.6, and SCS subjects had a mean difference of 1.8 ± 3.0. These results persisted throughout the study (Fig. 4).
Figure 4.
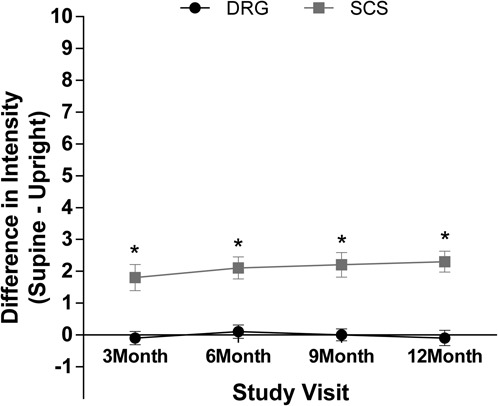
Postural variation in paresthesia intensity. Variation in the intensity of paresthesia was calculated as the difference in intensity during supine and upright positions, rated on an 11-point numerical rating scale. Wilcoxon test indicated that subjects using DRG stimulation had significantly less postural variation in paresthesia intensity than SCS subjects. *P < 0.001. DRG, dorsal root ganglion; SCS, spinal cord stimulation.
3.5. Other end points
3.5.1. Short-Form-36
Table 6 summarizes the SF-36 results. Both the DRG stimulation and SCS groups experienced improvements in SF-36 scores from baseline to 3 months (P < 0.05) and 12 months, with the one exception that the General Health scale was not significantly improved at 12 months in the SCS group (P > 0.05).
Table 6.
Change in Short-Form-36 scores from baseline through 12 months.
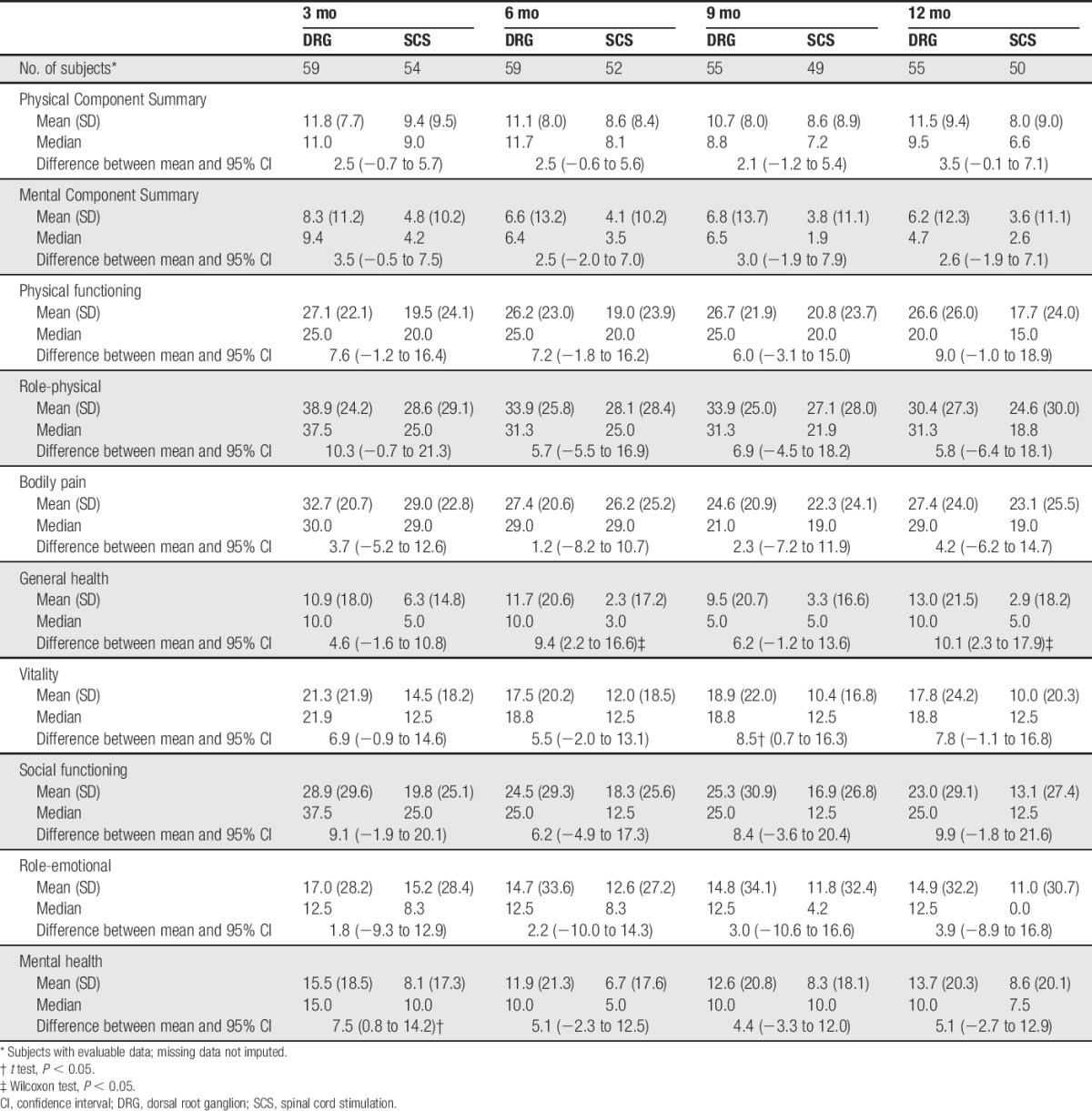
At 3 months, the change in the mental health dimension was statistically better for DRG stimulation subjects compared to SCS subjects (P = 0.0295). At 12 months, DRG subjects had statistically greater improvement on 3 scales: overall change in the physical component score (P = 0.04), general health (P = 0.03), and social functioning (P = 0.03) when compared to SCS subjects.
3.5.2. Profile of mood states
Both groups experienced improvements in all domains of the POMS from baseline to 3 months (P < 0.05). At 12 months, DRG subjects had statistically significant improvements in all scales (P < 0.05), and the SCS subjects had statistically significant improvements (P < 0.05) in all scales except for the depression and confusion scales compared to baseline.
Figure 5 presents the change in POMS scores through the 12-month visit. The changes in POMS scores from baseline to 3 months were statistically greater for DRG subjects than for SCS subjects for the Total Mood Disturbance scale (P = 0.0466) and the tension domain (P = 0.0430). Specifically, the Total Mood Disturbance at 3 months improved by a magnitude of 20.4 points (29.0 at baseline to 8.6 at 3 months) for DRG subjects, and only a magnitude of 14.7 points (25.6 at baseline to 10.9 at 3 months) for SCS subjects. These improvements in the Total Mood Disturbance and tension domain score for DRG subjects persisted to 12 months (P = 0.021 and P = 0.004, respectively). In addition, at 12 months, the depression (P = 0.004) and confusion (P = 0.020) domains also demonstrated statistically greater magnitudes of improvement for DRG subjects compared to the improvements for SCS subjects.
Figure 5.
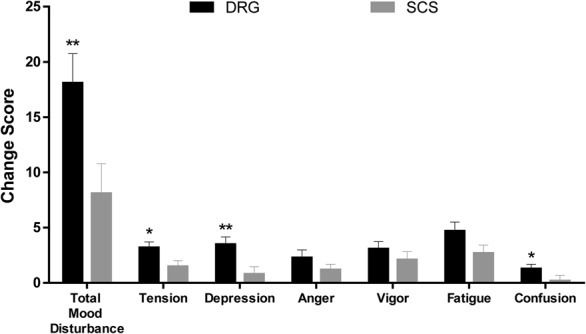
Change in profile of mood states (POMS) at 12 months. Change from baseline scores was calculated for each patient on each domain and the total score for the POMS. Mean change scores from baseline to 12 months are represented for both the DRG stimulation and the SCS groups. Error bars represent standard error of the mean. *Significant between-group difference with P < 0.05. **Significant between-group difference with P < 0.001. DRG, dorsal root ganglion; SCS, spinal cord stimulation.
3.5.3. Brief pain inventory
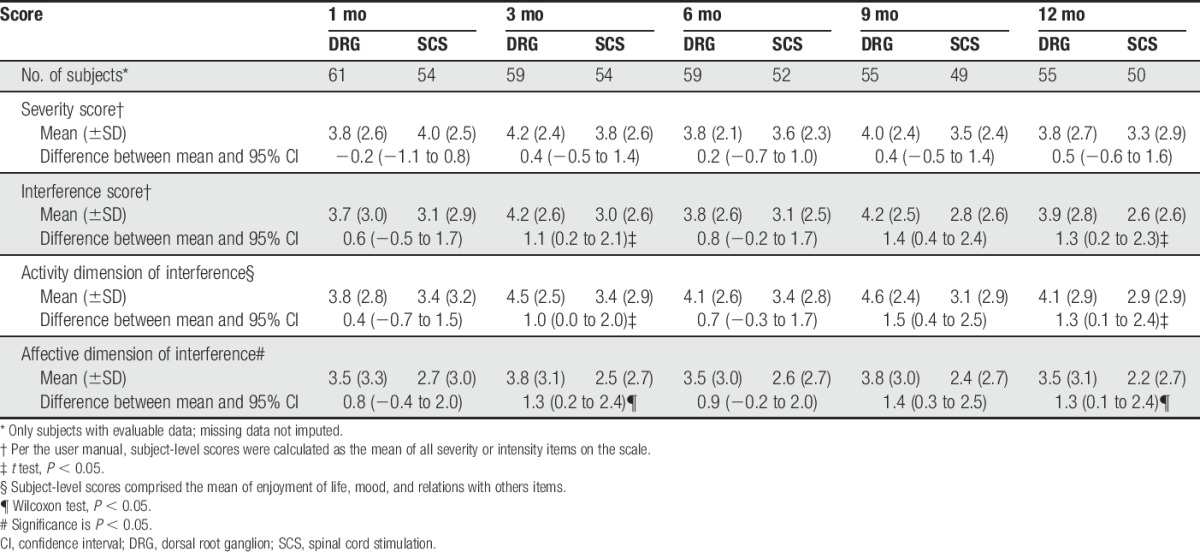
As shown in Table 7, both groups experienced improvements in all of the BPI scales from baseline to 3 months (P < 0.05) and 12 months (P < 0.05). Between the 2 groups, improvements from baseline on the interference scale (treatment 4.2, control 3.0), the activity scale (treatment 4.5, control 3.4), and the affective scale (treatment 3.8, control 2.5) were statistically greater (P < 0.05) for DRG subjects compared to SCS subjects at 3 months. These results persisted to 12 months.
Table 7.
Change from baseline in brief pain inventory through 12 months.
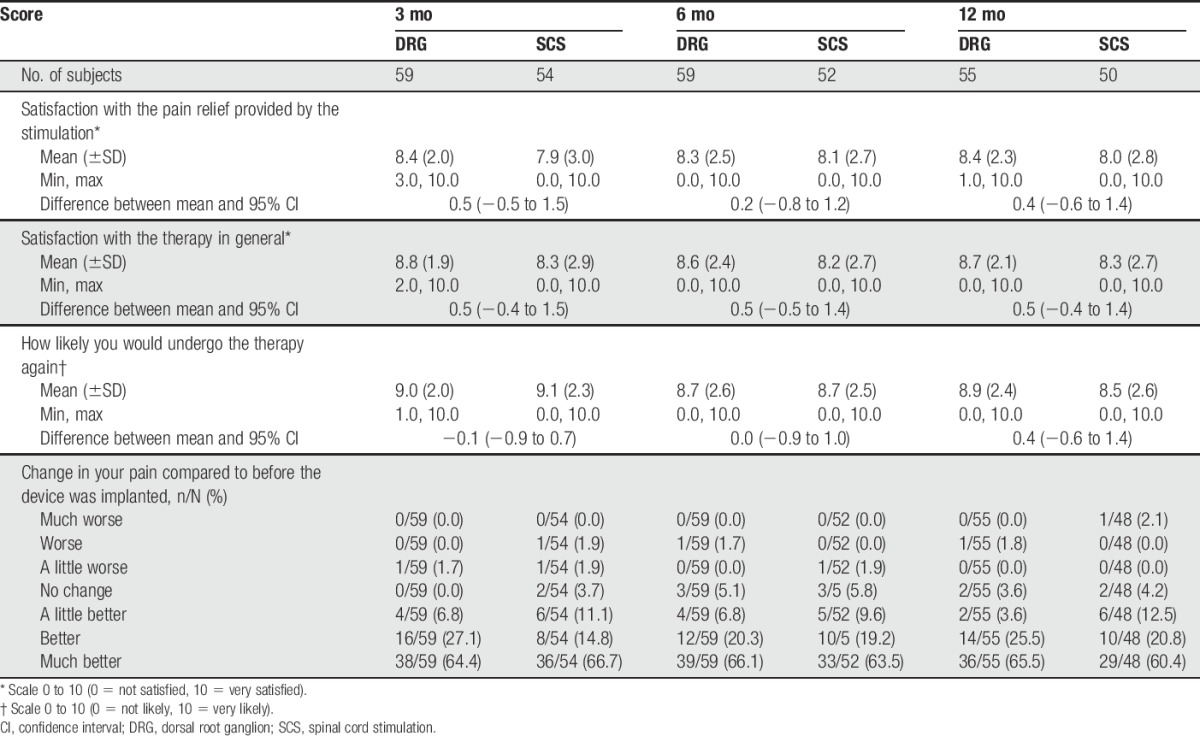
3.5.4. Subject satisfaction
The majority of patients in both groups reported high degrees of satisfaction (Table 8) for all 4 satisfaction items. However, no statistical significance was found between the groups for all items assessed (P > 0.05).
Table 8.
Subject satisfaction through 12 months.
3.5.5. Stimulation specificity
At 3 months, SCS subjects were 2.3 times more likely to report feeling paresthesia in one or more nonpainful areas as DRG subjects (35.2% vs 15.3%, P = 0.0142). At 12 months postimplant, SCS subjects were 7.1 times more likely to report feeling paresthesia in one or more nonpainful areas as DRG subjects (38.8% vs 5.5%, P < 0001). The percent of subjects who reported that they felt paresthesia in only their painful region(s) at 3 and 12 months was 84.7% and 94.5% in the DRG group, and 64.8% and 61.2% in the SCS group.
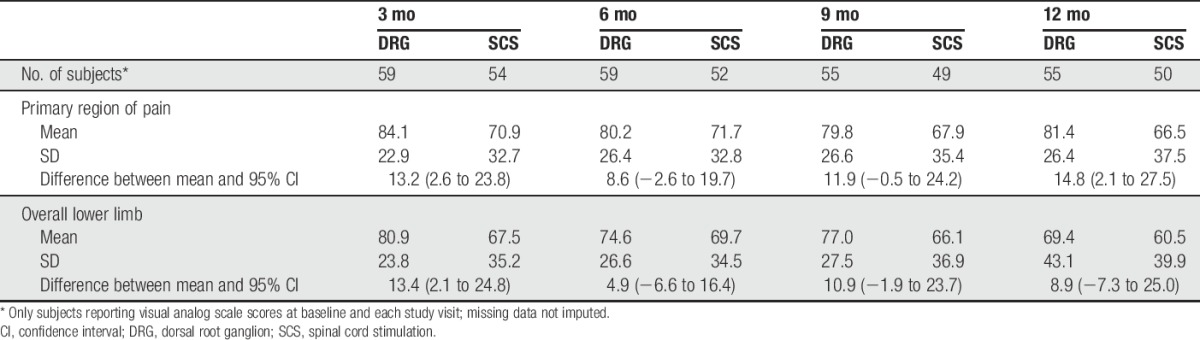
3.5.6. Percentage change in visual analog scale
As shown in Table 9, DRG stimulation demonstrated a greater mean percent reduction in VAS scores than SCS (84.1% vs 70.9%, respectively) with the significant reduction persisting to 6 months and 12 months. Subjects using DRG reported mean VAS of 80.6 mm at baseline, which reduced to 13.1 mm at 3 months and remained low, at 15.0 mm, at 12 months. The subjects using SCS reported a baseline mean VAS of 80.7, 3-month mean VAS of 23.8 mm, and 12-month mean VAS of 26.5 mm.
Table 9.
Percent change from baseline in visual analog scale scores through 12 months.
3.6. Safety analysis
A total of 21 SAEs occurred in 19 subjects (8 DRG subjects and 11 SCS subjects). The rates of SAEs were 10.5% (8/76) in the DRG arm and 14.5% (11/76) in the SCS arm. The difference in the rate of SAEs between groups was not statistically different (P = 0.62). Two of the SAEs in the control group were adjudicated as definitely related to the implant procedure. Both events were infections that required device explant. There were no unanticipated SAEs or stimulation-induced neurological deficits at any time during the study. None of the subjects died.
Table 10 presents the rates of related AEs. Fifty two procedure-related events were reported by 35 patients (46.1%) in the DRG arm, and 29 procedure-related events were reported by 20 patients (26.3%) in the SCS arm, yielding a statistically significant difference between the groups (P = 0.018). Possible contributors to the differential rate of procedure-related AEs are the procedure times and number of leads. Procedure times for permanent implant averaged 107.2 minutes (±51.2) for DRG subjects and 75.7 minutes (±32.2) for SCS subjects. In addition, 16.4% (10/61) of DRG subjects were implanted with 3 or 4 leads, while all SCS subjects had 1 or 2 leads implanted. For both groups, the most frequently occurring procedure-related AE was pain at the incision sites with 7 events reported by 6 patients (7.9%) in the DRG arm and 5 events reported by 5 patients (6.6%) in the SCS arm.
Table 10.
Rates of related adverse events.
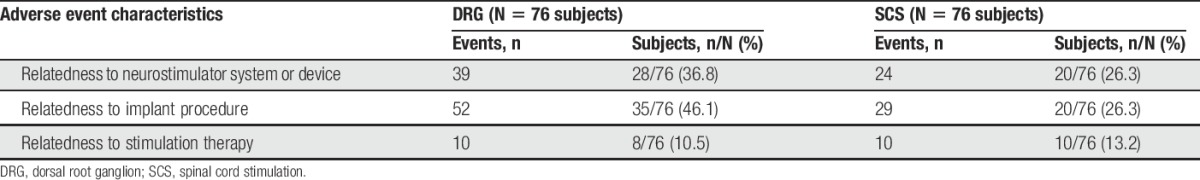
For device-related AEs, 39 events were reported by 28 patients (36.8%) in the DRG arm and 24 events were reported by 20 patients (26.3%) in the SCS arm. No statistical difference was found between the groups (P = 0.22). The most frequently occurring device-related AE in the DRG arm was implantable pulse generator (IPG) pocket pain with 10 events reported by 10 patients (13.2%). On the other hand, the most frequently occurring device-related AE in the SCS arm was loss of stimulation due to lead migration with 8 events reported by 8 (10.5%) patients.
There was also no statistical difference between the groups for stimulation-related AEs (P = 0.8025). Ten events were reported by 8 patients (10.5%) in the DRG arm, and 10 events were reported by 10 patients (13.2%) in the SCS arm. The most frequently occurring stimulation-related AE for both groups was overstimulation with 3 events reported by 3 patients (3.9%) in the DRG arm and 5 events reported by 5 patients (6.6%) in the SCS arm.
4. Discussion
This study represents the largest randomized controlled trial assessing DRG stimulation for the treatment of chronic, intractable pain associated with the diagnoses of CRPS or causalgia. Analysis of the primary end point revealed that subjects using DRG stimulation had a higher rate of treatment success (81.2%) compared with the treatment success rate for traditional SCS (56.7%). Furthermore, pain relief persisted through 12 months of follow-up and remained significantly lower for DRG subjects than for those using SCS. Subjects using DRG reported significantly less postural-related changes in paresthesia and showed larger improvements on measures of quality of life, functional status, and psychological disposition than subjects using SCS. The safety profile of the DRG stimulation device was similar to traditional SCS devices, with the exception of the rate of procedural events.
These results for DRG stimulation as a treatment of chronic neuropathic pain associated with CRPS and causalgia must be interpreted within the context of previous neurostimulation studies for this population. Treatment of chronic reflex sympathetic dystrophy with SCS, in combination with physical therapy, reduced pain to a greater degree than physical therapy alone14; mean VAS scores for implanted patients reduced to 3.5 cm on a 10-cm VAS scale after 6 months of SCS. A retrospective analysis of SCS for the treatment of CRPS reported a mean VAS of 5.6 cm over a mean follow-up time of 88 months.19 Mean VAS scores during SCS therapy in both these previous studies were higher, by a clinically meaningful margin10 than the VAS score of 13.1 mm and 15 mm reported by subjects treated with DRG stimulation in our study at 3 and 12 months. Similarly, Geurts et al.11 reported only a 50% pain reduction in an observational trial of SCS for CRPS.
A study using a heterogeneous population, including subjects with CRPS, reported that 68.4% of subjects were able to achieve ≥50% leg pain relief, and 60% of subjects achieved ≥50% pain relief for overall pain.21 A published case series of CRPS subjects reported that 71.4% of subjects achieved ≥50% pain relief after 6 months of DRG stimulation.28 In addition, a randomized trial comparing SCS to physical therapy for subjects with CRPS reported that 50% of subjects achieved at least 50% reduction in pain intensity.14 Here, we report an 84% reduction in pain for patients treated with DRG stimulation and that 81% of subjects achieved ≥50% pain relief. Furthermore, the optimal programming for DRG stimulation is still being developed; Table 2 shows that SCS and DRG parameters were quite different. Additional developments in optimized programming for DRG should improve clinical outcomes over time for this therapy. Taken together, we conclude that DRG stimulation provides better pain relief than traditional SCS.
Patients with CRPS and causalgia are difficult to treat with symptoms for 20% to 80% of CRPS I patients persisting for 1 year, even when treatment was considered successful.2 Surgical interventions such as joint denervation or neurolysis also have variable outcomes; approximately 20% of patients failed to report low pain intensity and improved activities of daily living 2 years after surgery.9 For patients with CRPS I or causalgia who do not achieve adequate pain management with conservative therapies, SCS provides an additional and reversible treatment option. Furthermore, DRG stimulation augments the patient experience by providing a therapy that is adaptable to each patient's individual pain profile through more precise anatomical targeting.
The pathways for sensory afferents into the central nervous system via the DRG are well documented.4,13 Anatomically, peripheral inputs associated with pain symptoms can be traced to relevant DRG at one or more spinal levels. Stimulation of the relevant DRG modifies pain signaling from the periphery for only the affected dermatomes. By contrast, SCS targets large dermatomal areas through stimulation of the dorsal column at anatomically defined spinal levels, and, as such, modifies ascending pathways for pain while also modulating collateral afferents in or near the medial lemniscus. Modulating pain signals from distal appendages with SCS typically requires that multiple dermatomes be captured–with paresthesias in the entire region. Our results showed that subjects treated with DRG stimulation had significantly less perceived stimulation sensation in nonpainful areas than subjects using SCS, while reporting better pain relief. This may indicate more precision targeting by virtue of the greater anatomical specificity with DRG stimulation.
The differences in collateral paresthesia may also be influenced by differences in programming parameters. Programming parameters were individualized for each subject's optimal experience. The resulting parameters were quite different between the 2 therapies (Table 2) with much lower amplitudes for DRG programming. This was expected from pilot work7 and because diffusion of energy by the cerebrospinal fluid is less influential at the DRG. The between-subjects design of this study prohibits a real comparison of the relationship between targeting, programming, and pain relief; more research is needed.
Chronic pain conditions, in general, are associated with disturbances in mood and physical and social functioning.1,22,24 The targeted pain relief provided by DRG stimulation in the ACCURATE study was also associated with additional benefits. After 3 months, subjects using DRG stimulation reported significantly greater improvements in total mood disturbance, as measured by the POMS, as well as larger improvements pain interference, affective disruption, and activity, as measured by the BPI. Moreover, by 12 months, subjects treated with DRG stimulation reported significantly larger improvements than SCS subjects for physical function, general health, and social function, as measured by the SF-36.
Despite the differences reported for treatment success, pain relief, and affective or functional outcomes, the majority of subjects were satisfied with their respective therapy, regardless of treatment group. While subjects using DRG stimulation reported a larger magnitude of change and there was a greater proportion of successful subjects with DRG stimulation, SCS subjects, as a group, did report significant improvements from baseline in all measured domains. The satisfaction results reported here reflect the improvements from preimplant baseline experienced by subjects.
The rate of AEs for DRG stimulation, through 12 months postimplant, was similar to that seen for the SCS-treated subjects in this study and in previous reports.17,20 Only 2 subjects had procedure-related SAEs; 2 infections in the SCS group that required explant. It is notable that the rate of nonserious procedure-related events was higher for the DRG stimulation group (46%) compared with the SCS group (26%). The higher rate of procedure-related events may be attributed to the differences in average procedure time and a greater number of leads placed for DRG some subjects, which may increase exposure to risk. It is expected that additional experience with DRG implantation will result in shorter procedure times and fewer procedure-related events.
There are limitations to this study that may affect the interpretation of the results. The calculated success rate was contingent upon subjects not only achieving 50% pain relief but also continuing in the study (dropouts were counted as failures). Therefore, the success rate could be influenced by factors associated with the lack of blinded treatments (eg, SCS subjects were less motivated to stay in the trial, uncontrolled differences in health care provider interactions). In addition, subjects were required to maintain a stable regimen of pain medications through 3 months only, and the long-term results after 3 months may be affected by medication changes. The SCS device also had limitations placed on the programming of the device so that the comparison between the devices was not confounded by unique SCS device programming features. In particular, the accelerometer function in the SCS device was disabled. If the accelerometer was enabled, the SCS group may have had less postural changes in perceived paresthesia intensity. In addition, the analysis of subjects who did and did not experience paresthesia when stimulation was on was confounded by the fact that the SCS device instruction for use requires the device to be programmed for subjects to receive paresthesia. In addition, the number of subjects who did not have paresthesia is very small, and this end point was not adequately powered to detect the difference in pain relief for subjects who reported feeling vs not feeling paresthesia.
In conclusion, CRPS I and causalgia, in their chronic forms, are difficult to treat with variable outcomes with conservative symptom management. Neuromodulation techniques, like SCS, may benefit many patients who have exhausted other therapy options. SCS, however, often has a limited ability to target discrete focal anatomical regions of pain, as is common in CRPS and causalgia. Dorsal root ganglion stimulation provides an effective alternative that provides precision stimulation targeting and improved patient outcomes.
Conflict of interest statement
All authors were paid by Spinal Modulation & St Jude Medical as investigators for the clinical trial. T. R. Deer is a consultant for Axonics, Bioness, Flowonix, Medtronic, Jazz, Nevro, St. Jude, and Saluda and has consulting or equity for Axonics and Bioness. T. R. Deer formerly had equity in Spinal Modulation and Nevro. R. M. Levy has served as a consultant for Bioness, BlueWind Medical, Boston Scientific, Flowonix, Medtronic, Microtransponder, Nevro, Saluda, Spinal Modulation, and St Jude Medical. R. M. Levy is or has been a minority shareholder in Saluda, Spinal Modulation, Bioness, Vertos, and Nevro. N. Mekhail formerly had a consultation agreement with spinal modulation to serve as medical monitor of the ACCURATE study. Currently, he is a consultant for St Jude Medical, Saluda medical, Stimwave, Medtronic neurological, and Flowonix inc. K. Amirdelfan is a consultant for St. Jude Medical, Nevro, Saluda, Nalu, and Biotronik. J. Pope is a consultant for Medtronic, NEVRO, St Jude, Flowonix, Jazz Pharmaceuticals, and Suture Concepts. T. Yearwood is a consultant for St Jude Medical, Boston Scientific, Nevro, Flowonix, and Neuronano; he serves as an officer for Meghan Medical. W. P. McRoberts serves or has served as a consultant for St Jude Medical, Medtronic, Nevro, Boston Scientific, Bioness, Vertiflex, and SPR. T. Davis has conducted research for Spinal Modulation, Vertiflex, Medtronic, Axsome, Nature Cell, and Halyard Health; has received fees for consulting, education, or speaking from St Jude Medical, Medtronic Restorative Therapies, Stryker, Vertiflex, DrChrono, and Tenex Health; and has ownership interests in Paradigm Spine <1%, LDR Holdings <1%, Alpha Diagnostics Neuromonitoring, and Broadway Surgical Institute. J. Scowcroft has served as a consultant for Boston Scientific. L. Kapural is a consultant for St Jude Medical, Nevro, Neuros, SPR Therapeutics, and Saluda. R. Paicius is a consultant for St Jude Medical, Nevro, and Boston Scientific. J. Kramer, Burton, Johnson, and Kristina Davis are employees of St. Jude Medical. The remaining authors have no conflicts of interest to declare.
This study was sponsored by Spinal Modulation, LLC, a wholly owned company of St. Jude Medical.
All authors contributed to the study design, data acquisition, and/or writing of this manuscript in accordance with the guidelines set forth by the International Coalition of Medical Journal Editors. T. R. Deer and R. M. Levy are co-primary authors with equal contributions to the work. In addition to the above, N. Mekhail served as an independent medical monitor of the study.
Acknowledgements
The authors thank Kaisa Kivilaid for statistical support and Angela Leitner for technical help on this project.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
References
- [1].Banks SM, Kerns RD. Explaining high rates of depression in chronic pain: a diathesis-stress framework. Psychol Bull 1996;119:95–110. [Google Scholar]
- [2].Bean DJ, Johnson MH, Kydd RR. The outcome of complex regional pain syndrome type 1: a systematic review. J Pain 2014;15:677–90. [DOI] [PubMed] [Google Scholar]
- [3].Blackwelder WC. Proving the null hypothesis in clinical trials. Controlled Clin Trials 1982;3:345–53. [DOI] [PubMed] [Google Scholar]
- [4].Caspary T, Anderson KV. Patterning cell types in the dorsal spinal cord: what the mouse mutants say. Nature reviews. Neuroscience 2003;4:289–97. [DOI] [PubMed] [Google Scholar]
- [5].Cleeland CS, Ryan KM. Pain assessment: global use of the brief pain inventory. Ann Acad Med 1994;23:129–38. [PubMed] [Google Scholar]
- [6].Curran S, Andrykowaski M, Studts J. Short form of the profile of mood states (POMS-SF): psychometric information. Psychol Assess 1995;7:80–3. [Google Scholar]
- [7].Deer TR, Grigsby E, Weiner RL, Wilcosky B, Kramer JM. A prospective study of dorsal root ganglion stimulation for the relief of chronic pain. Neuromodulation 2013;16:67–72. [DOI] [PubMed] [Google Scholar]
- [8].Deer T, Mekhail N, Provenzano D, Pope J, Krames E, Leong M, Levy R, Abejon D, Buchser E, Burton A, Buvanendran A, Candido K, Caraway D, Cousins M, DeJongste M, Diwan S, Eldabe S, Gatzinsky K, Foreman R, Hayek S, Kim P, Kinfe T, Kloth D, Kumar K, Rizvi S, Lad S, Liem L, Linderoth B, Mackey S, McDowell G, McRoberts P, Poree L, Prager J, Raso Lou, Rauck R, Russo M, Simpson B, Slavin K, Staats P, Stanton-Hicks M, Verrills P, Wellington J, Williams K, North R. The appropriate use of neurostimulation of the spinal cord and the peripheral nervous system for the treatment of chronic pain and ischemic diseases: the neuromodulation appropriateness consensus committee. Neuromodulation 2014;17:515–50. [DOI] [PubMed] [Google Scholar]
- [9].Dellon AL, Adonian E, Rosson GD. CRPS of the upper or lower extremity: surgical treatment outcomes. J Brachial Plex Peripher Nerve Inj 2009;4:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dworkin R, Turk D, McDermott M, Peirce-Sandner S, Burke L, Cowan P, Farrar J, Hertz S, Raja S, Rappaport B, Rauschkolb C, Sampaio C. Interpreting the clinical importance of group differences in chronic pain clinical trials: IMMPACT recommendations. PAIN 2009;146:238–44. [DOI] [PubMed] [Google Scholar]
- [11].Geurts JA, Smits H, Kemler MA, Brunner F, kessles AG, van Kleef M. Spinal cord stimulation for complex regional pain syndrome type I: a prospective cohort study with long-term follow-up. Neuromodulation 2013;16:523–9. [DOI] [PubMed] [Google Scholar]
- [12].Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007;8:326–31. [DOI] [PubMed] [Google Scholar]
- [13].Hogan QH. Labat lecture: the primary sensory neuron: where it is, what it does, and why it matters. Reg Anesth Pain Med 2010;35:306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kemler MA, Barendse GA, van Kleef M, de Vet HC, Rijks CP, Furnee CA, van den Wildenberg FA. Spinal cord stimulation in patients with chronic reflex sympathetic dystrophy. N Engl J Med 2000;343:618–24. [DOI] [PubMed] [Google Scholar]
- [15].Koopmeiners AS, Mueller S, Kramer J, Hogan QH. Effect of electrical field stimulation on dorsal root ganglion neuronal function. Neuromodulation 2013;16:304–11. [DOI] [PubMed] [Google Scholar]
- [16].Kramer J, Liem L, Russo M, Smet I, Van Buyten JP, Huygen F. Lack of body positional effects on paresthesias when stimulating the dorsal root ganglion (DRG) in the treatment of chronic pain. Neuromodulation 2015;18:50–7. [DOI] [PubMed] [Google Scholar]
- [17].Krishna K, Buchser E, Linderoth B, Meglio M, Van Buyten JP. Avoiding complications from spinal cord stimulation: practical recommendations from an international panel of experts. Neuromodulation 2007;10:24–33. [DOI] [PubMed] [Google Scholar]
- [18].Kumar K, Caraway DL, Rizvi S, Bishop S. Current challenges in spinal cord stimulation. Neuromodulation 2014;17:22–35. [DOI] [PubMed] [Google Scholar]
- [19].Kumar K, Rizvi S, Bnurs S. Spinal cord stimulation is effective in management of complex regional pain syndrome I: fact or fiction. Neurosurgery 2011;69:566–80. [DOI] [PubMed] [Google Scholar]
- [20].Kumar K, Wilson J, Taylor R, Gupta S. Complications of spinal cord stimulation, suggestions to improve outcome, and financial impact. J Neurosurg Spine 2006;5:191–203. [DOI] [PubMed] [Google Scholar]
- [21].Liem L, Russo M, Huygen FJ, Van Buyten JP, Smet I, Verrills P, Cousins M, Brooker C, Levy R, Deer T, Kramer J. One-year outcomes of spinal cord stimulation of the dorsal root ganglion in the treatment of chronic neuropathic pain. Neuromodulation 2015;2015:41–9. [DOI] [PubMed] [Google Scholar]
- [22].Modovan AR, Onac IA, Vantu M, Szntagotai A, Onac I. Emotional distress, pain catastrophizing an expectancies in patients with low back pain. J Cogn Behav Psychother 2009;9:83–93. [Google Scholar]
- [23].Oakley JC, Krames ES, Prager JP, Stamatos J, Foster AM, Weiner R, Rashbaum RR, Henderson J. A new spinal cord stimulation system effectively relieves chronic, intractable pain: a multicenter prospective clinical study. 2007;10:262–78. [DOI] [PubMed] [Google Scholar]
- [24].Robinson ME, Riley JL. The role of emotion in pain. In: Gatchel RJ, Turk DC, editors. Psychosocial factors in pain: clinical perspectives. New York: Guilford Press, 1999. pp. 74–88. [Google Scholar]
- [25].Schott GD. Mechanisms of causalgia and related clinical conditions. The role of the central and of the sympathetic nervous systems. Brain 1986;109:717–38. [DOI] [PubMed] [Google Scholar]
- [26].Taylor RS. Epidemiology of refractory neuropathic pain. Pain Pract 2006;6:22–6. [DOI] [PubMed] [Google Scholar]
- [27].User's manual for the SF-36v2 health survey. 2nd ed Lincoln, RI: Quality Metric Inc, 2007. [Google Scholar]
- [28].Van Buyten JP, Smet I, Liem L, Russo M, Huygen F. Stimulation of dorsal root Ganglia for the management of complex regional pain syndrome: a prospective case series. Pain Pract 2015;15:208–16. [DOI] [PubMed] [Google Scholar]
- [29].Ware JE, Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care 1992;30:473–83. [PubMed] [Google Scholar]