

Delivery of an interleukin-1 antagonist across the blood–brain barrier results in analgesia in the Seltzer model of neuropathic pain.
Keywords: Analgesic, Neuropathic pain, Interleukin-1, Blood–brain barrier, Antibody
Abstract
Neuropathic pain is a major unmet medical need, with only 30% to 35% of patients responding to the current standard of care. The discovery and development of novel therapeutics to address this unmet need have been hampered by poor target engagement, the selectivity of novel molecules, and limited access to the relevant compartments. Biological therapeutics, either monoclonal antibodies (mAbs) or peptides, offer a solution to the challenge of specificity as the intrinsic selectivity of these kinds of molecules is significantly higher than traditional medicinal chemistry–derived approaches. The interleukin-1 receptor system within the spinal cord has been implicated in the amplification of pain signals, and its central antagonism provides relief of neuropathic pain. Targeting the IL-1 system in the spinal cord with biological drugs, however, raises the even greater challenge of delivery to the central compartment. Targeting the transferrin receptor with monoclonal antibodies has proved successful in traversing the endothelial cell–derived blood–brain barrier and delivering proteins to the central nervous system. In this study, we describe a novel construct exemplifying an engineered solution to overcome these challenges. We have generated a novel anti–transferrin receptor-interleukin-1 receptor antagonist fusion that transports to the central nervous system and delivers efficacy in a model of nerve ligation–induced hypersensitivity. Approaches such as these provide promise for novel and selective analgesics that target the central compartment.
1. Introduction
Drugs based on biological molecules, such as monoclonal antibodies, peptides, and other proteins have been dramatically successful in the treatment of many diseases. This success is often because of an increased intrinsic selectivity and potency inherent in this class of drugs. Conversely, attrition of novel small molecule entities during the development phase is often because of poor physicochemical properties, selectivity (safety), and pharmacokinetics.37,33 These findings are equally applicable to new drugs for the treatment of chronic neuropathic pain, but with one additional challenge: Many novel drug targets for the treatment of this disease are located within the central nervous system (CNS), and thus inside the blood–brain barrier (BBB), an endothelial cell layer which is notoriously impermanent to biological drugs. Successful intervention in neuropathic pain using biological drugs therefore requires the development of new technologies which are able to transport biological payloads across the BBB.
Neuropathic pain, arising from damage or lesion of the somatosensory nervous system, has a complex pathophysiology and is difficult to treat. The unmet medical need is huge, with responder rates as low as 30% to 35% of patients.5 The discovery and development of novel analgesics have been unsuccessful, with no genuinely novel mechanisms launched for many decades. For targets located outside the CNS, biological interventions are showing promise,7 but targeting central pathways, which include second-order dorsal horn neurons, central neuroimmune interactions, or indeed manipulation of ascending and descending control, remains challenging. Bringing the selectivity of biological interventions to these targets may bring significant advantages, but the challenge of accessing the central compartment must first be addressed.
Interleukin (IL)-1 is a proinflammatory cytokine involved in both normal homeostasis and in pathological conditions.13 There is growing evidence for the role of IL-1–induced signalling in the development, maintenance, and propagation of pain. IL-1 expression is increased in conditions associated with pain and hyperalgesia in the periphery31 and the central compartment.1,2,11 When applied systemically, IL-1 is a potent hyperalgesic14 and if administered intrathecally (i.t.), IL-1 drives allodynia and hyperalgesia.30 In the CNS, IL-1 induces COX-2 expression, contributing to central hyperexcitability,32 and increases synaptic activity in superficial spinal cord.17
We have exploited the naturally occurring proinflammatory cytokine inhibitor, interleukin-1 receptor antagonist (IL-1RA),6 coupled to an engineered BBB transport solution to demonstrate analgesia in a mouse model of neuropathic pain. We have found that the neuropathic pain induced in this model is sensitive to central, but not peripheral, antagonism of the IL-1 receptor system. Central penetration IL-1RA was achieved using the anti–mouse transferrin receptor (TfR) antibody 8D318 engineered to further enhance central penetration. After subcutaneous (s.c.) administration, we have observed analgesia that is dependent on the dose of the IL-1RA fusion, and the affinity of the anti-TfR antibody for the TfR.
2. Materials and methods
2.1. Antibody and protein manipulation
2.1.1. Cloning, expression, and purification of 8D3 variants
DNA encoding the amino acid sequence of the VH and VL of the rat anti–mouse TfR antibody 8D34 was assembled by polymerase extension of overlapping oligonucleotides and cloned into expression vectors containing the appropriate light or heavy chain constant regions.27 Single alanine substitutions were introduced into the VH and VL complementarity-determining region 3 (CDR3) of 8D3 by site-directed mutagenesis. 8D3 variants were expressed as chimeric human IgG1 molecules with the S239D/A330L/I332E triple mutation (IgG1 TM).25 Antibodies were expressed in transiently transfected Chinese hamster ovary cells in serum-free media as described previously.10 Cultures were maintained in a humidified incubator at 37°C, 5% CO2 for 14 days after which the medium was harvested. Antibodies were purified from cell culture media using protein A affinity chromatography followed by size-exclusion chromatography. The concentration of IgG was determined by A280 using an extinction coefficient based on the amino acid sequence of the IgG.26
Plasmids enabling the expression of IL-1RA fused to the C-terminus of the IgG1 TM heavy chain through a (G4S)2 flexible linker were assembled by PCR amplification of the IL-1RA gene from complementary DNA obtained from Source Bioscience and subsequent PCR amplification with oligos that overlapped the IL-1RA gene and the IgG1 TM CH3 domain and incorporated the linker. Expression and purification of IgG1 TM-IL-1RA fusions were performed as described above.
2.1.2. Screening of 8D3 variants for reduced binding to mouse transferrin receptor
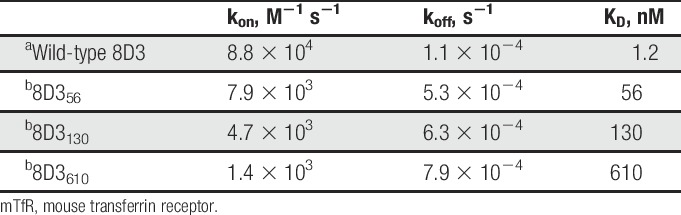
8D3 variants were screened for reduced affinity to mouse TfR (mTfR) using a bio-layer interferometry assay performed on an Octet RED384 System (ForteBio). Assays were performed in kinetics buffer (PBS containing 1 mg/mL BSA and 0.01% Tween-20). Purified IgGs were immobilised onto antihuman Fc capture biosensors at 20 μg/mL after which association and dissociation of mTfR (Sino Biological) were monitored at concentrations of 0.031 to 2 μM. Data were fitted using a 1:1 binding model to determine kinetic constants. Because of the slow off-rate of the complex between wild-type 8D3 and mTfR, it was not possible to accurately determine kinetic parameters for this interaction using the above assay. In this case, affinity was determined by surface plasmon resonance using a Biacore T100 system. Wild-type 8D3 IgG1 TM was diluted to 20 μg/mL in 10 mM sodium acetate of pH 5.0 and immobilised onto a CM3 Sensor Chip using the Amine Coupling Kit according to the manufacturer's instructions. Binding analysis was performed in HBS-P buffer at 1.6 to 25 nM mTfR with 120 seconds contact time and 600 seconds dissociation time. The surface was regenerated using 10 mM glycine of pH 2.5. A 1:1 binding model was used to fit the data.
2.1.3. In vitro potency testing of anti–transferrin receptor-IL-1RA constructs in cell-based cytokine release assay
The in vitro activity of the antibody–IL-1RA fusions was tested in a cellular cytokine secretion assay. Mouse NIH-3T3 cells were seeded on PDL-coated Greiner tissue culture–treated plates in 50 uL Dulbecco modified Eagle medium containing 10% FCS and 1% pen-strep and incubated for 18 hours at 37°C with 5% CO2. Anti–TfR-IL-1RA constructs were diluted in Dulbecco modified Eagle medium, applied to cells and incubated further for 30 minutes. NIH-3T3 cells were stimulated with recombinant mouse IL-1β (0.015 ng/mL, R&D Systems). After 18 hours, cell supernatants were collected and levels of secreted IL6 quantified using the MesoScale Discovery (MSD) kit according to the manufacturer's instructions. Levels of secreted IL6 were quantified by reference to standard curves generated using calibrator samples.
2.1.4. Peripheral kinetics and brain exposure
Noncompartmental (NCA) pharmacokinetic (PK) analysis was performed using Phoenix WinNonlin Professional [version 6.3; Pharsight (Certara), Sunnyvale, CA]. Nominal collection times were used for the PK data analyses, with below level of quantification values set to missing for calculation of the concentration means at nominal time points. The below level of quantification values were set as zero at predose, and missing after peak concentrations for the NCA.
The area under the concentration-time curve to the last measurable time point (AUC0-t) was calculated for plasma and brain using the linear trapezoidal method as implemented in WinNonlin Phoenix. In addition, systemic clearance (CL), terminal volume of distribution (Vz), and terminal half-life (t1/2) were determined for plasma. The Cmax and Tmax quoted are the observed values based on the mean concentration data at each time point.
All studies to measure antibody exposure in the periphery and brain were performed at Quotient Biosciences (Rushden, United Kingdom). Male C57B/6 mice, age 10 to 12 weeks were intravenously injected with anti-TfR variants or control IgG at 20 mg/kg or molar equivalent. Intravenous doses were administered into a tail vein at a constant dose volume of 10 mL/kg. Antibodies were supplied in D-PBS (Sigma). After dosing, 2 blood plasma samples were collected into individual Li-heparin containers from each of the 6 animals per time point per dose group. The first sample from each animal was collected from the lateral tail vein (ca 200 μL) into a Li-Hep microvette (BD Diagnostic Systems), whereas the second sample (ca 600 μL) was collected by cardiac puncture under isoflurane anaesthesia into a Li-Hep microtainer (BD Diagnostic Systems). After collection, blood samples were allowed to clot for 30 minutes and centrifuged at ×10,000 g for 2 minutes at 4°C and the resultant plasma drawn off. Plasma samples were flash frozen on dry ice for subsequent analysis. After final blood collection, the mice were perfused with D-PBS at a rate of 2 mL/min for 10 minutes until the extremities appeared white. Brains were excised and one hemisphere immediately processed, the other snap frozen in liquid nitrogen. To remove the spinal cord, the spinal column was exposed and severed at both the neck and at the base of the spine. To collect the spinal cord, D-PBS was rapidly forced through the section of spine through a needle and syringe. The excised spinal cord was dried with absorbent paper and samples weighed before homogenisation.
Spinal cord or brain hemisphere was homogenized in 5 volumes of ice-cold PBS containing 1% NP-40 and Complete protease inhibitor cocktail tablets (Roche Diagnostics). Homogenisation was performed in a 10 mL Potter-Elvehjem mortar type glass homogeniser with PTFE pestle, using 2 × 10 clockwise strokes with 5 seconds rest time. Homogenates were transferred to LoBind tubes (Eppendorf) and rotated at 4°C for 1 hour before centrifuging in a chilled benchtop centrifuge at 13,000 g for 20 minutes. The supernatant was isolated for brain antibody measurement.
2.1.5. Measurement of antibody concentrations in mouse brain, spinal cord, and plasma
Antibody concentrations in mouse plasma, brain, and spinal cord samples were measured through the MSD assay platform. The MSD method uses a plate-based sandwich immunoassay format where antihuman IgG capture antibody binds calibrator or samples, and a specific detection antibody labelled with SULFO-TAG emits light on electrochemical stimulation. Levels of anti-TfR variant and control antibody ± IL-1RA fusions in plasma, brain, and spinal cord samples were quantified by reference to standard curves generated using calibrator samples with a four-parameter nonlinear regression model.
2.1.6. Partial nerve ligation
All studies were performed using adult female C57Bl/6J mice weighing 18 to 22 g (Charles River, United Kingdom). Female mice were chosen as we have found them to be more suitable for behavioural studies. In addition, they are easier to keep group housed for longer-term studies thus removing the need to isolate them due to fighting. Eight to ten mice were used for each study group. Separate animals were used in each study. Animals were housed in groups of 5 to 6 per cage, in individually ventilated cages with free access to food and water under a 12-hour light/dark cycle (lights on 07:00-19:00). A total of 110 mice were used in these studies. Housing and procedure rooms were maintained at 21°C ± 2°C with a relative humidity of 65% ± 10%. All procedures were performed in accordance with the Animals (Scientific Procedures) Act 1986 and were approved by a local ethics committee. All mice underwent insertion of transponders under anaesthesia (3% isoflurane in oxygen) for identification purposes at least 5 days before the start of each study. Mechanical hyperalgesia was determined using an analgesiometer29 (Ugo Basile, Italy). An increasing force was applied to the dorsal surface of each hind paw in turn until a withdrawal response was observed. The application of force was halted at this point and the weight in grams recorded. Data were expressed as withdrawal threshold in grams for ipsilateral and contralateral paws. After the establishment of baseline readings, mice were divided into 2 groups with approximately equal ipsilateral/contralateral ratios which underwent surgery to partially ligate the sciatic nerve or served as sham-operated controls based on the previously described method of Seltzer et al.33 Operated mice were anaesthetised with isoflurane. After this, approximately 1 cm of the left sciatic nerve was exposed by blunt dissection through an incision at the level of the mid-thigh. A suture (9/0 Virgin Silk: Ethicon) was then passed through the dorsal third of the nerve and tied tightly. The incision was closed using Vetbond, and the mice were allowed to recover for at least 7 days before commencement of testing. Sham-operated mice underwent the same protocol but after exposure of the nerve, the mice were glued and allowed to recover. Mice were tested for baseline responses on day 7 and 10 postsurgery. Operated mice showing ipsilateral/contralateral ratios of greater than 80% were classed as nonresponders and were removed from the study. The remaining mice were then randomly allocated into treatment groups of 8 to 10 mice per group with approximately equal ipsilateral/contralateral ratios after which mice were treated with the compound under test. For the IL-1R antagonist Kineret (the commercial form of IL-1RA) study, all mice were anaesthetised using 3% isoflurane in oxygen and were then dosed intrathecally with one of the following treatments: PBS vehicle (5 μL per mouse) or Kineret (10, 13, or 100 μg per mouse). Sham-operated animals received PBS vehicle (5 μL per mouse). To investigate the effects of the transferrin fusions, animals received PBS vehicle (10 mL/kg bodyweight s.c.) or the relevant antibody IL-1RA fusion protein (100 mg/kg s.c.). Sham-operated mice received PBS vehicle (10 mL/kg bodyweight s.c.). Mice were retested for changes in mechanical hyperalgesia 4 hours postdose as described above. Mice were also retested at 1, 2, 3, and 4 days postdose.
2.1.7. Data analysis
Statistical analysis was performed in GraphPad Prism. Only animals which completed the study were included in the analysis. Results were analysed using 2-way analysis of variance. Pairwise comparisons, where appropriate, were made using Tukey test.
3. Results
3.1. Sciatic nerve ligation results in mechanical hyperalgesia that can be reversed by central exposure to IL-1RA
We investigated the partial nerve ligation model of Seltzer et al33 for the development of mechanical hyperalgesia. In rats, this procedure results in the development of a neuropathic pain phenotype that can be measured through increased sensitivity to mechanical pressure at the ipsilateral paw. It has been previously shown that mice respond in a similar manner8 and it was preferential for these studies to be performed in mice as the antibody 8D3 does not bind to rat TfR. In all studies, C57BL/6 mice subjected to partial ligation of the sciatic nerve exhibited mechanical hyperalgesia that manifested as a significant reduction in the ipsilateral but not contralateral sensitivity to mechanical force on day 7 and 10 postsurgery when compared with sham-operated controls. Having demonstrated a robust reproducible response in mice, we investigated the sensitivity of the model to the administration of drug molecules. When vehicle, PBS, was administered i.t., neither operated mice nor sham-operated mice showed any change in the level of mechanical hyperalgesia from predose levels. This indicated that animal handling and drug administration procedures did not alter response.
To test whether the mechanical hyperalgesia induced by partial ligation of the sciatic nerve was dependant on IL-1R signalling, we investigated the effect of peripheral and i.t. delivery of the IL-1R antagonist Kineret (the commercial form of IL-1RA). The administration of Kineret peripherally at a dose of 100 mg/kg demonstrated no reversal of the mechanical hyperalgesia. However, i.t. delivery of Kineret showed a dose-related reversal that peaked at 2 to 4 hours postinjection and then diminished over time, returning to baseline levels 2 days postdose. A dose of 100 μg of Kineret i.t. returned the ipsilateral/contralateral ratio from around 55 to approximately 80 at 2 hours postdose. This was maintained at 4 hours but had reduced to around 70 at 24 hours. A dose of 30 μg i.t. achieved a ratio of around 78 at 2 and 4 hours before declining to around 58 at 24 hours. A dose of 10 μg did not result in any significant analgesia at any time point (Fig. 1). This demonstrates that in this model, only central blockade of IL-1R signalling can result in dose-dependent analgesia and that a peripheral dose approximately 80-fold higher than the central dose exhibits no change in the response.
Figure 1.
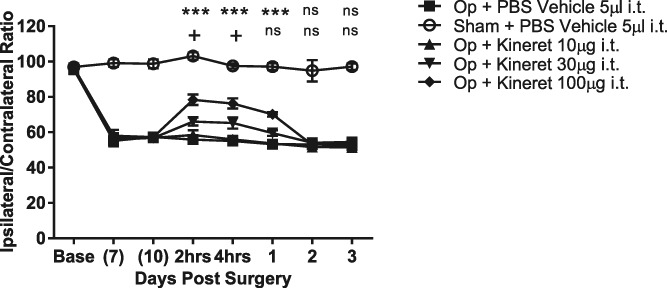
Effect of i.t. administered Kineret on the reversal of partial nerve ligation–induced mechanical hyperalgesia. Data are mean % ipsilateral/contralateral ratio ± SEM. N = 9 to 10 per group. +P < 0.05 Kineret (30 μg per mouse) vs PBS vehicle control; ***P < 0.001 Kineret (10 μg per mouse) vs PBS vehicle control. i.t., intrathecal; ns, not significant.
3.2. Identification and characterisation of a blood–brain barrier transmigrating anti–transferrin receptor antibody
To investigate whether IL-1RA could be delivered across the BBB in mice, the anti-TfR antibody 8D3 was chosen for its ability to enter the CNS.19–21,34,41,42 Despite previous evidence, we were unable to detect significant amounts of 8D3 within the brains of treated animals. It has been reported that the affinity of antitransferrin antibodies for TfR is important for their ability to cross the BBB.39 We therefore generated 8D3 variants with reduced affinity for TfR. Variants with reduced affinity were identified through alanine scanning mutagenesis of either the light or heavy chain CDR3s. Only modest reductions in affinity were achieved by this method, but pairing of the heavy and light chain alanine variants resulted in substantial further reductions in affinity relative to that of the parent antibodies, with KD values of up to 610 nM being observed. Three mutants which had KD values of 56, 130, and 610 nM, respectively, were selected for testing for brain exposure and ability to elicit a centrally mediated effect when coupled to IL-1RA. These mutants are referred to herein as 8D356, 8D3130, and 8D3610 on the basis of their respective affinities (Table 1).
Table 1.
Kinetic parameters for binding of wild-type 8D3 and variants to mTfR.
3.3. Pharmacokinetic properties of anti–transferrin receptor antibodies
3.3.1. Peripheral pharmokinetics
To confirm that variants of the anti-TfR antibody 8D3 had altered brain exposure dependent on their affinity for TfR, we conducted a series of PK and brain exposure studies in C57BL/6J mice. Studies were performed over a 2-week period with blood and brain homogenate samples taken at regular time points. The sampling procedure in these studies resulted in composite profiles for plasma exposure after a single intravenous dosing at 20 mg/kg, and all parameters were derived using the mean of each data point. A control group received the same dosage of an antibody to an irrelevant antigen of the same isotype as the anti-TfR antibodies (NIP228). Figure 2A shows the mean plasma data ± SE for the 5 antibodies profiled.
Figure 2.

Plasma and brain exposure of anti-transferrin variants in a mouse PK assay. (A) Plasma PK of anti-TfR variants and isotype control (NIP228) over a 2-week period. N = 6 per group (B) Brain exposure as a measure of % injected dose per gram of brain. (C) Spinal cord exposure as a measure of % injected dose per gram of spinal cord. PK, pharmokinetic; TfR, transferrin receptor.
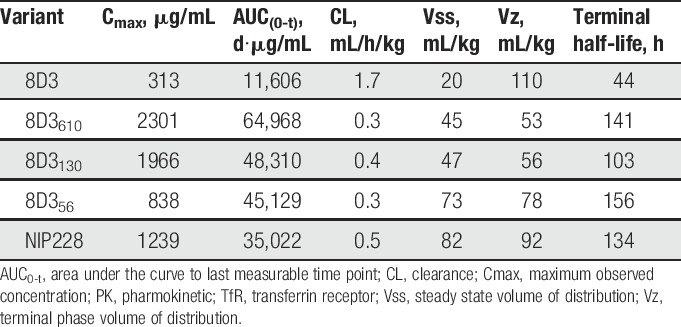
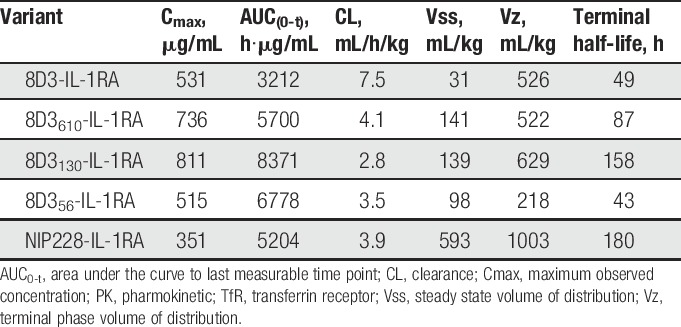
The pharmacokinetic parameters for the lower affinity variants of the anti-TfR antibody 8D3 (8D356, 8D3130, and 8D3610) were broadly similar, and within 2-fold of the isotype control antibody (Table 2). Their half-lives ranged from 4.3 to 6.5 days, differences that are driven by differences in Vβ and the rate of CL. However, in comparison, 8D3 had considerably altered systemic pharmacokinetic parameters that were driven by an approximately 4-6-fold higher CL than the lower affinity 8D3 variants or isotype control antibody.
Table 2.
Plasma PK parameters of anti-TfR variants and isotype control in hIgG1TM format.
3.3.2. Brain and spinal cord exposure of anti–transferrin receptor antibodies
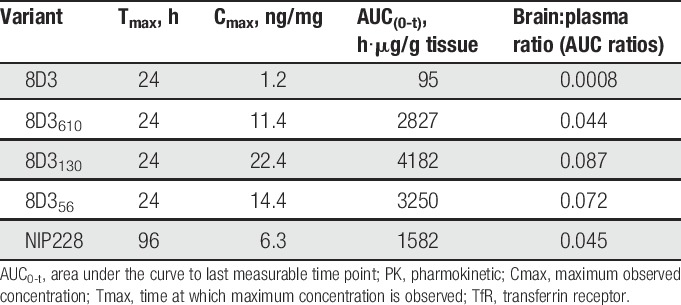
Measurement of each of the antibodies in homogenate of brain and spinal cord was performed to determine the central exposure. Brain and spinal cord samples were taken at 4, 24, 48, 96, 168, and 336 hours after intravenous administration and processed to homogenate for analysis through the MSD assay (Fig. 2B, C). For all 8D3 anti-TfR antibodies, Tmax occurred at 24 hours after administration (Table 3). The isotype control antibody reached Tmax much later at 4 days after administration. Brain Cmax and AUC(0-t) values observed for the anti-TfR antibodies indicated that there was a relationship between affinity and brain exposure similar to data that were previously reported by Yu et al39 and Couch et al.9 Lower affinity variants of 8D3 (8D356, 8D3130, and 8D3610) demonstrated significantly more brain and spinal cord exposure than 8D3 (Fig. 2B, C and Table 3). Pharmokinetic parameter analysis indicated that 8D3 possessed the poorest brain exposure, as described by the mean Cmax. This parameter was 18.7 times less than the best brain exposure of lower affinity variant—8D3130. In addition, AUC(0-t) for 8D3 was 44 times less than that achieved for 8D3130 (Table 3). Notably, 8D3 could only be detected in brain and spinal cord samples for the first 96 hours of the study (Fig. 2B, C). Assay readings for later time points were below the lower limit of quantification for the assay. All PK parameters were derived using mean data (Table 3).
Table 3.
Brain PK parameters for anti-TfR variants and isotype control in hIgG1TM format.
3.4. Pharmacokinetics for anti–transferrin receptor hIgG1-IL-1RA fusion proteins
3.4.1. Peripheral pharmokinetics
Peripheral PK analysis was performed on the antibody IL-1RA fusions over a 2-week period. The IL-1RA antibody fusions demonstrated significantly reduced plasma exposure, with a 1.8 and 2.7 times higher CL than the lower affinity anti-TfR antibodies (8D356, 8D3130, and 8D3610) (Fig. 3A and Table 4). There was also a 3.6 to 11.4 fold reduction in plasma exposure, measured by AUC(0-t), for all IL-1RA antibody fusions tested, when compared with anti-TfR antibodies without IL-1RA (Tables 2 and 4). This suggests that IL-1RA also played a significant role in plasma CL for these proteins. Cmax for all fusion proteins occurred at the first measured time point, 10 minutes after administration.
Figure 3.

Plasma, brain, and spinal cord exposure of anti-transferrin variant-IL-1RA fusion in mouse PK assay. (A) Plasma PK of anti-TfR-IL-1RA variants and isotype control (NIP228) over a 2 week period. N = 6 per group (B) Brain exposure as a measure of % injected dose per gram of brain. (C) Spinal cord exposure as a measure of % injected dose per gram of spinal cord. PK, pharmokinetic; TfR, transferrin receptor.
Table 4.
Plasma PK parameters for anti-TfR variants and isotype control hIgG1-IL-1RA fusion proteins.
3.4.2. Brain and spinal cord exposure
Brain and spinal cord exposure was measured for the antibodies fused to IL-1RA. Tmax for each of the antibody–IL-1RA fusions tested was at the first measured time point (4 hours), which is somewhat earlier than that observed for the antibodies alone at 24 hours (Fig. 3B, C and Table 5). 8D3 was only detectable in the brain and spinal cord for the first 24 hours of the study, after which it was below the lower limit of quantification for the assay (Fig. 3B, C). Brain exposure as measured by AUC(0-t) was between 22.6- and 69-fold higher for the lower affinity 8D3 variants compared with 8D3 (Table 5).
Table 5.
Brain PK parameters for anti-TfR variants and isotype control hIgG1-IL-1RA fusion proteins.
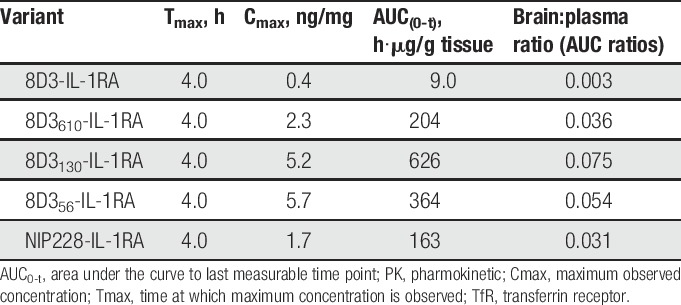
The lower central exposure of 8D3-IL-1RA was likely due to faster peripheral CL, but this fusion protein also had 12- to 25-fold lower brain:plasma AUC(0-t) ratio than the lower affinity 8D3 variant antibody IL-1RA fusions (Table 5).
3.5. In vitro activity of antibody–IL-1RA fusions
To test whether the fusion of IL-1RA to the antibodies resulted in any change in its ability to inhibit the action of IL-1, we measured potency of these fusion proteins in an IL-1β-driven in vitro–cell-based assay. Pretreatment of NIH-3T3 cells for 30 minutes with IL-1RA-anti–TfR fusions potently inhibited IL-1β-induced IL6 secretion (Fig. 4). 8D3-IL-1RA displayed a 3-fold increase in potency when compared with IL-1RA alone (Fig. 4). 8D3610-IL-1RA, 8D3130-IL-1RA, 8D356-IL-1RA, and the isotype control NIP228-IL-1RA displayed an ∼10-fold reduction in potency compared with IL-RA alone (Fig. 4). NIP228 isotype control with no IL-1RA fusion protein had no effect on IL-1β activity.
Figure 4.
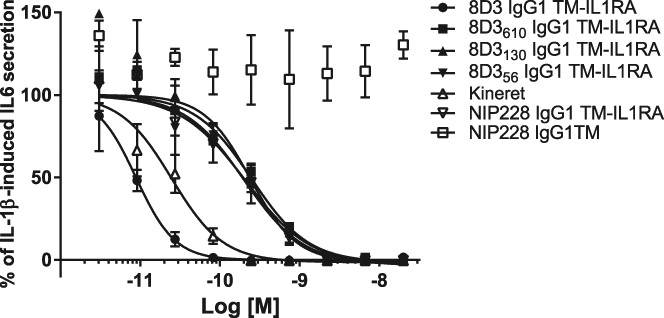
Effect of IL-1RA-anti–TfR fusions on IL-1β–induced IL6 secretion from NIH-3T3 cells in culture. Data are expressed as % IL-1β–induced IL6 mean ± SEM. Cells were pretreated with IL-1RA-anti–TfR fusions for 30 minutes before IL-1β stimulation. TfR, transferrin receptor.
3.6. Reversal of mechanical hyperalgesia by blood–brain barrier transmigrating anti–-TfR-IL-1RA fusions
We next tested whether fusions of IL-1RA to the anti-TfR antibodies were able to reverse the mechanical hyperalgesia. Antibodies were administered s.c. at doses of 100 mg/kg, and the mice monitored for 7 days postdose. Treatment with vehicle, PBS, or NIP228-IL-1RA had no effect on mechanical hypersensitivity in mice indicating that NIP228-IL-1RA was not able to access IL-1R in the central compartment. Administration of the 8D3-IL-1RA fusion also showed no reversal of the mechanical hyperalgesia throughout the study. However, reversal of the mechanical hyperalgesia was seen with the reduced affinity variants of 8D3 fused to IL-1RA. 8D3610-IL-1RA showed a significant reversal at all time points from 4 hours to 7 days postdose. The response was constant through the first 4 days and only started to decline on day 7. 8D3130-IL-1RA produced a significant reversal from 4 hours to 4 days postdose but no significance was seen at 7 days postdose. The magnitude of response was greater than that seen for 8D3610-IL-1RA during the first 4 days but the loss of effect at day 7 resulted in a greater response from 8D3610-IL-1RA at this time point. (Fig. 5).
Figure 5.
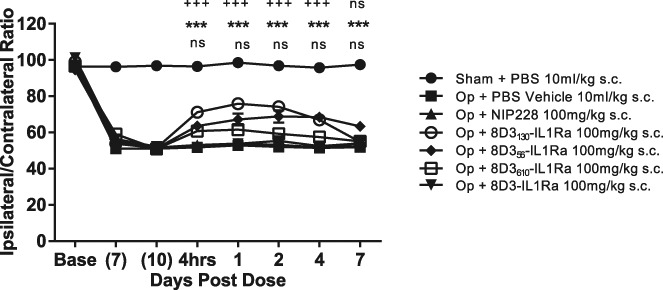
Effect of anti-transferrin-IL-1RA fusions on the reversal of partial nerve ligation induced mechanical hyperalgesia. N = 8 to 9 per group. **P < 0.01; ***P < 0.001 Op + NIP228 vs Op + 8D3610-IL-1RA; +++P < 0.001 Op + NIP228 vs Op + 8D3130-IL-1RA; ns, not significant (P > 0.5). s.c, subcutaneous.
To investigate the relationship between dose and the reversal of mechanical hyperalgesia, we administered 8D3130-IL-1RA at doses of 20, 50, and 100 mg/kg. The different dose levels showed differences in both the magnitude and duration of the response. The 20 mg/kg dose showed significant reversal of mechanical hyperalgesia for only 24 hours postdose, the 50 mg/kg for up to 4 days postdose, and the 100 mg/kg for up to 7 days postdose (Fig. 6).
Figure 6.
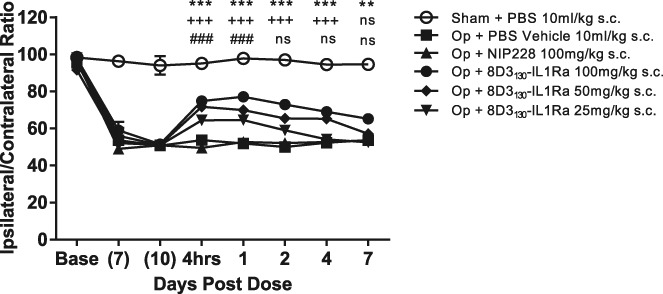
Dose response of 8D3130-IL-1RA fusions on the reversal of partial nerve ligation–induced mechanical hyperalgesia. N = 8 to 9 per group. **P < 0.01; ***P < 0.001 Op + NIP228 vs Op + 8D3130-IL-1RA (100 mg/kg); +++P < 0.001 Op + NIP228 vs Op + 8D3130-IL-1RA (50 mg/kg); ###P < 0.001 Op + NIP228 vs Op + 8D3130-IL-1RA (20 mg/kg); ns not significant (P > 0.5). s.c, subcutaneous.
4. Discussion
The expression of IL-1β by glia in the spinal cord and its direct action on spinal neurons potentiates nociceptive signalling through IL-1-receptor–dependent signalling and N-methyl-D-aspartate receptor phosphorylation. Furthermore, central administration of IL-1RA reduces inflammatory hyperalgesia by blocking this central mechanism of nociceptive processing.40 In rodent models of neuropathic pain including chronic constriction injury and spared nerve injury, central IL-1β levels correlate with nociceptive responses12 and blockade of IL-1 signalling with IL-1RA provides analgesia in part due to a reduction in ectopic neuronal discharge.16
Our results have confirmed that the symptoms of neuropathic pain can be relieved through central, but not peripheral, blockade of IL-1R signalling. Central IL-1R blockade was achieved by direct i.t. IL-1RA delivery or through peripheral delivery of an IL-1RA-anti–TfR fusion protein. However, such responses do not necessarily indicate the site of action as behind the BBB in the brain or spinal cord. Centrally delivered antibodies and other vectors may access the dorsal root ganglion (DRG) through the cerebrospinal fluid, which is known to reach the DRG.28,36 Furthermore, some of the changes that may contribute to neuropathic pain occur in the DRG including an increase in the expression of the IL-1β and IL-1R.22,35 We cannot rule out the possibility that peripherally administered IL-1RA-anti–TfR antibodies penetrate the DRG secondary to crossing the BBB into the spinal cord. However, our data indicate that CNS delivery of the IL-1R1 antagonist is a fundamental requirement in driving efficacy in this neuropathic pain model.
Antibodies have become an important class of drug molecules over recent years, demonstrating strong clinical efficacy in a variety of indications.3 However, there are few antibodies entering clinical development with a centrally driven mechanism of action. Development of effective antibody-based treatments for CNS disorders will require the codevelopment of technologies that enhance their BBB penetration. In vitro models of the BBB have proven useful for the measurement of small molecule transport and can help drive in vivo exposure prediction.38 However, such in vitro studies are far less common for antibodies with the majority of evidence supporting antibody-BBB delivery coming from in vivo studies.
IL-1RA is amenable to genetic manipulation and we have found it can be successfully fused at the C-terminus of the Fc domain of an IgG and maintaining sufficient activity for further study (Fig. 4). Such fusions do not influence the activity of the Fab domain of the antibody allowing molecules with differing antigen specificities or affinities to be generated all having near identical specific activity at IL-1R, with the exception of the fusion to 8D3. The high affinity antibody 8D3 when fused to IL-1RA had a higher in vitro IL-1 neutralising activity. We attribute this to be due to the high affinity anti-TfR antibody creating a higher local concentration of the fusion protein at the cell surface, thereby generating an apparently higher neutralising capability. As this fusion protein had undetectable activity in vivo, in contrast to its in vitro behaviour, we do not believe this anomalous behaviour had any bearing on the conclusion that a lower affinity for TfR enhances brain penetration. Thus the magnitude of any pharmacological response resulting from IL-1R blockade in vivo is dependent only on changes resulting from alteration in the antibody affinity for TfR (and not on any modulation of the IL-1RA).
Blood–brain barrier transport of antibodies through the TfR has been previously described.19,23,24,39 Transport of anti-TfR antibodies into the CNS is dependent on their affinity for TfR, with lower affinity antibodies demonstrating greater BBB transport.39 The variants of the antimouse transferrin receptor antibody 8D3 described in this article also have lowered affinities for TfR and demonstrate markedly differing abilities to cross the BBB (Fig. 2). The magnitude of brain exposure is similar to the findings of Yu et al.39 However, the temporal pattern is markedly different. Yu et al observed peak brain exposure at 4 to 5 hours after administration, whereas we see peak exposure 24 hours after administration. There are a number of possible reasons for these differences: Yu et al do not report the absolute affinities of their anti-TfR antibodies but only the differences relative to each other. We are not therefore able to say whether or not we are working in the same affinity range. The anti-TfR described by Yu et al39 may also have a distinct epitope to the 8D3 antibody we describe which could drive differing transport properties.
On fusion of IL-1RA to the antibodies, a slightly reduced peripheral half-life was noted, whereas the CNS exposure profile was markedly different (Fig. 3B, C). Peak brain exposure was observed at the first sample postinjection and decayed rapidly before plateauing at around 4 days. This suggests that IL-1RA affects the distribution of these antibodies and this effect is mostly likely due to binding of the IL-1RA component of the fusion to IL-1R in the peripheral and central compartments. The more marked change in the exposure profile in the brain may be due to the presence of IL-1R on endothelial cells of the BBB. The presence of both IL-1R and TfR on these cells may result in an avid interaction of the IL-1RA-anti–TfR fusions with the receptors on these cells and alter their ability to cross the BBB. An increase in avidity would increase the apparent affinity of these fusions for the endothelial cells and, as it has been demonstrated that affinity plays an important role in BBB transport, the result would likely be reduced transport across these cells. The more rapid CL from the blood would also reduce the further uptake of antibody across the BBB.
In agreement with previous studies,15,40 our data suggest a critical role for central IL-1β in nociceptive signalling during chronic pain states. Greater IL-1RA penetration of the CNS results in greater analgesia and prolonged exposure in the central compartment results in a longer duration of the analgesia induced by the IL-1RA fusion. Using a disease-relevant model of chronic pain, we have successfully shown for the first time that a biological peptide antagonist can be re-engineered, facilitating its CNS delivery and enhancing its analgesic properties.
Conflict of interest statement
All authors were employees of MedImmune at the time of the work and therefore have a theoretical conflict of interest through being employed by the organisation that both funded the work and has a potential commercial interest in the findings.
Acknowledgements
The authors thank all the staff of MedImmune's Biological Services unit for their invaluable help and technical assistance in performing the mechanical hyperalgesia studies. All work described in this manuscript was funded by MedImmune.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
References
- [1].Alexander GM, van Rijn MA, van Hilten JJ, Perreault MJ, Schwartzman RJ. Changes in cerebrospinal fluid levels of pro-inflammatory cytokines in CRPS. PAIN 2005;116:213–19. [DOI] [PubMed] [Google Scholar]
- [2].Apkarian AV, Lavarello S, Randolf A, Berra HH, Chialvo DR, Besedovsky HO, del Rey A. Expression of IL-1beta in supraspinal brain regions in rats with neuropathic pain. Neurosci Lett 2006;407:176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 2010;10:345–52. [DOI] [PubMed] [Google Scholar]
- [4].Boado RJ, Zhang Y, Wang Y, Pardridge WM. Engineering and expression of a chimeric transferrin receptor monoclonal antibody for blood-brain barrier delivery in the mouse. Biotech Bioeng 2009;102:1251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Burgess G, Williams D. The discovery and development of analgesics: new mechanisms, new modalities. J Clin Invest 2010;120:3753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Carter DB, Deibel MRJ, Dunn CJ, Tomich CS, Laborde AL, Slightom JL, Berger AE, Bienkowski MJ, Sun FF, McEwan RN. Purification, cloning, expression and biological characterization of an interleukin-1 receptor antagonist protein. Nature 1990;344:633–8. [DOI] [PubMed] [Google Scholar]
- [7].Chessell IP, Dudley A, Billinton A. Biologics: the next generation of analgesic drugs? Drug Discov Today 2012;17:875–9. [DOI] [PubMed] [Google Scholar]
- [8].Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL, Grahames CBA, Casula MA, Yiangou Y, Birch R, Anand P, Buell GN. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. PAIN 2005;114:386–96. [DOI] [PubMed] [Google Scholar]
- [9].Couch JA, Yu YJ, Zhang Y, Tarrant JM, Fuji RN, Meilandt WJ, Solanoy H, Tong RK, Hoyte K, Luk W, Lu Y, Gadkar K, Prabhu S, Ordonia BA, Nguyen Q, Lin Y, Lin Z, Balazs M, Scearce-Levie K, Ernst JA, Dennis MS, Watts RJ. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med 2013;5:183ra57. [DOI] [PubMed] [Google Scholar]
- [10].Daramola O, Stevenson J, Dean G, Hatton D, Pettman G, Holmes W, Field R. A high-yielding CHO transient system: coexpression of genes encoding EBNA-1 and GS enhances transient protein expression. Biotechnol Prog 2014;30:132–41. [DOI] [PubMed] [Google Scholar]
- [11].del Rey A, Apkarian AV, Martina M, Besedovsky HO. Chronic neuropathic pain-like behavior and brain-borne IL-1beta. Ann N Y Acad Sci 2012;1262:101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].del Rey A, Yau H, Randolf A, Centeno MV, Wildmann J, Martina M, Besedovsky HO, Apkarian AV. Chronic neuropathic pain-like behavior correlates with IL-1beta expression and disrupts cytokine interactions in the hippocampus. PAIN 2011;152:2827–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dinarello CA. Biologic basis for interleukin-1 in disease. Blood 1996;87:2095–147. [PubMed] [Google Scholar]
- [14].Ferreira SH, Lorenzetti BB, Bristow AF, Poole S. Interleukin-1 beta as a potent hyperalgesic agent antagonized by a tripeptide analogue. Nature 1988;334:698–700. [DOI] [PubMed] [Google Scholar]
- [15].Fiorentino PM, Tallents RH, Miller JH, Brouxhon SM, O'Banion MK, Puzas JE, Kyrkanides S. Spinal interleukin-1beta in a mouse model of arthritis and joint pain. Arthritis Rheum 2008;58:3100–9. [DOI] [PubMed] [Google Scholar]
- [16].Gabay E, Wolf G, Shavit Y, Yirmiya R, Tal M. Chronic blockade of interleukin-1 (IL-1) prevents and attenuates neuropathic pain behavior and spontaneous ectopic neuronal activity following nerve injury. Eur J Pain 2011;15:242–8. [DOI] [PubMed] [Google Scholar]
- [17].Kawasaki Y, Zhang L, Cheng J, Ji R. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci 2008;28:5189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kissel K, Hamm S, Schulz M, Vecchi A, Garlanda C, Engelhardt B. Immunohistochemical localization of the murine transferrin receptor (TfR) on blood-tissue barriers using a novel anti-TfR monoclonal antibody. Histochem Cell Biol 1998;110:63–72. [DOI] [PubMed] [Google Scholar]
- [19].Lee HJ, Engelhardt B, Lesley J, Bickel U, Pardridge WM. Targeting rat anti-mouse transferrin receptor monoclonal antibodies through blood-brain barrier in mouse. J Pharmacol Exp Ther 2000;292:1048–52. [PubMed] [Google Scholar]
- [20].Lee HJ, Boado RJ, Braasch DA, Corey DR, Pardridge WM. Imaging gene expression in the brain in vivo in a transgenic mouse model of Huntington's disease with an antisense radiopharmaceutical and drug-targeting technology. J Nucl Med 2002;43:948–56. [PubMed] [Google Scholar]
- [21].Lee HJ, Zhang Y, Zhu C, Duff K, Pardridge WM. Imaging brain amyloid of Alzheimer disease in vivo in transgenic mice with an abeta peptide radiopharmaceutical. J Cereb Blood Flow Metab 2002;22:223–31. [DOI] [PubMed] [Google Scholar]
- [22].Lee H, Lee K, Son S, Hwang S, Cho H. Temporal expression of cytokines and their receptors mRNAs in a neuropathic pain model. Neuroreport 2004;15:2807–11. [PubMed] [Google Scholar]
- [23].Moos T, Morgan EH. Evidence for low molecular weight, non-transferrin-bound iron in rat brain and cerebrospinal fluid. J Neurosci Res 1998;54:486–94. [DOI] [PubMed] [Google Scholar]
- [24].Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, Maier P, Rueger P, Stracke J, Lau W, Tissot A, Loetscher H, Ghosh A, Freskgard PO. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 2014;81:49–60. [DOI] [PubMed] [Google Scholar]
- [25].Oganesyan V, Gao C, Shirinian L, Wu H, Dall'Acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallog D Biol Crystallogr 2008;64:700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci 1995;4:2411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Persic L, Roberts A, Wilton J, Cattaneo A, Bradbury A, Hoogenboom HR. An integrated vector system for the eukaryotic expression of antibodies or their fragments after selection from phage display libraries. Gene 1997;187:9–18. [DOI] [PubMed] [Google Scholar]
- [28].Pettersson LME, Geremia NM, Ying Z, Verge VMK. Injury-associated PACAP expression in rat sensory and motor neurons is induced by endogenous BDNF. PLoS One 2014;9:e100730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther 1957;111:409–19. [PubMed] [Google Scholar]
- [30].Reeve AJ, Patel S, Fox A, Walker K, Urban L. Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain 2000;4:247–57. [DOI] [PubMed] [Google Scholar]
- [31].Ren K, Torres R. Role of interleukin-1beta during pain and inflammation. Brain Res Rev 2009;60:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 2001;410:471–5. [DOI] [PubMed] [Google Scholar]
- [33].Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. PAIN 1990;43:205–18. [DOI] [PubMed] [Google Scholar]
- [34].Shi N, Zhang Y, Zhu C, Boado RJ, Pardridge WM. Brain-specific expression of an exogenous gene after i.v. administration. Proc Natl Acad Sci U S A 2001;98:12754–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Uceyler N, Tscharke A, Sommer C. Early cytokine expression in mouse sciatic nerve after chronic constriction nerve injury depends on calpain. Brain Behav Immun 2007;21:553–60. [DOI] [PubMed] [Google Scholar]
- [36].Wang X, Wang C, Zeng J, Xu X, Hwang PYK, Yee W, Ng Y, Wang S. Gene transfer to dorsal root ganglia by intrathecal injection: effects on regeneration of peripheral nerves. Mol Ther 2005;12:314–20. [DOI] [PubMed] [Google Scholar]
- [37].Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, Pairaudeau G, Pennie WD, Pickett SD, Wang J, Wallace O, Weir A. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov 2015;14:475–86. [DOI] [PubMed] [Google Scholar]
- [38].Watson PM, Paterson JC, Thom G, Ginman U, Lundquist S, Webster CI. Modelling the endothelial blood-CNS barriers: a method for the production of robust in vitro models of the rat blood-brain barrier and blood-spinal cord barrier. BMC Neurosci 2013;14:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, Atwal J, Elliott JM, Prabhu S, Watts RJ, Dennis MS. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Trans Med 2011;3:84ra44. [DOI] [PubMed] [Google Scholar]
- [40].Zhang R, Li A, Liu B, Wang L, Ren K, Zhang H, Berman BM, Lao L. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. PAIN 2008;135:232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang Y, Pardridge WM. Delivery of beta-galactosidase to mouse brain via the blood-brain barrier transferrin receptor. J Pharmacol Exp Ther 2005;313:1075–81. [DOI] [PubMed] [Google Scholar]
- [42].Zhu C, Zhang Y, Pardridge WM. Widespread expression of an exogenous gene in the eye after intravenous administration. Invest Ophthalmol Vis Sci 2002;43:3075–80. [PubMed] [Google Scholar]