

Abstract
In 1974, the discovery of a mouse and a rat that spontaneously developed hyperglycemia led to the development of 2 autoimmune diabetes models: nonobese diabetic (NOD) mouse and Bio-Breeding rat. These models have contributed to our understanding of autoimmune diabetes, provided tools to dissect autoimmune islet damage, and facilitated development of early detection, prevention, and treatment of type 1 diabetes. The genetic characterization, monoclonal antibodies, and congenic strains have made NOD mice especially useful.
Although the establishment of the inbred NOD mouse strain was documented by Makino et al (Jikken Dobutsu. 1980;29:1–13), this review will focus on the not-as-well-known history leading to the discovery of a glycosuric female mouse by Yoshihiro Tochino. This discovery was spearheaded by years of effort by Japanese scientists from different disciplines and dedicated animal care personnel and by the support of the Shionogi Pharmaceutical Company, Osaka, Japan. The history is based on the early literature, mostly written in Japanese, and personal communications especially with Dr Tochino, who was involved in diabetes animal model development and who contributed to the release of NOD mice to the international scientific community. This article also reviews the scientific contributions made by the Bio-Breeding rat to autoimmune diabetes.
Key Words: nonobese diabetic mice, bio-breeding rats, type 1 diabetes
More than 100 years ago, the German pathologist Schmidt1 discovered the mononuclear cell infiltration in and around the islet of Langerhans in the pancreas of a patient who died of ketoacidosis. This was the first report that described islet inflammation associated with diabetes. The Swiss pathologist Von Meyenburg2 later termed this condition insulitis. Insulitis and islet destruction persist long after diabetes development. This was clearly illustrated in a report by Sutherland et al,3 in which his female type 1 diabetes (T1D) patient rejected a pancreas transplant from her nondiabetic, identical twin sister. The transplanted pancreas reversed diabetes, but shortly thereafter, the recurrence of disease became evident by hyperglycemia. This clinical finding took place approximately the same time as the discovery of spontaneously hypoinsulinemic diabetic rodents, nonobese diabetic (NOD) mice, and diabetic Bio-Breeding (BB) rats. The discovery of diabetic mice was first reported by Tochino et al at the monthly meeting of the Shionogi Laboratory in Osaka in June 1976 in a presentation entitled “Summary of Spontaneous Diabetic Mouse Arising From CTS (Cataract Shionogi) Mouse in Abrahi Farm”. In the same year, spontaneously diabetic BB rats were discovered in Ottawa, Canada.4
DISCOVERY OF MICE THAT SPONTANEOUSLY DEVELOP DIABETES
Development of a Mouse Model for Insulin-Dependent Diabetes
In early 1970s, Shionogi & Co Ltd, a leading Japanese pharmaceutical company, initiated efforts to developed a rodent model for insulin-dependent diabetes (IDDM) for the purpose of testing therapeutic compounds. Before that, Hiroshi Ohtori began development of a strain of mice that spontaneously developed cataracts at the Shionogi Abrahi Animal Research Facility from outbred ICR-JCL (ICR) mice obtained from the Nihon CREA, a Japanese experimental animal supplier. The ICR mice were of Swiss origin and imported to Japan from the United States. Ohtori first discovered cataracts in ICR mice in 1968,5 and by 1975, an inbred Cataract Shionogi (CTS) mouse line that spontaneously developed cataracts was established (Fig. 1). During the CTS mouse inbreeding process, Ohtori developed a hypothesis that cataracts might be associated with diabetes. To test the hypothesis, he established 2 separate sets of brother-sister matings starting from the sixth generation of CTS mice, which he divided into groups with either high or normal fasting plasma glucose levels. Toyohiko Yoshida in the Abrahi Facility started to be involved in this selective breeding program. As the generations progressed, the cataract trait disappeared in both higher and normal blood glucose colonies, thus indicating no relation between blood glucose and spontaneous cataract development. After Ohtori left the Abrahi Facility, Yoshida continued selective brother-sister breedings, which reached the 13th generation. By this time, 2 colonies of mice were clearly distinguishable by blood glucose: 1 colony showing fasting plasma glucose of approximately 100 mg/dL (normoglycemic) and the other exhibiting a slightly higher level of approximately 150 mg/dL (hyperglycemic) (Fig. 1).6
FIGURE 1.
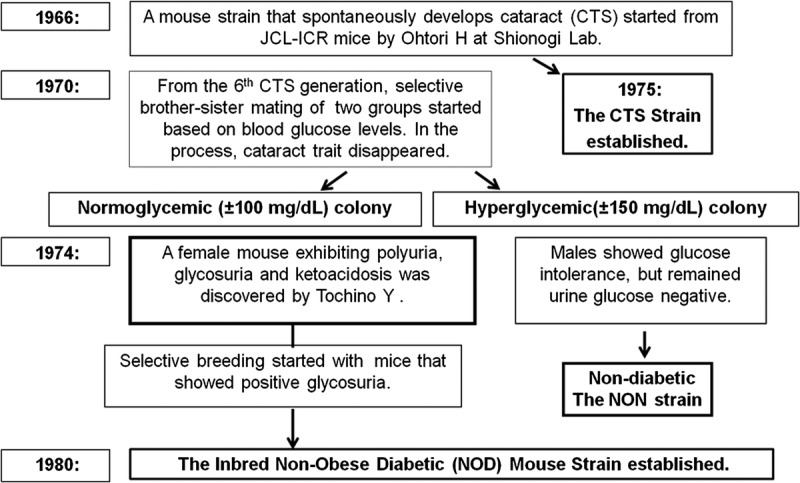
Discovery and development of the nonobese diabetic mouse.
At approximately this time, a special project team was set up at the Shionogi Laboratory in Osaka to develop a spontaneously diabetic animal model. The team was led by Tochino based on his expertise and knowledge on experimental diabetes in rodents and his long-term involvement in drug development. Jinsaku Maeda, an expert in animal husbandry with experience from the Abrahi Animal facility, also joined the team. In 1973, however, the project faced near-termination because of the lack of significant progress, and Maeda returned to the Abrahi Facility to care for the mouse colonies started by Ohtori after Yoshida left the facility. Mice in the hyperglycemic colony, particularly males, displayed higher plasma glucose and glucose intolerance but showed no sign of glycosuria. Maeda sent several pairs of mice from both normo- and hyperglycemic colonies to Tochino in Osaka for closer evaluation. In 1974, Yoshihiro Tochino discovered a female mouse that showed polyuria and positive urine glucose by TesTape (Fig. 1). Despite being from the normoglycemic colony, this mouse was hyperglycemic, rapidly lost weight, and died of ketoacidosis in less than 1 month without producing offspring. No biochemical or pathological examination was conducted. The remaining mice housed in the Osaka Laboratory did not develop polyuria or glycosuria.
The discovery of the glycosuric mouse was reported in 1975 at the Shionogi research meeting, and the normoglycemic colony from which this mouse derived was named “mt mice”, taking the initial of two key investigators, Jinsaku Maeda and Yoshihiro Tochino.
The discovery of the glycosuric female mouse traveled quickly to the Abrahi Laboratory, where Maeda found several incidences of newborn mouse death in the normoglycemic colony. The deaths were caused by drowning in wet bedding, but the incidence was not reported because the wet bedding was thought to be caused by water leakage from the drinking bottle (Tochino, personal communication).7 Thereafter, mice in wet cages were routinely tested. The TesTape examination detected additional mice positive for glycosuria in wet cages of the Ohtori/Yoshida normoglycemic colony but not in the hyperglycemic colony. By Tochino's request, Maeda separated polyuric/glycosuric mice to begin selective breeding with the premise to establish a diabetic mouse strain, and thus, the selective breeding of mice positive for urine glucose began (Fig. 2).
FIGURE 2.
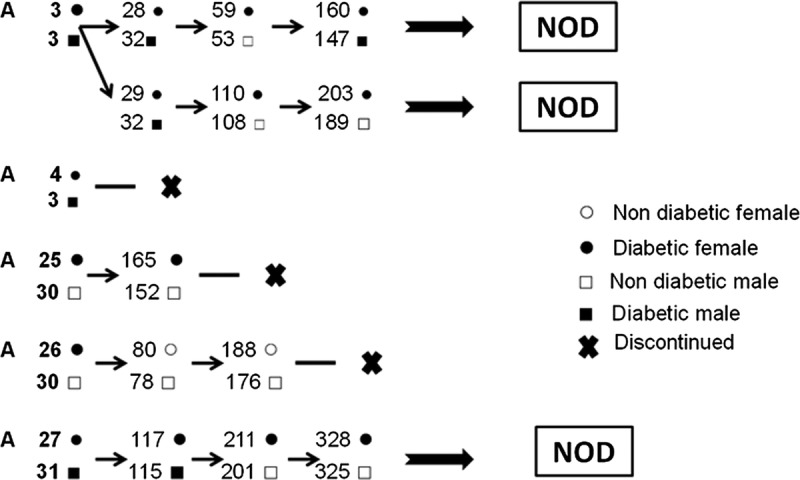
Initial breeding pedigree: selection of diabetic mice in the normoglycemic colony.
Detection of Insulitis in the Pancreas of Glycosuric mt Mice
Yoshihiro Muraoka, a pathologist at the Abrahi Laboratory, first discovered insulitis in the pancreas of mt mice from the 20th generation of the Ohtori/Yoshida colony. He examined pancreata from 41 females and 31 males before or near glycosuria development age but before the onset of diabetes. He detected the distinctive feature of insulitis, the infiltration of lymphocytes in and around islets. The presence of insulitis in mt mice was presented by Tochino, Kanaya, Makino, and Muraoka at the Kinki district meeting of the Japanese Diabetes Association in 1976. Detailed biochemical and histological findings were reported in the proceedings of the Kinki Symposium under the title “Mice With Spontaneous Glycosuria that May Be a Potential Animal Model for a Juvenile, Insulin-Dependent Diabetes” (Japanese) in 1979. This finding was subsequently reported by Makino et al.8
Development of the Nonobese Diabetic Mouse Strain
The Abrahi Animal Facility maintained various animal species for breeding and research purposes. Professor Kyoji Kondo from Nagoya University was appointed as a consultant in 1970. He was well known for the establishment of the type 2 diabetes model, the KK mouse strain in 1957, which exhibits elevated nonfasting blood glucose, glucose intolerance, and moderate obesity.9 Kondo, as a mentor of Susumu Makino for his PhD degree, requested that Makino 1) develop a mouse strain that spontaneously develops diabetes suitable for research, 2) establish a well-documented inbred strain of mice from mt mice descended from the Ohtori/Yoshida normoglycemic mouse colony, and 3) conduct a genetic analysis of diabetes in the developed mouse strain. When Maeda retired in 1975, Makino took over his mouse colonies, including the normoglycemic mt mice, which he continued to brother/sister mate beyond the 19th generation. Starting with the 20th generation of mt mice, Makino established the first generation of the NOD mouse breeding program using several female mt mice to start brother-sister matings under specific pathogen–free (SPF) conditions. These female mice were confirmed insulitis positive. The selection of breeding pairs was based on the presence of glycosuria as well as the specific pedigree of each mouse, and the incidence of diabetes in the offspring was carefully documented. By 1980, Makino's NOD mice reached the 20th generation, and the inbred strain of NOD mice that spontaneously develops insulitis and IDDM was firmly established.8
Release of NOD Mice for Diabetes Research
Susumu Makino presented the development of this unique NOD mouse strain at the first Non-Obese Diabetic Mouse Symposium held in 1981 in Japan. This presentation rapidly increased the demand for NOD mice from Japanese diabetes investigators, and by October 1981, approximately 260 glycosuria-positive mice and 900 glycosuria-negative or preglycosuric mice were shared to the Japanese scientific community. To fulfill increasing demands, approximately 100 mice were produced monthly by the Shionogi Abrahi Animal Facility. Although Makino's NOD colony was maintained under SPF conditions, these mass-produced NOD mice were maintained in non-SPF conditions. A significant change was noticed in the diabetes incidence between the 2 environmental conditions, which is now recognized as a low incidence of diabetes or a delayed appearance of glycosuria in NOD mice maintained in non-SPF conditions. Subsequently, the NOD Mouse Research Committee began to oversee the distribution of NOD mice.
Release of NOD Mice Abroad
The first international report of the discovery of mice that spontaneously develop IDDM and the development of an inbred NOD mouse strain was presented by Tochino et al at the Symposium on the Animal Model for Diabetes in Israel in 1981. In 1986, the book Insulitis and Type I Diabetes—Lessons From the NOD Mouse by Tarui et al was published by the New York Academic Press. By that time, sufficient clinical evidence had been accumulated to support the idea that IDDM in humans is a completely separate entity from non-IDDM. The information on the inbred NOD mouse strain spread rapidly worldwide. The request for NOD mice first arrived at Shionogi Co from Korea and subsequently from other countries. The first NOD mouse colony outside of Japan was established at the University of California at Los Angeles (UCLA) by Yoko Mullen. This colony was derived from 10 pairs of NOD mice provided by Shionogi Laboratory and primarily used for investigation of autoimmune-mediated destruction of transplanted islets. The UCLA colony was followed by the establishment of 2 more international colonies—the George Eisenbarth colony at the Joslin Diabetes Center, Harvard Medical School, by Masakazu Hattori for the investigation of autoimmune diabetes and the Jin Asamoto colony in Australia. The UCLA colony was subsequently expanded at Merck Research Laboratories by Linda Wicker (now at the University of Oxford) and contributed greatly to the immunological and immunogenetic investigation of autoimmune diabetes. The NOD/MrkTac available from TACONIC Biosciences is derived from the Wicker colony. The Asamoto colony in Australia gave rise to NOD/WEHI mice, which are known to exhibit insulitis, but not diabetes, unless treated by cyclophosphamide.10
As the requests for NOD mice from Japanese scientists continuously increased, the NOD Research Committee responsible for the NOD mouse distribution recognized the inability to fulfill these demands through the Abrahi Animal Facility. For the same reason, the committee also decided not to export NOD mice abroad. The existence of the NOD mouse had been mentioned in a 1982 article in Diabetes that described characteristics of diabetes and insulitis. The availability of a nondiabetic control, the NON mouse (Fig. 1) was also reported but noted as “not available for distribution.”11 In 1982 and 1983, Jackson Laboratory in Bar Harbor, Maine, requested a supply of NOD mice from Shionogi to establish a Jackson Laboratory colony. However, an agreement could not be reached, and the failure of this negotiation led to the decision by the American Diabetes Association to not accept papers from Japanese groups associated with NOD mouse investigations for publication in Diabetes. This pressure subsequently led to the agreement between the American Diabetes Association, the Japanese NOD mouse committee, and Shionogi Pharmaceuticals to release NOD mice through a commercial animal distributor (Nihon CREA) starting 1984.
Discovery and Development of BB Rats as Another Autoimmune Diabetes Model
In the middle of 1970, during the same time mice exhibiting spontaneous glycosuria were found in Japan, rats positive for urine glucose were also discovered in Ottawa, Canada, in a colony of SPF outbred Wistar rats and the diabetes-prone (DP) BB rat colony “BBdp” was established through selective breeding of animals that developed spontaneous hyperglycemia and ketoacidosis.4 Inbred DP animals designated BB/Wor rats were subsequently established from BBdp rats at the University of Massachusetts Medical School in Worcester, Massachusetts. Several other BB rat colonies originated from these earlier colonies. Another inbred rat strain, the DRBB/Wor (BBDR) rat, was developed from fifth-generation DPBB/Wor (BBDP) rats by selective inbreeding of rats that failed to become diabetic. The DRBB rats are diabetes resistant and do not become diabetic even under viral antibody–free (VAF) conditions. However, diabetes can be induced in BBDR rats under certain conditions, even when maintained under VAF conditions.12 Although all BB rats share the same major histocompatibility complex (MHC), RT1u, results obtained from one BB rat colony may not be transferable to another colony.12
DIABETES IN NOD MICE, BB RATS, AND HUMANS
Extensive investigations conducted in both rat and mouse diabetes models have greatly contributed to the understanding of autoimmune β-cell destruction and insulin-deficient diabetes in humans. A PubMed search using BB rats, diabetes returned 1771 publications in the period of 1978–2016; a similar search using NOD mice, diabetes returned 5033 publications in the period of 1980–2016. Each of these animal models has shown unique features and various similarities or differences in pathologic processes, diabetes characteristics, and the genes contributing to disease susceptibility between each other and humans. The following sections describe some interesting aspects of diabetes as well as pathologic observations in humans and both animal species (Table 1).
TABLE 1.
Characteristics of Diabetes and Insulitis in Humans, NOD Mice, and BB Rats

Characteristics of Insulitis and Diabetes in NOD Mice and BB Rats
Type 1 diabetes is an autoimmune disorder that develops spontaneously in humans and T1D models in 2 animal species. The incidence of diabetes is higher in females in NOD mice,8,13 equal in both sexes in BB rats,14 and is reported to have a slight prevalence in males in humans.15–17 Insulitis is detected in BB rats approximately 5 weeks after birth, followed by hyperglycemia between 60 and 120 days (median age of 96 days) in 60% to 80% of animals.18 Once hyperglycemia occurs, BB rats become severely polyuric, leading to a loss of more than 20 g of body weight overnight despite excessive drinking, and these rats will develop ketoacidosis within several days.14,19 Based on data from the NOD/ShiLtJ strain, NOD mice maintained under SPF conditions develop insulitis between 2 and 4 weeks in females and within 5 to 7 weeks in males. Diabetes develops in 90% to 100% of females by 30 weeks; in contrast, only 50% to 80% of males.20 In the NOD/MrkTac strain, diabetes is found in 60% of females at 12 weeks and approximately 40% of males by 24 weeks.21 Both NOD mice and BB rats exhibit other autoimmune phenomena such as inflammation of salivary glands and thyroiditis.21,22
Immune Cell Infiltration Into Pancreatic Islets: Insulitis
Diabetes is caused by selective destruction of β-cells in the islet of Langerhans after infiltration of autoreactive immune cells. The first histopathological change in the BBDP rat pancreas is immune cell infiltration into the islets,23 whereas in NOD mice, cellular infiltration starts in the periphery of islets (peri-insulitis), which is not usually observed in diabetic BB rats or humans.24 In the pancreas of nondiabetic mice, such as C57BL/6, immune cells are rarely found.
In NOD mice, cell infiltration starts with macrophages and dendritic cells (DCs) invading the exterior of the islets. This is followed by penetration into the islets and initiation of a cascade of immunopathological events. CD45+ cells are present around islets at 4 weeks and expand into the islet rapidly between 8 and 14 weeks. By the time insulitis is well established (12–14 weeks), lymphocytes take over as the majority cell type, and CD11b+ F4/80+ macrophages are found in lower numbers.25 Cytokines and soluble mediators produced by activated macrophages accelerate the recruitment of DCs and are essential for the activation of autoreactive CD4+ and CD8+ T cells.26–28 T cell tolerance to self-antigens is disturbed by increased prostaglandin (PGE)-2, leading to impairment of phagocytic activity and ability to present self-antigens of macrophages.29 Natural killer (NK) cells, which are normally associated with defense against viruses and intracellular pathogen-infected cells, also infiltrate into islets and contribute to β-cell apoptosis either directly through perforin- and granzyme-mediated cytotoxicity or indirectly through releasing high levels of proinflammatory cytokines.29,30 Similar types of cells, including NK cells, are also found in islets of BB rats.31,32 However, peri-insulitis is infrequently found throughout diabetes development.23
Although some similarities exist, the insulitis features of animal T1D models do not fully recapitulate the human disease process as monitored in the pancreas. The availability of pancreas samples from T1D patients is limited to only those obtained from postmortem collection, surgical biopsy, or organ donors with a history of T1D. In a study of islet inflammation in patients with T1D, approximately 200 specimens from recent-onset T1D were examined worldwide.33 Based on the study, the composition of islet infiltrates in T1D is predominantly CD8+ T cells, which persist throughout the inflammatory process.
Other immune cells that infiltrate human islets include CD4+ T cells, B cells, macrophages, and NK cells.34 Neutrophils are also found in small numbers in the surrounding exocrine tissue within or near inflamed islets.35 B lymphocytes are also found among islet infiltrates, more frequently in the later than the earlier stage of insulitis. The infiltrating CD20+ B cells are negative for the plasma cell marker, CD138, suggesting no local antibody production in the islets.36 Compared with rodents, insulitis in humans is detected much less frequently and in a relatively low (~10%) percentage of human T1D islets, and the number of infiltrating cells is also low. Insulitis appears more frequently in individuals who develop T1D at younger ages and is assessed soon after T1D onset.36,37
According to the recent proposal for comprehensive consensus,33 insulitis-positive in humans is defined as more than 3 islets containing more than 15 lymphocytes in the peri- and/or within-islet structure. This small number of lymphocytic infiltrates is significant because the islets of nondiabetic individuals rarely contain more than 5 lymphoid cells in a single cross section.16,33 Infiltrates are frequently dominated by CD8+ and CD4+ T cells with a variable frequency of B lymphocytes.36,38,39 The differences between human and rodent insulitis may reflect fluctuations over time or merely the much faster progression of diabetes in rodents. It should be noted that lymphocytic infiltration found in islets of a cadaveric donor pancreas may not be insulitis associated with T1D, but rather, this infiltration may be associated with extended hospitalization and/or a long brain death period before organ recovery.33,40
T Lymphocyte Involvement in Autoimmune Diabetes
T cells play critical roles in autoimmune diabetes, and autoreactive T cells are present in human as well as animal models of autoimmune diabetes.41,42 In experiments using the NOD mouse model, diabetes was adoptively transferred using purified T cells,43,44 and anti–T cell antibodies prevented the development of diabetes.32,45,46 In both NOD mice and BB rats, the induction of central as well as peripheral immune tolerance is defective,47,48 indicating the involvement of these defects in the thymic education of self-reactive T cells and regulatory T cells (Treg). In NOD mice, the thymus displays an abnormal architecture containing thymocytes that fail to efficiently express self-antigens. Consequently, the number of autoreactive T cells escaping out of the thymus into the periphery increases.29 However, the escape of autoreactive T cells alone cannot account for the development of T1D because autoreactive T cells are also found in normal mice and healthy individuals. The presentation of islet antigens to the thymus of T1D animal models by transplanting islets or by injecting autoantigens, such as insulin, can prevent diabetes development.49,50 In BB rats, neonatal thymectomy also prevented the development of diabetes.51
Regulatory T cells are an important subset of T helper cells that prevent autoimmunity. Nonobese diabetic mice have a normal Treg cell count, but the ability of these cells to regulate pathogenic T cells is limited.52 The induction and maintenance of FoxP3–expressing Treg cells require interleukin (IL)-2, which is encoded by the Idd3 susceptibility loci and expressed at reduced levels in NOD mice.21 This reduction leads to an imbalance between Treg and autoreactive T cells that can contribute to T1D development. The activation of autoreactive T cells occurs on MHC presentation of their complementary antigens by antigen-presenting cells and costimulatory signals. Early removal of pancreatic peripheral lymph nodes (pLNs), but not the spleen, in NOD mice prevents T1D, suggesting that the activation of islet-reactive T cells takes place first in the pancreatic pLN.53
The unique immune characteristic of diabetes-prone BB rats is the severe depletion of circulating T lymphocytes that express the maturation antigen RT. In contrast, diabetes-resistant DRBB rats have normal levels of circulating RT6+ T cells.22 Adoptive transfer of T cells from histocompatible DRBB rats into young DPBB rats increased the RT6+ T cell numbers and prevented T1D development,54 whereas depletion of RT6+ T cells in DRBB rats by treatment with cytotoxic monoclonal antibody and an immunostimulatory agent induced T1D in more than 90% of the treated DRBB rats.12
T cell–specific lymphopenia in DPBB rats results from spontaneous peripheral T cell apoptosis caused by the disruption of endoplasmic reticulum homeostasis, which occurs soon after their emigration from the thymus.55 This leads to greatly reduced CD4+ T cell numbers and the near absence of CD8+ T cells. The BB rat's T cell function is also poor, and their life span is greatly shortened.12,56 CD4+ART2+Treg cells are also severely impaired.57 Diabetes can be induced in BBDR rats within a predictable short time frame (2–3 weeks) by Treg depletion, toll-like receptor ligation, or virus infection.12 Dendritic cells of DPBB rats express lower levels of MHC class II antigens; NK and NK T cells are also low in numbers and/or function.58 Lymphopenia is not detected in T1D patients and NOD mice. However, a polyadenylation signal polymorphism in the human GIMAP5 gene is associated with increased levels of IA-2 autoantibodies in T1D patients59 and increased risk for the multisystem autoimmune disease, systemic lupus erythematosus.60
B Lymphocytes Involvement in Autoimmune Diabetes
The depletion of B cells by rituximab (anti-CD20) in patients with early-onset T1D preserved β-cell function for nearly 1 year as measured by serum C-peptide levels,61 indicating the involvement of B cells in islet destruction. Approximately 75% of newly produced B cells express autoreactive antigen receptors that must be silenced to prevent autoimmunity. Autoantigen–reactive B cells are usually eliminated by central tolerance, but if not silenced, they are maintained in the periphery in an anergic state. The anergic state is fragile and may be compromised by inflammatory mediators and other locally produced signals. Loss of anergy seems to be a B cell–intrinsic function in NOD mice, perhaps resulting from Idd susceptibility loci.62,63 In humans, the T1D risk allele, recently identified as PTPN22, was shown to permit potentially pathogenic B cells to escape tolerance.64
In NOD mice, B cells contribute to T1D development via mechanisms distinct from autoantibody production. It seems likely that in NOD mice, B cells promote β-cell destruction and disease progression primarily by presenting antigen to autoreactive T cells and through the secretion of proinflammatory cytokines. B cell–specific deletion by MHC class II I-Ag7 prevented T1D in NOD.65 In mixed bone marrow chimeras where only B cells are MHC class I deficient, T1D development was prevented.66 Thus, B cells likely play a role in T1D pathogenesis via interactions with both CD4+ and CD8+ T cells. Pancreas-infiltrating B cells have elevated levels of MHC I and II antigens. B7-1 and B7-2 are also upregulated and associated with enhanced T cell costimulation.67 These B cells are primed for destructive antigen-presenting cell function within the niche.66
B lymphocytes produce antibodies directed against various autoantigens. However, the production of autoantibodies seems not to be directly associated with β-cell destruction, but rather, secondary phenomenon in NOD mice. Furthermore, plasma cells are rarely found among the islet infiltrating cells.36
Mast cells and eosinophils are found in islets as well as in the peripancreatic lymph nodes in BB rats. In pLNs of 65-day-old diabetes-prone BBlyp/lyp rats, the eosinophil-recruiting chemokine (eotaxin) and the high affinity IgE receptor are upregulated fivefold over those in BBDR+/+ and 40-day-old BBDP rats.68 Mast cells are also found in islets of T1D patients.69
Genetics of Autoimmune Diabetes
Type 1 diabetes susceptibility genes were identified in humans as well as T1D rodents and designated as IDDM (Idd for mice and Iddm for rat). The MHC loci are the strongest susceptibility regions and correspond to human leukocyte antigen (HLA) DQB1 and HLA-DR loci (DR3 and DR4 serotypes) in humans. The second locus linked to T1D is the insulin locus and several other loci-encoding genes involved in immune signaling,70 which may predispose the individual to an exacerbated inflammatory and immune response to a given stimulus.
The MHC class I genes are expressed on all cells and involved in the presentation of intracellular antigens, whereas the MHC class II gene products are expressed on the surface of antigen-presenting cells and involved in the presentation of extracellular antigens. Variations in MHC class II molecules are the major genetic component of T1D in both humans and the NOD mouse.71,72 The effect of MHC loci on T1D risk is perhaps due to the nonoptimal presentation of self-antigens to naive lymphocytes during their maturation process in the thymus, leading to inefficient deletion of autoreactive T cells. Furthermore, the class II loci contribute to the loss of peripheral tolerance, leading to the activation of autoreactive CD4+ T cells.73–75 Although human T1D correlates more significantly with HLA class II haplotypes than class I, surprisingly, 85% of T1D patients have no family history, and T1D concordance in identical twins is only 30% to 50%, indicating involvement of multiple genes and other factors in T1D development. Several non–MHC-linked candidate genes or gene regions causing T1D are identified by human genome-wide association studies.76,77 Multiple lines of congenic strains of mice developed from the NOD mouse have contributed to testing of gene functions and gene-gene interactions to fulfill the need for in vivo testing, and striking correspondence was identified in T1D susceptibility genes between human and the NOD mouse.
The cytotoxic T cell–associated protein (CTLA-4) (Idd5.1), IL-2 (Idd3) pathways, and insulin (both as an autoantigen and an antigen expressed in the thymus) have all been implicated in T1D.78 The critical immune homeostasis regions are the Idd3 and Idd5.1 regions. The Idd3 region encodes the IL-2 and IL-21 genes. In NOD mice, IL-2 expression is abnormally low, but IL-21 expression is high. IL-2 is involved in the induction and maintenance of FoxP3-expressing Treg cells. The reduction of IL-2 causes an imbalance between Treg and pathogenic T cells, leading to autoreactivity. Hence, the Idd3 allele affects both self and alloimmune tolerance.79 The Idd5.1 region contains the CTLA-4 and inducible T cell costimulator genes. Nonobese diabetic mice have reduced expression of the ligand-independent isoform of CTLA-4 as compared with non-T1D control strains, indicating a decreased activation threshold of T cells.80 The expression level of mRNA encoding the soluble CTLA-4 isoform is reduced in humans having T1D susceptibility at CTLA4.80 Contrary, inducible T cell costimulator is expressed higher and heightens T cell costimulation,81,82 leading to heightened T cell stimulation and increased disease susceptibility. Idd7 contains genes that influence allelic exclusion of the T cell receptor during T cell development and thymic T cell deletion/selection.83 The Idd9 region in NOD mice is associated with increased B cell pathogenic activity,84 low numbers of NK T cells, and Treg cells.85,86
Diabetes in BB rats is also dependent on the MHC RT1u haplotype, which is an orthologue to human HLA-DQ. RT1u is also shared by other rat strains, such as the nondiabetic Wistar Furth and diabetic Tokushima rats, and the treatment of poly I:C can induce insulitis or diabetes in many non-BB RT1u rat strains.87,88 Lymphopenia (lyp) is a prerequisite for spontaneous development of diabetes in BBDP rats. The gene encoding GTPase immune-associated protein, Gimap-5, was identified as the lyp gene; a frameshift mutation results in the premature truncation of the Gimap5 protein.89 Gimap-5 encodes a mitochondrial protein necessary for post-thymic T cell survival and is integral to T cell development and survival.
A Marker Found in Serum and Genes Expressed in Islets and Peripancreatic Lymph Nodes in Type 1 Diabetes
Using a genomics-based bioassay, Kaldunski et al searched for the presence of unique disease markers/factors in sera of T1D patients. Their study identified a factor that induces a disease-specific, proinflammatory transcriptional signature in peripheral blood mononuclear cells, which were obtained from a third-party, healthy individual and incubated together with T1D sera.90,91 These transcriptional profiles were found only in recent-onset T1D sera. The detected genes were related to innate immunity and regulated by IL-1, indicative of an active, ongoing disease process in the serum donor. The profiles found in recent-onset T1D sera were significantly different from those obtained in sera of healthy individuals or long-standing T1D patients.91
The same team extended this line of study using diabetes-prone DRlyp/lyp and nondiabetic DR+/+ BB rats to link the genomic-based bioassay results of T1D sera to disease progression in the host pancreas/islets. As in T1D patients, sera from DRlyp/lyp rats induced transcription of cytokines, immune receptors, and signaling molecules in peripheral blood mononuclear cells of healthy DR+/+ BB rats. The signature was associated with the progression of diabetes and with the induction of many IL-1–regulated genes not induced in the sera from nondiabetic DR+/+ rats. An administration of IL-1 receptor antagonist to prediabetic DRlyp/lyp rats modulated the serum signature and delayed the onset of T1D.90
Changes in tissue-specific and age-dependent gene expression were detected in the islets and pancreatic lymph nodes of NOD mice during disease progression.92 Two candidate genes, deformed epidermal autoregulatory factor 1 (Deaf1) and adenosine A1 receptor 1 (Adora1), were differentially expressed and alternatively spliced in pancreatic lymph nodes and islets. The loss of Deaf1 function led to reduced antigen expression in peripheral tissue and in lymph node stromal cells, which was assumed to contribute a breakdown in peripheral tolerance. Reduced Adora1 function resulted in an early intrinsic α-cell defect and led to hyperglucagonemia and β-cell stress before diabetes onset. The investigators also reported that both genes were alternatively spliced in tissues of autoantibody-positive, prediabetic patients.
Autoantigens and Autoantibodies in Autoimmune Diabetes and its Animal Models
The presence of autoantibodies in the sera of T1D patients was discovered more than 2 decades ago.93,94 Since then, a vast amount of studies have revealed the presence of autoantibodies in T1D: 70% of newly diagnosed T1D patients have autoantibodies (vs 0.5% of the general population).95 Among the targets of autoantibodies, insulin and proinsulin are the only known β-cell–specific antigens, and insulin autoantibodies (IAAs) are detected in more than 59% of patients with either late preclinical or early-onset T1D. Insulin autoantibodies are also detected in NOD mice96 and BB rats.97 These unique β-cell antigens should play a key role in the initiation of autoimmunity. Insulin expression in the thymus indicates its involvement in T cell tolerance. An intrathymic injection of insulin or pancreatic islets prevented the onset of diabetes in NOD mice98,99 and BB rats.100
Autoantibodies directed to glutamic acid decarboxylase (GAD)-65, which are detected in sera of T1D patients and individuals with latent T1D, are an established marker for autoimmune diabetes. Glutamic acid decarboxylase 65 is predominantly expressed in islets of humans and rats, but not mice that express GAD67.101 In BB rats, immunoassay results from autoantibodies to GAD, insulin, and heat shock protein-65 showed that these protein levels did not exceed control levels and were much lower than the levels found in T1D humans.102 Glutamic acid decarboxylase 65 was recently proposed as a biomarker of β-cell loss, which is a predictor of autoimmune diabetes or rejection of islet allografts. However, the in vitro half-life of recombinant human GAD65 is 1.27 to 2.35 hours in serum or blood at 37°C. Glutamic acid decarboxylase 65 is detectable in vivo in rat serum for up to 48 hours after streptozotocin injection. At least 13 islets in rats and 1875 islets in humans must be simultaneously destroyed to give detectable GAD65 in circulation.103
The presence of islet cell cytoplasmic autoantibodies, GADA, insulinoma-2–associated autoantibodies (IA-2A), IAA, and /or zinc transporter 8 autoantibodies are present in 60% to 70% of newly diagnosed patients and prediabetic relatives.104 These sera also react to the IA-2 antigen obtained from rat β-cell line (RIN5AH). However, anti–IA-2 antibodies are not detected in sera from diabetic NOD mice and BB rats.26
There is an ongoing debate whether (1) autoantibodies are truly pathogenic, (2) autoantibodies are generated secondarily to molecules that were generated as a consequence of islet destruction, or (3) autoantibodies are markers of an underlying disease process. Most evidence favors the second interpretation. Despite the presence of autoantibodies, the pathogenic role of IAA has remained unclear, and the role of GAD-65, IA-2, and antibodies to other autoantigens in T1D is also unclear. It should be noted that there are individuals who develop autoantibodies but never progress to T1D. However, the presence of multiple autoantibodies is strongly predictive of T1D development in susceptible individuals, and nondiabetic individuals who show the presence of 3 or more circulating autoantibodies are in a greatly increased risk of progressing to T1D.16
Autoimmune Destruction and Regenerative Capacity of β-Cells
An advantage of animal models is to allow the investigation of metabolic changes and the pathologic change of pancreatic islets together in the same animal. Insulitis starts early in the life in both NOD mice and BB rats, but there is a considerable lapse of time from the onset of insulitis to persistent hyperglycemia. The several weeks between insulitis onset and diabetes development indicate that the process of β-cell destruction is generally slow. It is also possible that during this time, there is some level of β-cell repair or regeneration.
Immune cells infiltrating the islets generate an inflammatory microenvironment that is toxic to β-cells through both cell-mediated cytotoxicity and cytokines, leading to β-cell destruction. Proinflammatory cytokines, including IL-1β, tumor necrosis factor-α, and interferon (IFN)-γ released by activated lymphocytes, macrophages, and β-cells themselves105 cause functional damage in both insulin synthesis and secretion and in the subsequent apoptosis of β-cells.106–108 The activation of signaling pathways, such as nuclear factor-κB and p38 mitogen–activated protein kinases, also contribute to β-cell apoptosis.109
Beta-cells also should have regenerative capacity. For example, regenerated β-cells restore euglycemia in animals made diabetic by a near-total pancreatectomy.110,111 The regenerative activity involves both β-cell replication and neogenesis.112,113 An antiproliferative effect of inflammatory cytokines has been discovered in BB rats.114 This property, mediated at least by IFN-γ, is characterized by halting β-cell proliferation.115 This antiproliferative effect on β-cells can set the stage for β-cell destruction even before proceeding to β-cell apoptosis. In diabetes-prone BB rats, β-cell proliferation and/or replication are inhibited/halted, and islets are smaller. There is also no β-cell mass increase starting from 5 to 7 weeks, whereas a constant rate of β-cell proliferation is maintained in control Wistar rats until 11 weeks.115 At 7 to 8 weeks, BB rats exhibit periductal mononuclear cell infiltration, phagocytic macrophage infiltration, the presence of proinflammatory cytokines including IL-1β and IFN-γ, and counter-regulatory cytokines, such as IL-6 and IL-10.116,117 Antibody treatment directed against IFN-γ in 7-week-old BB rats restored β-cell proliferation and mass. However, the same treatment had no effect when given at a later time, and β-cell apoptosis progressed as examined at 11 weeks. The inhibition of β-cell proliferation caused reduced insulin synthesis and release, leading to insulin shortage and glucose intolerance.
Diabetes Susceptibility at the β-Cell Level
Lower expression of genes associated with anti-oxidative defense mechanisms seems to contribute to β-cell injury and to disease development in BB rats.118 Their β-cells are inherently susceptible to endoplasmic reticulum stress caused by environmental insults. These damaged β-cells release proinsulin and insulin, which in turn activates autoreactive T cells. Therefore, the islet is considered an important factor in disease development.119,120
Profiles of islet gene expression are distinctly different between diabetes prone DRlyp/lyp BB rats and healthy Fischer (F344) rats. DRlyp/lyp BB rats under-express genes involved in reactive oxygen species (ROS) metabolism, including glutathione S-transferase (GST) family members, superoxide dismutases (SODs), peroxidases, and peroxiredoxins. This pattern of gene under-expression is not observed in other tissue such as brain, liver, or muscle from the same strain of rats. The GST protein level in pancreatic tissue and in the serum of DRlyp/lyp BB rats is also lower than in F344 rats. An administration of antioxidant N-acetyl cysteine to DRlyp/lyp BB rats altered the abundance of peripheral eosinophils, reducing the severity of insulitis. This delays, but does not prevent development of diabetes. These results suggest that lower expression of genes related to antioxidative defense mechanisms in islets may contribute to T1D susceptibility.118
Influence of Environment on Type 1 Diabetes Development
Human T1D is a complex, multifactorial disease121; epidemiological data show that T1D onset often associates with viral infections, including mumps and rubella, and infections from rotavirus, parvovirus, and enterovirus. Second to viruses, nutrition also influences T1D.16 Analyzing the genetic complexity of human T1D and controlling the environmental factors are notoriously difficult for humans, whereas inbred autoimmune diabetic animal models are useful for these lines of investigations. Both NOD mice and BB rats raised under SPF conditions develop insulitis and diabetes at frequencies higher than those maintained in a conventional non-SPF environment. Of special note, viral infection of NOD mice often reduces the frequency of diabetes. The diabetes frequency also increases in BBDP rats, but not in BBDR rats, under VAF conditions.
The BBDR and BBDP rats share the same RT1u haplotype, but BBDR have a wild-type Gimps5 allele and are not lymphopenic.122 The DP lymphopenia gene region has been fixed on the BBDR background to develop diabetes-prone DRlyp/lyp rats. All DRlyp/lyp rats are lymphopenic, and 100% of these animals develop diabetes between 50 and 70 days. In contrast, DR+/+ and DRlyp/+ BB rats do not develop diabetes and lymphopenia.19 These nearly genetically identical strains are well suited for studying the influence of environmental factors. When placed under a diabetes-inducible condition, BBDR rats develop mild insulitis beginning at 10 days and rapidly increase cellular infiltration after 14 to 18 days, followed by β-cell destruction. Hyperglycemia occurs during the late stage of insulitis, followed by ketoacidosis several days later.12,123 Treg depletion by anti-ART2 antibody can also induce T1D in BBDR rats housed in a conventional but not in a VAF environment.124 The development of diabetes under these conditions is consistent and predictable in this animal model. Rats that develop diabetes are also susceptible to other autoimmune diseases seen in human T1D patients. Therefore, BBDR rats provide an invaluable model especially for studying complex environmental factors. For example, Kilham rat virus infection caused an outbreak of diabetes in a BBDR rat colony125 and provided an experimental tool elucidating mechanisms of T1D induction and diabetogenic factors.12
Other environmental factors, such as geographical location and dietary components, are known to associate with T1D development in humans. There is an inverse correlation between T1D incidence and proximity to the equator, which may indicate a correlation between ultraviolet radiation, temperature, and T1D incidence. Nonobese diabetic mice maintained at 23.7°C had lower T1D incidence than those maintained at 21°C.126 Vitamin D supplementation was shown to protect against T1D.127 Essential fatty acids and omega-3 reduced T1D incidence in children by influencing the generation and maintenance of Treg cells.128 Bio-Breeding rats are widely used to study gluten involvement in T1D.129
CONTRIBUTION OF NOD MICE AND BB RATS TO UNDERSTAND HUMAN TYPE 1 DIABETES
Mouse and rat animal models of autoimmune diabetes have enormously contributed to our understanding of T1D in humans (Fig. 3). Nonobese diabetic mice and their numerous congenic lines have provided guidance for understanding human T1D, in particular, dissection of gene involvement in immune responses that lead to autoimmunity and disease development. Involvement of IL-2 and Treg cells in diabetes seems especially important in the development of preventive measures for T1D and treatment of T1D patients. Nonobese diabetic mice have proved to be effective models for testing various strategies to diabetic interventions and will continue to provide opportunities to test innovative ideas and approaches to treat diabetes process before human application.
FIGURE 3.
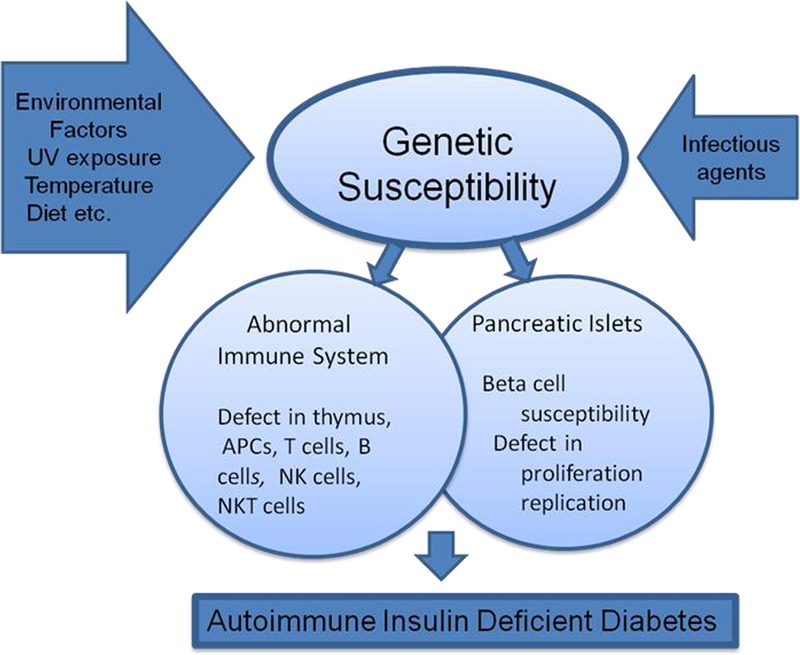
Mechanisms leading to autoimmune diabetes.
Bio-Breeding rats, which are larger than NOD mice, provide advantages for certain studies. Intrathymic injection of islets in BB rats first demonstrated the survival of islet allograft in the thymus without immunosuppression and prevention of diabetes recurrence. Because of their relatively large size, blood sampling can be repeated in a short interval, allowing the collection of larger amounts of blood without causing physiological derangement. In addition, repeated biopsy of the pancreas may be performed. The availability of nearly genetically identical strains, DRlyp/lyp, DR+/+, and DRlyp/+ rats, provides unique opportunity for various studies130 because diabetes develops within a short, predictable time frame after treating with a potential diabetogenic condition/factor. Therefore, this animal model allows investigators to study a specific stage of diabetes development and to test the role of environmental factors, virus infection, and many other factors or conditions that associate with autoimmune diabetes development as well as prevention of β-cell destruction.
Historically, rodent models have provided us the tools and guidance to increase our understanding of autoimmune diabetes. In the future, they will continue to provide further insight into autoimmunity and diabetes. However, we must recognize their limitations and acknowledge the differences between humans and rodents when interpreting results obtained using animal models.
ACKNOWLEDGMENTS
The author deeply appreciates the information and assistance gathered and provided by Dr Yoshihiro Tochino in preparation of this article. The author acknowledges Dr Tochino and her mentor, the late Dean Masakazu Abe, Tokyo Jikei University School of Medicine, for their contributions in establishing the first NOD mouse colony outside Japan. The establishment of the UCLA NOD mouse colony was supported by a grant from the Nora Eccles Treadwell Foundation. The author acknowledges special contributions by Dr Linda S. Wicker, The Wellcome Trust Centre for Human Genetics, University of Oxford, for her critical review; Dr Vay Liang W. Go, University of California, Los Angeles, and Editor-in-Chief, Pancreas, for his encouragement for writing this article; Dr Chris Gandhi for editorial service; and Mr Jeffrey Rawson, Dr Henry Lin, and Dr Fouad Kandeel, Beckman Research Institute of City of Hope, for their support in the preparation of this article.
Footnotes
This study was supported by a grant from the Nora Eccles Treadwell Foundation.
The author declares no conflict of interest.
REFERENCES
- 1.Schmidt M. Ueber die beziehung der langenhans'schen inseln des pankreas zum diabetes mellitus. München Med Wochenschr. 1902;49:51–54. [Google Scholar]
- 2.Von Meyenburg M. Ueber “insulitis” bei diabetes. Schweiz Med Wochenschr. 1940;21:554–557. [Google Scholar]
- 3.Sutherland DE, Goetz FC, Sibley RK. Recurrence of disease in pancreas transplants. Diabetes. 1989;38(Suppl 1):85–87. [DOI] [PubMed] [Google Scholar]
- 4.Nakhooda AF, Like AA, Chappel CI, et al. The spontaneously diabetic Wistar rat. Metabolic and morphologic studies. Diabetes. 1977;26:100–112. [DOI] [PubMed] [Google Scholar]
- 5.Ohotori H, Yoshida T, Inuta T. “Small eye and cataract”, a new dominant mutation in the mouse. Jikken Dobutsu (Experimental Animals). 1968;17:91–96. [Google Scholar]
- 6.Tarui S, Harada M, Hanafusa T, et al. History of studies on the NOD mouse (I)—breeding and etiopathogenesis. Diabetes Journals (Japanese). 1990;18:31–40. [Google Scholar]
- 7.Tarui S, Nonaka K, Hanafusa T, et al. History of studies on the NOD mouse (II)—The first–fourth symposium of NOD mouse. Diabetes Journals (Japanese). 1990;18:25–36. [Google Scholar]
- 8.Makino S, Kunimoto K, Muraoka Y, et al. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29:1–13. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida T, Umekawa T, Wakabayashi Y, et al. Anti-obesity and anti-diabetic effects of mazindol in yellow KK mice: its activating effect on brown adipose tissue thermogenesis. Clin Exp Pharmacol Physiol. 1996;23:476–482. [DOI] [PubMed] [Google Scholar]
- 10.Charlton B, Bacelj A, Slattery RM, et al. Cyclophosphamide-induced diabetes in NOD/WEHI mice. Evidence for suppression in spontaneous autoimmune diabetes mellitus. Diabetes. 1989;38:441–447. [DOI] [PubMed] [Google Scholar]
- 11.Coleman DL. Other potentially useful rodents as models for the study of human diabetes mellitus. Diabetes. 1982;31(Supp. 1 Pt 2):24–25. [DOI] [PubMed] [Google Scholar]
- 12.Bortell R, Yang C. The BB rat as a model of human type 1 diabetes. Methods Mol Biol. 2012;933:31–44. [DOI] [PubMed] [Google Scholar]
- 13.Makino S, Muraoka Y, Kishimoto Y, et al. Genetic analysis for insulitis in NOD mice. Jikken Dobutsu. 1985;34:425–431. [DOI] [PubMed] [Google Scholar]
- 14.Like AA, Rossini AA, Guberski DL, et al. Spontaneous diabetes mellitus: reversal and prevention in the BB/W rat with antiserum to rat lymphocytes. Science. 1979;206:1421–1423. [DOI] [PubMed] [Google Scholar]
- 15.Foulis AK. Pancreatic pathology in type 1 diabetes in human. Novartis Found Symp. 2008;292:2–13. [PubMed] [Google Scholar]
- 16.Atkinson MA. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2 pii: a007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Variation and trends in incidence of childhood diabetes in Europe. EURODIAB ACE Study Group. Lancet. 2000;355:873–876. [PubMed] [Google Scholar]
- 18.Mordes JP, Desemone J, Rossini AA. The BB rat. Diabetes Metab Rev. 1987;3:725–750. [DOI] [PubMed] [Google Scholar]
- 19.Dalberg U, Haase C, Hornum L, et al. The BB Rat. In: Eisenbarth GS, ed. Immunoendocrinology: Scientific and Clinical Aspects. New York, NY: Humana Press; 2011:183–197. [Google Scholar]
- 20.Leiter EH, Prochazka M, Coleman DL. The non-obese diabetic (NOD) mouse. Am J Pathol. 1987;128:380–383. [PMC free article] [PubMed] [Google Scholar]
- 21.Ridgway WM, Peterson LB, Todd JA, et al. Gene-gene interactions in the NOD mouse model of type 1 diabetes. Adv Immunol. 2008;100:151–175. [DOI] [PubMed] [Google Scholar]
- 22.Awata T, Guberski DL, Like AA. Genetics of the BB rat: association of autoimmune disorders (diabetes, insulitis, and thyroiditis) with lymphopenia and major histocompatibility complex class II. Endocrinology. 1995;136:5731–5735. [DOI] [PubMed] [Google Scholar]
- 23.Kolb H, Worz-Pagenstert U, Kleemann R, et al. Cytokine gene expression in the BB rat pancreas: natural course and impact of bacterial vaccines. Diabetologia. 1996;39:1448–1454. [DOI] [PubMed] [Google Scholar]
- 24.Dahlen E, Dawe K, Ohlsson L, et al. Dendritic cells and macrophages are the first and major producers of TNF-alpha in pancreatic islets in the nonobese diabetic mouse. J Immunol. 1998;160:3585–3593. [PubMed] [Google Scholar]
- 25.Magnuson AM, Thurber GM, Kohler RH, et al. Population dynamics of islet-infiltrating cells in autoimmune diabetes. Proc Natl Acad Sci U S A. 2015;112:1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoon JW, Jun HS. Cellular and molecular pathogenic mechanisms of insulin-dependent diabetes mellitus. Ann N Y Acad Sci. 2001;928:200–211. [DOI] [PubMed] [Google Scholar]
- 27.Jun HS, Santamaria P, Lim HW, et al. Absolute requirement of macrophages for the development and activation of beta-cell cytotoxic CD8+ T-cells in T-cell receptor transgenic NOD mice. Diabetes. 1999;48:34–42. [DOI] [PubMed] [Google Scholar]
- 28.Jun HS, Yoon CS, Zbytnuik L, et al. The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Exp Med. 1999;189:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jayasimhan A, Mansour KP, Slattery RM. Advances in our understanding of the pathophysiology of type 1 diabetes: lessons from the NOD mouse. Clin Sci (Lond). 2014;126:1–18. [DOI] [PubMed] [Google Scholar]
- 30.Young LH, Peterson LB, Wicker LS, et al. In vivo expression of perforin by CD8+ lymphocytes in autoimmune disease. Studies on spontaneous and adoptively transferred diabetes in nonobese diabetic mice. J Immunol. 1989;143:3994–3999. [PubMed] [Google Scholar]
- 31.MacKay P, Jacobson J, Rabinovitch A. Spontaneous diabetes mellitus in the Bio-Breeding/Worcester rat. Evidence in vitro for natural killer cell lysis of islet cells. J Clin Invest. 1986;77:916–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Like AA, Biron CA, Weringer EJ, et al. Prevention of diabetes in BioBreeding/Worcester rats with monoclonal antibodies that recognize T lymphocytes or natural killer cells. J Exp Med. 1986;164:1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan NG, Leete P, Foulis AK, et al. Islet inflammation in human type 1 diabetes mellitus. IUBMB Life. 2014;66:723–734. [DOI] [PubMed] [Google Scholar]
- 34.Dotta F, Censini S, van Halteren AG, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci U S A. 2007;104:5115–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez-Calvo T, Ekwall O, Amirian N, et al. Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes. Diabetes. 2014;63:3880–3890. Erratum in: Diabetes. 2016;65:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willcox A, Richardson SJ, Bone AJ, et al. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009;155:173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imagawa A, Hanafusa T, Tamura S, et al. Pancreatic biopsy as a procedure for detecting in situ autoimmune phenomena in type 1 diabetes: close correlation between serological markers and histological evidence of cellular autoimmunity. Diabetes. 2001;50:1269–1273. [DOI] [PubMed] [Google Scholar]
- 38.Coppieters KT, Dotta F, Amirian N, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. 2012;209:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arif S, Leete P, Nguyen V, et al. Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes. 2014;63:3835–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.In't Veld P, De Munck N, Van Belle K, et al. Beta-cell replication is increased in donor organs from young patients after prolonged life support. Diabetes. 2010;59:1702–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haskins K, McDuffie M. Acceleration of diabetes in young NOD mice with a CD4+ islet-specific T cell clone. Science. 1990;249:1433–1436. [DOI] [PubMed] [Google Scholar]
- 42.Di Lorenzo TP, Peakman M, Roep BO. Translational mini-review series on type 1 diabetes: Systematic analysis of T cell epitopes in autoimmune diabetes. Clin Exp Immunol. 2007;148:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wicker LS, Miller BJ, Mullen Y. Transfer of autoimmune diabetes mellitus with splenocytes from nonobese diabetic (NOD) mice. Diabetes. 1986;35:855–860. [DOI] [PubMed] [Google Scholar]
- 44.Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes. 1993;42:44–55. [DOI] [PubMed] [Google Scholar]
- 45.Maki T, Ichikawa T, Blanco R, et al. Long-term abrogation of autoimmune diabetes in nonobese diabetic mice by immunotherapy with anti-lymphocyte serum. Proc Natl Acad Sci U S A. 1992;89:3434–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simon G, Parker M, Ramiya V, et al. Murine antithymocyte globulin therapy alters disease progression in NOD mice by a time-dependent induction of immunoregulation. Diabetes. 2008;57:405–414. [DOI] [PubMed] [Google Scholar]
- 47.Markees TG, Serreze DV, Phillips NE, et al. NOD mice have a generalized defect in their response to transplantation tolerance induction. Diabetes. 1999;48:967–974. [DOI] [PubMed] [Google Scholar]
- 48.Kuttler B, Rosing K, Lehmann M, et al. Prevention of autoimmune but not allogeneic destruction of grafted islets by different therapeutic strategies. J Mol Med (Berl). 1999;77:226–229. [DOI] [PubMed] [Google Scholar]
- 49.Posselt AM, Barker CF, Friedman AL, et al. Prevention of autoimmune diabetes in the BB rat by intrathymic islet transplantation at birth. Science. 1992;256:1321–1324. [DOI] [PubMed] [Google Scholar]
- 50.Gerling IC, Serreze DV, Christianson SW, et al. Intrathymic islet cell transplantation reduces beta-cell autoimmunity and prevents diabetes in NOD/Lt mice. Diabetes. 1992;41:1672–1676. [DOI] [PubMed] [Google Scholar]
- 51.Like AA, Kislauskis E, Williams RR, et al. Neonatal thymectomy prevents spontaneous diabetes mellitus in the BB/W rat. Science. 1982;216:644–646. [DOI] [PubMed] [Google Scholar]
- 52.Brusko T, Atkinson M. Treg in type 1 diabetes. Cell Biochem Biophys. 2007;48:165–175. [DOI] [PubMed] [Google Scholar]
- 53.Gagnerault MC, Luan JJ, Lotton C, et al. Pancreatic lymph nodes are required for priming of beta cell reactive T cells in NOD mice. J Exp Med. 2002;196:369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burstein D, Mordes JP, Greiner DL, et al. Prevention of diabetes in BB/Wor rat by single transfusion of spleen cells. Parameters that affect degree of protection. Diabetes. 1989;38:24–30. [DOI] [PubMed] [Google Scholar]
- 55.Ramanathan S, Poussier P. BB rat lyp mutation and Type 1 diabetes. Immunol Rev. 2001;184:161–171. [DOI] [PubMed] [Google Scholar]
- 56.Jackson RA, Eisenbarth GS. Type I diabetes of man and the BB rat: monoclonal antibody-defined T-cell abnormalities. Diagn Immunol. 1983;1:240–244. [PubMed] [Google Scholar]
- 57.Greiner DL, Handler ES, Nakano K, et al. Absence of the RT-6 T cell subset in diabetes-prone BB/W rats. J Immunol. 1986;136:148–151. [PubMed] [Google Scholar]
- 58.Whalen BJ, Mordes JP, Rossini AA. The BB rat as a model of human insulin-dependent diabetes mellitus. Curr Protoc Immunol. 2001; Chapter 15:Unit 15.3. [DOI] [PubMed] [Google Scholar]
- 59.Shin JH, Janer M, McNeney B, et al. IA-2 autoantibodies in incident type I diabetes patients are associated with a polyadenylation signal polymorphism in GIMAP5. Genes Immun. 2007;8:503–512. [DOI] [PubMed] [Google Scholar]
- 60.Hellquist A, Zucchelli M, Kivinen K, et al. The human GIMAP5 gene has a common polyadenylation polymorphism increasing risk to systemic lupus erythematosus. J Med Genet. 2007;44:314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silveira PA, Dombrowsky J, Johnson E, et al. B cell selection defects underlie the development of diabetogenic APCs in nonobese diabetic mice. J Immunol. 2004;172:5086–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cox SL, Stolp J, Hallahan NL, et al. Enhanced responsiveness to T-cell help causes loss of B-lymphocyte tolerance to a β-cell neo-self-antigen in type 1 diabetes prone NOD mice. Eur J Immunol. 2010;40:3413–3425. [DOI] [PubMed] [Google Scholar]
- 64.Fousteri G, Liossis SN, Battaglia M. Roles of the protein tyrosine phosphatase PTPN22 in immunity and autoimmunity. Clin Immunol. 2013;149:556–565. [DOI] [PubMed] [Google Scholar]
- 65.Bonifacio E, Atkinson M, Eisenbarth G, et al. International Workshop on Lessons From Animal Models for Human Type 1 Diabetes: identification of insulin but not glutamic acid decarboxylase or IA-2 as specific autoantigens of humoral autoimmunity in nonobese diabetic mice. Diabetes. 2001;50:2451–2458. [DOI] [PubMed] [Google Scholar]
- 66.Marino E, Tan B, Binge L, et al. B-cell cross-presentation of autologous antigen precipitates diabetes. Diabetes. 2012;61:2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hussain S, Delovitch TL. Dysregulated B7-1 and B7-2 expression on nonobese diabetic mouse B cells is associated with increased T cell costimulation and the development of insulitis. J Immunol. 2005;174:680–687. [DOI] [PubMed] [Google Scholar]
- 68.Hessner MJ, Wang X, Meyer L, et al. Involvement of eotaxin, eosinophils, and pancreatic predisposition in development of type 1 diabetes mellitus in the BioBreeding rat. J Immunol. 2004;173:6993–7002. [DOI] [PubMed] [Google Scholar]
- 69.Geoffrey R, Jia S, Kwitek AE, et al. Evidence of a functional role for mast cells in the development of type 1 diabetes mellitus in the BioBreeding rat. J Immunol. 2006;177:7275–7286. [DOI] [PubMed] [Google Scholar]
- 70.Nejentsev S, Howson JM, Walker NM, et al. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature. 2007;450:887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wicker L, Wekerle H. Autoimmunity. Curr Opin Immunol. 1995;7:783–785. [DOI] [PubMed] [Google Scholar]
- 72.Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol. 2011;33:67–87. [DOI] [PubMed] [Google Scholar]
- 73.Corper AL, Stratmann T, Apostolopoulos V, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science. 2000;288:505–511. [DOI] [PubMed] [Google Scholar]
- 74.Latek RR, Suri A, Petzold SJ, et al. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity. 2000;12:699–710. [DOI] [PubMed] [Google Scholar]
- 75.Kwok WW, Domeier ML, Raymond FC, et al. Allele-specific motifs characterize HLA-DQ interactions with a diabetes-associated peptide derived from glutamic acid decarboxylase. J Immunol. 1996;156:2171–2177. [PubMed] [Google Scholar]
- 76.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Todd JA, Walker NM, Cooper JD, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wicker LS, Clark J, Fraser HI, et al. Type 1 diabetes genes and pathways shared by humans and NOD mice. J Autoimmun. 2005;25(Suppl):29–33. [DOI] [PubMed] [Google Scholar]
- 79.Pearson T, Weiser P, Markees TG, et al. Islet allograft survival induced by costimulation blockade in NOD mice is controlled by allelic variants of Idd3. Diabetes. 2004;53:1972–1978. [DOI] [PubMed] [Google Scholar]
- 80.Ueda H, Howson JM, Esposito L, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. [DOI] [PubMed] [Google Scholar]
- 81.Prevot N, Briet C, Lassmann H, et al. Abrogation of ICOS/ICOS ligand costimulation in NOD mice results in autoimmune deviation toward the neuromuscular system. Eur J Immunol. 2010;40:2267–2276. [DOI] [PubMed] [Google Scholar]
- 82.Hawiger D, Tran E, Du W, et al. ICOS mediates the development of insulin-dependent diabetes mellitus in nonobese diabetic mice. J Immunol. 2008;180:3140–3147. [DOI] [PubMed] [Google Scholar]
- 83.Serreze DV, Choisy-Rossi CM, Grier AE, et al. Through regulation of TCR expression levels, an Idd7 region gene(s) interactively contributes to the impaired thymic deletion of autoreactive diabetogenic CD8+ T cells in nonobese diabetic mice. J Immunol. 2008;180:3250–3259. [DOI] [PubMed] [Google Scholar]
- 84.Silveira PA, Chapman HD, Stolp J, et al. Genes within the Idd5 and Idd9/11 diabetes susceptibility loci affect the pathogenic activity of B cells in nonobese diabetic mice. J Immunol. 2006;177:7033–7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ueno A, Wang J, Cheng L, et al. Enhanced early expansion and maturation of semi-invariant NK T cells inhibited autoimmune pathogenesis in congenic nonobese diabetic mice. J Immunol. 2008;181:6789–6796. [DOI] [PubMed] [Google Scholar]
- 86.Yamanouchi J, Puertas MC, Verdaguer J, et al. Idd9.1 locus controls the suppressive activity of FoxP3+CD4+CD25+ regulatory T-cells. Diabetes. 2010;59:272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ewel CH, Sobel DO, Zeligs BJ, et al. Poly I:C accelerates development of diabetes mellitus in diabetes-prone BB rat. Diabetes. 1992;41:1016–1021. [DOI] [PubMed] [Google Scholar]
- 88.Hornum L, Lundsgaard D, Markholst H. PolyI:C induction of diabetes is controlled by Iddm4 in rats with a full regulatory T cell pool. Ann N Y Acad Sci. 2007;1110:65–72. [DOI] [PubMed] [Google Scholar]
- 89.MacMurray AJ, Moralejo DH, Kwitek AE, et al. Lymphopenia in the BB rat model of type 1 diabetes is due to a mutation in a novel immune-associated nucleotide (Ian)-related gene. Genome Res. 2002;12:1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kaldunski M, Jia S, Geoffrey R, et al. Identification of a serum-induced transcriptional signature associated with type 1 diabetes in the BioBreeding rat. Diabetes. 2010;59:2375–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang X, Jia S, Geoffrey R, et al. Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. J Immunol. 2008;180:1929–1937. [DOI] [PubMed] [Google Scholar]
- 92.Yip L, Fathman CG. Type 1 diabetes in mice and men: gene expression profiling to investigate disease pathogenesis. Immunol Res. 2014;58:340–350. [DOI] [PubMed] [Google Scholar]
- 93.Bottazzo GF, Florin-Christensen A, Doniach D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet. 1974;2:1279–1283. [DOI] [PubMed] [Google Scholar]
- 94.MacCuish AC, Irvine WJ, Barnes EW, et al. Antibodies to pancreatic islet cells in insulin-dependent diabetics with coexistent autoimmune disease. Lancet. 1974;2:1529–1531. [DOI] [PubMed] [Google Scholar]
- 95.Bingley PJ. Clinical applications of diabetes antibody testing. J Clin Endocrinol Metab. 2010;95:25–33. [DOI] [PubMed] [Google Scholar]
- 96.Maruyama T, Takei I, Yanagawa T, et al. Insulin autoantibodies in non-obese diabetic (NOD) mice and streptozotocin-induced diabetic mice. Diabetes Res. 1988;7:93–96. [PubMed] [Google Scholar]
- 97.Dean BM, Bone AJ, Varey AM, et al. Insulin autoantibodies, islet cell surface antibodies and the development of spontaneous diabetes in the BB/Edinburgh rat. Clin Exp Immunol. 1987;69:308–313. [PMC free article] [PubMed] [Google Scholar]
- 98.Charlton B, Taylor-Edwards C, Tisch R, et al. Prevention of diabetes and insulitis by neonatal intrathymic islet administration in NOD mice. J Autoimmun. 1994;7:549–560. [DOI] [PubMed] [Google Scholar]
- 99.Gerling IC, Atkinson MA, Leiter EH. The thymus as a site for evaluating the potency of candidate beta cell autoantigens in NOD mice. J Autoimmun. 1994;7:851–858. [DOI] [PubMed] [Google Scholar]
- 100.Koevary SB, Blomberg M. Prevention of diabetes in BB/Wor rats by intrathymic islet injection. J Clin Invest. 1992;89:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim J, Richter W, Aanstoot HJ, et al. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes. 1993;42:1799–1808. [DOI] [PubMed] [Google Scholar]
- 102.Mackay IR, Bone A, Tuomi T, et al. Lack of autoimmune serological reactions in rodent models of insulin dependent diabetes mellitus. J Autoimmun. 1996;9:705–711. [DOI] [PubMed] [Google Scholar]
- 103.Schlosser M, Walschus U, Kloting I, et al. Determination of glutamic acid decarboxylase (GAD65) in pancreatic islets and its in vitro and in vivo degradation kinetics in serum using a highly sensitive enzyme immunoassay. Dis Markers. 2008;24:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Verge CF, Gianani R, Kawasaki E, et al. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes. 1996;45:926–933. [DOI] [PubMed] [Google Scholar]
- 105.Maedler K, Sergeev P, Ris F, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110:851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Arnush M, Heitmeier MR, Scarim AL, et al. IL-1 produced and released endogenously within human islets inhibits beta cell function. J Clin Invest. 1998;102:516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.D'Hertog W, Overbergh L, Lage K, et al. Proteomics analysis of cytokine-induced dysfunction and death in insulin-producing INS-1E cells: new insights into the pathways involved. Mol Cell Proteomics. 2007;6:2180–2199. [DOI] [PubMed] [Google Scholar]
- 108.Eizirik DL, Darville MI. Beta-cell apoptosis and defense mechanisms: lessons from type 1 diabetes. Diabetes. 2001;50(Supp. 1):S64–69. [DOI] [PubMed] [Google Scholar]
- 109.Makeeva N, Myers JW, Welsh N. Role of MKK3 and p38 MAPK in cytokine-induced death of insulin-producing cells. Biochem J. 2006;393(Pt 1):129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Steil GM, Trivedi N, Jonas JC, et al. Adaptation of beta-cell mass to substrate oversupply: enhanced function with normal gene expression. Am J Physiol Endocrinol Metab. 2001;280:E788–796. [DOI] [PubMed] [Google Scholar]
- 111.Liu YQ, Montanya E, Leahy JL. Increased islet DNA synthesis and glucose-derived lipid and amino acid production in association with beta-cell hyperproliferation in normoglycaemic 60% pancreatectomy rats. Diabetologia. 2001;44:1026–1033. [DOI] [PubMed] [Google Scholar]
- 112.Dor Y, Brown J, Martinez OI, et al. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. [DOI] [PubMed] [Google Scholar]
- 113.Xu X, D'Hoker J, Stange G, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207. [DOI] [PubMed] [Google Scholar]
- 114.Blandino-Rosano M, Perez-Arana G, Mellado-Gil JM, et al. Anti-proliferative effect of pro-inflammatory cytokines in cultured beta cells is associated with extracellular signal-regulated kinase 1/2 pathway inhibition: protective role of glucagon-like peptide -1. J Mol Endocrinol. 2008;41:35–44. [DOI] [PubMed] [Google Scholar]
- 115.Perez-Arana G, Blandino-Rosano M, Prada-Oliveira A, et al. Decrease in {beta}-cell proliferation precedes apoptosis during diabetes development in bio-breeding/worcester rat: beneficial role of Exendin-4. Endocrinology. 2010;151:2538–2546. [DOI] [PubMed] [Google Scholar]
- 116.Seemayer TA, Tannenbaum GS, Goldman H, et al. Dynamic time course studies of the spontaneously diabetic BB Wistar rat. III. Light-microscopic and ultrastructural observations of pancreatic islets of Langerhans. Am J Pathol. 1982;106:237–249. [PMC free article] [PubMed] [Google Scholar]
- 117.Kolb-Bachofen V, Schraermeyer U, Hoppe T, et al. Diabetes manifestation in BB rats is preceded by pan-pancreatic presence of activated inflammatory macrophages. Pancreas. 1992;7:578–584. [DOI] [PubMed] [Google Scholar]
- 118.Bogdani M, Henschel AM, Kansra S, et al. Biobreeding rat islets exhibit reduced antioxidative defense and N-acetyl cysteine treatment delays type 1 diabetes. J Endocrinol. 2013;216:111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Graham KL, Sutherland RM, Mannering SI, et al. Pathogenic mechanisms in type 1 diabetes: the islet is both target and driver of disease. Rev Diabet Stud. 2012;9:148–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Maganti A, Evans-Molina C, Mirmira R. From immunobiology to β-cell biology: the changing perspective on type 1 diabetes. Islets. 2014;6:e28778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2:a007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mordes JP, Bortell R, Blankenhorn EP, et al. Rat models of type 1 diabetes: genetics, environment, and autoimmunity. ILAR J. 2004;45:278–291. [DOI] [PubMed] [Google Scholar]
- 123.Kruger AJ, Yang C, Lipson KL, et al. Leptin treatment confers clinical benefit at multiple stages of virally induced type 1 diabetes in BB rats. Autoimmunity. 2011;44:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Greiner DL, Mordes JP, Handler ES, et al. Depletion of RT6.1+ T lymphocytes induces diabetes in resistant biobreeding/Worcester (BB/W) rats. J Exp Med. 1987;166:461–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guberski DL, Thomas VA, Shek WR, et al. Induction of type I diabetes by Kilham's rat virus in diabetes-resistant BB/Wor rats. Science. 1991;254:1010–1013. [DOI] [PubMed] [Google Scholar]
- 126.Williams AJ, Krug J, Lampeter EF, et al. Raised temperature reduces the incidence of diabetes in the NOD mouse. Diabetologia. 1990;33:635–637. [DOI] [PubMed] [Google Scholar]
- 127.Badenhoop K, Kahles H, Penna-Martinez M. Vitamin D, immune tolerance, and prevention of type 1 diabetes. Curr Diab Rep. 2012;12:635–642. [DOI] [PubMed] [Google Scholar]
- 128.Issazadeh-Navikas S, Teimer R, Bockermann R. Influence of dietary components on regulatory T cells. Mol Med. 2012;18:95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Courtois P, Sener A, Scott FW, et al. Disaccharidase activity in the intestinal tract of Wistar-Furth, diabetes-resistant and diabetes-prone BioBreeding rats. Br J Nutr. 2004;91:201–209. [DOI] [PubMed] [Google Scholar]
- 130.Wallis RH, Wang K, Marandi L, et al. Type 1 diabetes in the BB rat: a polygenic disease. Diabetes. 2009;58:1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]