

Abstract
Objective
CADASIL is a genetic paradigm of cerebral small vessel disease caused by NOTCH3 mutations that stereotypically lead to the extracellular deposition of NOTCH3 ectodomain (Notch3ECD) on the vessels. TIMP3 and vitronectin are 2 extracellular matrix proteins that abnormally accumulate in Notch3ECD-containing deposits on brain vessels of mice and patients with CADASIL. Herein, we investigated whether increased levels of TIMP3 and vitronectin are responsible for aspects of CADASIL disease phenotypes.
Methods
Timp3 and vitronectin expression were genetically reduced in TgNotch3R169C mice, a well-established preclinical model of CADASIL. A mouse overexpressing human TIMP3 (TgBAC-TIMP3) was developed. Disease-related phenotypes, including cerebral blood flow (CBF) deficits, white matter lesions, and Notch3ECD deposition, were evaluated between 6 and 20 months of age.
Results
CBF responses to neural activity (functional hyperemia), topical application of vasodilators, and decreases in blood pressure (CBF autoregulation) were similarly reduced in TgNotch3R169C and TgBAC-TIMP3 mice, and myogenic responses of brain arteries were likewise attenuated. These defects were rescued in TgNotch3R169C mice by haploinsufficiency of Timp3, although the number of white matter lesions was unaffected. In contrast, haploinsufficiency or loss of vitronectin in TgNotch3R169C mice ameliorated white matter lesions, although CBF responses were unchanged. Amelioration of cerebrovascular reactivity or white matter lesions in these mice was not associated with reduced Notch3ECD deposition in brain vessels.
Interpretation
Elevated levels of TIMP3 and vitronectin, acting downstream of Notch3ECD deposition, play a role in CADASIL, producing divergent influences on early CBF deficits and later white matter lesions.
Cerebral small vessel disease (SVD) accounts for a quarter of ischemic strokes and is increasingly recognized as a leading cause of age- and hypertension-related cognitive decline and disability.1 CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy), the most common hereditary SVD, is caused by dominant mutations in NOTCH3,2,3 a heterodimeric receptor that is predominantly expressed in vascular smooth muscle cells and is a critical regulator of the developmental formation of small arteries.4 CADASIL is characterized by midlife onset of recurrent stroke and cognitive impairment, progressing to dementia and premature death at approximately 60 years of age.3 Currently, there are no therapies available to prevent or slow the pathogenesis of CADASIL.
CADASIL-associated NOTCH3 mutations stereotypically lead to the early extracellular deposition of NOTCH3 ectodomain (Notch3ECD) at the plasma membrane of vascular smooth muscle cells and pericytes and in extracellular deposits called granular osmiophilic material (GOM).5–7 Recently, we and others provided evidence that excess levels or multimerization of mutant Notch3ECD facilitate interactions with key components of the vascular extracellular matrix, including but not limited to TIMP3 and vitronectin, and promote their accumulation in Notch3ECD-containing deposits.6,8,9
TIMP3 is a member of the tissue inhibitor of metalloproteinases (TIMP) family, comprising 4 small proteins; as their name implies, these proteins are physiologic inhibitors of metalloproteinases, which degrade the extracellular matrix and promote shedding of cell surface molecules.10 Vitronectin is an adhesive glycoprotein that coordinates cell adhesion and cell migration with pericellular proteolysis and growth factor signaling.11 In the normal brain, TIMP3 and vitronectin are predominantly associated with the extracellular matrix of vessels.6,9
In this study, we sought to elucidate whether the increase in the levels of TIMP3 and vitronectin extracellular matrix proteins is pathogenic or a mere epiphenomenon.
Material and Methods
Mice
TgNotch3R169C (line 88) and TgNotch3WT (line 129) mice, which overexpress mutant and wild-type NOTCH3 receptors, respectively, at comparable levels, were maintained in a heterozygous state on an FVB/N background, as described previously.12 Timp3-null (Timp3−/−)13 and vitronectin-null (Vtn−/−) mice14 on a C57BL/6 background were obtained from B. Weber and H. Stoehr (Regensburg University, Germany) and E. Rondeau (Inserm, France), respectively. FVB/N mice were purchased from Charles River (Charles River Laboratories, Saint-Germain-Nuelles, France). The TgNotch3R169C/Timp3 double-mutant mice used in the study—TgNotch3R169C;Timp3+/+, TgNotch3R169C; Timp3+/−, non-Tg;Timp3+/+, and non-Tg;Timp3+/−—were littermates with the same hybrid background (88% FVB/ N:12% C57Bl/6). Likewise, the TgNotch3R169C/Vtn mice— TgNotch3R169C;Vtn+/+, TgNotch3R169C;Vtn+/−, and TgNotch 3R169C;Vtn−/−, as well as non-Tg;Vtn+/+, non-Tg;Vtn+/−, and non-Tg;Vtn−/−—were littermates on the same hybrid background (81% FVB/N:19% C57Bl/6).
TgBAC-TIMP3 mice were generated using a 182.9kb bacterial artificial chromosome (BAC) clone, hRP11 419C14, containing the entire human TIMP3 locus. The fidelity of the final product was determined by restriction enzyme fragment mapping, field inversion gel electrophoresis, polymerase chain reaction (PCR), and DNA sequencing. BAC DNA was prepared and purified using the Large-Construct Kit (Qiagen, Valencia, CA) and injected into C57BL/6 mouse oocytes. We established 2 heterozygous transgenic (Tg) lines (lines 2 and 29). The transgene in line 29, which was integrated into chromosome X, was associated with prenatal lethality in males. Hence, line 2 was used for subsequent experimentation. Mice were backcrossed 3 times onto an FVB/N background and maintained on this hybrid background (88% FVB/N: 12% C57Bl/6).
Genotyping analyses were performed by PCR using the following primer pairs: TgNotch3, 5′-TCA ACG CCT TCT CGT TCT TC-3′ (forward) and 5′-AAT ACC GTC GTG CTT TCG AG-3′ (reverse); Timp3 wild-type allele, 5′-TTC AGT AAG ATG CCC CAC G-3′ (forward) and 5′-TAC ATC TTG CCT TCA TAC ACG-3′ (reverse); Timp3 knockout allele, 5′-TTC AGT AAG ATG CCC CAC G-3′ (forward) and 5′-ATG TGG AAT GTG TGC GAG G-3′ (reverse); vitronectin wild-type allele, 5′-TCA GAA GTG TGC CAG TGC TT-3′ (forward) and 5′-GCT GAG CCA TCT TTC CAG TC-3′ (reverse); vitronectin knockout allele, 5′-GCC AGA GGC CAC TTG TGT AG-3′ (forward) and 5′-TCA GAA GTG TGC CAG TGC TT-3′ (reverse); and TgBAC-TIMP3, 5′-CCA GGA GAC AGC AAG TAG CC-3′ (forward) and 5′-GCT GCT GTT TAG GGA TCT GC-3′ (reverse). Mice were bred and housed in pathogen-free animal facilities (Transgénèse et Archivage d’Animaux Modèles (TAAM) or University Paris 7, site Villemin) and fed a standard diet ad libitum with free access to water. All experiments described in this study were conducted in full accordance with the guidelines of our local institutional animal care and use committee (Lariboisière-Villemin), with every effort made to minimize the number of animals used. Unless otherwise noted, experiments were performed in age-matched littermates to minimize confounding effects attributable to background heterogeneity.
RNA Preparation and Quantitative Reverse Transcription PCR Analyses
RNA was extracted from cerebral pial arteries dissected under a microscope, and quantitative reverse transcription (RT)-PCR was performed as previously described.15 The following primer pairs were used: murine Timp3, 5′-GCC ATT CTC AAT CCC TGG TA-3′ (forward) and 5′-ACC GCT CTC CGA TGA TGT AA-3′ (reverse); murine vitronectin, 5′-TGT TTG AGC ACT TTG CCT TG-3′ (forward) and 5′-GTG GGA TAA GGA GCC AGT GA-3′ (reverse); rat Notch3 transgene, 5′-CTC CCT GGC TTC TCT CTC-3′ (forward) and 5′-GGT CAG CCC CTA CCC ATT-3′ (reverse); β-actin, 5′-CTG CGT CTG GAC CTG GCT-3′ (forward) and 5′-ACG CAC GAT TTC CCT CTC A-3′ (reverse); and transgelin 5′-TCT AAT GGC TTT GGG CAG TT-3′ (forward) and 5′-GCA GTT GGC TGT CTG TGA AG-3′ (reverse).
In Vivo Analysis of Cerebrovascular Reactivity
SURGICAL PROCEDURE
Male mice were anesthetized with isoflurane (maintenance, 2%), tracheally intubated, and artificially ventilated with an oxygen–nitrogen mixture using a ventilator (Sar-830/P; CWE, Ardmore, PA). The femoral artery was cannulated for recording mean arterial pressure and collecting blood samples. A small craniotomy (2 × superfused with Ringer solution (37°C; pH 7.3–7.4). After surgery, isoflurane was gradually discontinued and anesthesia was maintained with urethane (750mg/kg−1) and chloralose (50mg/kg−1). Rectal temperature was maintained at 37°C, and arterial blood gases were measured. The level of anesthesia was monitored by testing corneal reflexes and motor responses to tail pinch. To minimize confounding effects of anesthesia on vascular reactivity, we kept the time interval between the administration of urethane–chloralose and the testing of cerebral blood flow (CBF) responses consistent among the different groups of mice studied. Arterial blood pressure, blood gases, and rectal temperature were monitored and controlled (Table).
TABLE.
| Genotype (age, mo) | No. | MAP, mmHg | pCO2, mmHg | pO2, mmHg | pH |
|---|---|---|---|---|---|
| nonTg (6) | 6 | 78 ± 6 | 34 ± 3 | 125 ± 5 | 7.33 ± 0.1 |
| TgNotch3R169C (6) | 6 | 74 ± 2 | 35 ± 2 | 126 ± 3 | 7.34 ± 0.10 |
| TgNotch3WT (6) | 5 | 79 ± 4 | 36 ± 3 | 127 ± 4 | 7.33 ± 0.09 |
| nonTg;Vtn+/+ (6) | 5 | 78 ± 4 | 34 ± 2 | 126 ± 5 | 7.33 ± 0.1 |
| TgNotch3R169C;Vtn+/+ (6) | 5 | 75 ± 3 | 35 ± 1 | 125 ± 7 | 7.34 ± 0.01 |
| nonTg;Vtn+/− (6) | 6 | 76 ± 2 | 35 ± 2 | 127 ± 6 | 7.34 ± 0.02 |
| TgNotch3R169C;Vtn+/− (6) | 6 | 74 ± 2 | 35 ± 2 | 127 ± 5 | 7.35 ± 0.02 |
| nonTg;Vtn−/− (6) | 7 | 75 ± 6 | 34 ± 1 | 127 ± 3 | 7.34 ± 0.02 |
| TgNotch3R169C;Vtn−/− (6) | 8 | 76 ± 4 | 35 ± 2 | 125 ± 6 | 7.35 ± 0.02 |
| nonTg;Vtn+/+ (12) | 5 | 72 ± 4 | 35 ± 1 | 123 ± 4 | 7.35 ± 0.1 |
| TgNotch3R169C;Vtn+/+ (12) | 7 | 76 ± 2 | 34 ± 2 | 126 ± 4 | 7.34 ± 0.01 |
| nonTg;Vtn+/− (12) | 7 | 76 ± 2 | 35 ± 2 | 125 ± 5 | 7.34 ± 0.02 |
| TgNotch3R169C;Vtn+/− (12) | 6 | 79 ± 4 | 36 ± 2 | 126 ± 5 | 7.33 ± 0.1 |
| nonTg;Timp3+/+ (6) | 6 | 79 ± 3 | 37 ± 2 | 124 ± 4 | 7.34 ± 0.08 |
| TgNotch3R169C;Timp3+/+ (6) | 6 | 77 ± 2 | 37 ± 3 | 126 ± 5 | 7.34 ± 0.15 |
| nonTg;Timp3+/− (6) | 5 | 75 ± 5 | 36 ± 2 | 125 ± 4 | 7.32 ± 0.1 |
| TgNotch3R169C;Timp3+/− (6) | 6 | 74 ± 6 | 36 ± 3 | 127 ± 5 | 7.33 ± 0.07 |
| nonTg (6) | 5 | 79 ± 3 | 34 ± 2 | 126 ± 5 | 7.32 ± 0.1 |
| TgBAC-TIMP3 (6) | 5 | 90 ± 3a | 37 ± 2 | 128 ± 4 | 7.33 ± 0.1 |
All mice used in these studies are males.
p < 0.05 versus nontransgenic (nonTg), 1-way analysis of variance followed by Tukey post hoc test.
MAP = mean arterial pressure.
CBF MONITORING
Relative CBF was continuously monitored at the site of the cranial window using a laser-Doppler probe (Moor Instruments, Axminster, UK) positioned stereotaxically 0.5 to 1mm from the cortical surface. CBF values were expressed as percentage increase relative to the resting level ([CBFstimulus − CBFresting]/CBFresting). Zero values for CBF were obtained after the heart was stopped by an overdose of isoflurane at the end of the experiment.16 Laser-Doppler flowmetry provides an accurate assessment of relative CBF changes, but it does not provide measurements of absolute flow.
CBF recordings were started after arterial pressure and blood gases had reached a steady state, as previously described.16 All pharmacological agents studied were dissolved in a modified Ringer solution.17 The increase in CBF produced by somatosensory activation was studied by stimulating the whiskers contralateral to the cranial window by side-to-side deflection for 60 seconds. Acetylcholine (10 μmol/l; Sigma-Aldrich, St Louis, MO) or the calcium ionophore A23187 (3 μmol/l; Sigma-Aldrich), both endothelium-dependent vasodilators, were topically superfused for 5 minutes, and the resulting changes in CBF were monitored. CBF responses to the smooth muscle–dependent relaxant adenosine (400 μmol/l; Sigma-Aldrich) were also examined. The lower limit of CBF autoregulation was determined at the end of the experiment, as previously described.18 Considering that basal cortical blood flow in TgNotch3R169C mice is weakly reduced (by 5–8% at 12 months of age),12 the amplitude of evoked CBF responses is unlikely to be affected by a ceiling effect.
Ex vivo Analysis of Myogenic Tone
Myogenic responses were analyzed on segments of the posterior cerebral artery, and myogenic tone was determined by increasing intraluminal pressure from 10 to 100mmHg using a pressure-servo control pump as previously described.12 Myogenic tone was expressed as the percentage of passive diameter ([passive diameter − active diameter]/passive diameter × 100).
Immunoblot Analysis
Protein extracts were prepared from cerebral pial arteries and subjected to immunoblotting using rabbit monoclonal anti–TIMP-3 (1:2,500, clone D74B10; Cell Signaling Technology, Danvers, MA), and mouse monoclonal anti–smooth muscle alpha actin (1:25,000, clone 1A4, Sigma-Aldrich) as previously described.6 Densitometric quantification of band intensity was performed using ImageJ (v10.2, NIH).
Immunohistochemistry
For myelin basic protein (MBP) staining, mice were deeply anesthetized with sodium pentobarbital (80mg/kg) and transcardially perfused with 50ml of phosphate buffer (PB) followed by 50ml of 4% paraformaldehyde in PB. The brain was removed, postfixed in 4% paraformaldehyde, and processed for cryopreservation. Free-floating cryosections (16 μm-thick sagittal slices, n = 4–8 sections/mouse) were immunostained with mouse monoclonal anti-MBP antibody (1:10,000, SMI94; BioLegend, London, UK) as described.19
For Notch3ECD staining, mice were overdosed with isoflurane, and brains were harvested, frozen in liquid nitrogen, and stored at −80°C. Acetone-fixed cryosections (12 μm thick, n = 9 sections/mouse) were incubated overnight at 4°C with rabbit polyclonal anti-Notch3ECD primary antibody raised against epidermal growth factor repeats 17 to 21 of rat Notch3 (1:16,000; A.J., unpublished data), followed by detection with Alexa 594-conjugated antirabbit secondary antibody (1:500; Life Technologies, Saint Aubin, France). Arteries were identified by immunostaining with fluorescein isothiocyanate–conjugated anti–smooth muscle α-actin primary antibody (1:1,000, clone 1A4, Sigma-Aldrich), and capillaries were identified by immunostaining with rat monoclonal antiperlecan antibody (1:500, clone A7L6; Millipore, Molsheim, France) followed by detection with Alexa 488–conjugated antirat secondary antibody (1:500; Life Technologies).
Quantification of Myelin Debris and Notch3ECD Deposits
Stained sections were imaged with an Eclipse 80i microscope (Nikon, Champigny sur Marne, France) at × 20 (MBP analysis), × 40 (capillary analysis), or × 60 (artery analysis) magnification. Images were captured using an Andor Neo sCMOS camera and NIS Elements BR v4.0 software (Nikon), with identical settings across compared groups. The entire procedure was performed with prefixed parameters under blinded conditions.
Myelin debris spots were counted over the whole corpus callosum using a custom-made, 3-step NIH ImageJ macro, as described.19 Results were expressed as the number of SMI94 hyperintense foci over the area of the corpus callosum.
Notch3ECD deposits were counted on maximal intensity projections of image stacks using ImageJ software (v1.49g; Fiji Distribution, NIH) following a semiautomated procedure that includes 3 main steps: (1) manual delineation of pial arteries on the smooth muscle α-actin channel and delineation of capillaries by automated segmentation on the perlecan channel, followed by measurement of vessel area; (2) background suppression on the Notch3ECD channel; and (3) automatic detection and counting of Notch3ECD deposits within vessel borders using a local maxima approach. Results were expressed as the number of Notch3ECD deposits over the vessel area.
Analysis and Quantification of GOM Deposits
Ultrathin sections of brain artery were prepared and observed in a CM100 electron microscope (Philips, Best, the Netherlands), as previously described.12 Electron micrograph images of the artery over its entire circumference were captured using a digital camera at ×9,700 magnification. The number of GOM deposits on the abluminal perimeter of smooth muscle cells was counted in a blinded manner. Results were expressed as the number of GOM deposits per 100 μm.
Statistics
Data are expressed as mean ± standard error of the mean. CBF autoregulation and myogenic tone were analyzed by 2-way repeated-measure analysis of variance (ANOVA) followed by Bonferroni post hoc test. Myelin debris, CBF responses, Notch3ECD aggregates, and GOM deposits were analyzed by 1-way ANOVA followed by Bonferroni or Tukey post hoc tests, with the exception of GOM deposits in part C of the last figure, which were analyzed using Student t test. Differences with p-values < 0.05 were considered statistically significant.
Results
Genetic Reduction of Timp3 or Vitronectin in the TgNotch3R169C Mouse Model
To investigate the potential role of TIMP3 and vitronectin in CADASIL pathogenesis, we adopted a genetic interaction approach using the TgNotch3R169C mouse model, a well-established preclinical model of CADASIL.12 Mice completely lacking TIMP3 exhibit various developmental defects, including vascular alterations such as dilated vessels in the eye and outward remodeling of the mesenteric arteries, as well as decreased arterial blood pressure.13,20,21 To avoid potentially confounding developmental effects induced by the loss of TIMP3, we generated and analyzed TgNotch3R169C mice lacking 1 allele of Timp3 (TgNotch3R169C;Timp3+/−) along with their control littermates. Vitronectin-null mice (Vtn−/−) show no overt phenotype if unchallenged.14 Hence, we generated and analyzed TgNotch3R169C mice lacking 1 (TgNotch3R169C;Vtn+/−) or 2 (TgNotch3R169C;Vtn−/−) alleles of vitronectin along with their control non-Tg littermates. Double-Tg mice were born at the expected Mendelian ratio, and were indistinguishable from the wild-type and TgNotch3R169C littermates, even with aging, suggesting a lack of overt deficits. We confirmed reduction of Timp3 and vitronectin mRNA expression in brain arteries by quantitative RT-PCR analysis of dissected vessels (Fig 1A, C). Notably, Timp3 mRNA reduction in Timp3+/− mice was likely underestimated, as mRNA decay is incomplete in this Timp3−/− line (A.J., unpublished data). Importantly, genetic reduction of Timp3 (Timp3+/−) or vitronectin (Vtn+/−, Vtn−/−) in TgNotch3R169C mice did not affect Notch3 transgene mRNA expression compared to their TgNotch3R169C littermates on a wild-type Timp3 (Timp3+/+) and vitronectin (Vtn+/+) background, respectively (see Fig 1B, D). Therefore, any phenotypic change in these double-mutant mice cannot be attributed to an alteration in Notch3 transgene expression.
FIGURE 1.
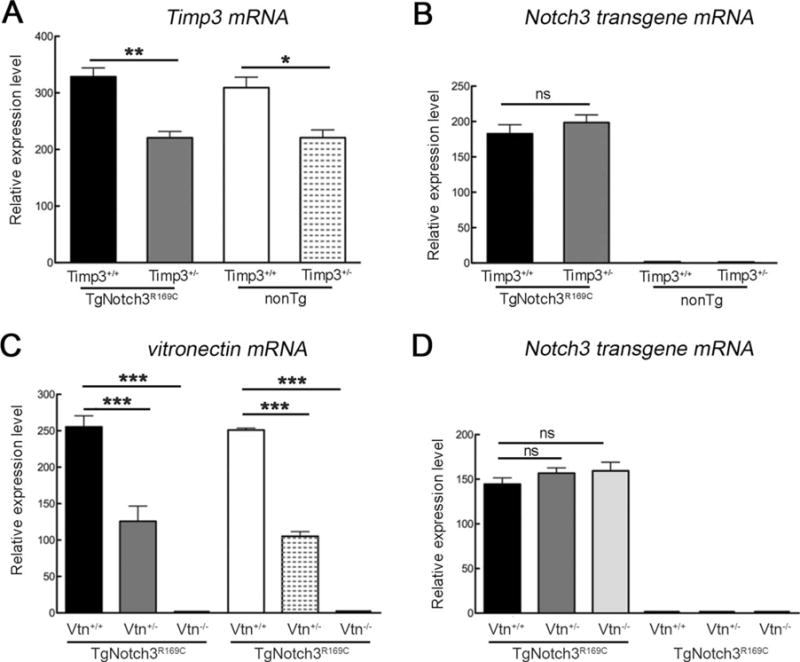
Genetic reduction of Timp3 or vitronectin in TgNotch3R169C mice does not affect Notch3 transgene mRNA expression. Expression levels of Timp3 (A), rat Notch3 transgene (B, D), and vitronectin (C) mRNA were analyzed by quantitative reverse transcription polymerase chain reaction in brain arteries from 6-month-old TgNotch3R169C;Timp3+/+, TgNotch3R169C;Timp3+/−, and control nontransgenic (nonTg) littermate mice (A, B), and TgNotch3R169C;Vtn+/+, TgNotch3R169C;Vtn+/−, TgNotch3R169C;Vtn−/−, and control nonTg littermate mice (C, D). Target mRNA levels were normalized to those of β-actin (n = 4–8 samples per genotype). Significance was determined by 1-way analysis of variance followed by Tukey post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001). ns = nonsignificant.
TgNotch3R169C Mice Exhibit a Profound Global Vasomotor Dysfunction That Is Rescued by Genetic Reduction of Timp3, but Not Vitronectin
We first sought to elucidate the effects of excess TIMP3 and vitronectin on cerebrovascular dysfunction elicited by R169C mutant NOTCH3. We previously reported that functional hyperemia—the process that serves to match local CBF with metabolic demands imposed by neuronal activity in the brain22—was disrupted in TgNotch3R169C mice as early as 6 months; also the lower limit of CBF autoregulation was significantly shifted to higher blood pressure, reflecting an inability of cerebral vessels to dilate in response to a decrease in arterial blood pressure.12 Using laser-Doppler flowmetry in mice equipped with an open cranial window over the somatosensory cortex, we monitored CBF increases to whisker stimulation (functional hyperemia) and CBF changes to stepwise reduction of arterial blood pressure (lower limit of CBF autoregulation) in a cohort of 6-month-old double-Tg mice and appropriate age-matched controls. As expected, we observed compromised functional hyperemia and autoregulation in TgNotch3R169C mice (Fig 2). These defects were retained in TgNotch3R169C;Timp3+/+ and TgNotch3R169C; Vtn+/+ mice with wild-type Timp3 and vitronectin alleles, obtained by crossing TgNotch3R169C mice with the corresponding heterozygous mice, indicating that minor differences in strain background between genotypes did not affect the TgNotch3R169C phenotype. Importantly, genetic reduction of Timp3 (TgNotch3R169C;Timp3+/−) restored functional hyperemia and the lower limit of autoregulation. In contrast, these deficits were unaffected by genetic reduction (TgNotch3R169C;Vtn+/−) or elimination (TgNotch3R169C; Vtn−/−) of vitronectin.
FIGURE 2.
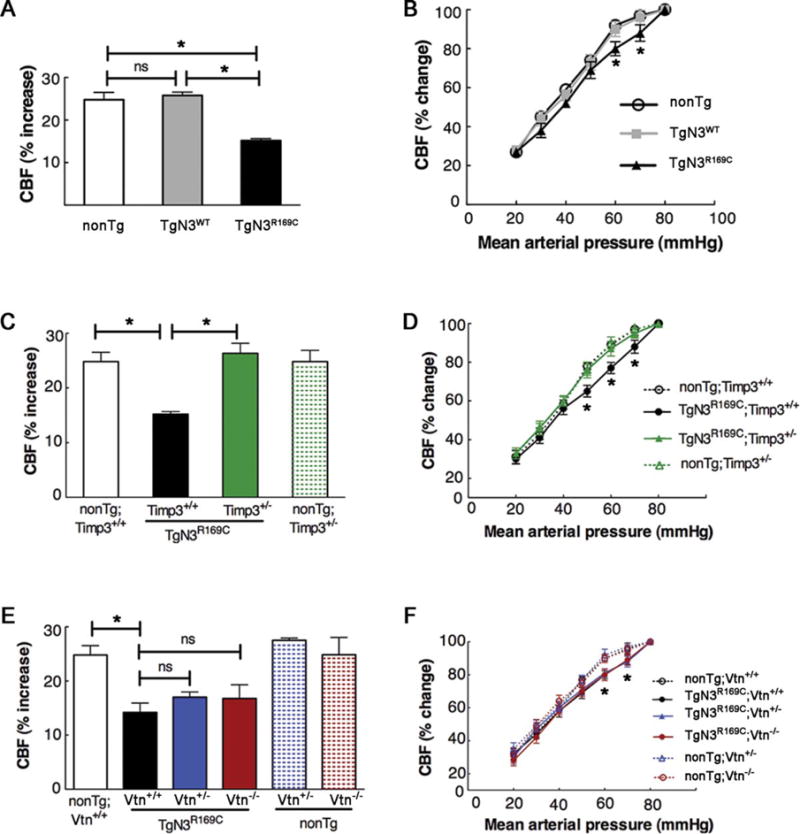
Timp3 haploinsufficiency protects against attenuated functional hyperemia and altered cerebral blood flow (CBF) autoregulation in TgNotch3R169C mice, whereas genetic reduction or ablation of vitronectin has no effect. CBF changes evoked by whisker stimulation (functional hyperemia; A, C, E) and controlled mean arterial pressure (MAP) reduction (B, D, F) were assessed in various 6-month-old transgenic mouse models (n = 5–8 males per genotype). (A, B) Single transgenic TgNotch3R169C (TgN3R169C) and TgNotch3WT (TgN3WT) mice, and nontransgenic (nonTg) littermate controls. (C, D) Double-mutant TgN3R169C; Timp3+/− mice, with Timp3 haploinsufficiency in the context of Notch3R169C overexpression; nonTg;Timp3+/+, nonTg;Timp3+/−, and TgNotch3R169C;Timp3+/+ mice were included as controls. (E, F) Double-mutant TgN3R169C;Vtn+/− and TgN3R169C;Vtn−/− mice, with vitronectin haploinsufficiency and complete ablation, respectively, in the context of Notch3R169C overexpression; non-Tg;Vtn+/+, nonTg;Vtn+/−, nonTg;Vtn−/−, and TgNotch3R169C;Vtn+/+ mice were included as controls. Functional hyperemia was strongly attenuated in TgN3R169C mice compared to TgN3WT and nonTg littermate mice, and TgN3R169C mice showed an impaired relationship between CBF and MAP; CBF was significantly different between TgN3R169C and TgN3WT or nonTg mice at MAP values of 60 to 70mmHg. CBF responses were similarly impaired in TgN3R169C mice expressing wild-type Timp3 (TgN3R169C;Timp3+/+) or wild-type vitronectin (TgN3R169C;Vtn+/+), whereas these responses were not altered in nonTg littermate mice with or without genetic reduction of Timp3 (nonTg;Timp3+/−) or vitronectin (nonTg;Vtn−/−). Importantly, TgN3R169C;Timp3+/− mice exhibited normalized CBF responses to whisker stimulation and a restored relationship between MAP and CBF comparable to that of their nonTg;Timp3+/− and nonTg;Timp3+/+ littermates (C, D). In contrast, neither TgN3R169C;Vtn+/− nor TgN3R169C;Vtn−/− mice (E, F) exhibited improved CBF responses. (A, C, E) One-way analysis of variance (ANOVA) followed by Tukey post hoc test; (B, D, F) 2-way repeated measures ANOVA followed by Bonferroni post hoc test (*p < 0.05). The main physiological variables for all mice are presented in the Table. ns = nonsignificant.
To explore the extent to which the rescue of the mutant NOTCH3 functional phenotype can be generalized, we assessed CBF responses to endothelial-dependent and smooth muscle–dependent vasodilators applied to the cranial window. The increase in CBF evoked by topical application of the endothelium-dependent vasodilators acetylcholine or A23187 (Ca2+ ionophore) was significantly reduced in TgNotch3R169C mice compared with TgNotch3WT and non-Tg littermates; CBF responses to the smooth muscle relaxant adenosine were likewise significantly reduced in TgNotch3R169C mice (Fig 3). Again, as expected, these cerebrovascular responses were similarly impaired in TgNotch3R169C;Timp3+/+ and TgNotch3R169C; Vtn+/+ mice. As was the case for hyperemic responses, CBF responses to vasodilators were not improved in TgNotch3R169C;Vtn+/− or TgNotch3R169C;Vtn−/− mice with a reduction or elimination of vitronectin. In striking contrast, these responses were completely normal in TgNotch3R169C;Timp3+/− mice, with reduced expression of Timp3. Collectively, these data suggest that TgNotch3R169C mice exhibit a global cerebrovascular dysfunction with a profound reduction in vasodilatory responses, and that decreasing levels of Timp3, but not vitronectin, protects TgNotch3R169C mice from this dysfunction.
FIGURE 3.
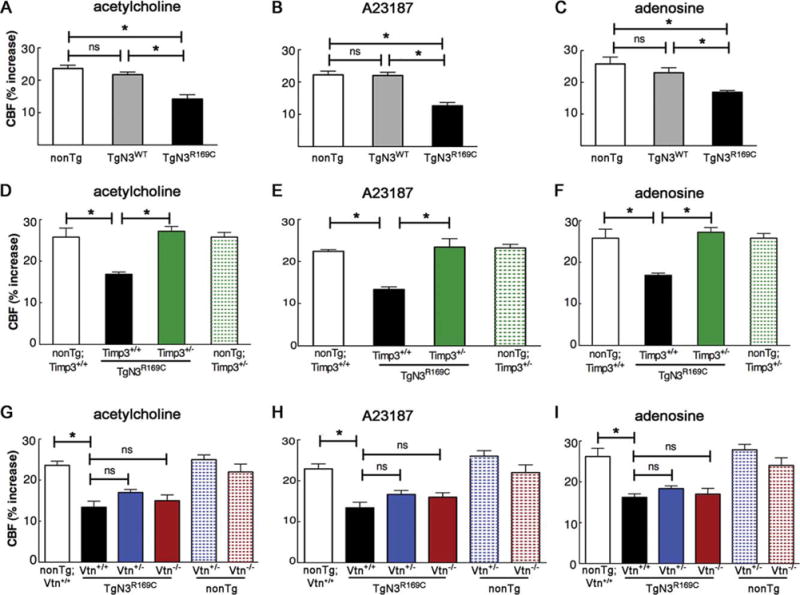
Cerebral blood flow (CBF) responses evoked by endothelium-dependent and endothelium-independent vasodilators are normalized in TgNotch3R169C mice by genetic reduction of Timp3, but not vitronectin. CBF increases evoked by the endothelium-dependent vasodilators acetylcholine (A, D, G) or calcium ionophore A23187 (B, E, H) and smooth muscle–dependent vasodilator adenosine (C, F, I) were analyzed in various 6-month-old transgenic mouse models (n = 5–8 males per genotype). (A–C) Single transgenic TgNotch3R169C (TgN3R169C) and TgNotch3WT (TgN3WT) mice, and nontransgenic (nonTg) littermate controls. (D–F) Double-mutant TgN3R169C;Timp3+/− mice, with Timp3 haploinsufficiency in the context of Notch3R169C overexpression; nonTg;Timp3+/+, nonTg;Timp3+/−, and TgNotch3R169C;Timp3+/+ mice were included as controls. (G–I) Double-mutant TgN3R169C;Vtn+/− and TgN3R169C;Vtn−/− mice, with vitronectin haploinsufficiency and complete ablation, respectively, in the context of Notch3R169C overexpression; nonTg;Vtn+/+, nonTg;Vtn+/−, nonTg;Vtn−/−, and TgNotch3R169C;Vtn+/+ mice were included as controls. All CBF responses were significantly attenuated in TgN3R169C mice with wild-type Timp3 and vitronectin (TgN3R169C, TgN3R169C;Timp3+/+, TgN3R169C;Vtn+/+). Responses of TgN3R169C;Vtn+/− and TgN3R169C;Vtn−/− mice were similarly impaired, whereas TgN3R169C;Timp3+/− mice had normal responses that were comparable to those of nonTg;Timp3+/+ and nonTg;Timp3+/− littermates. Significance was determined by 1-way analysis of variance followed by Tukey post hoc test (*p < 0.05). The main physiological variables for all mice are presented in the Table. ns = nonsignificant.
Genetic Reduction of Timp3 Rescues Myogenic Responses in TgNotch3R169C Mice
We next asked whether the protection provided by decreasing the amount of Timp3 acts at the level of brain vessels, as suggested by the predominant vascular expression pattern of TIMP3 in the brain.6 We previously reported that pressure-induced constriction (myogenic tone) of both pial and parenchymal arteries was markedly attenuated in TgNotch3R169C mice.12,23 Myogenic tone establishes the basal constriction of an artery, providing a state from which an artery can increase or decrease in diameter on demand. A decrease in the myogenic tone of cerebral arteries in TgNotch3R169C mice implies a diminution in the vasodilatory reserve that logically could represent a common mechanism for the global cerebrovascular dysfunction exhibited by these mice.24 To explore the contribution of Timp3 to this defect, we measured changes in diameter in response to changes in intraluminal pressure in pial arteries from TgNotch3R169C mice with normal or reduced expression of Timp3 at 6 months of age. Reducing Timp3 had no effect on the myogenic tone in arterial segments from non-Tg mice (non-Tg;Timp3+/−), but normalized myogenic tone in arteries from TgNotch3R169C mice (TgNotch3R169;Timp3+/−; Fig 4A). Hence, our findings suggest that excess TIMP3 acts at least at the level of isolated brain arteries to impair myogenic responses in TgNotch3R169C mice.
FIGURE 4.
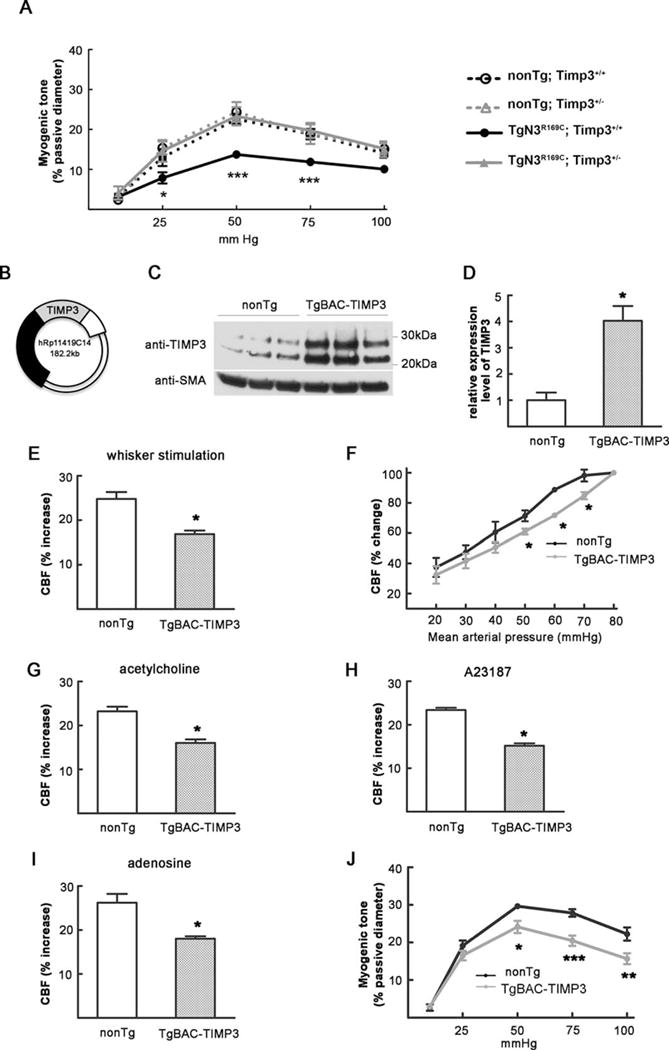
Timp3 haploinsufficiency protects against reduced myogenic tone in TgNotch3R169C mice and TIMP3 overexpression reproduces the cerebrovascular deficits of TgNotch3R169C mice. (A) Myogenic responses are normal in nontransgenic (non-Tg);Timp3+/− mice, are attenuated in TgN3R169C;Timp3+/+ mice, and are normalized in TgN3R169C;Timp3+/− mice (n = 7–8 mice per genotype). (B) Schematic representation of the bacterial artificial chromosome (BAC) containing the human TIMP3 locus. (C, D) Immunoblot analysis of brain arteries (C) shows approximately a 4-fold increase in TIMP3 levels in TgBAC-TIMP3 mice compared with nonTg littermates (D; n = 3 samples per group, each prepared with brain arteries dissected from 2 mice). (E–I) Cerebral blood flow (CBF) changes evoked by whisker stimulation (E), the endothelium-dependent vasodilators acetylcholine (G) or calcium ionophore A23187 (H), and smooth muscle–dependent vasodilator adenosine (I) are strongly reduced in 6-month-old TgBAC-TIMP3 mice (n = 5–6 mice per genotype). (F) TgBAC-TIMP3 mice show an impaired relationship between CBF and mean arterial pressure (MAP). CBF is statistically different between TgBAC-TIMP3 and nonTg at MAP values of 50 to 70mmHg. The main physiological variables for all mice are presented in the Table. (J) Myogenic responses are attenuated in TgBAC-TIMP3 mice compared with nonTg littermates (n = 7–8 per genotype). Significance was determined by Student t test (D, E, G–I) or 2-way repeated measures analysis of variance followed by Bonferroni post hoc test (A, F, J; *p < 0.05, **p < 0.01, ***p < 0.001). SMA = smooth muscle actin.
Genetic Overexpression of TIMP3 Mimics the Cerebrovascular Dysfunction Produced by Mutated Notch3 in TgNotch3R169C Mice
To further investigate the contribution of TIMP3 to cerebrovascular dysfunction, we tested whether elevated TIMP3 expression reproduced the cerebrovascular deficits of TgNotch3R169C mice. To this end, we developed a mouse overexpressing human TIMP3 using the endogenous human promoter on a BAC containing the human genomic TIMP3 locus. The full-length TIMP3 transcription unit is nested within intron V of the synpasin 3 gene, and with the exception of exons 5 to 7 of this gene, no other known or predicted genes lie on this BAC (see Fig 4B). Immunoblot analyses of pial arteries revealed that TIMP3 expression was increased ~4-fold over the endogenous murine TIMP3 in TgBAC-TIMP3 mice (see Fig 4C, D).
Remarkably, CBF responses to functional hyperemia, topical application of vasodilators, and decreases in blood pressure were similarly reduced in TgBAC-TIMP3 and TgNotch3R169C mice (see Figs (2 and 3), and 4E–I). Likewise, myogenic responses of brain arteries were attenuated in TgBAC-TIMP3 mice (see Fig 4J). Taken together, our findings indicate that elevated TIMP3 expression is responsible for the global impairment of cerebral vasodilatory responses and the attenuation of myogenic responses of brain arteries.
Reducing the Gene Dosage of Vitronectin, but Not Timp3, Ameliorates White Matter Lesions in TgNotch3R169C Mice
We next evaluated the impact of reducing Timp3 or vitronectin gene dosage on white matter lesions elicited by mutant NOTCH3. We previously reported that TgNotch3R169C mice develop vacuolation of the cerebral white matter tracts around the age of 20 months.12 Subsequent analyses revealed that white matter vacuolation was associated with segmental degradation of myelin fibers, as evidenced by immunolabeling brain sections with the SMI94 anti-MBP antibody. Notably, an assessment of the number of fluorescent foci (myelin debris) proved to reflect white matter pathology more accurately—and earlier—than vacuolation.19 Accordingly, we used SMI94 brain immunostaining to characterize a cohort of double-mutant mice and appropriate controls at 6, 12, and 20 months of age. TgNotch3R169C mice on a wild-type vitronectin or Timp3 background exhibited a significantly higher number of myelin debris spots at 6 months of age compared with age-matched wild-type non-Tg littermates. The number of these lesions almost doubled between 6 and 12 months and between 12 and 20 months of age (Figs 5 and 6). Importantly, genetic reduction of Timp3 (TgNotch3R169C;Timp3+/−) had no effect on white matter lesions at 6, 12, or 20 months of age (see Fig 5). In striking contrast, the severity of white matter lesions was significantly attenuated by genetic reduction of vitronectin (TgNotch3R169C;Vtn+/−) at 12 and 20 months of age (see Fig 6). Moreover, mutant mice lacking vitronectin (TgNotch3R169C;Vtn−/−) exhibited a significantly lower number of myelin debris spots at 20 months of age. Consistent with the observation that expression of vitronectin in TgNotch3R169C mice is altered at or above the age of 12 months,6 genetic reduction or elimination of vitronectin had no detectable effect on the number of myelin debris at 6 months of age. Thus, these data suggest that increased levels of vitronectin alone, but not TIMP3 alone, contribute to CADASIL-related white matter lesions.
FIGURE 5.
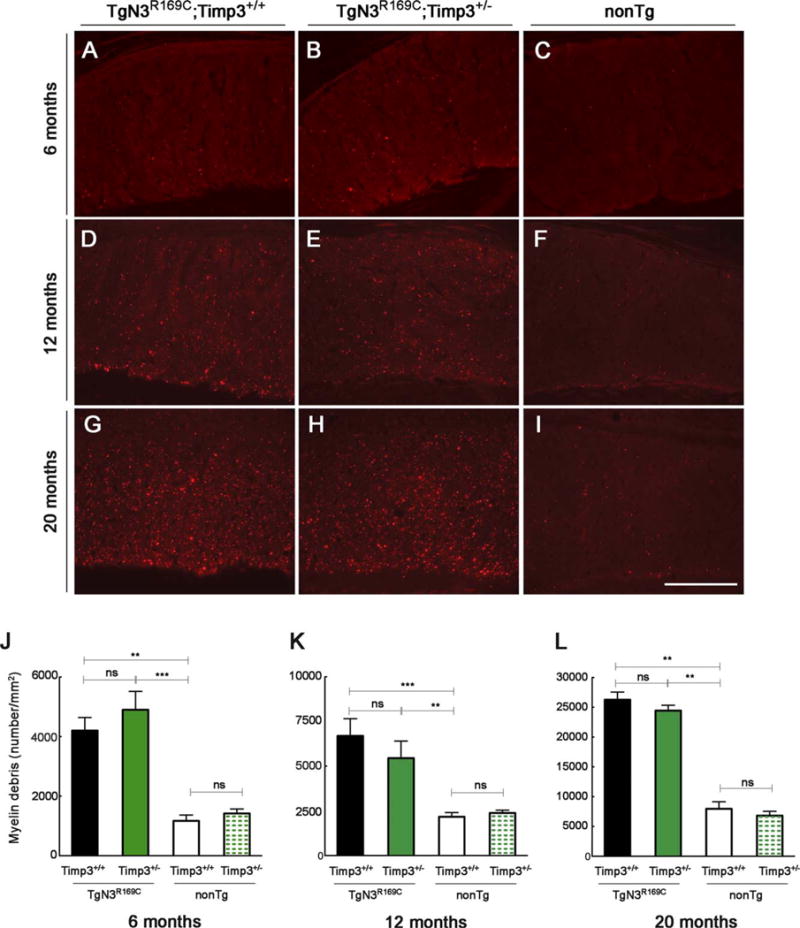
Genetic reduction of Timp3 does not ameliorate white matter lesions in TgNotch3R169C mice. (A–I) Representative images of immunostaining for myelin basic protein (SMI94 antibody) in the corpus callosum (genu) from TgN3R169C;Timp3+/+, TgN3R169C;Timp3+/−, and control nontransgenic (nonTg) littermates at (A–C) 6 months of age, (D–F) 12 months of age, and (G–I) 20 months of age. (J–L) Myelin debris spots per square millimeter of corpus callosum were quantified at 6 months of age (J; n = 5–10 mice), 12 months of age (K; n = 4–7 mice), and 20 months of age (L; n = 5–9 mice). Significance was determined by 1-way analysis of variance followed by Bonferroni post hoc test (**p < 0.01, ***p < 0.001). Scale bar = 140 μm. ns = nonsignificant.
FIGURE 6.
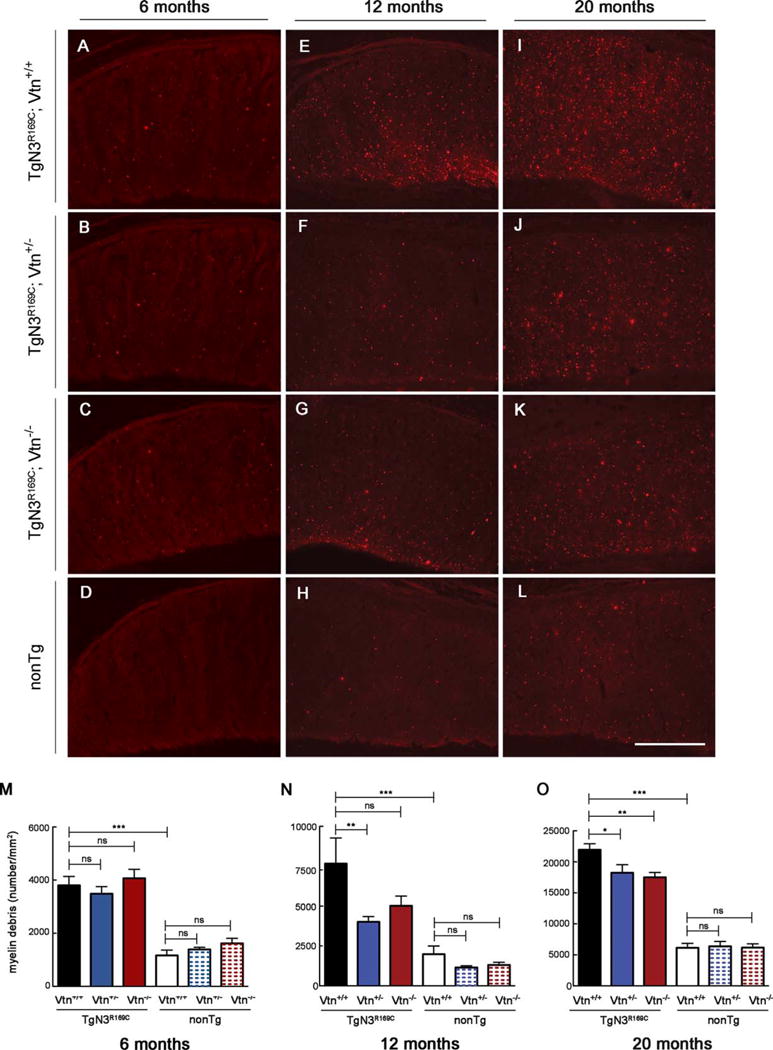
Genetic reduction of vitronectin decreases the number of white matter lesions in TgNotch3R169C mice. Representative images show immunostaining for myelin basic protein (SMI94 antibody) in the corpus callosum (genu) from TgN3R169C;Vtn+/+, TgN3R169C;Vtn+/−, TgN3R169C;Vtn−/−, and control nontransgenic (nonTg) littermates at (A–D) 6 months of age, (E–H) 12 months of age, and (I–L) 20 months of age. (M–O) Quantification of myelin debris spots per square millimeter of corpus callosum shows comparable amount of debris in TgN3R169C;Vtn+/+, TgN3R169C;Vtn+/−, and TgN3R169C;Vtn−/− at 6 months (M; n = 5–9 mice) but significantly less debris in TgN3R169C;Vtn+/− mice and a trend toward less debris in TgN3R169C;Vtn−/− mice compared with age-matched TgN3R169C;Vtn+/+ mice at 12 months (N; n = 6–9 mice). Both TgN3R169C;Vtn+/− and TgN3R169C;Vtn−/− mice have significantly fewer debris spots than age-matched TgN3R169C;Vtn+/+ mice at 20 months of age (O; n = 7–10 mice). Significance was determined by 1-way analysis of variance followed by Bonferroni post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001). Scale bar = 140 μm. ns = nonsignificant.
The above results show that reducing the gene dosage of vitronectin does not rescue the cerebrovascular dysfunction at 6 months of age but ameliorates the white matter lesions at 12 months of age. To further investigate the possibility that amelioration of white matter lesions occurs without restoration of CBF deficits, we assessed CBF responses at 12 months of age in TgNotch3R169C mice with normal (TgNotch3R169C;Vtn+/+) or half reduced expression (TgNotch3R169C;Vtn+/−) of vitronectin and appropriate controls. We found that the increases in CBF evoked by whisker stimulation, endothelium-dependent vasodilators, and adenosine as well as the lower limit of the autoregulation were similarly impaired in TgNotch3R169C;Vtn+/+ and TgNotch3R169C;Vtn+/− mice (data not shown). Collectively, these data support the conclusion that, in TgNotch3R169C mice, white matter pathology can develop in the presence of normal cerebrovascular reactivity and that phenotypic rescue of these lesions can occur without rescue of cerebrovascular deficits.
Vascular Notch3ECD Deposition is Unaffected by Genetic Reduction of Timp3 or Vitronectin
Our working hypothesis is that multimerization and extracellular deposition of mutant Notch3ECD leads to accumulation of TIMP3 and vitronectin. The present data suggest that both of these proteins exert biological effects. It is also possible that the mutant Notch3ECD is directly involved in the pathology, and that TIMP3 and/or vitronectin are involved in the development of Notch3ECD deposits. To test this possibility, we used quantitative immunofluorescence and transmission electron microscopy to assess the burden of Notch3ECD and GOM deposits in the brain vessels of double mutants. We analyzed TgNotch3R169C mice with normal or reduced expression of Timp3 at 6 months of age as we did in the rescue experiments (see Figs 2 and 3). We conducted similar analyses in TgNotch3R169C mice with normal, reduced, or no expression of vitronectin at 12 months of age (see Fig 6). The number of Notch3ECD deposits in arteries and capillaries was unaffected by reduction of Timp3 (Fig 7). From 6 to 12 months of age, the number of Notch3ECD deposits in brain arteries and capillaries from TgNotch3R169C mice on a wild-type background increased by 40 to 50%. Importantly, genetic reduction or elimination of vitronectin did not affect the number of Notch3ECD deposits in brain vessels. Similarly, the number of GOM deposits in brain arteries was unaffected by genetic reduction of Timp3 or vitronectin (Fig 8). Collectively, these data indicate that the protection provided by decreasing levels of Timp3 or vitronectin in TgNotch3R169C mice does not involve reduced deposition of Notch3ECD, supporting the idea that Notch3ECD deposition is upstream of the accumulation of TIMP3 and vitronectin.
FIGURE 7.
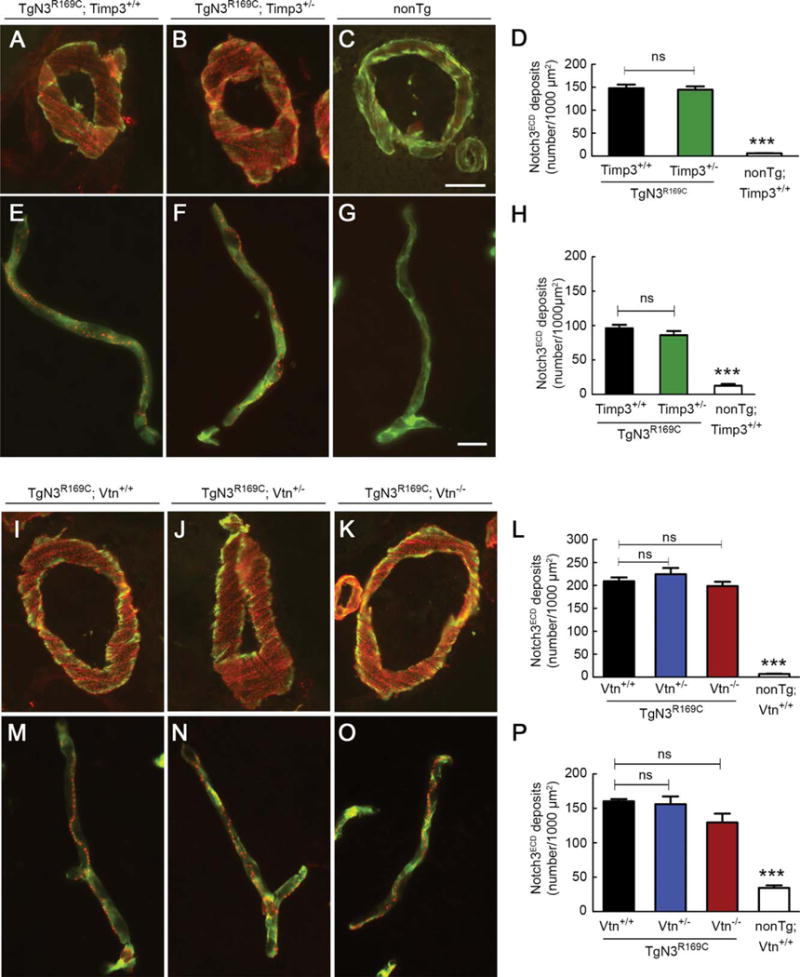
Notch3ECD deposition in brain vessels is unaffected by genetic reduction of Timp3 or vitronectin. Notch3ECD deposits were quantified in brain arteries and capillaries from mice with the indicated genotypes at 6 months of age (A–H) and 12 months of age (I–P). Representative images show brain arteries coimmunostained with anti-Notch3ECD antibody (red) and fluorescein isothiocyanate–conjugated anti–smooth muscle a-actin antibody (green; A–C, I–K) and brain capillaries coimmunostained with anti-Notch3ECD antibody (red) and antiperlecan antibody (green; E–G, M–0). Quantification of Notch3ECD deposit numbers per 1,000 μm2 in brain arteries (D, L) and brain capillaries (H, P) shows a significantly higher number of deposits in transgenic mice compared to nontransgenic (nonTg) mice, but a comparable number of deposits in TgN3R169C mice with normal or reduced expression of Timp3 (D, H) and normal, reduced, or no expression of vitronectin (L, P; n = 4–5 mice per genotype). Significance was determined by 1-way analysis of variance followed by Bonferroni post hoc test (***p < 0.001). Scale bars = 10 μm (A–C, I–K) and 15 μm (E–G, M–0). ns = nonsignificant.
FIGURE 8.
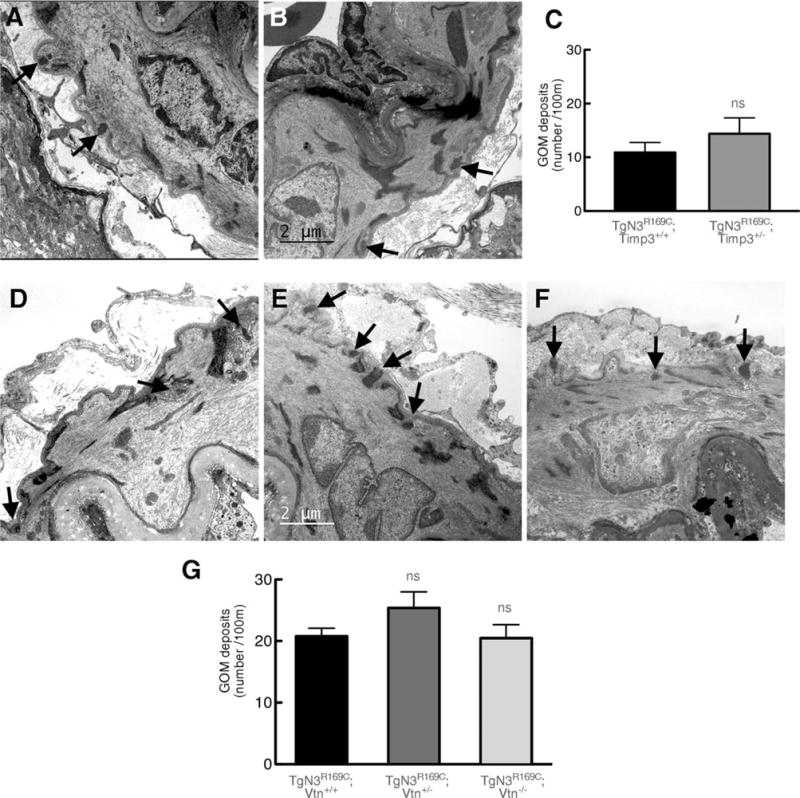
Genetic reduction of Timp3 or vitronectin does not affect the number of granular osmiophilic material (GOM) deposits. Shown are representative electron micrographs of segments of middle cerebral artery from TgN3R169C;Timp3+/+ (A) and TgN3R169C;Timp3+/− mice (B) aged 6 months, and from TgN3R169C;Vtn+/+ (D), TgN3R169C;Vtn+/− (E), and TgN3R169C;Vtn−/−(F) mice aged 12 months. Arrows point to GOM deposits. (C, G) Quantification shows that the number of GOM deposits is comparable between TgN3R169C;Timp3+/+ and TgN3R169C;Timp3+/− mice (C) and between TgN3R169C;Vtn+/+, TgN3R169C; Vtn+/− and TgN3R169C;Vtn−/− mice (G; n = 4 mice per genotype). Significance was determined by Student t test (C) or 1-way analysis of variance followed by Tukey post hoc test (G). Scale bar = 2 μm. ns = nonsignificant.
Discussion
Recently, we proposed the possibility that excess levels of extracellular matrix proteins, like TIMP3 or vitronectin, which are abnormally recruited in Notch3ECD-containing deposits, might contribute to the pathogenesis of CADASIL.6 In the present study, we tested this hypothesis using genetic-interaction approaches in a well-established preclinical mouse model of CADASIL. Our results provide genetic evidence that excess levels of TIMP3 and vitronectin contribute to 2 important pathophysiological events— compromised cerebrovascular reactivity and white matter lesions—and that Notch3ECD deposition is at the top of the pathogenic cascade. Although we cannot exclude interactive effects of increased TIMP3 and vitronectin, our results support the concept of divergent influences on early deficits in the control of CBF (TIMP3) and later increases in white matter lesions (vitronectin). The pathological weight of accumulated TIMP3 and vitronectin on cognitive decline and stroke remains to be determined; a major obstacle is the lack of a suitable model.25
Our work provides proof-of-concept that proteins detected in Notch3ECD-containing deposits play a pathophysiological role in CADASIL. The finding that white matter lesions are only partially rescued by manipulating vitronectin expression level suggests that other proteins might be in play. The protein content of Notch3ECD-containing deposits in brain vessels from mice or patients with CADASIL is as yet only partly understood.6,8,9,26 Thus, our study supports further investigation of these proteins, especially at the early stage of the disease.
An intriguing observation in CADASIL is that, despite the widespread distribution of Notch3ECD deposition in both the cerebral and peripheral vasculature, the clinical manifestations are essentially restricted to the brain.3 Our work suggests the attractive hypothesis that elevated levels of the proteins that contribute to the pathogenesis might be restricted to the cerebrovasculature. Interestingly, our preliminary analysis revealed that vitronectin, which is robustly expressed in the brain capillaries, is almost undetectable in capillaries of the heart or the kidney, organs commonly spared in CADASIL.
We found that reducing the gene dosage of Timp3 or vitronectin did not aggravate disease manifestations, but instead ameliorated them. The simplest interpretation of these results is that excess TIMP3 and vitronectin contribute to the disease through an increase in their biological activity. This interpretation is seemingly at odds with the dominant view that proteins trapped in aggregates lose their biological activity.27 However, there are several possible explanations, at least for TIMP3, for these apparently counterintuitive results. First, our previous biochemical studies revealed that not only insoluble cross-linked TIMP3 species but also soluble TIMP3 species accumulate in the brain vessels of TgNotch3R169C mice and patients with CADASIL, although the exact mechanism by which these soluble species accumulate is not entirely clear. Consistent with the observed accumulation of soluble species, reverse zymography analyses have provided evidence for elevated TIMP3 activity in these vessels.6 Thus, taken together with the mouse genetic results, these biochemical data are consistent with a pathological increase in TIMP3 biological activity. Second, TIMP3 might be sequestrated in Notch3ECD-containing deposits with 1 or more of its substrates. Given that TIMP3 acts as an inhibitor of proteases, a gain in TIMP3 activity in such a scenario could manifest as a loss in the activity of TIMP3 substrates. Third, it has been documented that proteins can dynamically or reversibly associate with aggregates, implying the availability of a pool of unassociated, active proteins.28 An alternative interpretation of our results is that elevated levels of TIMP3 or vitronectin might contribute to the disease through a toxic gain of function. For instance, TIMP3 or vitronectin might be sequestrated away from their normal subcellular location and gain novel biological activities.
Although we cannot formally exclude changes in other cellular components of the neurovascular unit,29 the global vasomotor dysfunction observed in TgNotch3R169C mice suggests a primary alteration in the vasomotor apparatus, namely vascular smooth muscle cells30,31; our data further suggest that excess TIMP3 is a critical contributor to this dysfunction. Several lines of evidence support this interpretation. First, vascular smooth muscle cells are the main source of NOTCH3 in the normal brain, and in patients and mice with CADASIL, and Notch3ECD accumulates at the plasma membrane and in the immediate vicinity of these cells.5,12 Second, myogenic responses of isolated brain arteries are strongly attenuated in TgNotch3R169C mice, and this defect is rescued by reducing expression of Timp3. Third, decreased myogenic responses are expected to decrease vasodilatory reserve and thus impair cerebral vasodilatory responses.24 Accordingly, TgNotch3R169C mice exhibit profoundly reduced vasodilation in response to diverse stimuli; importantly, reducing expression of Timp3 normalizes all these CBF responses and increasing TIMP3 expression reproduces these deficits.
Recent work provides insight into the potential mechanism by which increased TIMP3 activity might impair arterial tone and cerebrovascular function. Specifically, Dabertrand et al demonstrated that the diminished myogenic response in TgNotch3R169C mice is caused by an increase in the number of voltage-gated potassium (KV) channels in smooth muscle cells, and further showed that this defect could be rescued by promoting epidermal growth factor receptor (EGFR)-mediated KV channel endocytosis using the EGFR agonist heparin-binding epidermal growth factor (HB-EGF).23 Accordingly, increased levels of TIMP3, which inhibits metalloproteases, could lead to decreased shedding of HB-EGF,32 and thereby could lead to the suppression of HB-EGF–mediated KV channel endocytosis and thus an increase in KV channels in the plasma membrane.
Our study shows that reducing the levels of vitronectin dramatically attenuated the severity of white matter lesions in TgNotch3R169C model mice. Notably, this phenotypic rescue occurred without overt restoration of evoked CBF responses. These results raise 2 important questions. First, what are the precise mechanisms underlying the development of white matter injury in this CADASIL model? At first blush, our data suggest that dysregulation of CBF does not participate in the development of white matter injury, a finding that would fly in the face of conventional wisdom.33 Nonetheless, our prior and present data provide support for this interpretation. White matter lesions in TgNotch3R169C mice develop as early as 6 months, whereas resting blood flow in the white matter, as measured by quantitative autoradiography, is not significantly reduced until the age of 20 months.12 Moreover, restoration of evoked CBF responses by genetic reduction of Timp3 at 6 months of age is not associated with an attenuation of white matter lesion number. However, we recognize that additional long-term studies are needed to evaluate the CBF deficits and the benefit of reducing Timp3 expression with age. Moreover, it might also be worthwhile to assess resting flow in the hemispheric white matter in “rescued mice” at 20 months of age. It should also be stressed that the TgNotch3R169C mouse model might not recapitulate the full spectrum of white matter lesions encountered in CADASIL. Thus, future studies should seek to investigate other mechanisms that have been proposed to lead to white matter injury in cerebral SVD.33 Among these, alterations in the permeability of the blood–brain barrier within the hemispheric white matter are not supported by our previous structural and functional analyses of the TgNotch3R169C model.12,19 A second important, as yet unresolved, question is how vitronectin contributes to white matter pathology. In the brain, vitronectin is predominantly expressed in the vasculature,6 particularly in the capillaries (data not shown). Interestingly, both microarray analyses of purified vascular cells34 and RNA sequencing of different brain cell populations35 have shown that vitronectin is enriched in brain pericytes, a finding that is highly consistent with our immunohistochemical analysis (data not shown). The contribution of vitronectin, as well as the potential involvement of pericytes, in white matter pathology deserves further investigation.
In summary, our study identifies a new early stage disease mechanism in CADASIL—accumulation of TIMP3 and vitronectin—that provides a first missing link between Notch3ECD deposition and disease manifestations. Our work suggests that the optimal therapeutic strategy may require targeting Notch3ECD proteins, either by reducing their synthesis, by promoting their clearance, or by preventing the subsequent recruitment of these extracellular matrix proteins.
Acknowledgments
This work was supported by grants from the National Research Agency, France (ANR Genopath 2009-RAE09011HSA, ANR Blanc 2010-RPV11011HHA, A.J.), NIH (NIDDK R37DK053832, NHLBI -P01HL095488, NHLBI R01HL121706, M.T.N.), Totman Medical Research Trust (M.T.N.), and Leducq Foundation (Transatlantic Network of Excellence on the Pathogenesis of SVD of the Brain; A.J., M.T.N.). E.C. is a recipient of fellowships from Inserm (Poste Accueil).
We thank M. Monet-Leprêtre and S. Cleophax for excellent technical assistance, C. Diaz and J. P. Rio for providing assistance with electron microscopy, and TAAM-Orleans and Paris Diderot University–site Villemin for animal housing.
Footnotes
Author Contributions
All authors contributed to the design of the study, data analysis, and editing. M.T.N. contributed to drafting the paper. A.J. conceived the study and wrote the manuscript. E.C. and L.G. contributed equally to this work.
Potential Conflicts of Interest
A.J. has a patent “Gene Involved in CADASIL, Method of Diagnosis and Therapeutic Application” licensed to Athena diagnostics, and a patent “Immunological Treatment of CADASIL” pending.
References
- 1.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689–701. doi: 10.1016/S1474-4422(10)70104-6. [DOI] [PubMed] [Google Scholar]
- 2.Joutel A, Corpechot C, Ducros A, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 3.Chabriat H, Joutel A, Dichgans M, et al. Cadasil. Lancet Neurol. 2009;8:643–653. doi: 10.1016/S1474-4422(09)70127-9. [DOI] [PubMed] [Google Scholar]
- 4.Fouillade C, Monet-Leprêtre M, Baron-Menguy C, Joutel A. Notch signalling in smooth muscle cells during development and disease. Cardiovasc Res. 2012;95:138–146. doi: 10.1093/cvr/cvs019. [DOI] [PubMed] [Google Scholar]
- 5.Joutel A, Andreux F, Gaulis S, et al. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest. 2000;105:597–605. doi: 10.1172/JCI8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monet-Leprêtre M, Haddad I, Baron-Menguy C, et al. Abnormal recruitment of extracellular matrix proteins by excess Notch3 ECD: a new pathomechanism in CADASIL. Brain. 2013;136:1830–1845. doi: 10.1093/brain/awt092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamamoto Y, Craggs LJL, Watanabe A, et al. Brain microvascular accumulation and distribution of the NOTCH3 ectodomain and granular osmiophilic material in CADASIL. J Neuropathol Exp Neurol. 2013;72:416–431. doi: 10.1097/NEN.0b013e31829020b5. [DOI] [PubMed] [Google Scholar]
- 8.Kast J, Hanecker P, Beaufort N, et al. Sequestration of latent TGF-β binding protein 1 into CADASIL-related Notch3-ECD deposits. Acta Neuropathol Commun. 2014;2:96. doi: 10.1186/s40478-014-0096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joutel A, Haddad I, Ratelade J, Nelson MT. Perturbations of the cerebrovascular matrisome: a convergent mechanism in small vessel disease of the brain? J Cereb Blood Flow Metab. 2015 Apr 8; doi: 10.1038/jcbfm.2015.62. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Preissner KT, Reuning U. Vitronectin in vascular context: facets of a multitalented matricellular protein. Semin Thromb Hemost. 2011;37:408–424. doi: 10.1055/s-0031-1276590. [DOI] [PubMed] [Google Scholar]
- 12.Joutel A, Monet-Lepretre M, Gosele C, et al. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest. 2010;120:433–445. doi: 10.1172/JCI39733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janssen A, Hoellenriegel J, Fogarasi M, et al. Abnormal vessel formation in the choroid of mice lacking tissue inhibitor of metalloprotease-3. Invest Ophthalmol Vis Sci. 2008;49:2812–2822. doi: 10.1167/iovs.07-1444. [DOI] [PubMed] [Google Scholar]
- 14.Zheng X, Saunders TL, Camper SA, et al. Vitronectin is not essential for normal mammalian development and fertility. Proc Natl Acad Sci U S A. 1995;92:12426–12430. doi: 10.1073/pnas.92.26.12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cognat E, Baron-Menguy C, Domenga-Denier V, et al. Archetypal Arg169Cys mutation in NOTCH3 does not drive the pathogenesis in cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy via a loss-of-function mechanism. Stroke. 2014;45:842–849. doi: 10.1161/STROKEAHA.113.003339. [DOI] [PubMed] [Google Scholar]
- 16.Capone C, Faraco G, Peterson JR, et al. Central cardiovascular circuits contribute to the neurovascular dysfunction in angiotensin II hypertension. J Neurosci. 2012;32:4878–4886. doi: 10.1523/JNEUROSCI.6262-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Girouard H, Park L, Anrather J, et al. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2-derived radicals. Arterioscler Thromb Vasc Biol. 2006;26:826–832. doi: 10.1161/01.ATV.0000205849.22807.6e. [DOI] [PubMed] [Google Scholar]
- 18.Niwa K, Kazama K, Younkin L, et al. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol. 2002;283:H315–H323. doi: 10.1152/ajpheart.00022.2002. [DOI] [PubMed] [Google Scholar]
- 19.Cognat E, Cleophax S, Domenga-Denier V, Joutel A. Early white matter changes in CADASIL: evidence of segmental intramyelinic oedema in a pre-clinical mouse model. Acta Neuropathol Commun. 2014;2:49. doi: 10.1186/2051-5960-2-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basu R, Fan D, Kandalam V, et al. Loss of Timp3 gene leads to abdominal aortic aneurysm formation in response to angiotensin II. J Biol Chem. 2012;287:44083–44096. doi: 10.1074/jbc.M112.425652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basu R, Lee J, Morton JS, et al. TIMP3 is the primary TIMP to regulate agonist-induced vascular remodelling and hypertension. Cardiovasc Res. 2013;98:360–371. doi: 10.1093/cvr/cvt067. [DOI] [PubMed] [Google Scholar]
- 22.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 23.Dabertrand F, Krøigaard C, Bonev AD, et al. Potassium channelopathy-like defect underlies early-stage cerebrovascular dysfunction in a genetic model of small vessel disease. Proc Natl Acad Sci U S A. 2015;112:E796–E805. doi: 10.1073/pnas.1420765112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cipolla MJ. The cerebral circulation. 1st. San Rafael, CA: Morgan & Claypool Life Sciences; 2009. [PubMed] [Google Scholar]
- 25.Joutel A. Pathogenesis of CADASIL: transgenic and knock-out mice to probe function and dysfunction of the mutated gene, Notch3, in the cerebrovasculature. Bioessays. 2011;33:73–80. doi: 10.1002/bies.201000093. [DOI] [PubMed] [Google Scholar]
- 26.Arboleda-Velasquez JF, Manent J, Lee JH, et al. Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small-vessel disease. Proc Natl Acad Sci U S A. 2011;108:E128–E135. doi: 10.1073/pnas.1101964108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olzscha H, Schermann SM, Woerner AC, et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- 28.Kim S, Nollen EAA, Kitagawa K, et al. Polyglutamine protein aggregates are dynamic. Nat Cell Biol. 2002;4:826–831. doi: 10.1038/ncb863. [DOI] [PubMed] [Google Scholar]
- 29.Hall CN, Reynell C, Gesslein B, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunn KM, Nelson MT. Neurovascular signaling in the brain and the pathological consequences of hypertension. Am J Physiol Heart Circ Physiol. 2014;306:H1–H14. doi: 10.1152/ajpheart.00364.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hill RA, Tong L, Yuan P, et al. Regional blood flow in the normal and ischemic brain is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron. 2015;87:95–110. doi: 10.1016/j.neuron.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 33.Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daneman R, Zhou L, Agalliu D, et al. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PloS One. 2010;5:e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Chen K, Sloan SA, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]