

Abstract
Background
Intravenous immunoglobulin (IVIG) is a polyspecific pooled immunoglobulin G preparation and one of the commonly used therapeutics for autoimmune diseases including those of neurological origin. A recent report in murine model proposed that IVIG expands regulatory T (Treg) cells via induction of interleukin 33 (IL-33). However, translational insight on these observations is lacking.
Methods
Ten newly diagnosed Guillain-Barré syndrome (GBS) patients were treated with IVIG at the rate of 0.4 g/kg for three to five consecutive days. Clinical evaluation for muscular weakness was performed by Medical Research Council (MRC) and modified Rankin scoring (MRS) system. Heparinized blood samples were collected before and 1, 2, and 4–5 weeks post-IVIG therapy. Peripheral blood mononuclear cells were stained for surface CD4 and intracellular Foxp3, IFN-γ, and tumor necrosis factor alpha (TNF-α) and were analyzed by flow cytometry. IL-33 and prostaglandin E2 in the plasma were measured by ELISA.
Results
The fold changes in plasma IL-33 at week 1 showed no correlation with the MRC and MRS scores at weeks 1, 2, and ≥4 post-IVIG therapy. Clinical recovery following IVIG therapy appears to be associated with Treg cell response. Contrary to murine study, there was no association between the fold changes in IL-33 at week 1 and Treg cell frequency at weeks 1, 2, and ≥4 post-IVIG therapy. Treg cell-mediated clinical response to IVIG therapy in GBS patients was associated with reciprocal regulation of effector T cells-expressing TNF-α.
Conclusion
Treg cell expansion by IVIG in patients with autoimmune diseases lack correlation with IL-33. Treg cell frequency, but not plasma IL-33 levels, represents potential immunological biomarker to predict clinical response to IVIG therapy.
Keywords: IVIG, Guillain-Barré syndrome, Treg cells, IL-33, TNF-α, Clinical response, Regulatory T cells
Findings
Introduction
Intravenous immunoglobulin (IVIG) is a polyspecific pooled immunoglobulin G preparation and one of the commonly used immunotherapeutics for the treatment of autoimmune diseases including those of neurological origin [1–4]. Various reports demonstrate that IVIG exerts beneficial effects through several mutually non-exclusive mechanisms including expansion of CD4+Foxp3+ regulatory T cells (Treg cells) [5–10]. Some of these mechanisms are also being explored as biomarkers of IVIG response [11, 12]. Treg cells play an indispensable role in the maintenance of immune tolerance and suppress unnecessary deleterious immune responses, such as autoimmunity [13]. IVIG is shown to expand Treg cells and enhance their functions in experimental animal models and patients with autoimmune diseases [9, 14–17]. Recent studies suggested that IVIG-induced expansion of Treg cells requires the prime role of dendritic cells (DCs), and involves interaction of IVIG with C-type lectin receptors, such as dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) and dendritic cell immunoreceptor (DCIR), and is mediated through prostaglandin (PG) E2 secreted by DCs [15, 18]. Interestingly, based on the murine models of autoimmune diseases, i.e., antibody-mediated K/BxN arthritis and T cell-mediated experimental autoimmune encephalitis, a recent report proposed that induction of interleukin 33 (IL-33) in macrophages by IVIG through interaction of α2,6-sialylated crystallizable fraction (Fc) with SIGN-R1 or human DC-SIGN is essential for the expansion of Treg cells [19, 20]. However, translational insight on these observations in patients with autoimmune diseases is lacking.
Guillain-Barré syndrome (GBS) is an autoimmune disease of neurological origin affecting the peripheral nerves [21]. GBS is believed to be caused by effector T cells and autoantibodies to myelin components [22, 23]. Furthermore, GBS patients display reduced frequency of Treg cells that are required for the prevention of autoimmunity [24]. Amelioration of experimental autoimmune neuritis, an experimental model of GBS, was associated with up-regulation of Treg cells [25]. Currently IVIG is used as a first-line therapy for GBS [26]. Of note, sialylated IVIG has been shown to suppress anti-ganglioside antibody-mediated nerve injury in experimental GBS model and was associated with increased expression of IL-33 mRNA [27]. Sialylated IVIG also inhibited anti-ganglioside antibody-mediated complement deposition in vitro [28]. In view of these reports, we investigated the role of IL-33 towards clinical response to IVIG treatment and Treg cell expansion in GBS patients. We found that kinetics of peripheral Treg cell expansion and improvement of clinical symptoms in GBS patients following IVIG therapy lack correlation with the level of induction of IL-33 in the blood.
Materials and methods
Clinical evaluation and sample collection
A total of ten treatment-naïve patients (mean age of 68 years) exhibiting neurological signs of GBS were enrolled based on the diagnostic criteria (Table 1). The study was approved by relevant ethical committee (84-2012-08, CHU Limoges) and consent was obtained from the enrolled patients. The patients received IVIG at the rate of 0.4 g/kg for three (three patients) or five (seven patients) consecutive days. Clinical evaluation for the muscular weakness using Medical Research Council (MRC) and modified Rankin score (MRS) grading systems and collection of heparinized blood samples were done before and 1, 2, and 4–5 weeks after the initiation of IVIG treatment (post-IVIG) [29]. For all the patients, the MRC is a sum score of ten muscle groups tested bilaterally (min = 0, max = 100).
Table 1.
Demographics and clinical information of GBS patients
| Sl no. | Age (years) | Sex | MRC score (pre-IVIG) | MRS (pre-IVIG) |
|---|---|---|---|---|
| P1 | 61 | Male | 30 | 5 |
| P2 | 68 | Female | 80 | 4 |
| P3 | 57 | Male | 85 | 1 |
| P4 | 70 | Female | 68 | 4 |
| P5 | 82 | Male | 70 | 3 |
| P6 | 66 | Female | 78 | 2 |
| P7 | 85 | Male | 54 | 4 |
| P8 | 60 | Female | 70 | 4 |
| P9 | 68 | Male | 54 | 5 |
| P10 | 59 | Female | 13 | 4 |
Flow cytometry and ELISA
Plasma and peripheral blood mononuclear cells (PBMCs) were separated from the heparinized blood samples. PBMCs were stained for surface CD4 and intracellular Foxp3, and also interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) following stimulation with phorbol myristate acetate (50 ng/mL) and ionomycin (500 ng/mL, Sigma-Aldrich, France), along with GolgiStop (BD Biosciences, France), for 4 h. The intracellular staining was performed using Foxp3 staining kit (eBioscience, France) as per the manufacturer’s instructions. Stained cells were acquired on LSR II (BD Biosciences) flow cytometer and data was analyzed using BD FACS DIVA (BD Biosciences) and FlowJo (FlowJo LLC, USA) softwares. IL-33 was measured in the plasma by ELISA (R&D systems, France). The amount of PGE2 in the plasma samples was estimated by ELISA as described previously [15].
Statistical analysis
One-way ANOVA “Kruskal-Wallis test” was used for the analysis of IL-33 in the plasma. Pearson correlation (r) was used to determine the association between MRC or MRS scores and plasma levels of IL-33 and PGE2 and the frequency of Treg cells in the blood of GBS patients at different time points following IVIG treatment. Same assay was also used for determining the association between frequency of Treg cells and CD4+IFN-γ+ and CD4+TNF-α+ T cells.
Results and discussion
The overall rate of new incidence of GBS is 0.6–4.0/year/100,000 people. We limited our investigation only to newly diagnosed and treatment-naïve GBS patients to avoid possible modulatory effects of past IVIG therapy or other therapeutic agents on the Treg cells and other immune parameters. This explicates the difficulties in enrolling treatment-naïve patients for the investigation. As kinetics of IL-33 induction in the plasma of patients with autoimmune diseases following IVIG therapy was not explored earlier, we first analyzed plasma levels of IL-33 in ten GBS patients at various time points (pre-IVIG and weeks 1, 2, and ≥4 after initiation of IVIG therapy). Patients had variable pre-IVIG plasma IL-33 levels that significantly increased at week 1 in all the patients. This data thus confirms our recent observation in IVIG-treated inflammatory myopathy and anti-neutrophil cytoplasmic antibody-associated vasculitis patients [30]. At subsequent time points, IL-33 levels in the plasma of IVIG-treated GBS patients declined progressively but remain significantly higher than the pre-IVIG levels (Fig. 1a) indicating sustained effect of IVIG beyond its half-life on plasma IL-33 levels.
Fig. 1.
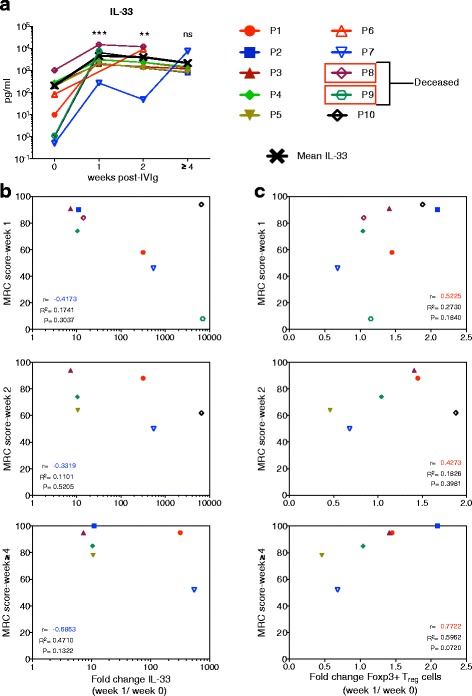
Correlation between changes in the plasma IL-33 and regulatory T (Treg) cell frequencies and clinical response to IVIG therapy in GBS patients. a Temporal changes in the amount of IL-33 (pg/ml) in the plasma of ten Guillain-Barré syndrome patients before (week 0) and weeks 1, 2, and ≥4 following initiation of IVIG therapy. Statistical significance as determined by one-way ANOVA. ***P < 0.001, **P < 0.01, ns = not significant. b Correlation between the clinical response (MRC score) at different time points (weeks 1, 2, and ≥4) after initiation of IVIG therapy and the fold changes in plasma IL-33 at week 1. c Correlation between the clinical response (MRC score) at different time points (weeks 1, 2, and ≥4) after initiation of IVIG therapy and the fold changes in circulating Treg cells at week 1. Each symbol represents individual patient. r = Pearson correlation. p = statistical significance
In order to explore the role of IL-33 in IVIG-mediated therapeutic effects, we analyzed whether IL-33 induction correlates with clinical response to IVIG therapy and Treg cell expansion [29]. Of note, the fold changes in plasma IL-33 level at week 1 showed no correlation with the MRC scores at weeks 1, 2, and ≥4 post-IVIG therapy (Fig. 1b). Also, the r values are consistently negative at all the time points. On the other hand, Treg cell response following IVIG therapy appears to be linked with clinical recovery from GBS and is consistent with the previous observations on essential role of Treg cells in IVIG-induced protection in mouse models [18, 31]. Yet, we observed no correlation between the fold-changes in Treg cells at week 1 and clinical score at weeks 1, 2, and ≥4 (Fig. 1c). These results suggest that Treg cells, but not IL-33, might predict clinical response to IVIG therapy.
Similar observations are also made with MRS parameters. We found that the fold changes in IL-33 level at week 1 displayed no correlation with MRS scores at weeks 1, 2, and ≥4 following initiation of IVIG therapy indicating that clinical improvement as analyzed by MRS is also not associated with changes in IL-33 levels in the blood. As in the case of MRC scores, changes in Treg cells were suggestive of clinical response to IVIG therapy by MRS parameters as well. However, the fold changes in Treg cells at week 1 and MRS scores at weeks 1, 2, and ≥4 post-IVIG therapy did not correlate (Table 2).
Table 2.
Correlation between MRS scores at different time points (weeks 1, 2, and ≥4) after initiation of IVIG therapy and the immunological parameters such as IL-33, Treg cells, and PGE2 at week 1. The values in the parenthesis denote statistical significance
|
r values (p values) |
MRS (week post-IVIG) | |||
|---|---|---|---|---|
| Week | 1 | 2 | ≥4 | |
| IL-33 | 1 | 0.57 (0.14) | −0.60 (0.20) | 0.71 (0.42) |
| Treg cell | 1 | −0.37 (0.3601) | −0.61 (0.1067) | −0.47 (0.0684) |
| PGE2 | 1 | −0.16 (0.6954) | −0.29 (0.4361) | −0.07 (0.8936) |
As data from the recent mouse study suggested that Treg cell expansion by IVIG is dependent on IL-33 [20], we analyzed correlation between IL-33 and Treg cells in these patients at various time points. Contrary to Fiebiger et al., we found no consistent association between the fold changes in IL-33 level at week 1 and Treg cell frequency at weeks 1, 2, and ≥4 after initiation of IVIG therapy (Fig. 2a), implying that Treg cell expansion by IVIG in patients with autoimmune diseases is not related to levels of IL-33 in the plasma.
Fig. 2.
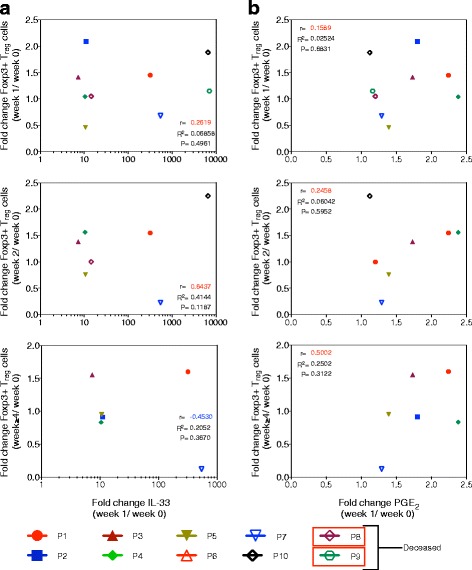
Correlation between changes in plasma IL-33 and PGE2 and IVIG-mediated circulating Treg cell expansion in GBS patients. a Correlation between the fold changes in circulating Treg cells at different time points (weeks 1, 2, and ≥4) after initiation of IVIG therapy and the fold changes in plasma IL-33 level at week 1. b Correlation between the fold changes in circulating Treg cells at different time points (weeks 1, 2, and ≥4) after initiation of IVIG therapy and the fold changes in plasma PGE2 level at week 1. Each symbol represents individual patient. r = Pearson correlation. p = statistical significance
Expansion of Treg cells in the periphery is mediated by the signals derived from professional antigen presenting cells such as DCs. Unlike other cytokines, IL-33 can be released into the microenvironment only upon injury to the cells and acts as alarmin to signal tissue damage to the immune system [32]. Our recent report shows that DC-SIGN-positive human innate cells derived either from the peripheral blood or from the spleen do not release IL-33 upon IVIG exposure [30]. Therefore, unlike murine models [19, 20], it is likely that damaged non-immune cells like endothelial cells or epithelial cells might have contributed to increased levels of IL-33 observed in the plasma of patients following IVIG therapy. Despite the lack of IL-33 induction, “IVIG-educated” DC-SIGN-positive human DCs induced Treg cell expansion, a process mediated via cyclooxygenase-2-dependent PGE2 and independent of Fc-fragments of IVIG [15]. Notably, there was a significant increase in the plasma levels of PGE2 in IVIG-treated GBS patients [33]. In the current study, although plasma PGE2 levels are enhanced in IVIG-treated patients, we found no correlation between the fold changes in PGE2 level at week 1 and Treg cell frequencies at week 1, 2, and ≥4 post-IVIG therapy (Fig. 2b). Similarly, fold changes in PGE2 level at week 1 did not correlate with MRC score at weeks 1, 2, and ≥4 post-IVIG therapy (Fig. 3). The reason for the non-significant correlation of values might rests in the low number of patients. A recent report shows that plasma PGE2 levels correlate with the prevention of IVIG resistance in Kawasaki disease [34]. Taken together, these results are suggestive of a role for PGE2-mediated expansion of Treg cells in the clinical recovery of patients following IVIG therapy.
Fig. 3.
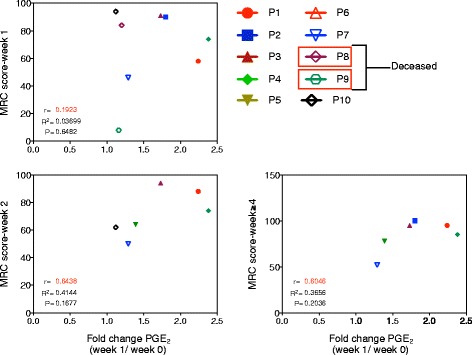
Clinical response to IVIG therapy and its correlation with plasma PGE2. Correlation between the clinical response (MRC score) at different time points (weeks 1, 2, and ≥4) after initiation of IVIG therapy and the fold changes in plasma PGE2 level at week 1. Each symbol represents individual patient. r = Pearson correlation. p = statistical significance
In the previous report [30], we analyzed IL-33 in the plasma of patients with rheumatic disorders as early as 48–72 h following initiation of IVIG therapy. With the exception of two, the remaining 14 patients had IL-33 levels below 500 pg/ml of plasma following IVIG therapy. Although the underlying pathologies in that report and current study are different, we found that at 1 week following initiation of IVIG therapy, IL-33 level in the plasma was more than 1000 pg/ml in nine GBS patients. Furthermore, in majority of the patients (seven), IVIG was given daily for five consecutive days. Therefore, lack of consistent association between the fold changes in IL-33 level and Treg cell frequency following IVIG therapy might not be due to loss of IL-33 subsequent to proteolytic cleavage and binding to ST2 receptor. Also, IL-33 levels observed at first week are maintained in majority of the patients even after 4 weeks post-IVIG. Whether polymorphisms in either ST2 receptor, IL-1RL1, and/or IL-33 itself, skew the plasma kinetics and/or downstream effects of IL-33 remains to be determined [35, 36].
Current literature suggests that IVIG can induce Treg cell expansion by several mutually non-exclusive mechanisms [9, 14, 16, 17, 31] that may vary among the pathologies and even among the patients with same underlying pathology. This might explain the lack of significant correlation between PGE2 and Treg cells. As we restricted our study only to treatment-naïve GBS patients, difficulty to get large number of patients provides an alternative explanation for the lack of significant positive correlation between changes in the IL-33 and PGE2, and Treg cells. Role of IL-33 in the microenvironment towards DC-mediated human Treg expansion requires further exploration.
In addition to expansion of Treg cells, IVIG is also known to enhance their functions and suppress effector T cell proliferation and production of cytokines such as IFN-γ and TNF-α [16, 31]. Interestingly, in IVIG-treated GBS patients, there was a significant negative correlation between the fold changes in Treg cells at week 1 and T cells expressing TNF-α at week 1 post-IVIG therapy. However, such consistent association was not observed with T cells expressing IFN-γ (Fig. 4a, b). Thus, Treg cell-mediated clinical response to IVIG therapy is associated with reciprocal regulation of effector T cells in GBS patients.
Fig. 4.
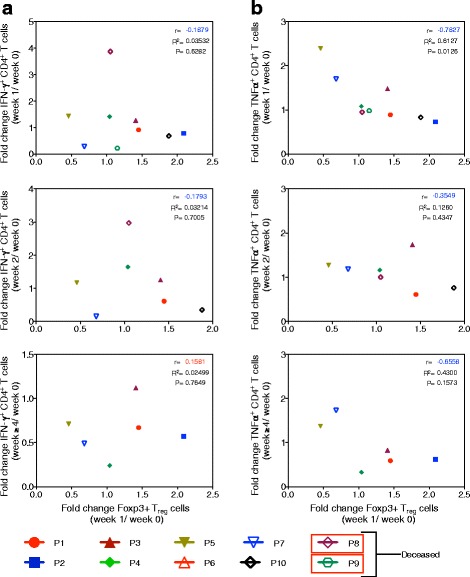
Treg cell response following IVIG therapy in GBS patients correlates negatively with effector T cells. a Correlation between the fold changes in circulating Treg cells at week 1 after initiation of IVIG therapy and the fold changes in IFN-γ+ CD4+ T cells at weeks 1, 2, and ≥4. b Correlation between the fold changes in circulating Treg cells at week 1 following initiation of IVIG therapy and the fold changes in TNF-α+ CD4+ T cells at weeks 1, 2, and ≥4. Each symbol represents individual patient. r = Pearson correlation. p = statistical significance
Conclusions
Because of its role as promoter of type 2 immune responses and regulator of innate and adaptive immune responses, IL-33 has been explored as therapeutic option in pre-clinical models of Alzheimer’s disease [37], stroke [38], cerebral malaria [39], and transplantation [40]. In addition, IL-33 was also reported to be essential to attenuate viral-induced encephalitis [41]. In line with these reports, enhanced expression of IL-33 in mice was suggested to mediate Treg cell expansion and protection by IVIG [19, 20, 27]. Translation of these results to human is complicated and requires thorough investigation. Indeed, we found that up-regulation of plasma IL-33 does not correlate with the clinical response to IVIG therapy and expansion of Treg cells in GBS patients. Treg cells on the other hand are negatively correlated to effector T cells expressing TNF-α. Thus, the mechanism of IL-33 induction by IVIG and its role in Treg cell expansion observed in mouse models might not be applicable to human. Small size of the patients’ cohort represents limitation of this study. Nevertheless, based on these findings, Treg cell frequency, but not plasma IL-33 levels, represents potential immunological biomarker to predict clinical response to IVIG therapy.
Acknowledgements
We thank Ms. A Karnam for the support.
Funding
Supported by Institut National de la Santé et de la Recherche Médicale, Université Pierre et Marie Curie, Université Paris Descartes (SVK and JB), Centre National de la Recherche Scientifique (SVK), European Community’s Seventh Framework Programme [FP7/2007-2013] under grant agreement 260338 ALLFUN (JB), and fellowship from Journées de Neurologie de Langue Française (MR). KNB is a J. C. Bose National fellow of Department of Science and Technology, New Delhi, India.
The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article as figures.
Authors’ contributions
MSM, ES-V, MD, PP, VKS, VS, MR, and JT performed research. FB, J-MV, and LM contributed analytical tools and evaluated the clinical scores. MSM, KNB, SVK, and JB analyzed the data. MSM and JB designed the research and wrote the paper. All authors read and approved the final manuscript.
Competing interests
This work is partially supported by financial support from CSL Behring, France.
Consent for publication
Not applicable
Ethics approval and consent to participate
The study was approved by relevant ethical committee (84-2012-08, CHU Limoges), and the consent was obtained from the enrolled patients.
Abbreviations
- DCIR
Dendritic cell immunoreceptor
- DCs
Dendritic cells
- DC-SIGN
Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin
- GBS
Guillain-Barré syndrome
- IFN-γ
Interferon gamma
- IL-33
Interleukin 33
- IVIG
Intravenous immunoglobulin
- MRC score
Medical research council score
- MRS
Modified Rankin score
- PGE2
Prostaglandin E2
- TNF-α
Tumor necrosis factor alpha
- Treg cells
Foxp3+ regulatory T cells
Contributor Information
Mohan S. Maddur, Email: mohanms.manu@gmail.com
Emmanuel Stephen-Victor, Email: esvkai@gmail.com.
Mrinmoy Das, Email: mdasmicro@gmail.com.
Praveen Prakhar, Email: praveenprakhar@gmail.com.
Varun K. Sharma, Email: varungenetics@gmail.com
Vikas Singh, Email: vikky.sinngh007@gmail.com.
Magalie Rabin, Email: magalie_rabin@hotmail.fr.
Jamma Trinath, Email: karunya.friend@gmail.com.
Kithiganahalli N. Balaji, Email: balaji@mcbl.iisc.ernet.in
Francis Bolgert, Email: francis.bolgert@gmail.com.
Jean-Michel Vallat, Email: jean-michel.vallat@unilim.fr.
Laurent Magy, Email: laurent.magy@unilim.fr.
Srini V. Kaveri, Phone: 00 33 1 44 27 82 03, Email: srini.kaveri@crc.jussieu.fr
Jagadeesh Bayry, Phone: 00 33 1 44 27 82 03, Email: jagadeesh.bayry@crc.jussieu.fr.
References
- 1.Perez EE, Orange JS, Bonilla F, Chinen J, Chinn IK, Dorsey M, El-Gamal Y, Harville TO, Hossny E, Mazer B, et al. Update on the use of immunoglobulin in human disease: a review of evidence. J Allergy Clin Immunol. 2017;139(3S):S1–46. doi: 10.1016/j.jaci.2016.09.023. [DOI] [PubMed] [Google Scholar]
- 2.Buttmann M, Kaveri S, Hartung HP. Polyclonal immunoglobulin G for autoimmune demyelinating nervous system disorders. Trends Pharmacol Sci. 2013;34:445–57. doi: 10.1016/j.tips.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Lunemann JD, Nimmerjahn F, Dalakas MC. Intravenous immunoglobulin in neurology—mode of action and clinical efficacy. Nat Rev Neurol. 2015;11:80–9. doi: 10.1038/nrneurol.2014.253. [DOI] [PubMed] [Google Scholar]
- 4.Bayry J, Hartung HP, Kaveri SV. IVIg for relapsing-remitting multiple sclerosis: promises and uncertainties. Trends Pharmacol Sci. 2015;36(7):419–21. doi: 10.1016/j.tips.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 5.Tha-In T, Bayry J, Metselaar HJ, Kaveri SV, Kwekkeboom J. Modulation of the cellular immune system by intravenous immunoglobulin. Trends Immunol. 2008;29:608–15. doi: 10.1016/j.it.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol. 2013;13:176–89. doi: 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]
- 7.Seite JF, Shoenfeld Y, Youinou P, Hillion S. What is the contents of the magic draft IVIG? Autoimmun Rev. 2008;7:435–9. doi: 10.1016/j.autrev.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 8.Lok KZ, Basta M, Manzanero S, Arumugam TV. Intravenous immunoglobulin (IVIG) dampens neuronal toll-like receptor-mediated responses in ischemia. J Neuroinflammation. 2015;12:73. doi: 10.1186/s12974-015-0294-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maddur MS, Othy S, Hegde P, Vani J, Lacroix-Desmazes S, Bayry J, Kaveri SV. Immunomodulation by intravenous immunoglobulin: role of regulatory T cells. J Clin Immunol. 2010;30(Suppl 1):S4–8. doi: 10.1007/s10875-010-9394-5. [DOI] [PubMed] [Google Scholar]
- 10.Maddur MS, Vani J, Hegde P, Lacroix-Desmazes S, Kaveri SV, Bayry J. Inhibition of differentiation, amplification, and function of human TH17 cells by intravenous immunoglobulin. J Allergy Clin Immunol. 2011;127:823–30. doi: 10.1016/j.jaci.2010.12.1102. [DOI] [PubMed] [Google Scholar]
- 11.Galeotti C, Kaveri SV, Bayry J. Molecular and immunological biomarkers to predict IVIg response. Trends Mol Med. 2015;21:145–7. doi: 10.1016/j.molmed.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Ritter C, Bobylev I, Lehmann HC. Chronic inflammatory demyelinating polyneuropathy (CIDP): change of serum IgG dimer levels during treatment with intravenous immunoglobulins. J Neuroinflammation. 2015;12:148. doi: 10.1186/s12974-015-0361-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 14.Bayry J, Mouthon L, Kaveri SV. Intravenous immunoglobulin expands regulatory T cells in autoimmune rheumatic disease. J Rheumatol. 2012;39:450–1. doi: 10.3899/jrheum.111123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trinath J, Hegde P, Sharma M, Maddur MS, Rabin M, Vallat JM, Magy L, Balaji KN, Kaveri SV, Bayry J. Intravenous immunoglobulin expands regulatory T cells via induction of cyclooxygenase-2-dependent prostaglandin E2 in human dendritic cells. Blood. 2013;122:1419–27. doi: 10.1182/blood-2012-11-468264. [DOI] [PubMed] [Google Scholar]
- 16.Kessel A, Ammuri H, Peri R, Pavlotzky ER, Blank M, Shoenfeld Y, Toubi E. Intravenous immunoglobulin therapy affects T regulatory cells by increasing their suppressive function. J Immunol. 2007;179:5571–5. doi: 10.4049/jimmunol.179.8.5571. [DOI] [PubMed] [Google Scholar]
- 17.Cousens LP, Tassone R, Mazer BD, Ramachandiran V, Scott DW, De Groot AS. Tregitope update: mechanism of action parallels IVIG. Autoimmun Rev. 2013;12:436–43. doi: 10.1016/j.autrev.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 18.Massoud AH, Yona M, Xue D, Chouiali F, Alturaihi H, Ablona A, Mourad W, Piccirillo CA, Mazer BD. Dendritic cell immunoreceptor: a novel receptor for intravenous immunoglobulin mediates induction of regulatory T cells. J Allergy Clin Immunol. 2014;133:853–63. doi: 10.1016/j.jaci.2013.09.029. [DOI] [PubMed] [Google Scholar]
- 19.Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature. 2011;475:110–3. doi: 10.1038/nature10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiebiger BM, Maamary J, Pincetic A, Ravetch JV. Protection in antibody- and T cell-mediated autoimmune diseases by antiinflammatory IgG Fcs requires type II FcRs. Proc Natl Acad Sci U S A. 2015;112:E2385–94. doi: 10.1073/pnas.1505292112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehmann HC, Hartung HP. Plasma exchange and intravenous immunoglobulins: mechanism of action in immune-mediated neuropathies. J Neuroimmunol. 2011;231:61–9. doi: 10.1016/j.jneuroim.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 22.Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barre syndrome. Cochrane Database Syst Rev. 2012;7:CD002063. doi: 10.1002/14651858.CD002063.pub5. [DOI] [PubMed] [Google Scholar]
- 23.van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barre syndrome. Lancet Neurol. 2008;7:939–50. doi: 10.1016/S1474-4422(08)70215-1. [DOI] [PubMed] [Google Scholar]
- 24.Chi LJ, Wang HB, Zhang Y, Wang WZ. Abnormality of circulating CD4 + CD25+ regulatory T cell in patients with Guillain-Barre syndrome. J Neuroimmunol. 2007;192:206–14. doi: 10.1016/j.jneuroim.2007.09.034. [DOI] [PubMed] [Google Scholar]
- 25.Li XL, Dou YC, Liu Y, Shi CW, Cao LL, Zhang XQ, Zhu J, Duan RS. Atorvastatin ameliorates experimental autoimmune neuritis by decreased Th1/Th17 cytokines and up-regulated T regulatory cells. Cell Immunol. 2011;271:455–61. doi: 10.1016/j.cellimm.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 26.Gilardin L, Bayry J, Kaveri SV. Intravenous immunoglobulin as clinical immune-modulating therapy. CMAJ. 2015;187:257–64. doi: 10.1503/cmaj.130375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang G, Massaad CA, Gao T, Pillai L, Bogdanova N, Ghauri S, Sheikh KA. Sialylated intravenous immunoglobulin suppress anti-ganglioside antibody mediated nerve injury. Exp Neurol. 2016;282:49–55. doi: 10.1016/j.expneurol.2016.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sudo M, Yamaguchi Y, Spath PJ, Matsumoto-Morita K, Ong BK, Shahrizaila N, Yuki N. Different IVIG glycoforms affect in vitro inhibition of anti-ganglioside antibody-mediated complement deposition. PLoS One. 2014;9(9):e107772. doi: 10.1371/journal.pone.0107772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maddur MS, Rabin M, Hegde P, Bolgert F, Guy M, Vallat JM, Magy L, Bayry J, Kaveri SV. Intravenous immunoglobulin exerts reciprocal regulation of Th1/Th17 cells and regulatory T cells in Guillain-Barre syndrome patients. Immunol Res. 2014;60:320–9. doi: 10.1007/s12026-014-8580-6. [DOI] [PubMed] [Google Scholar]
- 30.Sharma M, Schoindre Y, Hegde P, Saha C, Maddur MS, Stephen-Victor E, Gilardin L, Lecerf M, Bruneval P, Mouthon L, et al. Intravenous immunoglobulin-induced IL-33 is insufficient to mediate basophil expansion in autoimmune patients. Sci Rep. 2014;4:5672. doi: 10.1038/srep05672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ephrem A, Chamat S, Miquel C, Fisson S, Mouthon L, Caligiuri G, Delignat S, Elluru S, Bayry J, Lacroix-Desmazes S, et al. Expansion of CD4 + CD25+ regulatory T cells by intravenous immunoglobulin: a critical factor in controlling experimental autoimmune encephalomyelitis. Blood. 2008;111:715–22. doi: 10.1182/blood-2007-03-079947. [DOI] [PubMed] [Google Scholar]
- 32.Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16:676–89. doi: 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- 33.Maddur MS, Trinath J, Rabin M, Bolgert F, Guy M, Vallat JM, Magy L, Balaji KN, Kaveri SV, Bayry J. Intravenous immunoglobulin-mediated expansion of regulatory T cells in autoimmune patients is associated with increased prostaglandin E2 levels in the circulation. Cell Mol Immunol. 2015;12:650–2. doi: 10.1038/cmi.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuo HC, Wang CL, Yang KD, Lo MH, Hsieh KS, Li SC, Huang YH. Plasma prostaglandin E2 levels correlated with the prevention of intravenous immunoglobulin resistance and coronary artery lesions formation via CD40L in Kawasaki disease. PLoS One. 2016;11:e0161265. doi: 10.1371/journal.pone.0161265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Savenije OE, Mahachie John JM, Granell R, Kerkhof M, Dijk FN, de Jongste JC, Smit HA, Brunekreef B, Postma DS, Van Steen K, et al. Association of IL33-IL-1 receptor-like 1 (IL1RL1) pathway polymorphisms with wheezing phenotypes and asthma in childhood. J Allergy Clin Immunol. 2014;134:170–7. doi: 10.1016/j.jaci.2013.12.1080. [DOI] [PubMed] [Google Scholar]
- 36.Liu S, Zhou Z, Wang C, Guo M, Chu N, Li C. Associations between interleukin and interleukin receptor gene polymorphisms and risk of gout. Sci Rep. 2015;5:13887. doi: 10.1038/srep13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu AK, Hung KW, Yuen MY, Zhou X, Mak DS, Chan IC, Cheung TH, Zhang B, Fu WY, Liew FY, et al. IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc Natl Acad Sci U S A. 2016;113(19):E2705–13. doi: 10.1073/pnas.1604032113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korhonen P, Kanninen KM, Lehtonen S, Lemarchant S, Puttonen KA, Oksanen M, Dhungana H, Loppi S, Pollari E, Wojciechowski S, et al. Immunomodulation by interleukin-33 is protective in stroke through modulation of inflammation. Brain Behav Immun. 2015;49:322–36. doi: 10.1016/j.bbi.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 39.Besnard AG, Guabiraba R, Niedbala W, Palomo J, Reverchon F, Shaw TN, Couper KN, Ryffel B, Liew FY. IL-33-mediated protection against experimental cerebral malaria is linked to induction of type 2 innate lymphoid cells, M2 macrophages and regulatory T cells. PLoS Pathog. 2015;11(2):e1004607. doi: 10.1371/journal.ppat.1004607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai C, Lu FN, Jin N, Yang B, Gao C, Zhao B, Fu JZ, Hong SF, Liang HT, Chen LH, et al. Recombinant IL-33 prolongs leflunomide-mediated graft survival by reducing IFN-gamma and expanding CD4 + Foxp3+ T cells in concordant heart transplantation. Lab Invest. 2016;96(8):820–9. doi: 10.1038/labinvest.2016.54. [DOI] [PubMed] [Google Scholar]
- 41.Franca RF, Costa RS, Silva JR, Peres RS, Mendonca LR, Colon DF, Alves-Filho JC, Cunha FQ. IL-33 signaling is essential to attenuate viral-induced encephalitis development by downregulating iNOS expression in the central nervous system. J Neuroinflammation. 2016;13(1):159. doi: 10.1186/s12974-016-0628-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article as figures.