

Abstract
Mammalian membranous adenylyl cyclases (mACs) play an important role in transmembrane signaling events in almost every cell and represent an interesting drug target. Forskolin (FS) is an invaluable research tool, activating AC isoforms 1-8. However, there is a paucity of AC isoform-selective FS analogs. Therefore, we examined the effects of FS and six FS derivatives on recombinant ACs 1, 2 and 5, representing members of different mAC families. Correlations of the pharmacological properties of the different AC isoforms revealed pronounced differences between ACs 1, 2 and 5. Additionally, potencies and efficacies of FS derivatives changed for any given AC isoform, depending on the metal ion, Mg2+ or Mn2+. The most striking effects of Mg2+ and Mn2+ on the diterpene profile were observed for AC2 where the large inhibitory effect of BODIPY-FS in presence of Mg2+ was considerably reduced in presence of Mn2+. Sequence alignment and docking experiments confirmed an exceptional position of AC2 compared to ACs 1 and 5 with respect to the structural environment of the catalytic core and cation-dependent diterpene effects. In conclusion, mAC isoforms 1, 2 and 5 exhibit a distinct pharmacological diterpene profile, depending on the divalent cation present. mAC crystal structures and modeling/docking studies provided an explanation for the pharmacological differences between the AC isoforms. Our study constitutes an important step towards the development of isoform-specific diterpenes exhibiting stimulatory or inhibitory effects.
Keywords: Adenylyl cyclase, diterpenes, divalent cations, forskolin, molecular modelling
Graphical abstract
We show that the interactions of diterpenes of mammalian membranous adenylyl cyclases are determined by Mg2+ and Mn2+. Moreover, we report that AC2 exhibits a unique regulation by diterpenes.

Introduction
Mammalian membranous ACs (mACs) are integral plasma membrane proteins that play a central role in transmembrane signaling. Stimulation of G protein-coupled receptors is translated via Gs-proteins to AC-mediated conversion of adenosine 5′-triphosphate (ATP) to the second messenger cyclic adenosine 3′,5′-monophosphate (cAMP) [1, 2]. In mammals, nine membrane-bound AC isoforms (ACs 1-9) are expressed [1, 2]. The various AC isoforms exhibit tissue-specific expression and play a crucial role e.g. in cardiac contractility and the regulation of kidney and brain function [1]. The structure of mACs is characterized by two membrane-spanning domains and two cytosolic loops C1 and C2 [2]. C1 and C2 form the catalytic core and are relatively conserved among the different AC isoforms.
The catalytic core of AC also exhibits two regulatory sites for Mg2+ [3]. Mg2+ interacts with ATP, forming the biologically active chelate cation ATP complex and thus preparing the molecule for the nucleophilic attack by ACs [3, 4]. Since Mn2+ is very similar to Mg2+ in terms of its chemical properties, Mn2+ is often used as divalent cation in in vitro AC studies [5, 6]. However, differences in catalytic and regulatory properties of ACs have been noted depending on whether Mg2+ or Mn2+ served as the metal cofactor [6, 7].
Eight of the nine mammalian membranous AC subtypes (AC1–AC8) are activated by forskolin (FS), a lipophilic diterpene extracted from the roots of the Indian plant Coleus forskohlii [8]. The FS binding site is located at the interface of the catalytic units of ACs [2]. A FS-like molecule was supposedly identified in the cyst fluid of patients suffering from polycystic kidney disease [9]. A formidable challenge in the AC field is the development of isoform-specific FS analogs [10]. AC isoform-specific forskolin analogs would be of great therapeutic interest, e.g. in the treatment of addiction or heart failure and as spasmolytic or antithrombotic agents [11–13].
In previous studies, we characterized the effects of different diterpenes on ACs and investigated the interactions of AC with FS analogs in the presence of Mn2+ [14, 15]. However, the effect of Mg2+ in comparison to Mn2+ on the pharmacological properties of diterpenes is still unknown. This is an important issue since in vivo, Mg2+ is likely to act as cofactor for AC activity. In this paper, we examined the influence of Mg2+ and Mn2+ on the effects of FS and FS analogs on ACs 1, 2 and 5. These three mACs belong to different AC families [2]. We studied FS derivatives missing the OH-group at the 1- or 9-position of the diterpene ring structure, referred to as 1d(eoxy)- or 9d(eoxy)-FS, respectively (Fig. 1). The acetyl group at position 7 is critical for AC activation by interaction with Ser942 [14, 16]. Thus, we examined one derivative without the 7-acetyl-group (7-deacetyl-FS or 7DA-FS) and one where this acetyl-group switched from 7- to the 6-position (6-acetyl-7-deacetyl-FS or 6A7DA-FS). Additionally, we studied FS analogs with bulky substituents like the relatively water-soluble FS analog 7-deacetyl-7-[O-(N-methylpiperazino)-γ-butyryl]-FS (DMB-FS) [17] and boron-dipyrro-methene-FS (BODIPY-FS), which can also be used for fluorescent studies [18].
Fig. 1.
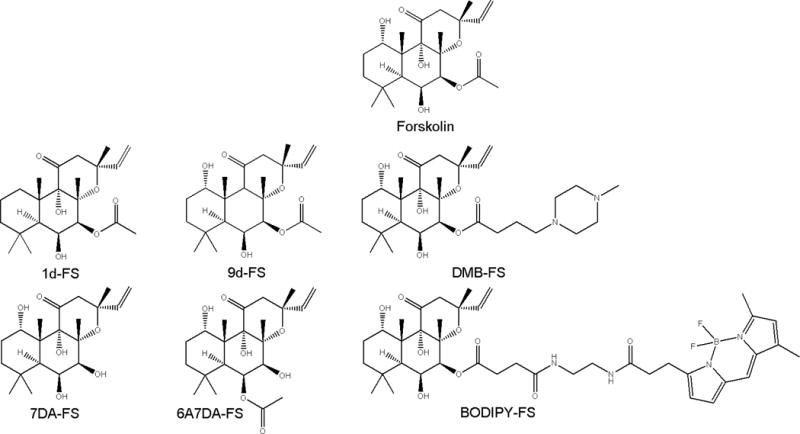
Structures of FS and FS analogs analyzed in this study.
Materials and Methods
Materials
Baculoviruses containing the cDNA for bovine AC1, rat AC2 and canine AC5 were kindly provided by Drs. A. G. Gilman (UT Southwestern Medical Center, Dallas, TX, USA) and R. K. Sunahara (University of Michigan Medical School, Ann Arbor, MI, USA) [19, 20]. Spodoptera frugiperda (Sf9) insect cells were from the American Type Cell Culture Collection (Rockville, MD, USA). FS was purchased from LC Laboratories (Woburn, MA, USA). DMB-FS was from Calbiochem (San Diego, CA, USA). BODIPY-FS was from Molecular Probes (Eugene, OR, USA). All other FS analogs were from Sigma-Aldrich (St. Louis, MO, USA). Stock solutions of FS and FS analogs (10 mM each) were prepared in DMSO and stored at −20°C. Dilutions of FS analogs were prepared in such a way that in all AC assays, a final DMSO concentration of 3% (v/v) was achieved. [α–32P]ATP (3,000 Ci/mmol) was purchased from PerkinElmer (Wellesley, MA, USA). Aluminum oxide (N Super 1) was purchased from MP Biomedicals (Eschwege, Germany). Data were analyzed by linear or non-linear regression using the Prism 5.01 program (GraphPad, San Diego, CA, USA).
Membrane preparation
Sf9 cell membrane preparation was performed as described [21]. For membrane preparation Sf9 cells (3.0 × 106 cells/mL) were infected with correspondent baculovirus encoding different mammalian ACs (1:100 dilutions of high-titer virus stocks) and cultured for 48 hours. Briefly, cells were harvested and cell suspensions were centrifuged for 10 min at 1,000 rpm at 4°C. Pellets were resuspended in 30 mL PBS buffer containing 137 mM NaCl, 2.6 mM KCl, 0.5 mM MgCl2, 0.9 mM CaCl2, 1.5 mM KH2PO4, 0.8 mM Na2HPO4, pH 7.4. After a second centrifugation step of 10 min at 1,000 g and 4°C, the pellets were suspended in 15 mL of lysis buffer (10 mM Tris/HCl, 1 mM EDTA, 0.2 mM phenylmethylsulfonyl fluoride, 10 μg/mL leupeptine and 10 μg/mL benzamide, pH 7.4). Thereafter, homogenization was performed with 20–25 strokes using a Dounce homogenizer. The resultant cell fragment suspension was centrifuged for 5 min at 500 g and 4°C to sediment nuclei. The cell membrane-containing supernatant suspension was centrifuged for 20 min at 30,000 g and 4°C. The supernatant fluid was discarded and cell pellets were again suspended in 20 mL lysis buffer. After a second high-speed centrifugation step, buffer consisting of 75 mM Tris/HCl, 12.5 mM MgCl2, and 1 mM EDTA, pH 7.4 was added to the membrane pellets. Aliquots of 1 mL of membrane suspension were prepared and stored at −80°C. The protein concentration for each membrane preparation was determined by the Lowry method using the Bio-Rad DC protein assay kit (Bio-Rad, Hercules, CA, USA).
AC activity assay
AC activity was determined essentially as described [6] with minor modifications. Briefly, just before experiments, Sf9 membranes with recombinant ACs were washed by adding assay buffer consisting of 50 mM triethanolamine and 1 mM EGTA, pH 7.4 and then centrifuged with 13,000 × g for 10 min at 4°C. Afterwards, membranes were resuspended with syringes in the sequence 21 G and 27 G and diluted in assay buffer to a protein concentration of 1 μg/μL. For the determination of the effects of FS and FS analogs on AC activity, reaction mixtures contained 7 mM Mn2+ or Mg2+, 40 μM ATP, 10 μM GTPγS, 100 μM cAMP, 0.4 mg/mL creatine kinase, 9 mM phosphocreatine, 100 μM IBMX and 0.3 μCi [α–32P]ATP. FS or FS analogs at various concentrations (100 nM – 300 μM) in the presence of 3% (v/v) DMSO were added to the assay tubes. After a pre-incubation time of 2 min at 30°C, reactions were initiated by the addition of 20 μL of membrane suspension. Reactions were conducted for 10 min at 30°C and were terminated by adding 20 μL of 2.2 N HCl. Denatured protein was precipitated by a 2 min centrifugation at room temperature and 13,500 × g. Sixty μL of the supernatant fluid were transferred onto columns filled with 1.4 g neutral alumina. [32P]cAMP was separated from [α–32P]ATP by elution of the product with 4 mL of 0.1 M ammonium acetate, pH 7.0. Samples were filled up with 10 mL double-distilled water, and [32P]cAMP was determined by liquid scintillation counting of Čerenkov radiation.
Docking FS derivatives to the isoform-specific mAC model
Docking experiments were performed by GOLD program [22]. The docking model was generated from the crystal structure of the catalytic AC subunits VC1 and IIC2 in complex with Gsα-GTPγS and 2′,5′-dideoxy-3′-AMP (PDB ID 1CJU), where co-crystallized FS was extracted and used as a reference ligand [3]. The charge assigned to the model protein residues and ligands were described previously [23]. Two Mg2+ or Mn2+ ions were assigned in the active sites of mACs. The binding pocket is defined as residues within 5 Å or 10 Å radius of the FS and ATP analogs/metal ions. The metal ion coordination geometries were determined by examining the angles between the protein coordinating atoms and Mg2+ or Mn2+ in A and B sites of the crystal structure. Bridging water molecule between side chain of Ser942 and oxygens at position 1 and 4 of FS were experimentally determined and thus, treated as part of the protein. All other water molecules were deleted from the protein structure before performing docking work. FS derivatives were built and energy minimized by SYBYL program (Tripos, St. Louis, MO, USA). The docking was performed by using default settings for the GA parameters with 10–15 docking trails performed for each ligand. The Goldscore and Chemscore scoring functions were used to rank binding poses. The models of C1 or C2 domain from ACs 1, 2 or 5 were built based on homology modelling from the sequence alignment and crystal structure of mAC (VC1:IIC2).
Data analysis
Catalytic activity of FS analogs on the AC isoforms was expressed in percent: Specifically, 0 % was defined as the basal AC activity without any diterpene and 100% was defined as the maximum stimulation obtained with 300 μM FS for each AC subtype. Emax-values were calculated in an analogous manner defining the AC activity with 300 μM of a given FS analog as maximum effect. Tables 1 and 2 were statistically analyzed using the Student’s t test to confirm statistical significance between the different metal ions with (*) p < 0.05, (**) p < 0.005 and (***) p < 0.0001. To analyze statistical comparisons between the examined AC isoforms under same conditions, ANOVA with the Bonferroni post-hoc test was applied. Differences were considered as statistically significant with (+) p < 0.05, (++) p < 0.01 and (+++) p < 0.001.
Table 1.
Potencies and efficacies of FS and FS analogs on recombinant ACs 1, 2 and 5 in the presence of 7 mM Mg2+.
| Mg2+ | AC1 | AC2 | AC5 | |||
|---|---|---|---|---|---|---|
| Diterpene | EC50 [μM] |
Efficacy [%] |
EC50 [μM] |
Efficacy [%] |
EC50 [μM] |
Efficacy [%] |
| FS(+++, xxx) | 5.5 ± 0.8* | 100 | 47.3 ± 8.1 | 100 | 4.6 ± 1.5* | 100 |
| DMB-FS(b, cc) | 2.5 ± 0.8 | 105 ± 9** | 80.3 ± 54.1 | 93.5 ± 1.7* | 26.2 ± 11.1 | 190.4 ± 42.6* |
| 6A7DA-FS(##, xx, a, b, ccc) | 6.5 ± 2.1 | 62.5 ±5.9** | EC50_1: 1.8 ± 5.5 EC50_2: 61.2 ± 28.6*** |
22.9 ± 1** | 52.1 ± 16.7* | 99.6 ± 11.3* |
| 7DA-FS(+++, ##, xxx) | 15.8 ± 4.2 | 95.0 ± 3.4* | 647 ± 8.5*** | 101.9 ± 17.2* | 262.5 ± 55.9* | 103.3 ± 11.1 |
| 9d-FS(+, c) | 16.8 ± 3.0 | 103.5 ± 17.1 | 242 ± 113 | 79.5 ± 10.9** | 77.8 ± 46.1 | 138.1 ± 27.0 |
| 1d-FS(c) | Ineffective | 11.1 ± 4.7 | Ineffective | 1.7 ± 3.4 | Ineffective | 14.7 ± 4.4* |
| BODIPY-FS(###, xxx, aaa, ccc) | 0.9 ± 0.2 | 18.7 ± 2.5 | 0.5 ± 0.04** | −115.9 ± 17.5*** | 23.6 ± 4.2** | 13.7 ± 1.7*** |
AC activities were determined as described in “Materials and Methods”. Reaction mixtures contained 7 mM Mg2+, [α–32P]ATP (0.3 μCi/tube), 10 μM GTPγS, 100 μM cAMP, 0.4 mg/mL creatine kinase, 9 mM phosphocreatine, 100 μM IBMX and diterpenes at concentrations from 100 nM – 300 μM. Data were analyzed by non-linear regression to determine the EC50-values. The efficacy for each analog was determined by dividing the maximal stimulation obtained for the analog by the maximum stimulation obtained by treatment with 300 μM FS expressed in percent. Significant impacts of the different metal ions on EC50- or Emax-values were analyzed using the Student’s t test. Statistical significance between the data of Table 1 and 2 is shown by (*) p < 0.05, (**) p < 0.005 and (***) p < 0.0001. Statistical differences between the examined AC isoforms were analyzed by ANOVA Bonferroni test. Comparison of EC50-values: AC1 vs. AC2 (+) p < 0.05, (++) p < 0.005 and (+++) p < 0.0001; AC1 vs. AC5 (#) p < 0.05, (##) p < 0.005 and (###) p < 0.0001; AC2 vs. AC5 (x) p < 0.05, (xx) p < 0.005 and (xxx) p < 0.0001. Comparison of Emax-values: AC1 vs. AC2 (a) p < 0.05, (aa) p < 0.005 and (aaa) p < 0.0001; AC1 vs. AC5 (b) p < 0.05, (bb) p < 0.005 and (bbb) p < 0.0001; AC2 vs. AC5 (c) p < 0.05, (cc) p < 0.005 and (ccc) p < 0.0001.
Table 2.
Effects of FS and FS analogs for recombinant ACs 1, 2 and 5 in the presence of 7 mM Mn2+.
| Mn2+ | AC1 | AC2 | AC5 | |||
|---|---|---|---|---|---|---|
| Diterpene | EC50 [μM] |
Efficacy [%] |
EC50 [μM] |
Efficacy [%] | EC50 [μM] |
Efficacy [%] |
| FS(+) | 3.3 ± 1.3 | 100 | 38.3 ± 14.9 | 100 | 17.8 ± 6.5 | 100 |
| DMB-FS(c) | 2.8 ± 1.6 | 65.4 ± 8.9 | 50.8 ± 31.2 | 46.0 ± 14.5 | 10.9 ± 1.0 | 81.4 ± 14.3 |
| 6A7DA-FS(aa, bb) | 3.0 ± 1.0 | 110 ± 2.2 | 16.7 ± 7.4 | 69.6 ± 3.5 | 13.4 ± 3.4 | 69.7 ± 7.0 |
| 7DA-FS(aa,cc) | 9.3 ± 1.7 | 108.8 ± 4.6 | 80.5 ± 46.8 | 61.7 ± 7.9 | 38.2 ± 6.4 | 124.2 ± 12.9 |
| 9d-FS(aa,cc) | 17.0 ± 3.3 | 98.3 ± 3.7 | 127 ± 31.8 | 29.5 ± 6.2 | 60.8 ± 12.3 | 108.1 ± 12.9 |
| 1d-FS(a) | Ineffective | 2.9 ± 0.7 | Ineffective | 0.4 ± 0.8 | Ineffective | 0.7 ± 0.3 |
| BODIPY-FS(xx, aaa, ccc) | 1.2 ± 0.2 | 73.1 ± 10.3 | 0.17 ± 0.07 | −18.9 ± 12.3 | 2.7 ± 1.1 | 68.5 ± 9.3 |
AC activities were determined as described in “Materials and Methods”. Reaction mixtures contained 7 mM Mn2+, [α–32P]ATP (0.3 μCi/tube), 10 μM GTPγS, 100 μM cAMP, 0.4 mg/mL creatine kinase, 9 mM phosphocreatine, 100 μM IBMX and diterpenes at concentrations from 100 nM – 300 μM. Data were analyzed by non-linear regression to determine the EC50-values. The efficacy for each analog was determined by dividing the maximal stimulation obtained for the analog by the maximum stimulation obtained by treatment with 300 μM FS expressed in percent. Statistical differences between the examined AC isoforms were analyzed by ANOVA Bonferroni test. Comparison of EC50-values: AC1 vs. AC2 (+) p < 0.05, (++) p < 0.005 and (+++) p < 0.0001; AC1 vs. AC5 (#) p < 0.05, (##) p < 0.005 and (###) p < 0.0001; AC2 vs. AC5 (x) p < 0.05, (xx) p < 0.005 and (xxx) p < 0.0001. Comparison of Emax-values: AC1 vs. AC2 (a) p < 0.05, (aa) p < 0.005 and (aaa) p < 0.0001; AC1 vs. AC5 (b) p < 0.05, (bb) p < 0.005 and (bbb) p < 0.0001; AC2 vs. AC5 (c) p < 0.05, (cc) p < 0.005 and (ccc) p < 0.0001.
Results
Effects of FS and FS analogs on recombinant ACs
FS robustly activated ACs 1, 2 and 5. Fig. 2 shows the concentration-response curves of the diterpenes on ACs 1, 2 and 5 in the presence of either 7 mM Mg2+ or 7 mM Mn2+. Table 1 summarizes the diterpene effects on the examined ACs under Mg2+ conditions. FS and FS analogs activated recombinant AC1 in the presence of Mg2+ in the order of potencies BODIPY-FS > DMB-FS ~ FS > 6A7DA-FS > 7DA-FS > 9d-FS. The order of efficacies was 9d-FS ~ DMB-FS ~ 7DA-FS > 6A7DA-FS > BODIPY-FS >> 1d-FS (ineffective).
Fig. 2. Effects of FS and FS analogs on ACs 1, 2 and 5 in the presence of Mg2+ or Mn2+.
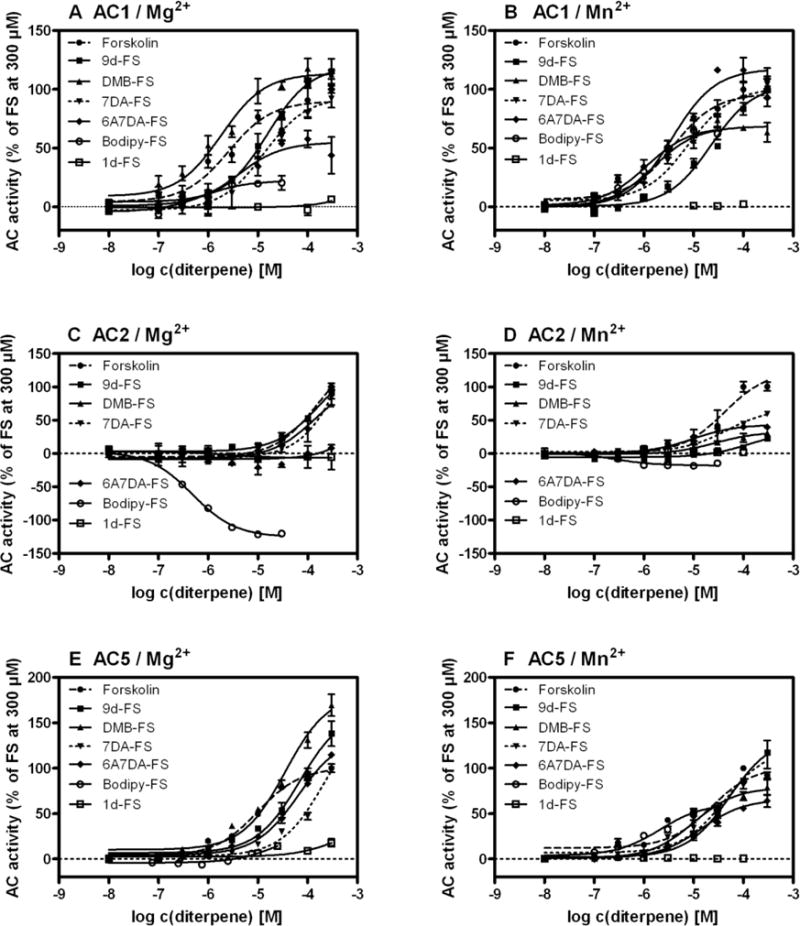
AC activity was determined as described in “Materials and Methods” for increasing concentrations of different diterpenes (100 nM – 300 μM). Tubes were incubated for 10 min at 30°C. A, concentration response curves of various diterpenes on AC1 in presence of Mg2+. B, diterpene effects on AC1 in presence of Mn2+. C, concentration response curves of different diterpenes on AC2 in presence of Mg2+. D, effects of FS and analogs on AC2 under Mn2+ conditions. E, effects of various diterpenes on AC5 in presence of Mg2+. F, diterpene effects on AC5 under Mn2+ conditions. Data shown are representative results (mean ± SD) of one of 3–5 experiments performed in duplicates or triplicates. The efficacy for each analog was determined by dividing the maximal stimulation obtained for the analog by the maximum stimulation obtained by treatment with 300 μM FS expressed in percent.
The pharmacological profile of AC2 differed considerably from the other ACs. When Mg2+ was the divalent cation, all diterpenes except BODIPY-FS showed lower potencies as compared to ACs 1 and 5 yielding the rank order BODIPY-FS >> FS ~ DMB-FS > 9d-FS > 7DA-FS. 6A7DA-FS exhibited unique effects at AC2. In particular, the concentration-dependent effect of 6A7DA-FS revealed an inhibition of AC activity at low concentrations (EC50_1, 1.8 μM), whereas 6A7DA-FS at high concentrations increased enzyme activity (EC50_2, 61.2 μM) (Fig. 3). The maximum inhibition of AC2 was determined at a concentration of 10 μM 6A7DA-FS yielding ~ −20% related to the maximum stimulation with 300 μM FS on AC2. Thereafter, AC activity increased to a level of ~ +20% related to the maximum stimulation with 300 μM FS. BODIPY-FS exhibited a very large inhibitory effect of −115.9 ± 17.5 % relative to the maximum stimulation with 300 μM FS on AC2. The order of efficacies was FS > DMB-FS ~ 7DA-FS > 9d-FS > 6A7DA-FS >> 1d-FS (ineffective) > BODIPY-FS.
Fig. 3. Concentration-response-curve of 6A7DA-FS on AC 2 in presence of Mg2+.
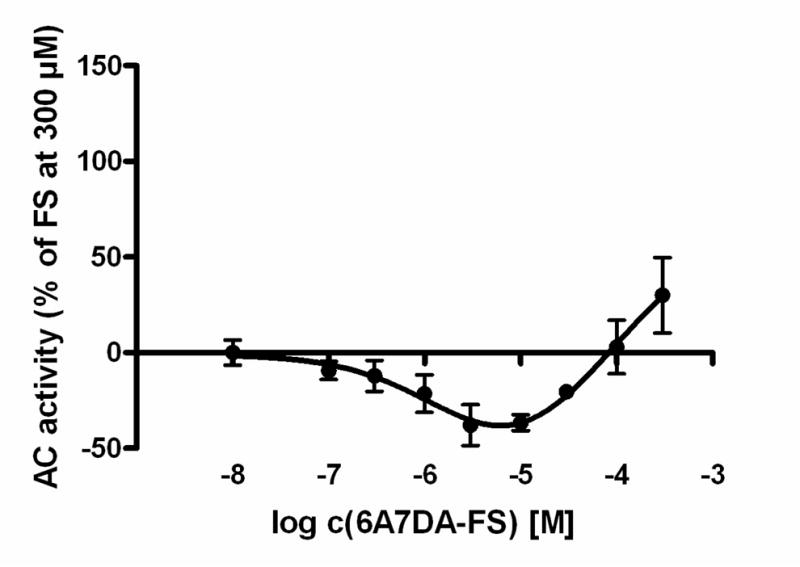
After a 10 min incubation period at 30°C, AC activity was determined as described in “Materials and Methods” for increasing concentrations of 6A7DA-FS (100 nM – 300 μM). Data shown are a combination of 3 independent representative experiments. AC activity was normalized in relation to maximum stimulation obtained by 300 μM FS.
The Mg2+-dependent rank order of potencies on AC5 was FS > BODIPY-FS > DMB-FS > 6A7DA-FS > 9d-FS > 7DA-FS. DMB-FS yielded a remarkably high enzyme activity of 190.4 ± 42.6 % related to the stimulation with 300 μM FS. The order of efficacies on AC5 was DMB-FS >> 9d-FS > 7DA-FS ~ FS ~ 6A7DA-FS > BODIPY-FS ~ 1d-FS.
The replacement of Mg2+ by Mn2+ had a profound influence on the effects of FS and FS analogs on ACs. Table 2 summarizes the effects of Mn2+ on AC activation, revealing changes in the pharmacological profiles of the AC isoforms. Although the rank order of potencies on AC1 did not change, the order of efficacies changed to 6A7DA-FS ~ 7DA-FS > FS ~ 9d-FS > BODIPY-FS > DMB-FS >> 1d-FS (ineffective).
The influence of the cation species is most clearly seen with the effect of 6A7DA-FS on AC2. In the presence of Mn2+, only stimulation on AC2 by 6A7DA-FS without inhibition at low concentrations was determined (Figs. 2 and 3). Additionally, the inhibitory effect of BODIPY-FS was much smaller under Mn2+ conditions as compared to the presence of Mg2+ and reached only −22.7 ± 11.7 % related to FS stimulation at 300 μM. AC2 utilizing Mn-ATP as substrate yielded the rank order of potencies BODIPY-FS >> 6A7DA-FS > FS > DMB-FS > 7DA-FS > 9d-FS. The order of efficacies on AC2 in presence of Mn2+ was FS > 6A7DA-FS > 7DA-FS > DMB-FS > 9d-FS > 1d-FS (ineffective) > BODIPY-FS.
When the effects of FS analogs on AC5 were assayed using Mn2+, the order of potencies showed moderate variations compared to Mg2+ conditions: BODIPY-FS > DMB-FS ~ 6A7DA-FS > FS > 7DA-FS > 9d-FS. The rank order of efficacies was 7DA-FS > 9d-FS > FS > DMB-FS > 6A7DA-FS ~ BODIPY-FS >> 1d-FS. Interestingly, maximal relative AC stimulation on AC5 was lower for all diterpenes in presence of Mn2+ than in presence of Mg2+ except for BODIPY-FS.
Statistical analysis using ANOVA and the Bonferroni post-hoc test to compare the corresponding EC50- or Emax-values of the different ACs under same conditions revealed an exceptional position of AC2 (Tables 1 and 2). Linear regression analysis illustrates the differences in regulation of AC isoforms by diterpenes and, again, highlights the exceptional properties of AC2 (Figs. S1–S3). The correlations do not only illustrate differences in the diterpene profiles of the different AC isoforms, they also point to the high impact of the divalent cations on catalysis. In general, the impact of Mg2+ or Mn2+ on the efficacies is smaller than the effects on the potencies of FS analogs.
To highlight the AC-sensitivity to divalent cations, we compared the pharmacological parameters of each AC isoform in the presence of Mg2+ with those in presence of Mn2+ and statistically analyzed using the Student’s t test. Striking effects on the diterpene profile depending on whether Mg2+ or Mn2+ serves the role of cation cofactor were observed for potencies and even more for efficacies at AC2 and AC5 (Table 1).
Docking of FS derivatives to ACs
Sequence alignment reveals that the ATP- and FS pocket residues are highly conserved among mAC isoforms (Fig. 4). The distribution of hydrophobic and polar residues in the pocket provides an ideal environment for FS binding. We examined bovine AC1, rat AC2 and dog AC5. Fig. S4 shows high sequence similarity in amino acid sequence of the C1 domains of various species and the C2 domains of various species. From the crystal structure of the mAC catalytic core (VC1:IIC2) it can be deduced that the binding pockets easily accommodate ATP analog/metal ions and FS analogs, respectively, between the C1 and C2 binding cleft [2–4].
Fig. 4. Sequence alignment of C1 and C2 domains of AC1, AC2 and AC5.

Red, blue, and cyan amino acid symbols correspond to functional residues that interact with metal ions, ATP and forskolin, respectively. Residues on yellow and gray background indicate the region within 5 Å and 10 Å of the substrate- and FS binding sites, respectively. VC1-d and VC1-h represent the C1 domain of AC5 from dog and human, respectively.
We conducted docking studies with model structures of AC1, AC2, and AC5 isoform-specific catalytic cores (Fig. 4) using the GOLD program. The overall sequence identities in the two cyclase-homology domains (CHDs) among the three isoforms are at least 56%. The sequence identities for residues within the docking perimeters are even higher (Fig. 4). Based on the sequence alignment and the crystal structure of mAC, putative AC1-, AC2-, and AC5 isoform-specific catalytic cores were generated. The homology modelling structures were superimposed and show high similarities in the substrate- and FS binding pockets (Fig. 5). Two residues, Tyr293 and Lys896, of AC2, are different from AC1 and AC5 within the 5 Å FS ligand docking zone. From the multiple sequence alignment, nine additional residues out of 101 amino acids in the 10 Å docking zone are different among these isoforms (Fig. 5).
Fig. 5. Detailed-view of the ATP- and FS binding sites in mAC.
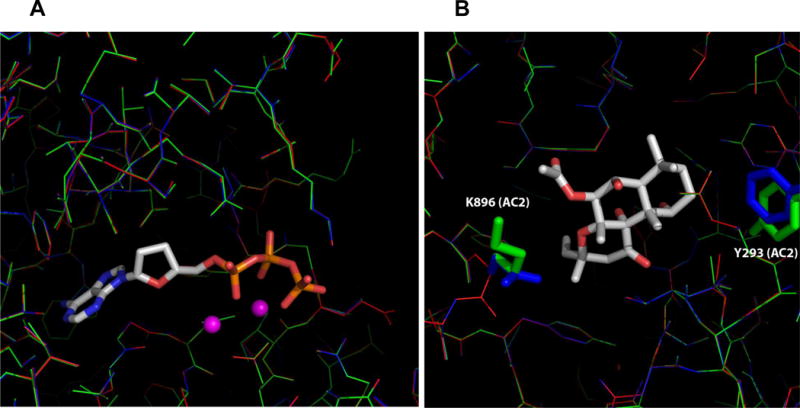
A, ATP substrate binding pocket. B, FS binding pocket. Two non-conserved residues, Tyr-293 and Lys-896, of AC2 and corresponding residues of AC1/AC5 within the 5 Å FS binding pocket are shown in stick mode. The color schemes for AC1, AC2, and AC5 model structure are blue, green, and red, respectively. ATP and FS are shown as stick models. Metal ions are shown as spheres.
In general, FS and FS analogs exhibited better predicted binding propensities in a larger docking perimeter (10 Å) than in a smaller one (5 Å) (data not shown). AC1 and AC5 showed better binding tendencies of FS and FS analogs than AC2 (Fig. 6). These data fit to experimentally determined ligand potencies (Tables 1 and 2 and Fig. 2). The docking profiles of ACs 1 and 5 were similar both in the presence of Mg2+ and Mn2+ (Figs. 6A and D). In contrast, AC2 exhibited a substantially different docking profile from other mAC isoforms as reflected by poor correlations of the parameters for AC2 with AC1 or AC5 in the presence of Mg2+ and Mn2+ (Figs. 6B, C, E, and F).
Fig. 6. Correlation of the docking profiles of diterpenes for mAC isoform models compared to each other.
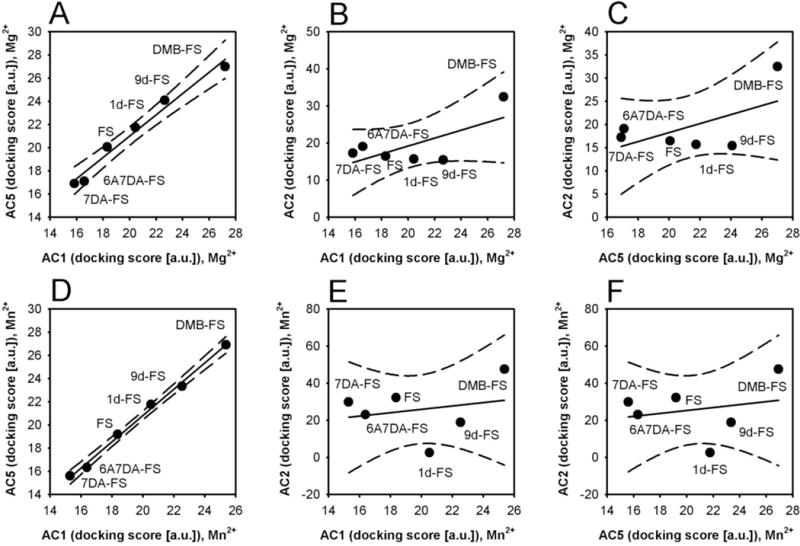
A, correlation of AC1 vs. AC5 in presence of Mg2+ (r2 = 0.97; slope = 0.92 ± 0.07). B, correlation of AC2 vs. AC1 in the presence of Mg2+ (r2 = 0.49; slope = 1.07 ± 0.6). C, correlation of AC2 vs. AC5 in presence of Mg2+ (r2 = 0.35; slope = 0.97± 0.7). D, correlation of AC1 vs. AC5 in the presence of Mn2+ (r2 = 0.99; slope = 1.13 ± 0.04). E, correlation of AC2 vs. AC1 in the presence of Mn2+ (r2 = 0.06; slope = 0.93 ± 1.9). F, correlation of AC2 vs. AC5 in the presence of Mn2+ (r2 = 0.05; slope = 0.79 ± 1.6). Comparisons were analyzed by linear regression; the dashed lines indicate 95% confidence intervals. a.u.; arbitrary unit.
The docking profiles of AC1 and AC5 were similar with Mg2+ and Mn2+ as reflected by high correlation coefficients (Fig. S5). The rank order of binding preference was DMB-FS > 9d-FS > 1d-FS > FS > 6A7DA-FS ~ 7DA-FS. In contrast, the effect of metal ions on docking was more significant for AC2. In the presence of Mn2+, the prediction for binding of FS and 7DA-FS to AC2 increased, but decreased for 1d-FS.
Finally, we docked FS and FS analogs into the pocket from the currently available X-ray crystal structure (C1 from AC5 and C2 from AC2) in the presence of Mg2+ or Mn2+. DMB-FS possesses many possible docking motifs with the diterpene core being more consistently docked into the FS pocket than its flexible piperazino tail. The root mean square deviations (RMSD) [the goodness of alignment of docking FS analogs compared to the FS in the mAC structure] between the diterpene core of DMB-FS and FS is less than 1.2 Å. This result is in agreement with the findings regarding the crystal structure of mAC with DMB-FS [4]. The correlation of docking propensities of FS and FS analogs with Mg2+ or Mn2+ is excellent (r2 = 0.98; slope = 1.03), indicating that the effect of different metal ions on interaction of diterpenes with VC1:IIC2 is minimal (Fig. S5). The ranked preferences are DMB-FS > 9d-FS > FS > 1d-FS > 7DA-FS > 6A7DA-FS.
Discussion
Structure-activity relationships of diterpenes for mAC isoforms
FS and its analogs interact with the catalytic subunit of mAC isoforms 1-8 [2–4]. Due to its diterpene structure, interactions between FS and AC are predominantly hydrophobic, involving aliphatic and aromatic side chains within the FS binding pocket [2–4, 24]. In a previous study we investigated the interactions of AC isoforms 1, 2 and 5 with FS and its analogs to systematically characterize the properties of the different AC subtypes [15]. The previous experiments were performed in the presence of 10 mM Mn2+, and reactions were conducted for 20 min at 37°C to allow a direct comparison of the mAC data with purified AC catalytic subunits C1/C2. In this study we changed the assay conditions to an incubation period of 10 min at 30°C in presence of 7 mM Mn2+ or Mg2+, respectively. With these modifications we aimed to adapt the conditions of the Sf9 cell system to the biological system we used for the examination of ACs in renal tissue [25]. This reduction of time and temperature was necessary to ensure constant biological activity of tissue AC during measurement. However, compared with Pinto et al. [15] we observed changes in Emax-values and potencies for different diterpenes [15]. Considering the highly lipophilic nature of diterpenes, it is not surprising that changes in temperature also have an impact of how these ligands interact with their target proteins.
Crystallographic, enzymatic and fluorescence spectroscopy studies revealed a crucial role of the hydrogen bond between the 1-OH group of FS and the backbone oxygen of Val506 of C1 for catalysis [4, 14]. The 9-OH group of FS resides in a binding region without any hydrogen bonding partners [24]. 9d-FS effectively stimulated ACs 1 and 5, corroborating the notion that interactions between the 9-OH group of the diterpene and the C1 domain of AC1 or AC5 are not important for catalysis. However, the smaller stimulatory effects of 9d-FS on AC2, particularly in presence of Mn2+, indicate a less favourable interaction of 9d-FS with this particular AC isoform.
The absence of the acetyl-group at position 7 was tolerated well by all ACs, as is evident from effective AC stimulation. However, it resulted in a decrease in potency compared to FS. These results are consistent with the data of Zhang et al. (1997) showing that the interaction of the 7-acetyl-group with Ser942 is important but not absolutely essential for FS activity [24]. The available crystal structures of mAC in complex with FS reveal a relatively large space around the C6/C7 positions of FS within the FS binding pocket [4, 24]. AC2, different from AC1 and AC5, bears the rather large side chain of Lys896 to occupy this space (Fig. 4). The switch of the acetyl group from position 7 to 6 did not largely reduce stimulations of ACs 1 and 5. However, the efficacy of 6A7DA-FS on AC2 was much lower that the efficacy of FS, i.e. the 6-acetyl substitution partially prevents this isoform from effective catalysis. Spatial constraints caused by Lys896 may account for this difference between AC2 compared to ACs 1 and 5. In addition, 6A7DA-FS exhibits a biphasic effect on AC2 activity in the presence of Mg2+, but not in the presence of Mn2+. Thus, this FS analog does not only possess different binding affinities to mAC isoforms, but it also exhibits distinct ability to activate AC2 in the presence of different metal ions. During the binding of FS derivatives to AC2, the protein may undergo a conformational change to accommodate the ligand, which, in turn, adjusts its capability to activate AC2 catalysis.
The bulky FS analog BODIPY-FS showed considerable inhibitory effects on AC2 in presence of Mn2+ and profound inhibitory effects in the presence of Mg2+. These data corroborate the notion that AC2 possesses a high basal (constitutive) activity [15]. The BODIPY-group resides outside the FS binding pocket where several amino acids of AC2 form a distinct environment around the BODIPY-substituent compared to the other ACs [15]. Thus, BODIPY-FS is a particularly valuable tool to study activation/inhibition mechanisms of AC and the possible physiological importance of high basal catalysis.
In a previous study, we found that efficacies of diterpenes at VC1:IIC2 were lower compared to the corresponding efficacies on recombinant ACs 1, 2 and 5, except for AC2 with Mn2+ [14]. Thus, although the functional groups of the crucial amino acids are similar in all examined AC isoforms [16], non-homologous AC regions seem to modify diterpene binding. Residues outside the binding site may influence isoform selectivity [20] or some side chains of the transmembrane domains may be involved in AC catalysis by binding the nucleotide substrate, stabilizing the transition state and neutralizing the negative charge of the PPi leaving group [26]. Thus, the structural environment influences the conformational change of the catalytic core from the inactive “open” to the active “closed” state, leading to characteristic properties of all seven diterpenes on the different AC isoforms. In other words, diterpenes do not stabilize a single active mAC state but multiple isoform-specific active state. These data indicate that in principle, it is possible to identify FS analogs with very specific and AC isoform-selective effects.
Comparison of Mg2+ and Mn2+
Tesmer et al. [3] demonstrated the binding of two metal ions in a crystal structure of the VC1:IIC2 substrate complex. The first metal ion (metal A) is coordinated to Asp396 and Asp440 and a water molecule [3]. This ion serves as Lewis acid and enhances the intracellular nucleophilic attack on the 3′-OH of ATP by the α-phosphate. The second metal ion (metal B) is chelated by Asp396, Asp440 and the carbonyl oxygen of Ile397 [26]. In the closed conformation of the enzyme, it is coordinated to the β- and γ-phosphates of the substrate and stabilizes the transition state [20]. Mg2+ or Mn2+ can satisfy this cation requirement.
An early study suggested that Mg2+ and Mn2+ influence the binding of FS [5]. In support of this study, comparison of potencies and efficacies determined with FS and its analogs in relation to the divalent metal ion showed significant influence of the metal cofactor on enzyme activity. In previous studies with mACs, we observed differential impacts of Mg2+ and Mn2+ on binding of inhibitors such as MANT-nucleotides to the catalytic site [6, 20, 27]. In most cases, the exchange of Mn2+ against Mg2+ increased potencies of inhibitors. Additionally, the determination of kinetic parameters showed a preference of Mn2+ at AC isoforms, supporting the view that the metal ions interact differentially with ACs [6, 20, 28]. This is particularly evident from the distinctive influence on basal activities of the various AC subtypes using Mg2+ or Mn2+ [29]. Electron density for the B-site metal ion is well defined with Mg2+ and Mn2+, but density for the A-site is weaker than that for the B-site in the Mg2+-bound complex [20]. Weak electrostatic interactions are also possible between the substrate and Mn2+, whereas the smaller Mg2+ is less accessible [6]. Thus, the occupancy of Mg2+ at the A-site appears to be lower than that of Mn2+ and the active site of the enzyme adopts a slightly more open conformation with Mg2+ than in presence of Mn2+ [20, 30].
Limitations of our study
Small molecule docking predictions for metalloenzymes can be a challenge. In addition to the ligand and protein pocket structure, interaction energies and optimization docking algorithms are affected by other factors such as ligand flexibility, water positions, ionic strength, and metal ion coordination geometry. Our docking results show that divalent cations affect FS analog binding profiles, predicting a larger impact on binding to AC2 than to AC1 and AC5 catalytic core model proteins. Ligands exhibit mAC isoform-specific inhibition due to non-consensus residues (Ala409 of AC5 and Ile1006 of AC2) in the substrate binding site [20, 27]. Two residues, Tyr293 and Lys896 of AC2, are different from AC1 and AC5 within the 5 Å FS binding pocket, probably contributing to the low diterpene potency at AC2.
Although this systematic docking analysis was performed with modelled structures of mAC isoforms, residues of the ligand interaction region within the catalytic core are highly conserved among these isoforms. The size and shape of the FS binding pocket and the distribution of the hydrophobic and polar residues throughout the pocket may be changed by additional domains from specific mAC isoforms. Nevertheless, our analysis provides at least a first glance into AC isoform-specific binding environments for FS and FS analogs. A more detailed analysis of the binding modes of diterpenes will require crystallographic resolution of the structures of various mAC isoforms. A further step to validate the findings of our docking studies will be the generation of mutations of the identified amino acid side chains of Tyr293 and Lys896.
Conclusions
AC isoforms 1, 2 and 5 exhibit distinct diterpene profiles, depending on the divalent cation present. AC2 exhibits a unique cation-dependency. Existing mAC crystal structure and current modelling/docking studies provided an explanation for the pharmacological differences between the AC isoforms. These observations constitute an important step towards the development of isoform-specific diterpenes exhibiting stimulatory or inhibitory effects.
Supplementary Material
Acknowledgments
We would like to thank Mrs. Susanne Brüggemann for expert technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft grant [Se 529/5-2] to R.S.
Abbreviations
- mAC
mammalian membranous adenylyl cyclase
- cAMP
cyclic adenosine 3′,5′-monophosphate
- FS
forskolin
- 1d-FS
1-deoxy-forskolin
- 9d-FS
9-deoxy-forskolin
- 7DA-FS
7-deacetyl-forskolin
- 6A7DA-FS
6-acetyl-7-deacetyl-forskolin
- DMB-FS
7-deacetyl-7-[O-(N-methylpiperazino)-γ-butyryl]-forskolin
- BODIPY-FS
boron-dipyrro-methene-forskolin
- GTPγS
guanosine 5′-[γ-thio]triphosphate
- RMSD
root mean square deviations
- Sf9
Spodoptera frugiperda
Contributor Information
Miriam Erdorf, Department of Pharmacology and Toxicology, University of Regensburg, Germany.
Tung-Chung Mou, Center for Biomolecular Structure and Dynamics, University of Montana, Missoula, Montana, USA.
Roland Seifert, Institute of Pharmacology, Medical School of Hannover, Germany.
References
- 1.Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–74. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- 2.Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Intervent. 2002;2:168–84. doi: 10.1124/mi.2.3.168. [DOI] [PubMed] [Google Scholar]
- 3.Tesmer JJ, Sunahara RK, Johnson RA, Gösselin G, Gilman AG, Sprang SR. Two-metal-ion catalysis in adenylyl cyclase. Science. 1999;285:756–60. doi: 10.1126/science.285.5428.756. [DOI] [PubMed] [Google Scholar]
- 4.Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with GSα–GTPγS. Science. 1997;278:1907–16. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- 5.Cech SY, Broaddus WC, Maguire ME. Adenylate cyclase: the role of magnesium and other divalent cations. Mol Cell Biochem. 1980;33:67–92. doi: 10.1007/BF00224572. [DOI] [PubMed] [Google Scholar]
- 6.Gille A, Lushington GH, Mou TC, Doughty MB, Johnson RA, Seifert R. Differential inhibition of adenylyl cyclase isoforms and soluble guanylyl cyclase by purine and pyrimidine nucleotides. J Biol Chem. 2004;279:19955–69. doi: 10.1074/jbc.M312560200. [DOI] [PubMed] [Google Scholar]
- 7.Wald H, Popovtzer MM. Effect of divalent ions on basal and hormone-activated renal adenylate cyclase/cyclic AMP system. Miner Electrolyte Metab. 1984;10:133–40. [PubMed] [Google Scholar]
- 8.Seamon KB, Daly JW. Forskolin: its biological and chemical properties. Adv Cyclic Nucleotide Protein Phosphorylation Res. 1986;20:1–150. [PubMed] [Google Scholar]
- 9.Putnam WC, Swenson SM, Reif GA, Wallace DP, Helmkamp GM, Jr, Grantham JJ. Identification of a forskolin-like molecule in human renal cysts. J Am Soc Nephrol. 2007;18:934–43. doi: 10.1681/ASN.2006111218. [DOI] [PubMed] [Google Scholar]
- 10.Onda T, Hashimoto Y, Nagai M, Kuramochi H, Saito S, Yamazaki H, et al. Type-specific regulation of adenylyl cyclase. Selective pharmacological stimulation and inhibition of adenylyl cyclase isoforms. J Biol Chem. 2001;276:47785–93. doi: 10.1074/jbc.M107233200. [DOI] [PubMed] [Google Scholar]
- 11.Metzger H, Lindner E. The positive inotropic-acting forskolin, a potent adenylate cyclase activator. Arzneimittelforschung. 1981;31:1248–50. [PubMed] [Google Scholar]
- 12.Toya Y, Schwencke C, Ishikawa Y. Forskolin derivatives with increased selectivity for cardiac adenylyl cyclase. J Mol Cell Cardiol. 1998;30:97–108. doi: 10.1006/jmcc.1997.0575. [DOI] [PubMed] [Google Scholar]
- 13.Pierre S, Eschenhagen T, Geisslinger G, Scholich K. Capturing adenylyl cyclases as potential drug targets. Nature Rev Drug Discov. 2009;8:321–35. doi: 10.1038/nrd2827. [DOI] [PubMed] [Google Scholar]
- 14.Pinto C, Hübner M, Gille A, Richter M, Mou TC, Sprang SR, et al. Differential interactions of the catalytic subunits of adenylyl cyclase with forskolin analogs. Biochem Pharmacol. 2009;78:62–9. doi: 10.1016/j.bcp.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinto C, Papa D, Hübner M, Mou TC, Lushington GH, Seifert R. Activation and inhibition of adenylyl cyclase isoforms by forskolin analogs. J Pharmacol Exp Ther. 2008;325:27–36. doi: 10.1124/jpet.107.131904. [DOI] [PubMed] [Google Scholar]
- 16.Tang WJ, Hurley JH. Catalytic mechanism and regulation of mammalian adenylyl cyclases. Mol Pharmacol. 1998;54:231–40. doi: 10.1124/mol.54.2.231. [DOI] [PubMed] [Google Scholar]
- 17.Laurenza A, Khandelwal Y, De Souza NJ, Rupp RH, Metzger H, Seamon KB. Stimulation of adenylate cyclase by water-soluble analogues of forskolin. Mol Pharmacol. 1987;32:133–9. [PubMed] [Google Scholar]
- 18.Takahashi N, Nemoto T, Kimura R, Tachikawa A, Miwa A, Okado H, et al. Two-photon excitation imaging of pancreatic islets with various fluorescent probes. Diabetes. 2002;51:S25–8. doi: 10.2337/diabetes.51.2007.s25. [DOI] [PubMed] [Google Scholar]
- 19.Tang WJ, Gilman AG. Type-specific regulation of adenylyl cyclase by G protein βγ subunits. Science. 1991;254:1500–3. doi: 10.1126/science.1962211. [DOI] [PubMed] [Google Scholar]
- 20.Mou TC, Gille A, Fancy DA, Seifert R, Sprang SR. Structural basis for the inhibition of mammalian membrane adenylyl cyclase by 2′(3′)-O-(N-methylanthraniloyl)-guanosine 5′-triphosphate. The J Biol Chem. 2005;280:7253–61. doi: 10.1074/jbc.M409076200. [DOI] [PubMed] [Google Scholar]
- 21.Seifert R, Lee TW, Lam VT, Kobilka BK. Reconstitution of β2-adrenoceptor-GTP-binding-protein interaction in Sf9 cells–high coupling efficiency in a β2-adrenoceptor-GSα fusion protein. Eur J Biochem. 1998;255:369–82. doi: 10.1046/j.1432-1327.1998.2550369.x. [DOI] [PubMed] [Google Scholar]
- 22.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52:609–23. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 23.Suryanarayana S, Pinto C, Mou TC, Richter M, Lushington GH, Seifert R. The C1 homodimer of adenylyl cyclase binds nucleotides with high affinity but possesses exceedingly low catalytic activity. Neurosci Lett. 2009;467:1–5. doi: 10.1016/j.neulet.2009.09.049. [DOI] [PubMed] [Google Scholar]
- 24.Zhang G, Liu Y, Ruoho AE, Hurley JH. Structure of the adenylyl cyclase catalytic core. Nature. 1997;386:247–53. doi: 10.1038/386247a0. [DOI] [PubMed] [Google Scholar]
- 25.Erdorf M, Seifert R. Pharmacological characterization of adenylyl cyclase isoforms in rabbit kidney membranes. Naunyn-Schmiedeberg’s Arch Pharmacol. 2011;383:357–72. doi: 10.1007/s00210-011-0600-7. [DOI] [PubMed] [Google Scholar]
- 26.Tesmer JJ, Sprang SR. The structure, catalytic mechanism and regulation of adenylyl cyclase. Curr Opin Struct Biol. 1998;8:713–9. doi: 10.1016/s0959-440x(98)80090-0. [DOI] [PubMed] [Google Scholar]
- 27.Mou TC, Gille A, Suryanarayana S, Richter M, Seifert R, Sprang SR. Broad specificity of mammalian adenylyl cyclase for interaction with 2′,3′-substituted purine- and pyrimidine nucleotide inhibitors. Mol Pharmacol. 2006;70:878–86. doi: 10.1124/mol.106.026427. [DOI] [PubMed] [Google Scholar]
- 28.Göttle M, Geduhn J, König B, Gille A, Höcherl K, Seifert R. Characterization of mouse heart adenylyl cyclase. J Pharmacol Exp Ther. 2009;329:1156–65. doi: 10.1124/jpet.109.150953. [DOI] [PubMed] [Google Scholar]
- 29.Pieroni JP, Harry A, Chen J, Jacobowitz O, Magnusson RP, Iyengar R. Distinct characteristics of the basal activities of adenylyl cyclases 2 and 6. J Biol Chem. 1995;270:21368–73. doi: 10.1074/jbc.270.36.21368. [DOI] [PubMed] [Google Scholar]
- 30.Mou TC, Masada N, Cooper DM, Sprang SR. Structural basis for inhibition of mammalian adenylyl cyclase by calcium. Biochemistry. 2009;48:3387–97. doi: 10.1021/bi802122k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.