

ABSTRACT
A therapeutic vaccine for human Chagas disease is under development by the Sabin Vaccine Institute Product Development Partnership. The aim of the vaccine is to significantly reduce the parasite burden of Trypanosoma cruzi in humans, either as a standalone product or in combination with conventional chemotherapy. Vaccination of mice with Tc24 formulated with monophosphoryl-lipid A (MPLA) adjuvant results in a Th1 skewed immune response with elevated IgG2a and IFNγ levels and a statistically significant decrease in parasitemia following T. cruzi challenge. Tc24 was therefore selected for scale-up and further evaluation. During scale up and downstream process development, significant protein aggregation was observed due to intermolecular disulfide bond formation. To prevent protein aggregation, cysteine codons were replaced with serine codons which resulted in the production of a non-aggregated and soluble recombinant protein, Tc24-C4. No changes to the secondary structure of the modified molecule were detected by circular dichroism. Immunization of mice with wild-type Tc24 or Tc24-C4, formulated with E6020 (TLR4 agonist analog to MPLA) emulsified in a squalene-oil-in-water emulsion, resulted in IgG2a and antigen specific IFNγ production levels from splenocytes that were not significantly different, indicating that eliminating putative intermolecular disulfide bonds had no significant impact on the immunogenicity of the molecule. In addition, vaccination with either formulated wild type Tc24 or Tc24-C4 antigen also significantly increased survival and reduced cardiac parasite burden in mice. Investigations are now underway to examine the efficacy of Tc24-C4 formulated with other adjuvants to reduce parasite burden and increase survival in pre-clinical studies.
KEYWORDS: antigen, chagas disease, pre-clinical testing, production, vaccine
Introduction
Chagas disease, also known as American trypanosomiasis, is a serious neglected tropical disease caused by the protozoan parasite Trypanosoma cruzi, which is endemic in parts of the Western Hemisphere extending from the southern United States to southern Argentina. The parasites which cause Chagas disease are transmitted to humans and other mammals by blood-sucking “kissing bugs” of the subfamily Triatominae, but can also be spread via blood transfusion, organ transplantation, and congenital transmission.1 In 2013, the number of people estimated to be infected with T. cruzi was approximately 9.4 million, mostly in Latin America.2 Additional projections suggest that overall 60–100 million people are at risk of infection in the Western Hemisphere.3-5 Human Chagas disease occurs in 2 important phases: the acute and the chronic stage. During the acute stage, parasites are present in the blood at high levels and those infected are either asymptomatic or exhibit an acute self-limiting febrile illness which typically resolves within 4–8 weeks in 90% of cases.6,7 Individuals with chronic Chagas disease, however, have very low levels of circulating parasites and the majority of these people have no clinical symptoms, but 20–30% will develop clinical symptoms characterized by neuronal cell loss, microvascular dysfunction, and myocardial damage8-10 – alterations that affect the nervous, digestive, and cardiovascular systems. To date, there are limited treatment options for chronic infection. Current drugs such as benznidazole and nifurtimox work effectively against parasite replication when given during the acute stage of infection, but can cause adverse side effects ranging from anorexia, weight loss, excitability, and nausea in the case of nifurtimox,11 or dermatitis, muscular pain, neuralgias, and potentially bone marrow disorders such as thrombocytopenia purpura or agranulocytosis in the case of benznidazole.12 In addition, both drugs require long treatment schedules (60 d for benznidazole and 90 d for nifurtimox), making treatment logistically difficult, and increasing the risk for drug resistance.5,11,13-16 These drugs also have the potential to cause anemia in pregnant mothers and insufficient weight gain in their children.16 A recent multicenter randomized study of benznidazole on patients with chronic Chagas cardiomyopathy showed a significant reduction in the number of circulating parasites, but there was no reduction in the progression of cardiac symptoms over a 5 y period, indicating that drug alone is ineffective against disease progression during the chronic stage of disease.17
To circumvent the problems with chemotherapeutic treatment of Chagas disease and to achieve protection from cardiac complications, a Chagas vaccine might offer a more effective solution. At the very minimum 2 vaccination strategies for human Chagas disease are conceivable – preventative vaccination and therapeutic vaccination.18 The therapeutic approach could rely on a standalone immunization or vaccination linked to chemotherapy with benznidazole or nifurtimox. Several vaccines for the treatment of Chagas disease are currently under development, including vaccines composed of peptides, plasmid DNA, or recombinant proteins.19 Specifically, vaccination of mice with a synthetic peptide containing predicted overlapping antigenic epitopes from T. cruzi mucin-like associated surface protein increased survival in mice with a reduction in parasite load in the heart.20 Immunization of adenovirus carrying amastigote surface protein-2 and trans-sialidase antigens,21 and plasmids expressing TcG1, TcG2, or TcG4, membrane associated glycosylphosphatidylinositol (GPI) proteins expressed on the surface of T. cruzi22 resulted in reduced parasite burden in tissue, reduced heart injury and increased survival. Our laboratory has been developing a therapeutic Chagas vaccine based on the Tc24 protein, a T. cruzi parasite excretory-secretory protein, by itself or combined with additional antigens and in the presence of one or more adjuvants. The major target product profile of the vaccine would include the prevention or delay of Chagasic cardiomyopathy in individuals with T. cruzi infection, possibly combined with antiparasitic chemotherapy (vaccine-linked chemotherapy).18 The Tc24 antigen was originally expressed in E. coli as a fusion protein and shown to induce significant protection in Balb/c mice against a lethal dose of T. cruzi parasites.23 Data suggests that amino acids 109–124 contain a putative T cell epitope which may be responsible for the protection.24 A DNA vaccine containing the Tc24 coding sequence has likewise shown efficacy when introduced into T. cruzi infected mice and dogs.25-28 Based on the fact that no human DNA vaccine has yet been registered or licensed, we are pursuing Chagas vaccine development through a recombinant protein-based approach. Recently, a study has been published where vaccination of mice with wild-type Tc24 (Tc24-WT) using MPLA as the adjuvant, induced a Th1-biased immune response, which provided partial protection after T. cruzi challenge.29 In another study, Tc24 delivered using a nanoparticle delivery system significantly reduced peak parasitemia, cardiac parasite burden and inflammatory cell infiltrate.30 This initial Tc24-WT antigen was expressed in E. coli and purified using a 2-step chromatography method. However, significant aggregation was observed during purification and storage of the protein, which would make reproducible scale-up of the protein and quality control more difficult and could impact immunogenicity and efficacy of the molecule as well.
In an attempt to improve production and control feasibility while at the same time reducing or eliminating aggregation, cysteine-to-serine mutations were introduced into the Tc24 DNA sequence during gene synthesis. Cysteine mutagenesis has been performed to increase expression levels and solubility, reduce mis-folding and aggregation, enhance stability, and improve the bioactivity of the target protein.31-36 Two modified expression cassettes were designed as follows: the first construct, Tc24-C2, had mutations at residues C4S and C66S, and the second, Tc24-C4, at C4S, C66S, C74S, and C124S. The C4S and C66S mutations were selected for the Tc24-C2 construct over cysteine residues C74 and C124 due to their location on the surface of the molecule as determined by X-ray crystallography.37 After expression of both modified proteins, Western blot analysis of Tc24-C2 revealed fewer protein aggregates than Tc24-WT, while the 4 cysteine-to-serine modifications in Tc24-C4 eliminated detectable protein aggregation as determined by Western blot analysis and dynamic light scattering. Tc24-WT, Tc24-C2, and Tc24-C4 were each produced by fermentation at the 10L scale and purified by ion exchange (IEX) and size exclusion chromatography (SEC). The purified Tc24-WT, Tc24-C2, and Tc24-C4 were formulated with a synthetic TLR-4 receptor agonist in squalene emulsions for immunologic testing in mice. Tc24-C2 and Tc24-C4 were shown to be immunologically equivalent to Tc24-WT, indicating that the cysteine to serine modifications did not adversely impact the antigenicity of the molecule. In addition, a significant reduction in cardiac tissue parasite burden was observed in mice vaccinated with either formulated Tc24-WT or Tc24-C4. Based on these data and the reduced aggregation of Tc24-C4 compared to Tc24-C2 and Tc24-WT, the recombinant Tc24-C4 protein has been selected as the lead vaccine candidate antigen and will be developed and produced for pre-clinical assessment for the treatment and prevention of human Chagas disease.
Results
Here we present our efforts on the cloning, expression, characterization, and initial pre-clinical investigation of our lead Tc24 Chagas disease vaccine candidate. During early feasibility of expression and purification experiments we observed significant aggregation with both the His-tagged as well as the tag-less Tc24-WT proteins. Since treatment of either protein with reducing agents resulted in a significant reduction in aggregation, we had strong evidence that the aggregation observed was due to the formation of disulfide bridges between Tc24 molecules. The aggregation observed was resolved by expression of modified Tc24 molecules in which the codons representing cysteine residues were replaced with codons representing serine residues. Expression and purification of the resulting monomeric Tc24 provided a means to continue development and pre-clinical testing of our Tc24 based Chagas vaccine.
Expression and purification of Tc24-WT
Our initial Chagas vaccine antigen candidate was based on wild-type Tc24 plasmid DNA vaccines previously amplified by PCR from T. cruzi isolated from human cases in Yucatan, Mexico.25 The same Tc24-WT sequence, with the addition of a hexahistidine tag at the C-terminus (Tc24-WT + His), was also expressed in E. coli BL21 (DE3) as a soluble recombinant protein using the T7 expression system. Purification of Tc24-WT + His was performed using a 2-step purification scheme encompassing Immobilized Metal Affinity Chromatography followed by Q Sepharose XL anion exchange chromatography to remove endotoxin. The yield from this process was approximately 56 mg of protein per liter of culture which represents a recovery of approximately 40% compared to the starting material (142 mg Tc24-WT + His per liter of biomass). The low yield per liter was due to the low biomass generated from the shake flask cultures (4–5 g per liter). Analysis of both the in-process as well as the final purified protein by non-reducing SDS-PAGE and Western blot revealed significant aggregation of the protein (Fig. 1A). Aggregation of this nature poses significant hurdles for both scale up as well as quality control of any candidate vaccine and thus needed to be addressed. Interestingly, we did observe that upon reduction of Tc24-WT + His prior to SDS-PAGE and Western analysis, there was a significant decrease in protein aggregation (Fig. 1B), indicating that intermolecular disulfide bonds between Tc24-WT + His monomers were at least partially responsible for the aggregation observed. We observed very similar aggregation with the non-tagged version of wild-type Tc24 protein.
Figure 1.
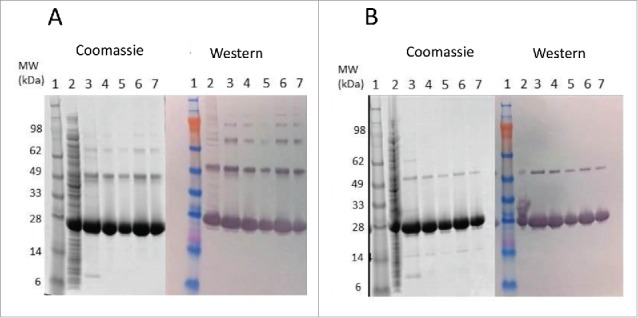
SDS-PAGE and western blot analysis of Tc24-WT+His in-process samples. Samples were separated on 4–12% Bis-Tris gels under Non-Reduced (A) and Reduced (B) conditions. Lane 1: SeeBlue Plus Molecular Weight marker (10 µl). Lane 2: Starting Material (7.5 µl). Lane 3: IMAC eluate (7.5 µl). Lanes 4–5 QXL eluate (4 µl and 2 µl load). Lanes 6–7: Final Tc24-WT+His (4 µl and 2 µl load). Western blot detection was performed using an in house polyclonal mouse anti-Tc24 antibody at 1:2,500 followed by a goat anti-mouse IgG alkaline phosphatase conjugated secondary antibody at 1:7,500).
To reduce or eliminate these disulfide bonds, cysteine-to-serine codon exchanges were introduced at either 2 or 4 cysteine encoding codons within the Tc24-WT DNA sequence: Tc24-C2 contained 2 codon replacements (C4S, C66S) and Tc24-C4 was derived by replacing all 4 cysteine residues with serine residues (C4S, C66S, C74S, and C124S) (Fig. 2). The C4 and C66 residues were selected for the Tc24-C2 construct since these 2 residues likely are surface exposed based on high resolution crystallography.37 Since the final vaccine candidate was to be free of any purification tags, all work performed from this point forward utilized a tag-free version of Tc24-WT, Tc24-C2, and Tc24-C4 which were cloned, expressed, and subsequently purified through ion exchange chromatography and size exclusion chromatography as described in Materials and Methods. Yield and purity of all 3 proteins are shown in Table 1. Although the yield of target protein per mg biomass was higher for Tc24-WT compared to Tc24-C2 and Tc24-C4, our fermentations for Tc24-C2 and Tc24-C4 yielded significantly more biomass which lead to a higher overall yield when compared to Tc24-WT. More importantly, Western blot analysis of all 3 protein preparations suggested an inverse correlation between the number of cysteine residues removed and the level of aggregation observed. Tc24-C2 protein still showed some evidence of aggregation whereas almost none was seen with Tc24-C4 (Fig. 3A–C). Thus, elimination of all 4 cysteine residues in Tc24 proved to be a promising strategy to developing a scalable process for the production of Tc24.
Figure 2.

Tc24 amino acid sequence alignment. Amino acid sequence alignment of wild-type (Tc24-WT) and the cysteine-mutated Tc24 constructs (Tc24-C2 and Tc24-C4). Cysteine residues are highlighted in red and mutated serine residues in green.
Table 1.
Comparison of yield and purity for Tc24 constructs.
| Construct | Biomass yield (g/L) | Tc24 Expression Level (mg/g biomass) | Theoretical Yield (g of Tc24 per L culture) | Starting purification load (mg) | Final purified Tc24 (mg) | Overall recovery (% of load) | Purity (%) |
|---|---|---|---|---|---|---|---|
| Tc24-WT | 36 | 45 | 1,630 | 183 | 83 | 45 | 93 |
| Tc24-C2 | 64 | 68 | 4,354 | 155 | 85 | 55 | 98 |
| Tc24-C4 | 90 | 56 | 5,062 | 192 | 100 | 52 | 98 |
Figure 3.
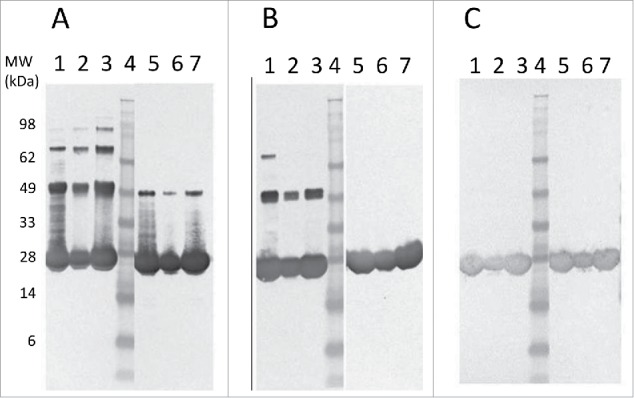
Western blot comparison of Tc24-WT (A), Tc24-C2 (B), and Tc24-C4 (C) purified proteins. Lanes 1–3: Non-Reduced. Lane 4: SeeBlue Plus Molecular Weight Marker. Lanes 5–7 Reduced. Lanes 1 and 5: Sample before size-exclusion chromatography (SEC) 8 µg load. Lanes 2 and 6: Post SEC low load (3 µg). Lanes 3 and 7: Post SEC high load (8 µg). Detection was performed using mouse polyclonal antibody against Tc24 expressed in Pichia pastoris as primary antibody diluted 1:2,500 in PBST and an alkaline phosphatase conjugated goat anti-mouse secondary antibody diluted 1:7,500 in PBST.
Scalable purification process for Tc24-C4
Based on the reduced aggregation observed with Tc24-C4, it was selected for further scale-up and pre-clinical testing. SDS-PAGE analysis of reduced and non-reduced in process samples of Tc24-C4 from crude E. coli lysate to final purified protein is shown in Fig. 4. After ion exchange (QXL) chromatography, where the protein eluted at 80 mM NaCl, Tc24-C4 had a purity of >94% (reduced) and >96% (non-reduced). After a subsequent size exclusion chromatography step, Tc24-C4 had a purity of >99% (reduced) and >98% (non-reduced) (Fig. 4). The final recovery of Tc24-C4 was 52% of the starting amount with a yield of 1,768 mg per liter of fermentation harvest (Table 1). The high yield and purity for Tc24-C4 allowed for initial pre-clinical immunogenicity testing of the candidate antigen.
Figure 4.
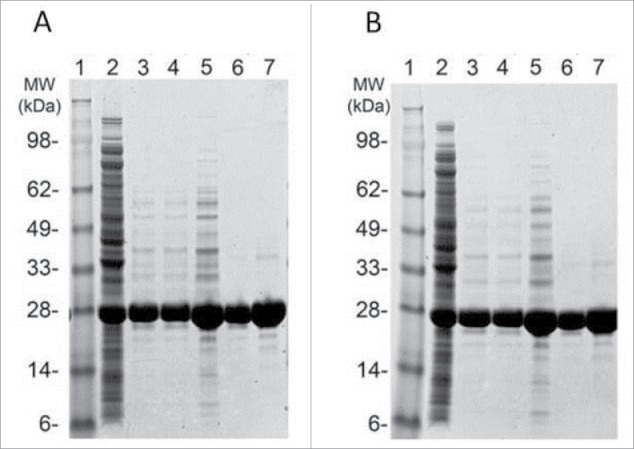
SDS-PAGE analysis of in-process and final Tc24-C4 protein samples. Protein samples were separated on a 4–12% Bis-Tris SDS PAGE gel under reducing (A) and non-reducing (B) conditions. Lanes 1: SeeBlue Plus 2 molcular weight marker. Lanes 2: Cell Lysate (3 µg). Lane 3: QXL1 elution (3 µg). Lane 4: QXL2 elution (3 µg). Lane 5: Concentrated QXL (8 µg). Lane 6: SEC elution (3 µg). Lane 7: SEC elution (8 µg).
Protein stability
Tc24-WT, Tc24-C2 and Tc24-C4, plus an alkylated version of Tc24-WT + His were compared in a short-term stability study. After 10 d at 4°C, Western analysis was performed. As anticipated, aggregation was observed with Tc24-WT + His, which could be eliminated by alkylation of the cysteines. Tc24-C2 and Tc24-C4 preparations exhibited enhanced stability and reduced aggregation which correlated with the number of cysteines removed from their sequence (Fig. 5). The effect of the cysteine induced aggregation was even more noticeable when monitoring the average size and size distribution (polydispersity) of the proteins in solution by dynamic light scattering where a significant reduction in both hydrodynamic radius and polydispersity were observed for the alkylated Tc24-WT as well as the Tc24-C2 and Tc24-C4 proteins (Fig. 6). These data support the conclusion that elimination of disulfide bond formation results in a more consistent, monodispersed recombinant protein.
Figure 5.
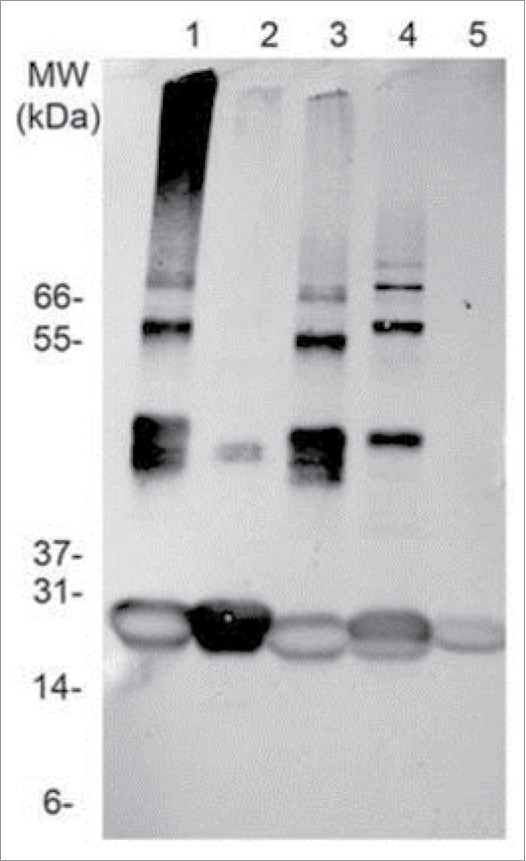
Stability assessment of Tc24. Western blot of different Tc24 constructs, taken after storing the proteins for 10 d at 4°C in PBS. Lane 1: Tc24-WT+His, Lane 2:Tc24-WT+His, alkylated, Lane 3: Tc24-WT, Lane 4: Tc24-C2, Lane 5: Tc24-C4. A Ponceau-stained Mark 12 protein ladder was used as a MW reference. Detection was performed using mouse polyclonal antibody against Tc24 expressed in Pichia pastoris as primary antibody diluted 1:2,500 in PBST and an alkaline phosphatase conjugated goat anti-mouse secondary antibody diluted 1:7,500 in PBST.
Figure 6.
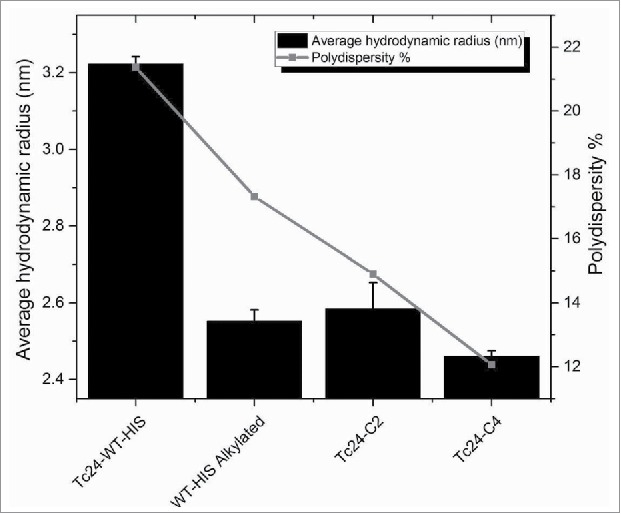
Hydrodynamic radius and polydispersity of Tc24 antigens after 3 d at 4°C. Tc24-C4 was noticeably the most monodispersed product.
Structural equivalence of the Tc24 constructs
Circular dichroism was performed to investigate the impact of the cysteine to serine modifications on the secondary structure of Tc24. Tc24-WT + His (+/− alkylation) and Tc24-C4 demonstrated an identical secondary structure characteristic of α-helical proteins (Fig. 7a). Specifically, based on comparison with 2 reference sets using CONTIN, SELCON, and CDSSTR data fitting programs,38 the protein was predicted to fold into 70% α helix, 7% Beta sheet and 23% turns and loops. In addition, the tertiary folding of the proteins was compared by thermal melt analysis (Fig. 7b), where Tc24-WT + His and Tc24-C4 displayed similar melting behavior, suggesting that the removal of the cysteine residues had no detrimental impact on protein folding. Mass spectrometry performed on purified Tc24 C4 confirmed the identity of the molecule and determined the purity to be 99.75%.
Figure 7.
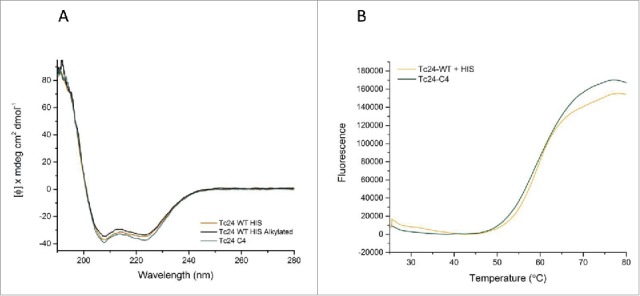
Structural comparison of Tc24 constructs. A) Circular Dichroism (CD). Far UV CD spectrum of different constructs of Tc24 were taken on a Jasco J-1500. All tested Tc24 protein have a virtual identical CD profile with overlapping spectra. Negative peaks at 222 nm and 208 nm and a positive peak at 193 nm indicate that Tc24 is an α-helical protein. B) Thermal melting profile of Tc24-WT and Tc24-C4 measured using Protein Thermal Shift™ kit (Life Technologies).
Immunogenicity, antigenicity, and efficacy of Tc24 constructs
Multiple studies in mouse models have shown that vaccine constructs that induce antigen specific IFNγ production from splenocytes and increased antigen specific IgG2a protect mice infected with T. cruzi, resulting in decreased parasite burdens and reduced cardiac pathology.21,26,29,39,40 To determine whether the cysteine to serine modifications had any impact on the immunogenicity of the protein, we measured IFNγ and IgG2a production in mice vaccinated with formulated Tc24-WT + His, Tc24-WT, Tc24-C2 and Tc24-C4. IFNγ secretion was measured from splenocytes harvested from vaccinated mice and re-stimulated in vitro with homologous or heterologous proteins (Fig. 8). The results showed that IFNγ secretion was similar between groups, regardless of the protein used for re-stimulation, and that only the combination of antigen and adjuvant induced significant IFNγ secretion when compared to antigen alone (p < 0.0001) or adjuvant alone (p = 0.0007) (Fig. 8, Panel C). These data indicate that the cysteine to serine modifications did not significantly affect the antigen-specific cellular immunogenicity of Tc24-C4 in naïve mice. As a correlate of Th1 skewed immune responses, Tc24-specific IgG2a antibody titers were also measured from the serum of vaccinated mice (Fig. 9). Terminal antibody titers observed for mice which had been vaccinated with formulated non-tagged Tc24-WT and Tc24-C4 antigens were not significantly different, further indicating that mutation of the cysteine residues did not significantly impact immunogenicity in naïve mice. Although terminal Tc24 specific IgG2a antibody titers in mice vaccinated with formulated Tc24-C4 were significantly lower (1:25,600) compared to mice vaccinated with formulated Tc24-WT + His protein (> 1:100,000), this may be due to His specific antibodies induced in mice vaccinated with this construct. Nonetheless, when comparing terminal antibody titers between mice vaccinated with either formulated Tc24-WT + His and formulated Tc24-WT no-His there was no statistically significant differences in titers, indicating that any His specific antibodies present did not significantly impact measurement of Tc24-WT specific IgG2a. These data suggest that although the His-tagged version of Tc24 may be more antigenic, mutation of all 4 cysteine residues did not appear to abrogate B-cell specific antigenic epitopes when compared to untagged Tc24-WT. Although reduced compared to Tc24-WT + His, antibody titers were still considered robust at the levels observed. This conclusion is supported by the experiments shown in Fig. 10 (Panel A), in which mice infected with T. cruzi parasites developed serum antibodies against native Tc24 protein that recognized Tc24-WT and Tc24-C4 at equal levels indicating that Tc24 epitopes responsible for the humoral immune response were not disrupted by the C4 mutation. Importantly, the Tc24-C4 no His antigen combined with E6020 was protective, significantly increasing survival (p = 0.0322) when compared to mice that received only PBS treatment (Panel B). While adjuvant alone was also able to significantly increase survival (p = 0.0372), the combination of Tc24-C4 plus E6020 SE adjuvant was required to significantly reduce cardiac parasite burdens (p = 0.0147) (Panel C), indicating better protection was afforded by the complete vaccine.
Figure 8.
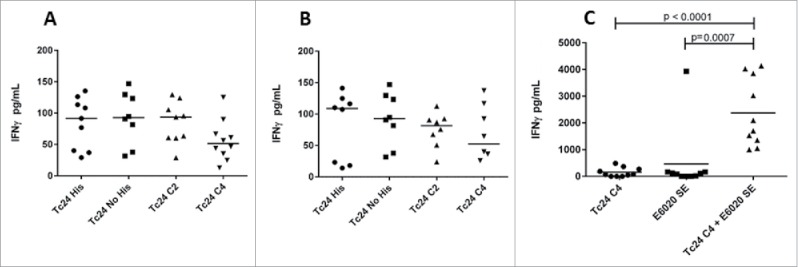
IFNγ measurements after homologous or heterologous stimulation of vaccinated mice by ELISA. IFNγ concentrations in supernatants from splenocytes harvested from mice vaccinated with the indicated protein combined with E6020 emulsified in AddaVaxTM and re-stimulated in vitro with homologous recombinant protein (A) or mice vaccinated with Tc24-WT No His combined with E6020 emulsified with AddaVaxTM and re-stimulated in vitro with heterologous protein as indicated (B). No statistically significant differences in antigen specific IFNγ secretion were observed. (C) IFNγ secretion from mice vaccinated with Tc24 C4 combined with E6020 in a stable squalene emulsion was significantly increased compared to mice vaccinated with Tc24 C4 (protein only) or E6020 SE alone (adjuvant only control).
Figure 9.
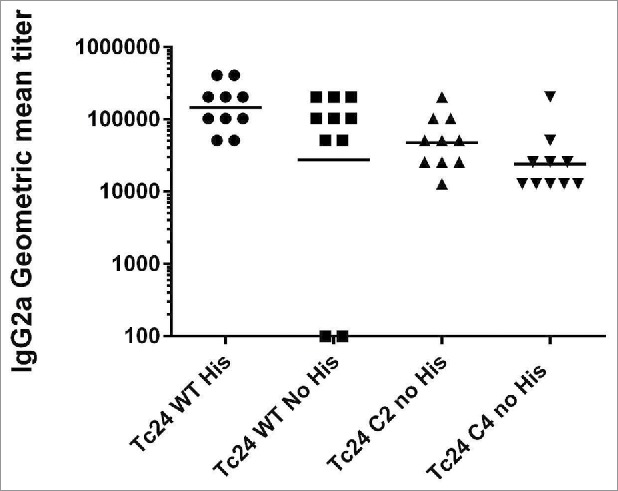
Measurement of IgG2a in vaccinated mice by ELISA. Anti-Tc24 IgG2a antibody titers in terminal serum were measured by ELISA for wild-type and mutant Tc24 constructs. Antibody titers in mice vaccinated with Tc24-C2 no His or Tc24-C4 no His were not significantly different from mice vaccinated with Tc24-WT no His.
Figure 10.
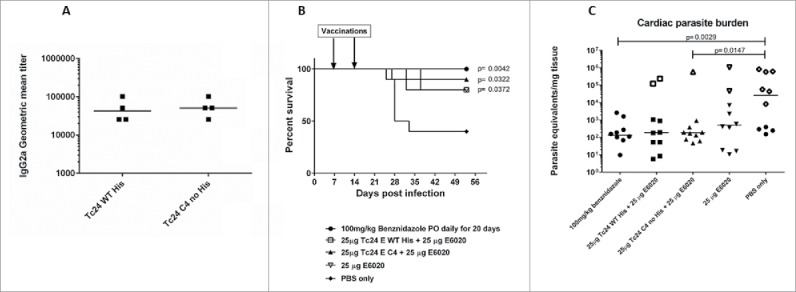
Specificity of antibodies from infected mice against wild-type and mutant Tc24 constructs, survival and tissue parasite burdens of acutely infected mice vaccinated therapeutically. Antibodies from mice infected with T. cruzi parasites were evaluated for specificity against either Tc24–WT (His-tagged) or Tc24-C4 by ELISA (Panel A). Plates were coated with either antigen and bound antibodies were detected using labeled goat anti-mouse IgG2a secondary antibodies. Geometric mean titers were calculated. Acutely infected mice were vaccinated therapeutically with either Tc24-WT (His-tagged) or Tc24-C4 combined with E6020 SE. Survival was monitored daily (Panel B) and tissue parasite burdens were measured by quantitative PCR from cardiac tissue (Panel C). Group 1 (●) treated with benznidazole; Group 2 (□) vaccinated with Tc24-WT His + E6020 SE; Group 3 (▲) vaccinated with Tc24-C4 + E6020 SE; Group 4 (▽) vaccinated with E6020 SE; Group 5 (♦) vaccinated with PBS only. Open symbols in panel C indicate mice that did not survive until the end of the study.
Discussion
Tc24, a T. cruzi 24 kDa parasite excretory-secretory protein has been selected as a lead antigen for a therapeutic Chagas vaccine under development by the Sabin Vaccine Institute Product Development Partnership (Sabin PDP). The protein expresses at both a high yield and solubility in the E. coli T7 expression system. However, aggregation of the wild-type protein became apparent which had to be resolved in order to move this Chagas vaccine development program forward. We had evidence that the aggregation observed was mostly due to the formation of disulfide bridges between Tc24 monomers since alkylation of the wild type protein almost entirely prevented aggregation. The approach selected to minimize or eliminate the aggregation observed was to modify the cysteine residues present in the molecule to serine residues which would eliminate aberrant disulfide bond formation without introducing local structural changes within the molecule. Mutagenesis of cysteine residues has been shown to reduce aggregation of Cu, Zn-superoxide dismutase,31 as well as improve the solubility of human renal dipeptidase,32 human galectin-2,34 and simvastatin synthase LovD35 expressed in E. coli. Expression of the gingipain adhesion/hemagglutinin domain (Hgp44) on the surface of Lactococcus lactis, as a vaccine candidate for periodontitis, was improved without eliminating antigenic properties.36 Changing the Tc24-WT protein in such a fashion did have the desired impact - mutation of 2 cysteine residues partially eliminated the aggregation, while elimination of all 4 cysteine residues almost completely abolished protein aggregation. This change made it possible to develop a scalable production process with greater yield and higher quality due to the elimination of aggregation and less protein lost during purification. However, further physicochemical analyses had to be performed to rule out potential structural and antigenic changes due to the mutations.
Biophysical analysis tools such as circular dichroism, light scattering, and thermal shift assays are routinely used for the critical assessment of a protein's stability and biological activity. In particular, during the identification of the optimal buffer system, the ideal pH range and during the screening for the best excipients. In this study, we used SDS-PAGE, Western blot, protein alkylation, and dynamic light scattering to show that cysteine mutagenesis eliminated the problem of protein aggregation which would have made quality control and reproducible production of the molecule very difficult. Just as important, we wanted to verify that the structure of the mutated protein was similar to the wild type protein in order to maintain equivalent antigenicity. Many other proteins have likewise been modified by cysteine mutagenesis in order to reduce aggregation and improve solubility while maintaining conformation and/or biological activity or antigenicity including human renal dipeptidase,32 simvastatin synthase,35 and a 44 kDa gingipain adhesion/hemagglutinin domain (Hgp44) which shows promise as a candidate antigen for Porphyromonas gingivalis induced periodontitis.36 Therefore we hypothesized that the cysteine mutations would not significantly impact protein structure. Using circular dichroism41,42 and thermal melt analysis,43 we show that removal of the disulfide bonds does not affect the biophysical structure of the protein significantly. CD analysis demonstrated identical secondary structures characteristic of α-helical proteins for Tc24-WT + His and Tc24-C4 (70% α helix, 7% β sheet and 23% turns). Thermal melt analysis showed that the tertiary folding between Tc24-WT + His and Tc24-C4 was comparable.
Next, we proceeded to show that Tc24-C4 had retained the immunogenic and antigenic properties of the parent molecule through in vitro and in vivo assessment. Protection studies have shown that induction of antigen specific Th1 responses, characterized by increased IFNγ production from T cells and increased serum IgG2a, results in reduced blood and tissue parasite burdens as well as decreased cardiac pathology in both acute and chronic infections.21,22,26,29,39,40,44 In the studies described here, we showed that mice vaccinated with either formulated wild-type or mutant Tc24 proteins increase serum IgG2a production and develop antigen specific Th1 immune responses upon homologous re-stimulation as evidenced by IFNƴ production from splenocytes re-stimulated in vitro. The magnitude of the IFNγ responses from mice primed in vivo with wild-type or mutant proteins were similar, indicating that the cysteine to serine mutations did not negatively impact the overall immunogenicity of the mutant proteins (Fig. 8). Additionally, when splenocytes from mice vaccinated with either the formulated Tc24-C2 or Tc24-C4 mutants were re-stimulated in vitro with wild-type proteins (heterologous re-stimulation), IFNγ production was not significantly reduced compared to homologous re-stimulation, indicating that the cysteine to serine mutation did not significantly alter the native T cell epitopes responsible for inducing the cellular response.
Serum IgG2a antibody titers to the wild-type protein in mice vaccinated with formulated Tc24-WT, the Tc24-C2 or Tc24-C4 constructs were robust, indicating that formulated untagged wild type and TC24-C4 proteins induce a similar humoral response (Fig. 9). Despite the 2 mice vaccinated with formulated Tc24-WT no His that did not appear to develop antigen specific antibody titers, the geometric mean antibody titer for the group was not significantly different from the geometric mean titer of mice vaccinated with formulated Tc24-WT + His. When comparing geometric mean titers of mice vaccinated with either formulated Tc24-C2 no His or Tc24-C4 no His to those vaccinated with formulated Tc24-WT no His, there were no statistically significant differences. Also, the fact that splenocytes from those mice secrete robust levels of IFNγ upon re-stimulation in vitro (Fig. 8) confirms that a Th1 skewed antigen specific immune response was triggered by vaccination. Taken together, the IFNγ and IgG2a data indicate that mice vaccinated with formulated Tc24-C4 are likely to develop both cellular and humoral immune responses against the native Tc24 protein upon challenge with T. cruzi parasites. This was confirmed by evaluation of serum antibody titers in mice acutely infected with T. cruzi, which showed that infected mice exposed to native Tc24 antigen developed IgG2a antibodies that recognized Tc24-WT no His and Tc24-C4 antigens equally (Fig. 10, Panel A) Further, protective efficacy of the Tc24-C4 no His protein was confirmed in acutely infected mice vaccinated therapeutically. Vaccination with either Tc24-WT no His or Tc24-C4 combined with E6020 SE significantly increased survival (Fig. 10, Panel B). Interestingly, vaccination with E6020 SE alone significantly increased survival as well, likely due to a general boosting of the immune response to native T. cruzi antigens released from the parasites during infection. However, it is important to note that only mice vaccinated with Tc24-C4 no His combined with E6020 SE had significantly reduced cardiac tissue parasite burdens (Fig. 10, Panel C). This indicates that while there is some protective effect from adjuvant alone, the presence of additional exogenous Tc24-C4 no His antigen enhances the protective effect, further reducing cardiac parasite burdens and likely tissue damage. Taken together, these data indicate that the Tc24-C4 no His antigen is both immunogenic in naïve mice, and protective in acutely infected mice. We are investigating whether the reduced aggregation observed for the Tc24-C4 protein could result in reduced B cell surface receptor engagement and subsequently less antibody production compared to the aggregated antigen. However, since aggregated protein therapeutics has been associated with adverse effects in humans,45,46 achieving a robust level of immunogenicity with the candidate vaccine to induce protection must be balanced with minimizing protein aggregates to minimize adverse effects.
By successfully eliminating protein aggregation, which would have complicated manufacture and quality control of the molecule, while simultaneously demonstrating only minor alterations on immunogenicity, we are in the process of performing technology transfer to a contract manufacturing operation for the scaled-up expression of Tc24-C4 in order to provide sufficient quantities of antigen to support ongoing efficacy studies and future Phase 1 clinical trials.
Material and methods
Construction and expression of recombinant Tc24 in E. coli
DNA coding for the full-length, wild-type T. cruzi 24 kDa excretory-secretory protein (Tc24-WT)25 was codon optimized for expression in E. coli and chemically synthesized. Constructs for Tc24-C2 and Tc24-C4 were derived from the wild-type Tc24 DNA sequence with the first 2 (C4S, C66S) or all 4 cysteine residues modified to serine residues (C4S, C66S, C74S and C124S), respectively (Fig. 2). The synthesized DNAs for Tc24-WT, Tc24-C2 and Tc24-C4 were sub-cloned in frame into the E. coli expression vector pET41a (EMD Millipore) using the restriction sites NdeI and XhoI with the GST fusion deleted. Tc24-WT was initially cloned in frame with a hexahistidine tag at the C-terminus (Tc24-WT + His). An identical Tc24-WT construct was also generated without the C-terminal His-tag in order to optimize the manufacturing process for a tag-free product. The correct insert sequence and reading frame in all recombinant plasmids were confirmed by double-stranded DNA sequencing. The sequence-confirmed recombinant plasmid DNAs were transformed into E. coli BL21 (DE3) (EMD Millipore) and recombinant protein expression was induced with 1 mM Isopropyl-β-D-1-thiogalactopyranoside (IPTG). The clone with the highest expression for each construct was chosen to create research seed stocks.
Small scale expression and purification of His-tagged Tc24-WT
Six 2.5 L Tunair shake flasks containing 1 L sterile LB medium (BD Difco) each with 30 μg/mL kanamycin were inoculated with 20 mL of an overnight seed culture of Tc24-WT + His and incubated at 37°C with agitation until the OD600 reached 0.7–0.8. The temperature was then reduced to 30°C and IPTG was added to a final concentration of 0.5 mM. After 5 hours of induction, cells were collected by centrifugation (12,227 x g, 4°C, 30 min) and biomass was collected and stored at −80°C.
Frozen biomass of Tc24-WT + His was thawed and re-suspended in extraction Buffer A1 (30 mM Tris-HCl, 500 mM NaCl, 20 mM Imidazole, pH 8.0) at a ratio of 20 mL buffer per gram of wet cell paste. For small scale expression, homogenization was performed using an EmulsiFlex-C3 high pressure homogenizer (Avestin, Inc.). The suspension was passed through the homogenizer 3 times at 15,000 psi and incubated on ice during and between passes. After extraction, the homogenate was centrifuged twice (31,000 x g, 4°C, 30 min) in order to remove insoluble debris. The supernatant was then filtered consecutively through 0.45 µm and 0.22 μm filters before being loaded, at 3 mL/min, onto 3 tandem 5 mL HiTrap IMAC Sepharose 6 FF columns (GE Healthcare), equilibrated with buffer A1. The columns were washed with 10 column volumes (CVs) buffer A1 and protein was eluted using a linear gradient over 20 CVs of buffer B1 (30 mM Tris-HCl, 500 mM NaCl, 500 mM Imidazole, pH 8.0). Peak fractions containing the Tc24-WT + His were pooled and buffer exchanged using a Sephadex G-25 (Fine) XK50 desalting column (GE Healthcare) into Q Sepharose XL (QXL) Buffer A (50 mM Tris-HCl, 30 mM NaCl, pH 7.5). The protein was loaded at 5 mL/min onto 3 tandem 5 mL HiTrap QXL columns (GE Healthcare), washed with 10 CV's of QXL buffer A, and eluted over a linear gradient of 0–100% B with QXL Buffer B (50 mM Tris-HCl, 1 M NaCl, pH 7.5). Fractions containing Tc24 were pooled and a Sephadex G25 (Fine) XK50 desalting column was used to buffer exchange into 1X PBS, pH 7.4. Protein aliquots were stored at −80°C.
Large scale expression and purification of non-tagged wild-type and mutant Tc24 proteins in E. coli
One L LB medium was added to a 2.5 L Tunair baffled shake flask and autoclaved. After media sterilization and cooling, kanamycin was added to a final concentration of 50 µg/mL. An aliquot from a frozen glycerol seed stock of Tc24-WT, Tc24-C2, or Tc24-C4, was used to inoculate the shake flask for expansion of the seed culture. After overnight incubation at 37°C and agitation at 210–240 rpm, an aliquot of the seed culture was used to inoculate a fermentation vessel containing 10 L E. coli BSM medium (5.0 g/L K2HPO4, 3.5 g/L KH2PO4, 3.5 g/L (NH4)2HPO4, 40.0 g/L glycerol, 4.0 mL/L 25% MgSO4 x 7 H2O (added after sterilization and cooling)) with 1 mL/L K-12 Bacterial Trace Salts (composed of 5.0 g/L NaCl, 1.0 g/L ZnSO4 x 7H2O, 4.0 g/L MnCl2 x 4 H2O, 4.75 g/L FeCl3 6H2O, 0.4 g/L CuSO4 5H2O, 0.575 g/L H3BO3, 0.5 g/L Na2MoO4 x 2 H2O, 7.5 mL/L 10 N H2SO4), 10 mL/L of 15 g/L CaCl2 x 2 H2O, 1 mL/L kanamycin (100 mg/mL). The amount of seed used for inoculation of the fermenter was adjusted in order to have a starting cell density (OD600) of 0.05. At the time the cell density reached approximately 0.5, the temperature was reduced to 30°C. When the cell density reached an OD600 of 0.6–1.0, IPTG was added to a final concentration of 1 mM to ensure full induction due to the high cell density achieved during fermentation. Approximately 5 hours after the start of induction, fed-batch medium (50% glycerol (v/v), 20 mM MgCl2) was added to the fermenter at a rate of 3 mL/L/hr. Agitation was set at 500 rpm, 1 vvm air flow, pH of 7.2, and DO of 30% was maintained through fermentation. After approximately 18 hours of induction, the culture was removed from the fermenter and biomass was collected by centrifugation (12,227 x g, 4°C, 45 min). The cell paste was collected and stored at −80°C prior to downstream processing.
Each gram of biomass was re-suspended in 20 mL of 50 mM Tris-HCl, pH 8.0 and mixed on a stir plate until homogeneous. Tc24-WT and Tc24-C2 were homogenized using an EmulsiFlex C3 (Avestin, Inc.) with 3–5 passes at an average pressure of 15,000 psi. The protein solution was kept cold using a heat exchanger. After extraction, the solution was centrifuged twice (31,000 x g, 4°C, 30 min). Tc24-C4 was homogenized on a larger scale using an EmulsiFlex C-55 (Avestin, Inc.) with 3–5 passes at an average pressure of 15,000 psi. The protein solution was kept cold using a heat exchanger. After extraction, the solution was centrifuged 3 times (17,700 x g, 4°C, 45 min). For Tc24-WT, Tc24-C2, and Tc24-C4, clarified supernatant was filtered using a 0.45 µm filter unit followed by a 0.2 µm filter unit and then loaded onto a Tricorn 10/200 column (GE Healthcare) packed with Q Sepharose XL resin (QXL, bed height 19.5 cm; column volume 15.3 mL) that was pre-equilibrated with approximately 5 CVs of extraction buffer. The column was washed with 2–3 CVs of 50 mM Tris-HCl pH 8.0 followed by an 8–9 CV wash with 50 mM Tris-HCl, pH 8.0, 75 mM NaCl. Tc24 was eluted from the column with 20 CVs of 50 mM Tris-HCl, pH 8.0, 125 mM NaCl. The remaining protein was removed from the column and discarded by stripping with 5 CVs of 50 mM Tris-HCl, pH 8.0, 1 M NaCl followed by 5 CVs 0.5 N NaOH. The QXL column was re-equilibrated with 50 mM Tris-HCl, 200 mM NaCl pH 8.0. To the QXL eluate, 5 M NaCl was added to bring the final concentration to approximately 200 mM. The NaCl adjusted QXL elution was passed through the re-equilibrated QXL column (negative capture) to remove residual endotoxin. The QXL flow-through was concentrated greater than 45-fold by using a Centricon Plus-70 (Millipore) with a 10 kDa molecular weight cut off, and the final volume was adjusted to approximately 4.5 mL using 50 mM Tris-HCl, 200 mM NaCl, pH 8.0. The concentrated Tc24 protein was loaded onto an SEC column (Sephacryl S-200 HR HiPrep 26/60 (GE Healthcare), previously equilibrated with 2–3 CVs 1X PBS pH 7.4. The protein was eluted using 2 CVs of 1X PBS pH 7.4. The final concentration of Tc24 protein was determined spectrophotometrically (A280) using 22.46 mM−1cm−1 as the molar extinction coefficient and 23.58, 23.61, or 23.64 kDa as the molecular weight for Tc24-WT, Tc24-C2, and Tc24-C4, respectively. Aliquots were stored at −80°C.
SDS-PAGE and Western blot analysis
For in-process and purified protein samples, SDS-PAGE and Western Blot analysis were carried out using non-reduced and reduced 4–12% Bis-Tris gradient gels (Life Technologies) in 2-(N-morpholino) ethanesulfonic acid (MES) running buffer (Life Technologies) and lithium dodecyl sulfate (LDS) sample loading buffer (Life Technologies). The reducing agent 2-mercaptoethanol (Sigma Aldrich) was added to the sample loading buffer at a concentration of 5% (v/v) prior to sample preparation. Electrophoresis was performed at 200 Volts for 35 min at room temperature and gels were then stained with 0.1% Coomassie Blue R-350 and de-stained with successive washes of 5% methanol in 10% acetic acid. Western blots were performed using in-house generated anti-Tc24 polyclonal mouse antisera at a dilution of 1:2,500 in 1X PBST (phosphate buffered saline + 0.05% Tween 20). Initial blocking of the nitrocellulose membrane was performed using 5% dry milk in 1X PBST. A goat anti-mouse (IgG) secondary antibody conjugated to alkaline phosphatase was diluted 1:7,500 in 1X PBST and used for detection. Incubations were carried out for one hour at room temperature followed by 3–5 washes using PBST. Color development was accomplished using a 5-Bromo-4-chloro-3-indolyl phosphate/Nitro Blue Tetrazolium (BCIP/NBT) membrane phosphatase substrate solution (KPL Laboratories).
For the alkylation experiments, proteins were separated on 4–20% Tris Glycine gels and transferred to nitrocellulose membranes. Membranes were blocked as described above and the primary Tc24 antibody was used at a dilution of 1:5,000 followed by a goat anti-mouse (IgG) secondary antibody conjugated to alkaline phosphatase (KPL Laboratories) used at a dilution of 1:5,000 in PBST. Incubations were carried out for one hour at RT followed by 3 washes in PBST. Color development was accomplished using a BCIP/NBT membrane phosphatase substrate solution (KPL Laboratories). For Quantitative SDS-PAGE analysis, 3–5 dilutions of protein samples were separated on 12% Bis-Tris gels along with 5 dilutions of a calibrator protein (either BSA or purified Tc24) followed by Coomassie Blue staining. After staining, gels were scanned using a GE ImageScanner II with LabScan v6.0 and ImageQuantTL v8.1 software.
Endotoxin analysis
Endotoxin assays were conducted using the Charles River Endosafe PTS System. Each sample was tested after dilution from 1:10 to 1:5,000 with endotoxin-free water using an Inhibition/Enhancement cartridge which contains a known amount of endotoxin spiked in each sample channel. This spike test checks for matrix interference that can result in either inhibition or enhancement of endotoxin detection. The lowest sample dilution from the Inhibition/Enhancement test which resulted in a spike recovery within the range 50%–200% was then used for final testing using an Endosafe®-PTS™ test cartridge with the Endosafe®PTS™ system to determine the endotoxin level present within the sample.
Mass spectrometry
Approximately 5 μg of Tc24-C4 was reduced with DTT, alkylated with iodoacetamide, and digested with trypsin for 5.5 hours. Approximately 10 ng (1X) and 50 ng (5X) protein samples were analyzed on a NanoAcquity UPLC system in-line with a Synapt mass spectrometer (Waters Corp; Milford, MA). The 5X load was performed to identify potential impurities present in the sample. Protein identification and quantification was performed using a Protein-Lynx Global Server (PLGS v3.0; Waters Corp). Peptides identified from the 1X load covered 49.76% of the Tc24-C4 sequence which confirmed identity. Peptides from the 5X load showed that the Tc24-C4 protein was 99.75% pure with only 2 minor E. coli contaminants identified.
Alkylation of Tc24-WT + His
Tc24-WT + His was diluted to 1 mg/mL in a buffer containing 100 mM Tris-HCl, pH 8.3. Dithiothreitol (DTT) was added to a final concentration of 5 mM and the sample was incubated for 30 min at 45°C to reduce the disulfide bonds. The protein mixture was then cooled to room temperature followed by the addition of iodoacetamide to 15 mM. The alkylation of the cysteine residues was continued in the dark for 30 min at room temperature. The unreacted iodoacetamide was then quenched by the addition of DTT to a final concentration of 10 mM during a 15 min incubation period. The protein was then dialyzed into 1 x PBS.
Dynamic light scattering
Dynamic light scattering (DLS) analysis was performed on a DynaPro Plate Reader (Wyatt Technology). Samples of Tc24 (Tc24-WT, Tc24-C2, and Tc24-C4, 0.5 mg/mL each) were prepared in PBS (pH 7.4) and filtered through a 0.02 µm inorganic membrane filter (Anotop 10, Whatman). Samples were measured 10 times over 5s in a UV-transparent 384 black plate (Aurora Biotechnologies). Dynamics Software package version 7.1.7.16 (Wyatt Technology) was used for data analysis.
Circular dichroism
Samples for Circular dichroism (CD) experiments were prepared by diluting purified Tc24 (Tc24-WT + His (+/− alkylated), and Tc24-C4) to a final concentration of 0.185 mg/mL. CD spectra were recorded with a Jasco J-1500s spectrophotometer, scanning from 280 nm to 185 nm at 100 nm/min with a bandwidth of 1 nm and response time of 1 sec. Experiments were performed using one quartz cuvette with a path length of 0.1 cm, keeping a constant temperature of 25°C. The average value was determined after 5 scans and the spectrum of the matching ‘buffer alone’ sample served as the control. The secondary structure of the Tc24 was predicted using the software CDPro38 by comparing with 2 reference sets (SP43and SMP56) and using 3 data fitting programs (CONTIN, SELCON3, and CDSSTR).
Protein thermal shift studies
Samples for Protein Thermal Shift Studies were prepared by diluting purified Tc24-WT + His and Tc24-C4 to a final concentration of 0.5 mg/mL. The assay was executed using Protein Thermal Shift™ reagents, ViiA™ 7 qPCR Instrument and Protein Thermal Shift™ Software v2.0 (Life technologies) according to manufacturers' protocol.
Mice, immunization, infection and treatment
Female BALB/c mice, 5–6 weeks old (Taconic Farms, Inc.) were used in all experiments. After one week of acclimation, pre-immune blood samples were taken from all mice. For immunogenicity studies, mice were then vaccinated subcutaneously (SC) with 25 μg of the selected Tc24 protein construct combined with 5 μg E6020 (Eisai Co. Ltd, a synthetic Toll-like receptor 4 (TLR-4) agonist47) emulsified in AddaVax™, a squalene-oil-in-water emulsion (InvivoGen) prior to immunization. The level of endotoxin, a potent agonist for TLR-4, was measured for each antigen and determined to be below 20 EU/mg of protein prior to use. Two weeks after prime vaccination, mice were boosted with the same vaccine formulation. Two weeks after boost, serum was collected and spleens were harvested for the evaluation of antigen specific immune responses.
T. cruzi H1 strain parasites, previously isolated from a human case in Yucatan, Mexico25 were maintained by serial passage in mice. To obtain immune serum from infected mice, naïve mice were infected with 500 trypomastigotes by intraperitoneal injection. Parasitemia was confirmed by microscopic examination of blood collected by tail vein microsampling approximately 21 d post infection. At 53 d post infection, mice were humanely euthanized by CO2 inhalation and terminal serum samples were collected to evaluate antibody responses to proteins. To determine protective efficacy of candidate vaccines, mice were infected with 5,000 T. cruzi H1 parasites as described, then vaccinated subcutaneously with 25 µg of the selected antigen combined with 25 µg of E6020 in a stable squalene emulsion (Eisai Co. Ltd.) on days 7 and 14 of infection. As a positive control for cure, one group of infected mice was treated orally with 100mg/kg benznidazole (Sigma-Aldrich) re-suspended in distilled water once daily for 20 days, starting 7 d after infection.48 Blood was collected from the tail vein twice weekly starting 7 d after infection and mice were monitored daily. Any moribund mice were humanely euthanized following AVMA Guidelines on euthanasia. At 53 d post infection, all remaining animals were humanely euthanized and heart tissue was collected for tissue parasite quantification. Total DNA was isolated from blood and cardiac tissue using a DNeasy blood and tissue kit (Qiagen) and 4 ng of DNA from blood or 50ng of DNA from cardiac tissue was used in quantitative real-time PCR.30,48-50 Parasite equivalents were calculated based on a standard curve. Parasite burdens were compared using a Mann-Whitney test and p values less than 0.05 were considered significant. For survival after infection, groups were compared to the infected untreated controls using a Log-Rank (Mantel Cox) test and p values less than 0.05 were considered significant. Animal experiments were performed in compliance with the National Institutes for Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978) and were approved by the BCM Institutional Animal Care and Use Committee (IACUC).
Interferon gamma (IFNγ) production
Splenocytes, from vaccinated mice were mechanically dissociated from spleens using a nylon screen, rinsed with DMEM medium supplemented with 5% FBS and 100 IU Penicillin/100 µg Streptomycin (complete DMEM), and pelleted by centrifugation. Red cells were lysed by incubation of the cell pellet with 1 mL ammonium-chloride-potassium (ACK) lysis solution for 5 minutes. Cells were washed once with cDMEM, then counted using a Cellometer Auto 2000 automated cell counter (Nexcelom). Antigen specific IFNγ release into the supernatant from splenocytes was quantified using an eBioscience kit following the manufacturer's protocol. Background IFNγ release was determined from non-stimulated (media only) control cells and this value was subtracted from IFNγ measured from protein re-stimulated cells to determine the antigen specific IFNγ release from splenocytes. Responses to antigen specific stimulation were compared between treatment groups using a Mann-Whitney test and Prism Graph Pad software. P values ≤ 0.05 were considered statistically significant.
Serum IgG2a antibody response
Serum antibodies specific to the Tc24-WT + His protein were measured by ELISA. 96-well NUNC High/binding ELISA plates were coated with 1.25 µg/mL recombinant Tc24-WT + His protein and then blocked with 0.1% BSA in PBST for at least 2 hrs. Serum samples serially diluted in 1 x PBS, 0.05% Tween 20 were added in duplicate and plates were incubated for 2 hrs. at room temperature. IgG2a antibody was detected with an HRP-conjugated anti-IgG2a antibody using TMB Substrate and 1 M HCl for color development. Absorbance was measured at 450 nm using a Spectra Max Plate Reader and SoftMax Pro software. Tc24-WT + His specific IgG2a titers were calculated by first subtracting the background OD450 (wells with no serum added) from each individual well followed by calculating the average OD450 from replicate wells for each sample. The positive cutoff titer, or titer representing the lower limit of detection in the ELISA assay, was identified to be 1:1,600. This is based on the observation that 1:1,600 was the lowest dilution of serum tested from experimental animals which resulted in an average OD450 value at least 3 standard deviations greater than the same dilution tested from naive animals. A positive cutoff of 2 to 3 times the SD of naïve (negative) control serum group is accepted practice.51 We chose to use 3 standard deviations rather than 2 in order to increase the specificity of the assay and to ensure that true responders were detected. For each sample, the titer was determined as the lowest dilution with an average OD450 above the positive cutoff. Geometric mean titers for each group were plotted using Graph Pad Prism software. Kruskal-Wallis ANOVA and Dunn's multiple comparisons tests were applied using Graph Pad Prism software. P values ≤ 0.05 were considered statistically significant.
Disclosure of potential conflicts of interest
Several of the authors are investigators and patent holders on vaccines against Chagas diseases and are in part supported by grants to develop these vaccines.
Acknowledgments
The authors would like to express their gratitude to Nathaniel Wolf, MLS for his assistance in the preparation and submission of this manuscript.
Funding
This work was supported by funding from the Carlos Slim Foundation, the Global Health Innovation Technology Fund (G2014-002), and the Southwest Electronic Energy Medical Research Institute (SWEMRI).
References
- [1].Haberland A, Saravia SG, Wallukat G, Ziebig R, Schimke I. Chronic Chagas disease: from basics to laboratory medicine. Clin Chem Lab Med 2013; 51:271-94; PMID:23045386; http://dx.doi.org/ 10.1515/cclm-2012-0316 [DOI] [PubMed] [Google Scholar]
- [2].Bonney KM. Chagas disease in the 21st century: a public health success or an emerging threat? Parasite 2014; 21:11; PMID:24626257; http://dx.doi.org/ 10.1051/parasite/2014012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Garcia MN, Murray KO, Hotez PJ, Rossmann SN, Gorchakov R, Ontiveros A, Woc-Colburn L, Bottazzi ME, Rhodes CE, Ballantyne CM, et al.. Development of chagas cardiac manifestations among Texas blood donors. Am J Cardiol 2015; 115:113-7; PMID:25456877; http://dx.doi.org/ 10.1016/j.amjcard.2014.09.050 [DOI] [PubMed] [Google Scholar]
- [4].Reithinger R, Tarleton RL, Urbina JA, Kitron U, Gurtler RE. Eliminating Chagas disease: challenges and a roadmap. Bmj 2009; 338:b1283; http://dx.doi.org/ 10.1136/bmj.b1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sanchez-Burgos G, Mezquita-Vega RG, Escobedo-Ortegon J, Ramirez-Sierra MJ, Arjona-Torres A, Ouaissi A, et al.. Comparative evaluation of therapeutic DNA vaccines against Trypanosoma cruzi in mice. FEMS Immunol Med Microbiol 2007; 50:333-41; PMID:17521394; http://dx.doi.org/ 10.1111/j.1574-695X.2007.00251.x [DOI] [PubMed] [Google Scholar]
- [6].Rassi A Jr, Rassi A, Little WC. Chagas' Heart Disease. Clin Cardiol 2000; 23:883-9; PMID:11129673; http://dx.doi.org/ 10.1002/clc.4960231205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rassi A Jr, Rassi A, Marin-Neto JA. Chagas Disease. Lancet 2010; 375:1388-402; PMID:20399979; http://dx.doi.org/ 10.1016/S0140-6736(10)60061-X [DOI] [PubMed] [Google Scholar]
- [8].Andrade ZA. Pathogenesis of Chagas' Disease. Res Immunol 1991; 142:126-9; PMID:1907753; http://dx.doi.org/ 10.1016/0923-2494(91)90021-A [DOI] [PubMed] [Google Scholar]
- [9].Bestetti RB, Muccillo G. Clinical course of Chagas' heart disease: a comparison with dilated cardiomyopathy. Int J Cardiol 1997; 60:187-93; PMID:9226290; http://dx.doi.org/ 10.1016/S0167-5273(97)00083-1 [DOI] [PubMed] [Google Scholar]
- [10].Hotez PJ, Bottazzi ME, Franco-Paredes C, Ault SK, Periago MR. The neglected tropical diseases of Latin America and the Caribbean: a review of disease burden and distribution and a roadmap for control and elimination. PLoS neglected tropical diseases 2008; 2:e300; PMID:18820747; http://dx.doi.org/ 10.1371/journal.pntd.0000300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Castro JA, de Mecca MM, Bartel LC. Toxic side effects of drugs used to treat Chagas' disease (American trypanosomiasis). Hum Exp Toxicol 2006; 25:471-9; PMID:16937919; http://dx.doi.org/ 10.1191/0960327106het653oa [DOI] [PubMed] [Google Scholar]
- [12].Pinazo MJ, Munoz J, Posada E, Lopez-Chejade P, Gallego M, Ayala E, del Cacho E, Soy D, Gascon J. Tolerance of benznidazole in treatment of Chagas' disease in adults. Antimicrob Agents Chemother 2010; 54:4896-9; PMID:20823286; http://dx.doi.org/ 10.1128/AAC.00537-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fabbro DL, Streiger ML, Arias ED, Bizai ML, del Barco M, Amicone NA. Trypanocide treatment among adults with chronic Chagas disease living in Santa Fe city (Argentina), over a mean follow-up of 21 years: parasitological, serological and clinical evolution. Revista Soc Bras Med Trop 2007; 40:1-10; PMID:17486245; http://dx.doi.org/ 10.1590/S0037-86822007000100001 [DOI] [PubMed] [Google Scholar]
- [14].Campos MC, Leon LL, Taylor MC, Kelly JM. Benznidazole-resistance in Trypanosoma cruzi: evidence that distinct mechanisms can act in concert. Mol Biochem Parasitol 2014; 193:17-9; PMID:24462750; http://dx.doi.org/ 10.1016/j.molbiopara.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pinazo MJ, Guerrero L, Posada E, Rodriguez E, Soy D, Gascon J. Benznidazole-related adverse drug reactions and their relationship to serum drug concentrations in patients with chronic chagas disease. Antimicrob Agents Chemother 2013; 57:390-5; PMID:23114763; http://dx.doi.org/ 10.1128/AAC.01401-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Le Loup G, Pialoux G, Lescure FX. Update in treatment of Chagas disease. Curr Opin Infect Dis 2011; 24:428-34; PMID:21857512; http://dx.doi.org/ 10.1097/QCO.0b013e32834a667f [DOI] [PubMed] [Google Scholar]
- [17].Morillo CA, Marin-Neto JA, Avezum A, Sosa-Estani S, Rassi A Jr, Rosas F, Villena E, Quiroz R, Bonilla R, Britto C, et al.. Randomized Trial of Benznidazole for Chronic Chagas' Cardiomyopathy. New England journal of Medicine 2015; 373:1295-306; PMID:26323937; http://dx.doi.org/ 10.1056/NEJMoa1507574 [DOI] [PubMed] [Google Scholar]
- [18].Dumonteil E, Bottazzi ME, Zhan B, Heffernan MJ, Jones K, Valenzuela JG, Kamhawi S, Ortega J, Rosales SP, Lee BY, et al.. Accelerating the development of a therapeutic vaccine for human Chagas disease: rationale and prospects. Expert Rev Vaccines 2012; 11:1043-55; PMID:23151163; http://dx.doi.org/ 10.1586/erv.12.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Beaumier CM, Gillespie PM, Strych U, Hayward T, Hotez PJ, Bottazzi ME. Status of vaccine research and development of vaccines for Chagas disease. Vaccine 2016; 34(26):2996-3000 [DOI] [PubMed] [Google Scholar]
- [20].Serna C, Lara JA, Rodrigues SP, Marques AF, Almeida IC, Maldonado RA. A synthetic peptide from Trypanosoma cruzi mucin-like associated surface protein as candidate for a vaccine against Chagas disease. Vaccine 2014; 32:3525-32; PMID:24793944; http://dx.doi.org/ 10.1016/j.vaccine.2014.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pereira IR, Vilar-Pereira G, Marques V, da Silva AA, Caetano B, Moreira OC, Machado AV, Bruna-Romero O, Rodrigues MM, Gazzinelli RT, et al.. A human type 5 adenovirus-based Trypanosoma cruzi therapeutic vaccine re-programs immune response and reverses chronic cardiomyopathy. PLoS Pathog 2015; 11:e1004594; PMID:25617628; http://dx.doi.org/ 10.1371/journal.ppat.1004594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bhatia V, Garg NJ. Previously unrecognized vaccine candidates control Trypanosoma cruzi infection and immunopathology in mice. Clin Vaccine Immunol 2008; 15:1158-64; PMID:18550728; http://dx.doi.org/ 10.1128/CVI.00144-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Taibi A, Espinoza AG, Ouaissi A. Trypanosoma cruzi: analysis of cellular and humoral response against a protective recombinant antigen during experimental Chagas' disease. Immunol Lett 1995; 48:193-200; PMID:8867851; http://dx.doi.org/ 10.1016/0165-2478(95)02465-4 [DOI] [PubMed] [Google Scholar]
- [24].Taibi A, Plumas-Marty B, Guevara-Espinoza A, Schoneck R, Pessoa H, Loyens M, Piras R, Aguirre T, Gras-Masse H, Bossus M, et al.. Trypanosoma cruzi: immunity-induced in mice and rats by trypomastigote excretory-secretory antigens and identification of a peptide sequence containing a T cell epitope with protective activity. J Immunol 1993; 151:2676-89 [PubMed] [Google Scholar]
- [25].Dumonteil E, Escobedo-Ortegon J, Reyes-Rodriguez N, Arjona-Torres A, Ramirez-Sierra MJ. Immunotherapy of Trypanosoma cruzi infection with DNA vaccines in mice. Infect Immun 2004; 72:46-53; PMID:14688079; http://dx.doi.org/ 10.1128/IAI.72.1.46-53.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Limon-Flores AY, Cervera-Cetina R, Tzec-Arjona JL, Ek-Macias L, Sanchez-Burgos G, Ramirez-Sierra MJ, Cruz-Chan JV, VanWynsberghe NR, Dumonteil E. Effect of a combination DNA vaccine for the prevention and therapy of Trypanosoma cruzi infection in mice: role of CD4+ and CD8+ T cells. Vaccine 2010; 28:7414-9; PMID:20850536; http://dx.doi.org/ 10.1016/j.vaccine.2010.08.104 [DOI] [PubMed] [Google Scholar]
- [27].Quijano-Hernandez IA, Bolio-Gonzalez ME, Rodriguez-Buenfil JC, Ramirez-Sierra MJ, Dumonteil E. Therapeutic DNA vaccine against Trypanosoma cruzi infection in dogs. Ann N Y Acad Sci 2008; 1149:343-6; PMID:19120245; http://dx.doi.org/ 10.1196/annals.1428.098 [DOI] [PubMed] [Google Scholar]
- [28].Quijano-Hernandez IA, Castro-Barcena A, Vazquez-Chagoyan JC, Bolio-Gonzalez ME, Ortega-Lopez J, Dumonteil E. Preventive and therapeutic DNA vaccination partially protect dogs against an infectious challenge with Trypanosoma cruzi. Vaccine 2013; 31:2246-52; PMID:23499599; http://dx.doi.org/ 10.1016/j.vaccine.2013.03.005 [DOI] [PubMed] [Google Scholar]
- [29].Martinez-Campos V, Martinez-Vega P, Ramirez-Sierra MJ, Rosado-Vallado M, Seid CA, Hudspeth EM, Wei J, Liu Z, Kwityn C, Hammond M, et al.. Expression, purification, immunogenicity, and protective efficacy of a recombinant Tc24 antigen as a vaccine against Trypanosoma cruzi infection in mice. Vaccine 2015; 33:4505-12; PMID:26192358; http://dx.doi.org/ 10.1016/j.vaccine.2015.07.017 [DOI] [PubMed] [Google Scholar]
- [30].Barry MA, Wang Q, Jones KM, Heffernan MJ, Buhaya MH, Beaumier CM, Keegan BP, Zhan B, Dumonteil E, Bottazzi ME, et al.. A therapeutic nanoparticle vaccine against Trypanosoma cruzi in a BALB/c mouse model of Chagas disease. Hum Vaccin Immunother 2016; 12:976-87; PMID:26890466; http://dx.doi.org/ 10.1080/21645515.2015.1119346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cozzolino M, Amori I, Pesaresi MG, Ferri A, Nencini M, Carri MT. Cysteine 111 affects aggregation and cytotoxicity of mutant Cu,Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J Biol Chem 2008; 283:866-74; PMID:18006498; http://dx.doi.org/ 10.1074/jbc.M705657200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].O'Dwyer R, Razzaque R, Hu X, Hollingshead SK, Wall JG. Engineering of cysteine residues leads to improved production of a human dipeptidase enzyme in E. coli. Appl Biochem Biotechnol 2009; 159:178-90; PMID:18931951; http://dx.doi.org/ 10.1007/s12010-008-8379-9 [DOI] [PubMed] [Google Scholar]
- [33].Peng D, Wang F, Li N, Zhang Z, Song R, Zhu Z, Ruan L, Sun M. Single cysteine substitution in Bacillus thuringiensis Cry7Ba1 improves the crystal solubility and produces toxicity to Plutella xylostella larvae. Environ Microbiol 2011; 13:2820-31; PMID:21895913; http://dx.doi.org/ 10.1111/j.1462-2920.2011.02557.x [DOI] [PubMed] [Google Scholar]
- [34].Wang H, He L, Lensch M, Gabius HJ, Fee CJ, Middelberg AP. Single-site Cys-substituting mutation of human lectin galectin-2: modulating solubility in recombinant production, reducing long-term aggregation, and enabling site-specific monoPEGylation. Biomacromolecules 2008; 9:3223-30; PMID:18942878; http://dx.doi.org/ 10.1021/bm800801b [DOI] [PubMed] [Google Scholar]
- [35].Xie X, Pashkov I, Gao X, Guerrero JL, Yeates TO, Tang Y. Rational improvement of simvastatin synthase solubility in Escherichia coli leads to higher whole-cell biocatalytic activity. Biotechnol Bioeng 2009; 102:20-8; PMID:18988191; http://dx.doi.org/ 10.1002/bit.22028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yamamoto I, Ishihara K, Muramatsu K, Wada Y, Kiwaki M, Kushiro A, Okuda K. Expression of Porphyromonas gingivalis gingipain antigen Hgp44 domain on surface of Lactococcus lactis. Bull Tokyo Dent Coll 2013; 54:233-41; PMID:24521549; http://dx.doi.org/ 10.2209/tdcpublication.54.233 [DOI] [PubMed] [Google Scholar]
- [37].Wingard JN, Ladner J, Vanarotti M, Fisher AJ, Robinson H, Buchanan KT, Engman DM, Ames JB. Structural Insights into Membrane Targeting by the Flagellar Calcium-binding Protein (FCaBP), a Myristoylated and Palmitoylated Calcium Sensor in Trypanosoma cruzi. J Biol Chem 2008; 283:23388-96; PMID:18559337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sreerama N, Woody RW. Computation and analysis of protein circular dichroism spectra. Methods Enzymol 2004; 383:318-51; PMID:15063656; http://dx.doi.org/ 10.1016/S0076-6879(04)83013-1 [DOI] [PubMed] [Google Scholar]
- [39].Gupta S, Garg NJ. Prophylactic efficacy of TcVac2 against Trypanosoma cruzi in mice. PLoS Negl Trop Dis 2010; 4:e797; PMID:20706586; http://dx.doi.org/ 10.1371/journal.pntd.0000797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gupta S, Garg NJ. Delivery of antigenic candidates by a DNA/MVA heterologous approach elicits effector CD8(+)T cell mediated immunity against Trypanosoma cruzi. Vaccine 2012; 30:7179-86; PMID:23079191; http://dx.doi.org/ 10.1016/j.vaccine.2012.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Colombo N, Cabrele C. Synthesis and conformational analysis of Id2 protein fragments: impact of chain length and point mutations on the structural HLH motif. J Pept Sci 2006; 12:550-8; PMID:16733829; http://dx.doi.org/ 10.1002/psc.764 [DOI] [PubMed] [Google Scholar]
- [42].Valencia A, Serrano L, Caballero R, Anderson PS, Lacal JC. Conformational alterations detected by circular dichroism induced in the normal ras p21 protein by activating point mutations at position 12, 59, or 61. Euro J Biochemistry / FEBS 1988; 174:621-7; http://dx.doi.org/ 10.1111/j.1432-1033.1988.tb14143.x [DOI] [PubMed] [Google Scholar]
- [43].Indu S, Kochat V, Thakurela S, Ramakrishnan C, Varadarajan R. Conformational analysis and design of cross-strand disulfides in antiparallel β-sheets. Proteins 2011; 79:244-60; PMID:21058397; http://dx.doi.org/ 10.1002/prot.22878 [DOI] [PubMed] [Google Scholar]
- [44].Gupta S, Garg NJ. TcVac3 induced control of Trypanosoma cruzi infection and chronic myocarditis in mice. PloS One 2013; 8:e59434; PMID:23555672; http://dx.doi.org/ 10.1371/journal.pone.0059434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang W, Singh SK, Li N, Toler MR, King KR, Nema S. Immunogenicity of protein aggregates–concerns and realities. Int J Pharm 2012; 431:1-11; PMID:22546296; http://dx.doi.org/ 10.1016/j.ijpharm.2012.04.040 [DOI] [PubMed] [Google Scholar]
- [46].Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: influence of aggregation. J Immunotoxicol 2014; 11:99-109; PMID:23919460; http://dx.doi.org/ 10.3109/1547691X.2013.821564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Singh M, Kazzaz J, Ugozzoli M, Baudner B, Pizza M, Giuliani M, Hawkins LD, Otten G, O'Hagan DT. MF59 oil-in-water emulsion in combination with a synthetic TLR4 agonist (E6020) is a potent adjuvant for a combination Meningococcus vaccine. Hum Vaccines Immunother 2012; 8:486-90; PMID:22832252; http://dx.doi.org/ 10.4161/hv.19229 [DOI] [PubMed] [Google Scholar]
- [48].Cencig S, Coltel N, Truyens C, Carlier Y. Parasitic loads in tissues of mice infected with Trypanosoma cruzi and treated with AmBisome. PLoS Negl Trop Dis 2011; 5:e1216; PMID:21738811; http://dx.doi.org/ 10.1371/journal.pntd.0001216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Caldas S, Caldas IS, Diniz Lde F, Lima WG, Oliveira Rde P, Cecilio AB, Ribeiro I, Talvani A, Bahia MT. Real-time PCR strategy for parasite quantification in blood and tissue samples of experimental Trypanosoma cruzi infection. Acta Trop 2012; 123:170-7; PMID:22609548; http://dx.doi.org/ 10.1016/j.actatropica.2012.05.002 [DOI] [PubMed] [Google Scholar]
- [50].Gangisetty O, Reddy DS. The optimization of TaqMan real-time RT-PCR assay for transcriptional profiling of GABA-A receptor subunit plasticity. J Neurosci Methods 2009; 181:58-66; PMID:19406150; http://dx.doi.org/ 10.1016/j.jneumeth.2009.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Crowther JR. The ELISA guidebook. Methods Molecular Biol 2000; 149:Iii–iv, 1-413; PMID:11028258 [DOI] [PubMed] [Google Scholar]