

ABSTRACT
Mapping out regions of PrP influencing prion conversion remains a challenging issue complicated by the lack of prion structure. The portion of PrP associated with infectivity contains the α-helical domain of the correctly folded protein and turns into a β-sheet-rich insoluble core in prions. Deletions performed so far inside this segment essentially prevented the conversion. Recently we found that deletion of the last C-terminal residues of the helix H2 was fully compatible with prion conversion in the RK13-ovPrP cell culture model, using 3 different infecting strains. This was in agreement with preservation of the overall PrPC structure even after removal of up to one-third of this helix. Prions with internal deletion were infectious for cells and mice expressing the wild-type PrP and they retained prion strain-specific characteristics. We thus identified a piece of the prion domain that is neither necessary for the conformational transition of PrPC nor for the formation of a stable prion structure.
KEYWORDS: amino acid deletion, infection, prion disease, structure
Mammalian prions consist of β-sheet-rich assemblies of the PrP protein.1,2 However resolution of their structures remains elusive due to the insolubility and heterogeneity of these aggregates. While the correctly folded protein (PrPC) contains 3 helices,3 biophysical data indicate that there is no more α-Helical content in prions (PrPSc).4 Different structural models of PrPSc were proposed, most of them postulating an alternation of β strand and loops or turns.5-7 It is thus conceivable that some stretches of the protein especially those included in the unstructured regions are not absolutely indispensable for mammalian prion. To support this hypothesis, we may recall that deletions inside the loop joining the 2 rungs of β-sheets of the solenoid were compatible with production of functional HET-s prions in Podospora Anserina.8 Whether completeness of the “90–231” segment of PrP associated with the infectivity2,9 is required for mammalian prions was not clearly answered. Indeed although many inside deletions were done, so far they failed to generate prion entities still able to convert the wild-type PrPC.10-12 We knew from our previous work that the sequence specificity of the C-terminal part of PrP helix H2 was not essential for prions, even if this sequence is highly conserved in mammalian PrP. Indeed, insertion of 8 extra amino acids in the last turns of the helix did not impair prion conversion.13 This observation suggested that the C-terminal residues of H2 were not involved in the backbone of the prion structure but might rather be, or be included into an unstructured or poorly structured part of PrPSc. Other studies indicating that sequence changes in this area appear to be compatible with prion conversion support this hypothesis.14-16 It was thus appealing to delete the region to determine the impact on PrPC structure and prion replication. We performed a series of deletions (Fig. 1) and found that removal of the last 5 residues of the helix H2 did not impair prion conversion.17 This was the first clear-cut demonstration that a stretch of residues within the prion-associated domain of PrP is dispensable to generate bona fide prions.
FIGURE 1.

Map of deletions performed in the prion-associated domain of ovine PrP. The sequence of the C-terminal part of PrP (residues 85 to 234) is indicated. Amino acids included in α helices or β-strands are in black, while those located in unstructured areas are in blue. The 2 cysteines of the disulfide bridge linking H2 to H3 are in bold and asparagines of the 2 glycosylation sites are underlined. Deletions are indicated by red lines.
The Overall Structure of PrPC is Maintained Even After Removal of One-Third of Helix H2
Structural integrity of the PrP deletion mutants was first assessed by perturbation analysis based on amide chemical shifts, which are sensitive to conformational changes. Perturbations, though wider spread with the Δ190–197 than the Δ193–196 deletion, remain localized in the H2-H3 hairpin (Fig. 2A). This was confirmed by comparison of 3D NMR structures of wild-type and mutant PrPs (Fig. 2B). The Δ193–196 deletion shortened H2 by one turn and a half, as expected, but the overall structure of the protein was preserved, which is consistent with the ability of the mutant protein to convert into prion.17 Surprisingly, the Δ190–197 deletion that removes 8 highly conserved amino acids and about one third of H2 did not substantially alter the structure of the rest of the protein. The strong lock provided by the 182C-217C disulfide bond helped maintaining the relative position of the truncated H2 with respect to H3, despite the tension induced by shortening of the H2-H3 connection. The scaffold formed by aromatic residues was slightly rearranged, but key interactions that drive stacking of H1 onto H3 was conserved. The main conclusion was that the 190–197 segment was not essential for the integrity of PrP structure. Therefore, failure in converting Δ190–197 PrPC in cells or in cell-free conversion assay by protein misfolding cyclic amplification (PMCA) was not associated with a direct effect of the deletion on the structure of the protein. This would be rather associated with the extended size of the deletion that prevents the conversion process of PrPC or the establishment of a stable misfolded PrPSc structure.
FIGURE 2.
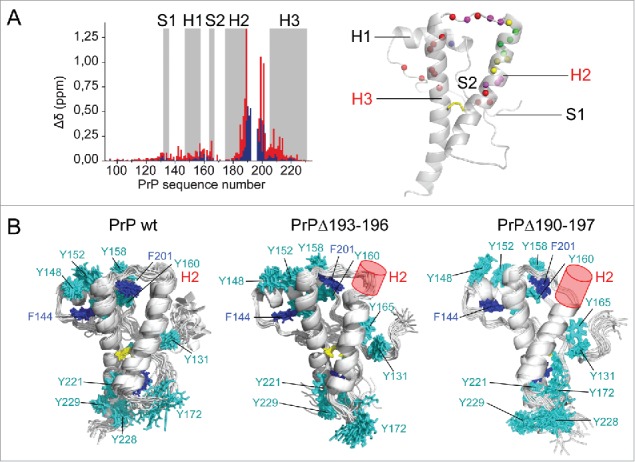
Structural analysis of deletion mutants. (A) Perturbation analysis was performed by measuring amide1H,15N chemical shift deviations (Δδ) for PrPΔ193–196 (blue) and PrPΔ190–197 (red). The results are mapped on the PrP structure (in cartoon). Colored spheres represent amide nitrogen atoms with Δδ > 0.1 ppm in blue and red for each mutant, in magenta if deviations are observed in both. Yellow and green spheres indicate deleted residues in the mutants. (B) NMR structure ensembles of wild-type PrP and deletion mutants are shown in cartoon, without the disordered N-terminus. The disulfide bond (yellow), Phe (blue) and Tyr (cyan) side chains are represented in sticks. Deletions are indicated with a red cylinder.
The C-Terminus of PrP Helix 2 Is Not Required for Prion Conversion
Ectopic expression of PrP from different mammals is known to confer prion susceptibility to RK13 cells.18 Populations of stably transfected RK13 cells were selected to express a series of ovine PrP with increasing H2 C-terminal deletions. Mutant PrPC were mainly glycosylated and correctly routed to the cell surface. Cells were exposed to prions and analyzed for proteinase K resistant PrPSc content (PrPres) on subsequent passages of the cultures. Ovine PrPC deleted of amino acids TTTT (Δ193–196) or TTTTK (Δ193–197) were successfully converted into PrPSc upon infection by each of the prion strains assayed: 127S, LA21K fast, T1Ov and T2Ov. The 127S and LA21K fast prions are derived from sheep scrapie isolates and rapidly induce a prion disease in tg338 mice overexpressing ovine PrP.19 T1Ov and T2Ov are 2 prion strains isolated on adaptation of a human sporadic CJD case to tg338 mice.20 We found that PrPΔ193–196 and PrPΔ193–197 conferred to RK13 cells the same degree of susceptibility to 127S infection than the wild-type protein.17 The levels of PrPres accumulated in cells also compared at least up to 12 passages of the cultures. The size distribution of cell-formed PrPres aggregates was assessed by sedimentation velocity and found to be the same for the wild-type and mutant proteins (Fig. 3). However we noted the presence of an additional, more N-terminally truncated PrPres fragment in cells expressing the deleted PrPs. This might reflect a stronger cell processing of ΔPrPSc or the production of some variant structures. However, PrPres species with the expected size were always predominant. Populations of cells infected by either T1Ov or T2Ov also produced high amounts of mutant PrPres from the first passage onwards and at least for 8 passages post infection. This was rather unexpected, as populations of RK13 cells expressing the wild-type PrPC were not found susceptible to T1Ov or T2Ov. Only one subclone selected for its substantially increased susceptibility to prions was found to be really permissive to these agents.20 Removal of one additional residue (V192) dramatically reduced replication of 127S prion in RK13 cells but had a weaker impact on T1Ov and T2Ov. Extending further the deletion to generate Δ190–197 conferred resistance to the 3 prion strains. Unpublished results indicate that this is the larger size of the deletion rather than the specific absence of the amino acids 190 and 191 that prevented the conversion.
FIGURE 3.
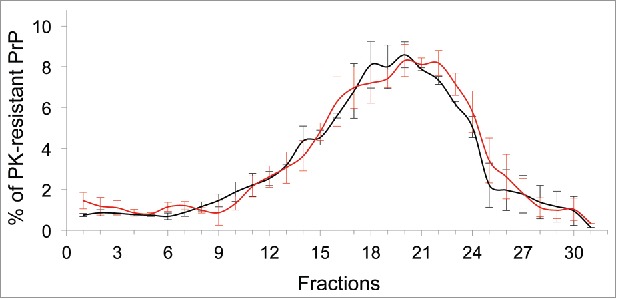
Size distribution of wild-type and mutant PrPres aggregates accumulated in infected cells. Lysates of 127S-infected cells were solubilized in detergents, centrifuged on a continuous 10–25% iodixanol gradient (Optiprep, Axys-shield) and fractionated to separate PrPres assemblies by sedimentation velocity19. Thirty fractions were recovered, PK-treated and analyzed of PrPres content by immunoblotting. The graph shows quantification of PrPres signals from the top to the bottom of the gradient for wild-type (black line) and Δ193–196 mutant (red line).
Altogether we have shown that the 193–196/7 H2 C-terminal portion is not necessary for the efficient conversion of PrPC into a self-perpetuating protease-resistant form. However we noticed that removal of these residues can introduce some effects on PrPres presentation and can even favor the replication of certain prion strains that are difficult to propagate in this cellular context, such as T1Ov and T2Ov. Also PrPins19313, a mutant with an insertion of 8 extra amino acids modifying the H2 end was found to be convertible into PrPSc following T1Ov infection, while wild-type PrPC was not. Whether modification or removal of the last turns of helix H2 facilitates somehow the unfolding of PrPC and thus its conversion by certain prion strains, remains to be determined.
Prions with an Internal Deletion Are Infectious and Transfer the Strain-Specific Information
We further showed that PrPSc lacking residues 193–196 or 193–197 were infectious for naïve homologous and wild-type PrP expressing cells. ΔPrPSc were also efficient seeds for PMCA. They produced a stereotyped prion disease upon inoculation to tg338 mice, which expressed the wild-type ovine PrP. It is commonly thought that prion strain-specific characteristics are encoded within differences in PrPSc structures or assemblies. ΔPrPSc induced a phenotype in tg338 mice that was superimposable to the parental prions used for cell culture infections. In particular, PrPres electrophoretic signature and neuroanatomical deposition in the infected mouse brain were conserved. Altogether these observations indicate that the strain-specific information was not lost through the propagation of prions on mutant PrPs. This suggested that the structural determinants of prion strains were maintained despite removal of the internal residues.
Conclusion and Perspectives
We have shown that a short portion inside the “90–231” segment of PrP is not essential to establish a stable, self-propagating prion structure and to allow PrPC to undertake the conformational change. Moreover removal of residues corresponding to the H2 C-terminus in PrPC does not impair the encoding of prion strain-specific information suggesting that these deletions have little impact, if any, on prion structure. In other words it is unlikely that residues 193–197 are included into β-sheet structures that form the backbone of prions. One interesting possibility would be that the region in between the 2 glycosylation sites remains unstructured in PrPSc, therefore size of this loop would not be critical for prions as it tolerates both insertions or deletions. Are there or not other parts of the infectivity-associated domain of PrP that are dispensable for prion structure? The answer to this question is important but represents a real challenge as the introduction of significant sequence changes, particularly in the globular domain, can alter PrPC structure or routing to the cell surface and thereby may prevent conversion even though the area might not be crucial for the structure of prions. New approaches such as the “cell-based mb-PMCA”21 might overcome some of these limitations.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
- [1].Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry 1991; 30(31):7672-80; PMID:1678278 [DOI] [PubMed] [Google Scholar]
- [2].Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95(23):13363-83; PMID:9811807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. NMR structure of the mouse prion protein domain PrP(121–231). Nature 1996; 382(6587):180-2; PMID:8700211; http://dx.doi.org/ 10.1038/382180a0 [DOI] [PubMed] [Google Scholar]
- [4].Baron GS, Hughson AG, Raymond GJ, Offerdahl DK, Barton KA, Raymond LD, Dorward DW, Caughey B. Effect of glycans and the glycophosphatidylinositol anchor on strain dependent conformations of scrapie prion protein: improved purifications and infrared spectra. Biochemistry 2011; 50(21):4479-90; PMID:21539311; http://dx.doi.org/ 10.1021/bi2003907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Requena JR, Wille H. The structure of the infectious prion protein: experimental data and molecular models. Prion 2014; 8(1):60-6. Epub 2014/March/04; PMID:24583975; http://dx.doi.org/ 10.4161/pri.28368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Groveman BR, Dolan MA, Taubner LM, Kraus A, Wickner RB, Caughey B. Parallel in-register intermolecular beta-sheet architectures for prion-seeded prion protein (PrP) amyloids. J Biol Chem 2014; 289(35):24129-42. Epub 2014/July/17; PMID:25028516; http://dx.doi.org/ 10.1074/jbc.M114.578344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Silva CJ, Vazquez-Fernandez E, Onisko B, Requena JR. Proteinase K and the structure of PrPSc: The good, the bad and the ugly. Virus Res 2015; 207:120-6; PMID:25816779; http://dx.doi.org/ 10.1016/j.virusres.2015.03.008 [DOI] [PubMed] [Google Scholar]
- [8].Ritter C, Maddelein ML, Siemer AB, Luhrs T, Ernst M, Meier BH, Saupe SJ, Riek R. Correlation of structural elements and infectivity of the HET-s prion. Nature 2005; 435(7043):844-8. Epub 2005/June/10; PMID:15944710; http://dx.doi.org/ 10.1038/nature03793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. Embo J 1996; 15(6):1255-64; PMID:8635458 [PMC free article] [PubMed] [Google Scholar]
- [10].Supattapone S, Bosque P, Muramoto T, Wille H, Aagaard C, Peretz D, Nguyen HO, Heinrich C, Torchia M, Safar J, et al.. Prion protein of 106 residues creates an artifical transmission barrier for prion replication in transgenic mice. Cell 1999; 96(6):869-78. Epub 1999/April/02; PMID:10102274; http://dx.doi.org/ 10.1016/S0092-8674(00)80596-6 [DOI] [PubMed] [Google Scholar]
- [11].Vorberg I, Chan K, Priola SA. Deletion of beta-strand and alpha-helix secondary structure in normal prion protein inhibits formation of its protease-resistant isoform. J Virol 2001; 75(21):10024-32. Epub 2001/October/03; PMID:11581371; http://dx.doi.org/ 10.1128/JVI.75.21.10024-10032.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Taguchi Y, Mistica AM, Kitamoto T, Schatzl HM. Critical significance of the region between Helix 1 and 2 for efficient dominant-negative inhibition by conversion-incompetent prion protein. PLoS Pathog 2013; 9(6):e1003466. Epub 2013/July/05; PMID:23825952; http://dx.doi.org/ 10.1371/journal.ppat.1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Salamat K, Moudjou M, Chapuis J, Herzog L, Jaumain E, Beringue V, Rezaei H, Pastore A, Laude H, Dron M. Integrity of helix 2-helix 3 domain of the PrP protein is not mandatory for prion replication. J Biol Chem 2012; 287(23):18953-64; PMID:22511770; http://dx.doi.org/ 10.1074/jbc.M112.341677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Geissen M, Mella H, Saalmuller A, Eiden M, Proft J, Pfaff E, Schätzl HM, Groschup MH. Inhibition of prion amplification by expression of dominant inhibitory mutants-a systematic insertion mutagenesis study. Infect Disord Drug Targets 2009; 9(1):40-7. Epub 2009/February/10; PMID:19200014; http://dx.doi.org/ 10.2174/1871526510909010040 [DOI] [PubMed] [Google Scholar]
- [15].Hafner-Bratkovic I, Bester R, Pristovsek P, Gaedtke L, Veranic P, Gaspersic J, Mancek-Keber M, Avbelj M, Polymenidou M, Julius C, et al.. Globular domain of the prion protein needs to be unlocked by domain swapping to support prion protein conversion. J Biol Chem 2011; 286(14):12149-56. Epub 2011/February/18; PMID:21324909; http://dx.doi.org/ 10.1074/jbc.M110.213926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shirai T, Saito M, Kobayashi A, Asano M, Hizume M, Ikeda S, Teruya K, Morita M, Kitamoto T. Evaluating prion models based on comprehensive mutation data of mouse PrP. Structure 2014; 22(4):560-71. Epub 2014/February/25; PMID:24560805; http://dx.doi.org/ 10.1016/j.str.2013.12.019 [DOI] [PubMed] [Google Scholar]
- [17].Munoz-Montesino C, Sizun C, Moudjou M, Herzog L, Reine F, Chapuis J, Ciric D, Igel-Egalon A, Laude H, Béringue V, et al.. Generating Bona Fide Mammalian Prions with Internal Deletions. J Virol 2016; 90(15):6963-75; PMID:27226369; http://dx.doi.org/ 10.1128/JVI.00555-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Vilette D. Cell models of prion infection. Vet Res 2008; 39(4):10; PMID:18073097; http://dx.doi.org/ 10.1051/vetres:2007049 [DOI] [PubMed] [Google Scholar]
- [19].Tixador P, Herzog L, Reine F, Jaumain E, Chapuis J, Le Dur A, Laude H, Béringue V, et al.. The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog 2010; 6(4):e1000859; PMID:20419156; http://dx.doi.org/ 10.1371/journal.ppat.1000859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chapuis J, Moudjou M, Reine F, Herzog L, Jaumain E, Chapuis C, Quadrio I, Boulliat J, Perret-Liaudet A, Dron M, et al.. Emergence of two prion subtypes in ovine PrP transgenic mice infected with human MM2-cortical Creutzfeldt-Jakob disease prions. Acta Neuropathol Commun 2016; 4(1):10; PMID:26847207; http://dx.doi.org/ 10.1186/s40478-016-0284-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Moudjou M, Chapuis J, Mekrouti M, Reine F, Herzog L, Sibille P, Laude H, Vilette D, Andréoletti O, Rezaei H, et al.. Glycoform-independent prion conversion by highly efficient, cell-based, protein misfolding cyclic amplification. Sci Rep 2016; 6:29116; PMID:27384922; http://dx.doi.org/ 10.1038/srep29116 [DOI] [PMC free article] [PubMed] [Google Scholar]