

Abstract
Herein we disclose a scalable organo-catalytic direct arylation approach for the regio- and atroposelective synthesis of non-C2-symmetric 2,2′-dihydroxy-1,1′-binaphthalenes (BINOLs). In the presence of catalytic amounts of axially chiral phosphoric acids, phenols and naphthols are coupled with iminoquinones via a cascade process that involves sequential aminal formation, sigmatropic rearrangement, and rearomatization to afford enantiomerically enriched BINOL derivatives in good to excellent yields. Our studies suggest that the (local) symmetry of the initially formed aminal intermediate has a dramatic impact on the level of enantioinduction in the final product. Aminals with a plane of symmetry give rise to BINOL derivatives with significantly lower enantiomeric excess than unsymmetrical ones featuring a stereogenic center. Presumably asymmetric induction in the sigmatropic rearrangement step is significantly more challenging than during aminal formation. Sigmatropic rearrangement of the enantiomerically enriched aminal and subsequent rearomatization transfers the central chirality into axial chirality with high fidelity.
Biaryl compounds that exhibit axial chirality (i.e., hindered rotation about the C–C bond) are common among natural products, pharmaceuticals, ligands, and catalysts (Figure 1A). The ease of racemization in enantiopure biaryls depends on the magnitude of the rotational barrier, which is determined by both the size and number of substituents at the ortho positions flanking the aryl–aryl bond.1 During the past two decades, both C2- and non-C2-symmetric axially chiral biaryl compounds1a–c (e.g., BINAP, BINOL, BINAM, NOBIN, and their derivatives; Figure 1A) have played key roles as ligands for transition metals in the development of catalytic enantioselective transformations.2 In addition to their role as “privileged chiral catalysts”,2j recently it was recognized that controlling the chirality of functionalized biaryl structures will have enormous implications in the future development of pharmaceuticals.3 In view of the importance of biaryls, it is surprising that relatively few methods are available for their atroposelective synthesis in an operationally simple and scalable fashion.4 Current strategies5 include classical resolution of racemic biaryls, desymmetrization of preformed prochiral biaryls, dynamic kinetic resolution of rapidly racemizing preformed chiral biaryls,5c,i transition-metal-catalyzed aryl–aryl coupling,4b,5d de novo construction of an aromatic ring,5b,j and central-to-axial chirality exchange5f,h,k (Figure 1B).
Figure 1.

Organocatalytic atroposelective direct arylation of hydroxyarenes to afford non-C2-symmetric BINOLs.
As part of an ongoing program in the Kürti group to develop new and practical transition-metal-free direct arylation methods for the preparation of highly functionalized symmetrical and unsymmetrical biaryls,6a,5h,6b we recently successfully exploited quinone and iminoquinone monoacetals as arylating agents to access both BINOL- and NOBIN-type functionalized biaryls that are atropoisomeric but non-C2-symmetric from phenols and naphthols under organocatalytic conditions (1 + 2 → 5; Figure 2A).6b We also briefly explored the possibility of using chiral BINOL-derived phosphoric acids as catalysts to obtain the biaryl products in an enantiomerically enriched form. Unfortunately, although moderate to good isolated yields were achieved, the level of enantioinduction was very poor (3–10% enantiomeric excess, ee), which we partially attributed to interference by the MeOH liberated during the formation of the mixed-acetal intermediate (1 + 2 → 3; Figure 2A);6b addition of 3 Å molecular sieves (MS) did not improve the ee. We observed similar interference by proton donors (i.e., H2O) during the catalytic synthesis of BINAM derivatives;5h in that case the addition of 4 Å MS was advantageous.
Figure 2.

Development of our catalytic atroposelective direct arylation approach to non-C2-symmetric BINOL derivatives (A,6b B) and a recent report by Tan and co-workers (C).5k
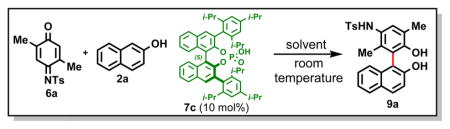
On the basis of these findings,6b we decided to redesign the quinone monoacetal coupling partner in a way that avoids the generation of a proton source (i.e., MeOH), thereby removing this potentially detrimental factor from the catalytic cycle.7 We selected readily available N-sulfonyl-protected iminoquinones as substrates, as these were expected to undergo acid-catalyzed in situ aminal formation8 and subsequent [3,3]-rearrangement/rearomatization (Figure 2B). In contrast, a 1,4-addition mechanism was recently proposed by Tan and co-workers for a related reaction (Figure 2C).5k We were pleased to observe that a 10 mol % loading of chiral phosphoric acid 7b catalyzed the coupling of 6a with 2a under mild conditions and led to the formation of functionalized biaryl 9a in excellent isolated yield with 49% ee. Encouraged by this initial result, we conducted a survey of structurally diverse BINOL-derived chiral phosphoric acid catalysts2d,9 and solvents to find the optimum conditions that maximize the enantiomeric excess of the product (Table 1; see the Supporting Information (SI) for details).
Table 1.
Catalyst Screen for the Atroposelective Synthesis of 9a from 6a and 2a
| ||||
|---|---|---|---|---|
| Entrya | Solvent | Time (h) | erb | ee (%) |
| 1 | CH2Cl2 | 48 | 88.5:11.5 | 77 |
| 2 | toluene | 48 | 86:14 | 72 |
| 3 | 1,2-dichloroethane (DCE) | 48 | 94:6 | 88 |
| 4 | CHCl3 | 48 | 90.5:9.5 | 81 |
|
| ||||
| 5 | chlorobenzene | 84 | 97:3 | 94 |
|
| ||||
| 6 | 1,3-di-CF3-benzene | 60 | 93:7 | 86 |
|
| ||||
| 7c | 1,2-dichloroethane (DCE) | 24 | 94:6 | 88 |
|
| ||||
| 8c | chlorobenzene | 48 | 96:4 | 92 |
| 9d | 1,2-dichloroethane (DCE) | 8 | 91:9 | 82 |
| 10c,e | 1,2-dichloroethane(DCE) | 100 | 88.5:11.5 | 77 |
Reaction conditions: 2a (0.075 mmol), 6a (0.05 mmol), cat. (10 mol %), solvent (1 mL). Reactions were stopped when all of the 6a was consumed.
Enantiomeric ratios were determined by HPLC analysis.
Reacted at 50 °C.
Reacted at 80 °C.
Using 5 mol % 7c.
On the basis of the optimization studies described in Table 1, we selected DCE as the preferred solvent, chiral phosphoric acid 7c (at 10 mol % loading) as the preferred catalyst, and either 25 or 50 °C as the optimum reaction temperature. At first we evaluated the coupling of iminoquinone 6a with 14 structurally different hydroxyarenes, including 11 naphthols (Table 2, entries 1–11) and three monocyclic phenols (Table 2, entries 12–14).
Table 2.
Exploration of the Substrate Scope by Coupling of 6a with Structurally Diverse Hydroxyarenes

|
Reaction conditions: 2 (0.225 mmol), 6a (0.15 mmol), cat. 7c (10 mol %), DCE or chlorobenzene (3 mL), rt or 50 °C, unless indicated otherwise.
Reactions were stopped when all of the 6a was consumed.
Determined by HPLC analysis.
Compound 9ea was obtained in 92% yield with 94% ee when the reaction was performed on a 3 mmol scale in chlorobenzene.
At 50 °C over 2 h the reaction proceeded in 97% yield to afford 9na with 21% ee.
2a:6a ratio = 2.5:1; the absolute configuration of 9ea’ is (R,R).
For 2-naphthols, the biaryl products were formed in good to excellent yields, and the observed enantioselectivities ranged between 78 and 96% ee. No clear pattern showing how electron-withdrawing and electron-donating groups on the naphthalene ring influence the level of enantioinduction can be discerned.
The nearly perfect enantioselectivity (99% ee) obtained during the formation of terphenyl compound 9ea’ from 2,3-dihydroxynapthalene (Table 2, entry 15) is remarkable and suggests that significant substrate stereocontrol occurs as the second biaryl linkage is established (see the discussion in the SI).
With one exception (Table 2, entry 14), monocyclic phenols afforded good levels of enantioinduction (entries 12 and 13). In a few cases (entries 1 and 5), using chlorobenzene as the solvent instead of DCE improved the enantioselectivity.
Next, we explored how structural changes (i.e., symmetry as well as size and nature of the substituents) in the iminoquinone (6b–j) influence the yield and enantioselectivity of the biaryl product (Table 3). Among the unsymmetrical iminoquinones 6b–e that were coupled with 2,3-dihydroxynaphthalene (2e), the presence of a large substituent (i.e, i-Pr) at the ortho position of the N-Ts imine moiety led to a somewhat lower isolated yield and ee for 9ed (entry 18) relative to isomeric 9eb and 9ec (entries 16 and 17) with a smaller Me substituent at the ortho position. Presumably, the larger i-Pr group slows aminal formation and lowers the enantioselectivity of this step. The nature of the acyl/sulfonyl group on the N atom also appears to be important: the ee increases as more electron-withdrawing groups are used (i.e., Ts ≥ Ms > Ac > p-NO2-benzoyl; compare 9aa in Table 1 with entries 16, 23, and 24 in Table 2).
Table 3.
Expansion of the Substrate Scope by Coupling of Structurally Diverse Iminoquinones

|
Reaction conditions: 2 (0.225 mmol), 3a (0.15 mmol), cat. 1c (10 mol %), DCE or chlorobenzene (3 mL), rt or 50 °C.
Reactions were stopped when all of the 6b–j was consumed.
Determined by HPLC analysis.
Compound 9aj’ was found to racemize easily: even at room temperature overnight, the initial 25% ee decreased to 12% ee.
The most dramatic drop in the level of enantioinduction occurred when symmetrical (6f) and pseudosymmetrical (6g) iminoquinones were utilized as coupling partners (Table 3, entries 20 and 21). In fact, the poor ee’s observed for biaryls 9ef and 9eg provided us with a very valuable clue that helped us establish whether indeed aminal formation as opposed to 1,4-addition is involved in the key stereochemistry-determining step.
On the basis of several experimental findings (Figure 3), it appears that in catalytic enantioselective processes where sequential chirality-transfer steps are involved, the highest level of enantioinduction will most likely take place in those cases where the catalyst does not “miss/skip” an opportunity to transfer chiral information. One way to “lose” or “skip” an opportunity for chirality transfer is when one or more symmetrical intermediates are formed along the pathway (see Figure 3A,C). Naturally, symmetrical (i.e., prochiral) intermediates can be desymmetrized using chiral catalysts, but the level of enantioinduction in these desymmetrizations must be very high, which is often difficult to achieve. In particular, organocatalytic asymmetric versions of the Claisen rearrangement are challenging, and there are only a few highly enantioselective examples in the literature.10
Figure 3.

Symmetries of intermediates in chirality transfer processes have a dramatic impact on the final products’ ee.
In light of the enantioinduction levels for biaryls 9ea (96% ee) and 9eg (21% ee), we can make a convincing mechanistic case for the involvement of sequential aminal formation/[3,3]-rearrangement. Figure 4 clearly shows that if a direct 1,4-addition were operational, the influence of the highlighted extra methyl group could not account for the dramatic loss of enantioselectivity.
Figure 4.
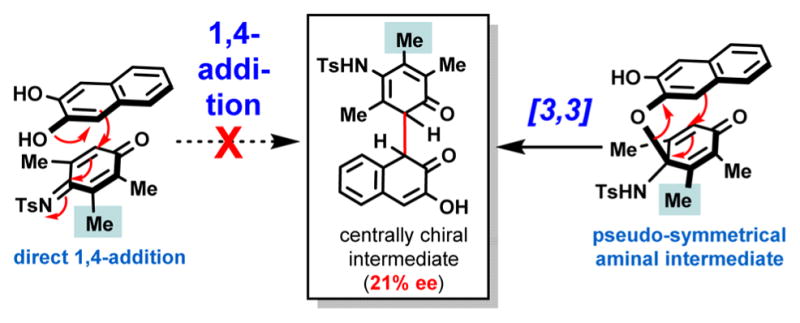
Case is made for the aminal-formation/[3,3]-rearrangement sequence as opposed to a direct 1,4-addition.
In conclusion, we have successfully developed a practical organocatalytic atroposelective synthesis of non-C2-symmetric BINOL derivatives starting from readily available hydroxyarenes and iminoquinones. The nearly two dozen axially chiral and structurally diverse functionalized biaryl products represent new chemical space and are expected to find broad utility in asymmetric catalysis, drug discovery, and materials science.
Supplementary Material
Acknowledgments
Financial support from the National Natural Science Foundation of China (81373303, 81473080, 81573299, and 21502230) is gratefully acknowledged. This project was also supported by the Jiangsu Province Natural Science Foundation (BK20150688), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT1193). L.K. gratefully acknowledges the generous financial support from Rice University, NIH (R01 GM-114609-01), NSF (CAREER:-SusChEM CHE-1455335), the Robert A. Welch Foundation (Grant C-1764), ACS-PRF (Grant 51707-DNI1), Amgen (2014 Young Investigators’ Award for L.K.), and Biotage (2015 Young Principal Investigator Award).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b01458.
Procedures and characterization data (PDF)
Crystallographic data for 9ea’ (CIF)
References
- 1.(a) Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Garner J, Breuning M. Angew Chem, Int Ed. 2005;44:5384. doi: 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]; (b) Wolf C. Dynamic Stereochemistry of Chiral Compounds: Principles and Applications. Chapter 3 Royal Society of Chemistry; Cambridge, U.K: 2007. [Google Scholar]; (c) Bringmann G, Gulder T, Gulder TAM, Breuning M. Chem Rev. 2011;111:563. doi: 10.1021/cr100155e. [DOI] [PubMed] [Google Scholar]; (d) Zask A, Murphy J, Ellestad GA. Chirality. 2013;25:265. doi: 10.1002/chir.22145. [DOI] [PubMed] [Google Scholar]
- 2.(a) Chen Y, Yekta S, Yudin AK. Chem Rev. 2003;103:3155. doi: 10.1021/cr020025b. [DOI] [PubMed] [Google Scholar]; (b) Kocovsky P, Vyskocil S, Smrcina M. Chem Rev. 2003;103:3213. doi: 10.1021/cr9900230. [DOI] [PubMed] [Google Scholar]; (c) Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. Springer; New York: 2004. [Google Scholar]; (d) Akiyama T. Chem Rev. 2007;107:5744. doi: 10.1021/cr068374j. [DOI] [PubMed] [Google Scholar]; (e) Blaser H-U, Federsel H-J, editors. Asymmetric Catalysis on Industrial Scale: Challenges, Approaches, and Solutions. 2. Wiley-VCH; Weinheim, Germany: 2010. [Google Scholar]; (f) Corey EJ, Kürti L. Enantioselective Chemical Synthesis: Methods, Logic and Practice. Direct Book Publishing; Dallas, TX: 2010. [Google Scholar]; (g) Ojima I, editor. Catalytic Asymmetric Synthesis. 3. Wiley; Hoboken, NJ: 2010. [Google Scholar]; (h) Busacca CA, Fandrick DR, Song JJ, Senanayake CH. Adv Synth Catal. 2011;353:1825. [Google Scholar]; (i) Magano J, Dunetz JR. Chem Rev. 2011;111:2177. doi: 10.1021/cr100346g. [DOI] [PubMed] [Google Scholar]; (j) Zhou Q-L, editor. Privileged Chiral Ligands and Catalysts. Wiley-VCH; Weinheim, Germany: 2011. [Google Scholar]
- 3.(a) Clayden J, Moran WJ, Edwards PJ, LaPlante SR. Angew Chem, Int Ed. 2009;48:6398. doi: 10.1002/anie.200901719. [DOI] [PubMed] [Google Scholar]; (b) LaPlante SR, Edwards PJ, Fader LD, Jakalian A, Hucke O. ChemMedChem. 2011;6:505. doi: 10.1002/cmdc.201000485. [DOI] [PubMed] [Google Scholar]; (c) LaPlante SR, Fader LD, Fandrick KR, Fandrick DR, Hucke O, Kemper R, Miller SPF, Edwards PJ. J Med Chem. 2011;54:7005. doi: 10.1021/jm200584g. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wallace TW. Org Biomol Chem. 2006;4:3197. doi: 10.1039/b608470m. [DOI] [PubMed] [Google Scholar]; (b) Kozlowski MC, Morgan BJ, Linton EC. Chem Soc Rev. 2009;38:3193. doi: 10.1039/b821092f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bencivenni G. Synlett. 2015;26:1915. [Google Scholar]; (d) Ma G, Sibi MP. Chem -Eur J. 2015;21:11644. doi: 10.1002/chem.201500869. [DOI] [PubMed] [Google Scholar]; (e) Wencel-Delord J, Panossian A, Leroux FR, Colobert F. Chem Soc Rev. 2015;44:3418. doi: 10.1039/c5cs00012b. [DOI] [PubMed] [Google Scholar]; (f) Loxq P, Manoury E, Poli R, Deydier E, Labande A. Coord Chem Rev. 2016;308:131. [Google Scholar]
- 5.(a) Bringmann G, Breuning M, Pfeifer RM, Schenk WA, Kamikawa K, Uemura M. J Organomet Chem. 2002;661:31. [Google Scholar]; (b) Tanaka K. Chem - Asian J. 2009;4:508. doi: 10.1002/asia.200800378. [DOI] [PubMed] [Google Scholar]; (c) Gustafson JL, Lim D, Miller SJ. Science. 2010;328:1251. doi: 10.1126/science.1188403. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shen X, Jones GO, Watson DA, Bhayana B, Buchwald SL. J Am Chem Soc. 2010;132:11278. doi: 10.1021/ja104297g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Cozzi PG, Emer E, Gualandi A. Angew Chem, Int Ed. 2011;50:3847. doi: 10.1002/anie.201008031. [DOI] [PubMed] [Google Scholar]; (f) Guo F, Konkol LC, Thomson RJ. J Am Chem Soc. 2011;133:18. doi: 10.1021/ja108717r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) De CK, Pesciaioli F, List B. Angew Chem, Int Ed. 2013;52:9293. doi: 10.1002/anie.201304039. [DOI] [PubMed] [Google Scholar]; (h) Li GQ, Gao H, Keene C, Devonas M, Ess DH, Kürti L. J Am Chem Soc. 2013;135:7414. doi: 10.1021/ja401709k. [DOI] [PubMed] [Google Scholar]; (i) Mori K, Ichikawa Y, Kobayashi M, Shibata Y, Yamanaka M, Akiyama T. J Am Chem Soc. 2013;135:3964. doi: 10.1021/ja311902f. [DOI] [PubMed] [Google Scholar]; (j) Link A, Sparr C. Angew Chem, Int Ed. 2014;53:5458. doi: 10.1002/anie.201402441. [DOI] [PubMed] [Google Scholar]; (k) Chen YH, Cheng DJ, Zhang J, Wang Y, Liu XY, Tan B. J Am Chem Soc. 2015;137:15062. doi: 10.1021/jacs.5b10152. [DOI] [PubMed] [Google Scholar]
- 6.(a) Gao H, Ess DH, Yousufuddin M, Kürti L. J Am Chem Soc. 2013;135:7086. doi: 10.1021/ja400897u. [DOI] [PubMed] [Google Scholar]; (b) Gao H, Xu QL, Keene C, Yousufuddin M, Ess DH, Kürti L. Angew Chem, Int Ed. 2016;55:566. doi: 10.1002/anie.201508419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Intriguingly, when the coupling between 2a and 6a to form biaryl 9a was conducted in the presence of catalyst 7c (10 mol %) and 1 equiv of MeOH (in DCE as the solvent), no detrimental impact on either the isolated yield (97%) or the enantiomeric excess (86% ee) was observed. However, when 20 equiv of MeOH was added, the reaction slowed dramatically and the isolated yield and ee dropped to 35% and 62%, respectively (at 50% conversion).
- 8.Li G, Fronczek FR, Antilla JC. J Am Chem Soc. 2008;130:12216. doi: 10.1021/ja8033334. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kampen D, Reisinger CM, List B. Top Curr Chem. 2010;291:395. doi: 10.1007/978-3-642-02815-1_1. [DOI] [PubMed] [Google Scholar]; (b) Terada M. Synthesis. 2010;2010:1929. [Google Scholar]; (c) Parmar D, Sugiono E, Raja S, Rueping M. Chem Rev. 2014;114:9047. doi: 10.1021/cr5001496. [DOI] [PubMed] [Google Scholar]
- 10.(a) Uyeda C, Roetheli AR, Jacobsen EN. Angew Chem, Int Ed. 2010;49:9753. doi: 10.1002/anie.201005183. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rodrigues TCAF, Silva WA, Machado AHL. Curr Org Synth. 2015;12:795. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.