

Abstract
This study investigates the respirability and efficacy of peptide-micelle hybrid nanoparticles as carriers for inhalational therapy of pulmonary arterial hypertension (PAH). CARSKNKDC (CAR), a cell penetrating and lung homing peptide, conjugated DSPE-PEG micelles containing fasudil, an investigational anti-PAH drug, were prepared by solvent evaporation method and characterized for various physicochemical properties. The pharmacokinetics and pharmacological efficacy of hybrid particles containing fasudil were evaluated in healthy rats and monocrotaline induced PAH rats, respectively. CAR-micelles containing fasudil had an entrapment efficiency of ∼58%, showed controlled release of the drug, and were monodispersed with an average size of ∼14nm. NMR scan confirmed the drug's presence in the core of peptide-micelle hybrid particles. Compared with plain micelles, CAR peptide increased the cellular uptake by ∼1.7-fold and extended the drug half-life by ∼5-fold. The formulations were more prone to accumulate in the pulmonary vasculature than in the peripheral blood, which is evident from the ratio of the extent of reduction of pulmonary and systemic arterial pressures. On the whole, this study demonstrates that peptide-polymer hybrid micelles can serve as inhalational carriers for PAH therapy.
Keywords: micelle, targeted drug delivery, pulmonary delivery/absorption, toxicity, nanotechnology, controlled release, encapsulation, phospholipids, in-vitro models, formulation
Introduction
Block copolymers, composed of hydrophilic and hydrophobic segments, band together in water to form micelles that are thermodynamically favorable molecular aggregates. This molecular banding or assembly follows a specific pattern: the hydrophobic segments orient inward to form the core and hydrophilic parts position outward in the aqueous phase. These donut-like nanostructures are easy to prepare with sizes ranging between 10 and 100 nm, encapsulate a variety of substances,1 and alter the pharmacokinetics and bio-distribution of therapeutic agents. Micellar carriers also improve efficacy, promote specificity, and enhance safety of encapsulated therapeutic agents.2 Of the various micellar formulations, micelles made of polyethylene glycol-distearoyl-phosphoethanolamine conjugate (DSPE-PEG) have been used to deliver a number of drugs including calcitonin, sirolimus, polymyxin B and indisulam.3-6 DSPE-PEG micelles are preferred over micelles made of conventional amphiphilic polymers because the former are more stable in-vivo and can form micelles at very low concentrations. As the critical micelle concentration (CMC) of DSPE-PEG is very low (∼10-6M), DSPE-PEG offers many advantages over amphiphiles with higher CMCs. In fact, high CMC amphiphiles tend to dissociate into monomers and make micellar core leaky. PEG chains in DSPE-PEG grant the micelles a covert property that empowers DSPE-PEG micelles to evade macrophageal uptake and extend drug circulation time in the blood. PEG chains also allow functionalization with a number of groups such as maleimide, carboxylic acid, amine, and folate. Functionalized micelles can be easily modified with various targeting devices including peptides and antibodies. Importantly, DSPE-PEG is an US FDA approved pharmaceutical additive.7
Micelles made of DSPE-PEG have been used in the delivery of various drugs such as rifampicin, budesonide, paclitaxel and beclomethasone for the treatment of a number of respiratory diseases. In terms of stability, targeting efficiency and prolongation of drugs' half-lives, these micelles are superior to their conventional counterparts such as microparticles and liposomes.8-11 For example, intratracheally administered paclitaxel-loaded DSPE-PEG micelles showed a greater accumulation in rat lungs compared with microparticles, solid lipid nanoparticles or lipid nanocapsules containing paclitaxel.5 Local concentration elevates when a drug is administered directly to the lung because of lung's reduced enzymatic activity and drug's avoidance of first-pass-loss. Thus, we and others have shown that respiratory diseases such as asthma, tuberculosis, and pulmonary hypertension can be treated safely and efficaciously by delivering drugs directly to the lung. Local lung delivery is preferred over systemic administration because only a fraction of the systemically administered drug enters and concentrates in the lung.
Inhalable nanocarriers are used to enhance drug concentration in the lung and treat various respiratory diseases. We have, recently, shown that nanocarriers such as liposomes, magnetic nanoparticles and erythrosomes can extend drug half-life when administered as aerosolized droplets into the lung.12,13 However, the pulmonary administration of drug encapsulated in unmodified nanocarriers results in elevated levels of drug in the blood that generally stems from rapid migration of the formulations from the alveolar region to the peripheral blood. Indeed, short residence time, fast clearance, and non-specific distribution are major impediments toward the success of inhalational delivery of nanocarrier based formulations.
Particles can be modified or engineered to extend lung residence time, retard respiratory clearance, and improve site specificity. One of these approaches is to modify particle surface with peptides that has a propensity to bind with cell surface molecules or penetrate the cells. Thus, we hypothesize that intratracheal delivery of surface engineered peptide-micelle hybrid particles containing fasudil extends lung residence time and amplifies site specificity of the drug for superior therapeutic efficacy. To test this, we chose fasudil as our model drug that has been used in the treatment of PAH.14 We propose to encapsulate fasudil in the micelles and modify micellar surface with a cyclic peptide, CARSKNKDC (CAR). This peptide has a propensity to accumulate in PAH lungs owing to its affinity for cell surface heparan sulfate.15 Thus, we prepared fasudil containing CAR-micelles, optimized them for physicochemical properties, and evaluated drug absorption in healthy rats. We also studied the pharmacological efficacy in a widely used rat model of PAH and investigated the safety of the particles for inhalational delivery.
Experimental Section
Materials
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene-glycol)-5000] (DSPE-PEG5000) and DSPE-PEG2000-maleimide were purchased from Avanti Polar Lipids, Inc. (Alabaster, Alabama). CAR peptide was purchased from LifeTein, LLC. (South Plainfield, NJ). Fasudil hydrochloride and monocrotaline (MCT) were from LC labs, Inc. (Woburn, MA) and Sigma-Aldrich, Inc. (St. Louis, MO), respectively. All other chemicals including chloroform, methanol, phosphate buffered saline, acetonitrile and dimethyl sulfoxide (DMSO) were of analytical grade and obtained from various vendors in the United States. All chemicals were used without further purification.
Preparation of CAR conjugated micelles containing fasudil
Fasudil loaded micelles were prepared by solvent evaporation and hydration method as reported earlier.16 To prepare various formulations, DSPE-PEG5000 concentration was varied (5-30 mg/ml) and the fasudil concentration was kept constant at 10 mg/ml. DSPE-PEG2000-maleimide (0.5%) was incorporated to conjugate CAR peptide with the micelles. Briefly, fasudil and pegylated lipids were dissolved in methanol (2 mL) and mixed in a round bottom flask followed by removal of organic solvent using a rotary evaporator (Buchi Rotavaporator® R-114; BUCHI Labortechnik AG, Switzerland) at 40°C. The thin film of drug-micelle material mixture was further dried under vacuum to remove the residues of the organic solvents. The dried film was hydrated in 2 mL distilled water by incubating the mixture in water bath for 20 min at 40°C. Un-encapsulated fasudil was removed by syringe filtration (0.22 μm polyvinylidene fluoride filters) followed by centrifugation at 10,000 rpm for 15 min. Micelles containing fasudil were incubated with a solution of CAR peptide in PBS for 30 min in the dark. Unconjugated peptide was removed as described above and the purified micelles were then freeze dried.
Physicochemical characterization
The particle size, polydispersity index (PDI), and zeta potential of micelles were measured by a Malvern® Zetasizer (Malvern® Instruments Limited, Worcestershire, UK). The amount of fasudil encapsulated in micelles was determined by disrupting a known amount of formulation (5 mg) in methanol and measuring the released drug at 320 nm using a UV spectrophotometer. The entrapment efficiency was calculated using the following equation: Entrapment efficiency (%) = (Amount of drug in micelles/Amount of drug originally added) × 100.
Freeze-dried fasudil loaded micelles were tested for stability after storage at 4°C for 21 days. Samples withdrawn on Day 0, 7, 14 and 21 were evaluated for changes in vesicle size and entrapment efficiency as described above. Further, an eight-stage Marc-II Andersen Cascade Impactor (Westech Instruments Inc., Marietta, Georgia) were used to determine mass median aerodynamic diameter (MMAD) and fine particle fraction (FPF) of the micelles.17,18 First, the pre-weighed glass-fiber filter papers were put on the plates of impactor; a HandiHaler® (Pfizer) fired the dry micellar formulation, at a flow rate of 28.3 L/min, into the impactor. The weight of micelles deposited on each stage was determined by subtracting the weight of filter paper before and after firing of the formulation. A logarithmic plot of % cumulative amount of micelles deposited on each stage versus effective cut-off diameter (μm) of each stage was used to calculate the particle MMAD. The total amount of micelles deposited on stage 3 was considered as FPF. An aliquot of micelles collected from stage 1 were suspended in PBS and analyzed for changes in size and entrapment efficiency as described above. The stability of micelles during nebulization was also assessed. Briefly, a suspension of micelles was loaded in a Hamilton Syringe® attached with a PennCentury™ Microsprayer® (Model IA-1B, PennCentury, PA) and sprayed five times followed by collecting and analyzing the sample for particle size, polydispersity index, and entrapment efficiency.
NMR spectroscopic studies
Formulations were scanned in a NMR spectrophotometer to confirm the encapsulation of fasudil in the micelles. 1H NMR scans of fasudil, DSPE-PEG5000, and freeze-dried micelles containing fasudil in deuterated water (D2O) and methanol (CD3OD) were recorded in an AVANCE™ III HD NMR Spectrometer operating at 400 MHz (Bruker, Billerica, MA). NMR spectra of fasudil in CD3OD, DSPE-PEG5000 in D2O, and freeze-dried micelles containing fasudil in D2O and CD3OD were obtained and evaluated for characteristic resonance peaks.
In-vitro release studies
Drug release studies were performed in a simulated lung fluid (SLF) according to our previous publication using Moss formula plus 0.1% Tween 80 at pH 7.4.17 Briefly, 5 mg of freeze-dried micelles were suspended in 1 mL PBS and loaded onto dialysis bags (MWCO 3.5 kD). The dialysis bag was immersed in 1.5 L SLF to maintain the sink conditions and stirred at 500 rpm at 37±1 °C. An aliquot of the samples were withdrawn at various time points and replaced with an equal volume of SLF. The drug amount was then assayed in an UV spectrophotometer at 320 nm.
Cellular uptake and cytotoxicity of micelles
Rat pulmonary arterial smooth muscle (PASM) cells were used for cellular uptake and cytotoxicity studies.12 Briefly, rat PASM cells were seeded on a cover slip placed in a 12-well plate. Prior to the uptake study, cells were activated with tissue growth factor-beta (TGF-β, 10 ng/mL) for 12 hrs, and then treated with plain and CAR conjugated micelles for 60 min. CAR peptide was tagged with green fluorescent 5-carboxyfluorescein (FAM) moiety to visualize the micelles under a microscope; plain micelles were loaded with fluorescein isothiocyanate-dextran (FITC-dextran). Cells were washed thoroughly, fixed and stained red for actin cytoskeleton by sequential incubation in monoclonal anti-β-actin primary antibody (Sigma-Aldrich, St. Louis, MO) and Alexa Fluor® 594 goat anti-mouse IgG antibody (Invitrogen, Grand Island, NY). Cellular uptake was evaluated under a fluorescence microscope (Olympus IX-81, Center Valley, PA).
The cellular uptake of CAR-micelles was further quantitated using a flow cytometer. Following the treatment, the cells were washed, trypsinized and suspended in PBS. The flow cytometer, (BD Accuri™ C6, BD Biosciences, San Jose, CA) was set at FL-3 channel to count 10,000 cells per sample and measure the fluorescence intensity. Cells that received no treatment were used as a control.
The cell viability assay was performed using rat PASM cells by an MTT assay as reported previously.12 Briefly, cells (5×104 cells/well) were seeded in a 96-well plate and treated with (i) saline (ii) 0.1% sodium dodecyl sulfate (SDS), (iii) plain fasudil, (iv) plain CAR, (v) plain mixture of lipids, (vi) plain micelles, (vii) CAR-micelles, or (vii) 100 μM fasudil. Following 24 hrs incubation, cells were washed and incubated with 100 μl MTT for an additional 4 hrs. The formazan crystals were dissolved in DMSO by moderate shaking for an hr. Finally, the absorbance was recorded at 570 nm using a Synergy™MX Microplate Reader (Biotek, Winnoski, VT). The cell viability was calculated using the following equation: Percent Cell viability = (ODsample – ODblank) / (ODcontrol – ODblank) × 100; OD is optical density, sample is test compound, control is saline and blank is no treatment.
Determination of drug-retention using isolated perfused rat lung (IPRL) system
We have measured the fraction-drug-retained in the lung after administration of plain and micellar drug using an IPRL system (Hugo Sachs Elektronik – Harvard Apparatus GmbH, March-Hugstetten, Germany). To remove the rat lung from the body, we first anesthetized adult male Sprague Dawley (SD) rats (∼250-300 g) using ketamine plus xylazine and opened the rat neck to expose the trachea, insert a cannula, and provide positive pressure ventilation. The chest cavity was then opened to insert pulmonary arterial and left ventricular cannulae that served as the inlet and outlet for the perfusion medium, respectively. The perfusion medium (made of CaCl2, NaCl, KCl, MgSO4, NaH2PO4, Glucose, NaHCO3 and Ficoll) was circulated through the lungs at pH 7.4 and 37°C. The lung-heart bloc was resected from the chest cavity and housed in the humidified artificial thoracic chamber, maintained at negative pressure, of the IPRL setup. Plain or micellar fasudil (6 mg/kg) was administered directly to the lung via the tracheal cannula using a PennCentury™Microsprayer; the lungs were perfused with the above perfusion medium for 2 hours; the perfusate was collected at various time points. The samples were analyzed for fasudil content by a reverse-phase HPLC method (Varian ProStar 320, Walnut Creek, CA) as reported previously.12
To quantitate the drug amount remaining in the isolated lungs, we removed the isolated lungs from the thoracic system after two hours of perfusion and flash-frozen in liquid nitrogen, homogenized, and centrifuged. The drug was extracted from the supernatant by precipitation with acetonitrile (1:1 v/v) followed by centrifugation for 10 min at 10,000 rpm. The supernatant was analyzed for fasudil content by the HPLC method as described above.
Pharmacokinetics of fasudil loaded CAR-micelles
We evaluated the pharmacokinetics of plain fasudil and fasudil-loaded CAR-micelles in adult male SD rats (∼250-300 g) as described previously.12 A cocktail of ketamine and xylazine was used to anesthetize the animals prior to the following treatments: aqueous solution of plain fasudil via (i) intravenous (IV), and (ii) intratracheal (IT), and CAR-micelles (suspended in PBS 1X, pH 7.4) via (iii) IV and (iv) IT routes. The dose, 6 mg/kg fasudil, was chosen based on the published IC50 values and the dose used in human and animals.19,20 An IV bolus was injected via the penile vein and IT dose was instilled using a PennCentury™ Microsprayer® for rats. Blood samples were collected into citrated microcentrifuge tubes via tail-vein milking method; the plasma was separated by centrifuging the blood at 5,000 rpm for 5 minutes and stored at -20°C until further analysis. The concentration of fasudil in plasma samples was determined as described above.13 A standard curve of fasudil in plasma (0.05-25 μg/ml) was prepared to quantify the drug concentration in samples.
Acute efficacy studies in monocrotaline (MCT) induced PAH rats
The hemodynamic efficacy was studied in an MCT model as reported previously.13 Briefly, MCT (Sigma-Aldrich Inc., St. Louis, MO) at a dose of 50 mg/kg was injected subcutaneously to male SD rats weighing ∼250-300 g. Rats were housed for 4 weeks for the development of PAH. Before the treatments, PAH rats were anesthetized by a cocktail of ketamine and xylazine. Two catheters, one in pulmonary artery (via jugular vein and right ventricle) and the other one in right carotid artery, were placed and sutured properly for the measurement of mean pulmonary arterial pressure (MPAP) and mean systemic arterial pressure (MSAP). The fluid filled catheters were attached with the MEMSCAP SP844 physiological pressure transducers (MEMSCAP AS, Scoppum, Norway) which were connected with the bridge amplifiers linked to a PowerLab 16/30 system. Data recording and acquisition was performed by a LabChart Pro 7.0 software (ADInstruments, Inc., Colorado Springs, CO). Four groups of rats received the following treatments: (i) plain fasudil IV, (ii) plain fasudil IT, (iii) plain micelles IT and (iv) CAR-micelles IT at a fixed dose of 3 mg/kg drug. Drug administrations were performed as described above. All animals were monitored until the pressures were back to the base values and anesthesia was maintained throughout the experiment.
All animal studies were performed in accordance with NIH Guidelines for the care and use of Laboratory Animals under a protocol approved by TTUHSC Animal Care and Use Committee (AM-10012).
Short-term toxicity studies of formulations
The safety of CAR-micelles for lung delivery was tested by bronchoalveolar lavage (BAL) study.12 Rats were divided into three groups to receive the following treatments: (i) saline (negative control), (ii) CAR-micelles (equivalent to 6 mg/kg fasudil) and (iii) 0.1% SDS. After 12 hrs, lungs of sacrificed rats were excised, wet lung weights were recorded, and the lungs were lavaged by instilling 5 mL saline. This procedure was repeated thrice followed by centrifugation of BAL fluid at 500×g for 10 min. The supernatant was analyzed for injury markers, lactate dehydrogenase (LDH) and alkaline phosphatase (ALP), by using commercial kits (Pointe Scientific, Canton, MI). Enzyme activities were reported as IU/L.
Data analysis
The data are presented as mean ± standard deviation and were analyzed by one-way ANOVA followed by Tukey's post-hoc test using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA). Differences in p values less than 0.05 were considered statistically significant. Pharmacokinetic analysis was performed by standard non-compartmental analysis (WinNonlin®, Pharsight Corp., Cary, NC) to calculate the area under the plasma concentration vs. time curve (AUC0→inf), maximum plasma concentration (Cmax), and elimination half-life (t1/2).
Results and Discussion
Formulation and characterization of fasudil containing CAR-micelles
Fasudil was loaded in DSPE-PEG5000 micelles by the solvent evaporation and hydration method. Fasudil is freely soluble in water (∼200 mg/ml), but moderately soluble in methanol. When fasudil was encapsulated during the hydration step, the drug entrapment was ∼3%; thus to maximize entrapment, the drug was added along with the lipids prior to film formation step. PEG5000-functionalized lipids were used to prepare the micelles because longer PEG chains form a protective sheath around the drug and shield the therapeutic agent against degradation in the biological fluid.21 The evaporation of the solvent yielded a transparent drug-polymer matrix that tangled the drug with the polymer chains. The water molecules forms hydrogen bond with the PEG blocks and give rise to the corona; the lipid segments along with the drug orient away from the water and form the glassy core of the micelles.22 Because of this core shell structure of micelles, the PEG block reduces the clearance by macrophages and extends the residence time in the lungs. Also, DSPE block, which constitutes the core, made the formulations biodegradable because of DSPE's propensity to be degraded by phospholipase-A2 secreted by mammalian cells.23 Further, the CMC of DSPE-PEG5000 is in the micromolar range (∼10-6 M) that adds significant stability to the micellar formulation upon dilutions in the physiological fluids.24
The micelles were homogenous (PDI = ∼0.17-0.43) with an average size of 12-14 nm with a rather narrow size distribution (Table 1). Because of their small size, the particles are less likely to be phagocytosed by alveolar macrophages.12,13 Smaller size will allow a large number of particles to form nebulized droplets and deliver the drug into the deep lung. After landing on the lung surface, the nanosized micelles may float over the lung lining fluid and undergo dissolution.25 Stability of colloidal system depends on the zeta potential value; colloidal dispersions with large positive or negative zeta potential show enhanced stability against aggregation; strong steric hindrance keep particles separated and prevent coalescence. The zeta potential of DSPE-PEG5000 micelles was about -30 mV, a value that ensures colloidal stability of the formulation.9 Drug loading increased with the increase in polymer concentration; elevated lipid concentration allows micelles to entangle a large fraction of the drug in the core. However, with the increase in polymer concentration, the micellar formulation loses its homogenous characters and thus we set polymer concentration at 25 mg/ml for micelles used in various in vitro and in vivo studies.
Table 1.
Physicochemical characteristics of fasudil loaded CAR-micelles. Data represent mean ± standard deviation (n=3).
| DSPE-PEG5000 (mg/ml) | Size (nm) | Zeta potential (mV) | PDI | Entrapment Efficiency (%) |
|---|---|---|---|---|
| 5 | 12.11±0.32 | -18.32±0.12 | 0.179±0.04 | 38.13±1.22 |
| 10 | 12.33±0.11 | -18.77±0.46 | 0.188±0.07 | 38.99±1.38 |
| 15 | 12.47±0.21 | -18.98±0.55 | 0.171±0.08 | 46.77±2.39 |
| 20 | 13.22±0.14 | -24.31±1.39 | 0.176±0.04 | 48.31±2.11 |
| 25 | 14.18±1.03 | -29.86±1.27 | 0.178±0.01 | 57.91±1.28 |
| 30 | 14.47±1.07 | -29.94±2.04 | 0.433±0.09 | 59.11±3.88 |
Micelles were freeze-dried and later evaluated for the influence of lyophilzation on the formulations. Freeze drying and subsequent reconstitution of fasudil-loaded micelles did not noticeably change in the size, homogeneity and entrapment efficiency. Also lyophilization aids in enhancing formulation stability that can be stored for months. We stored the micelles at 4°C and evaluated them periodically for the changes in size and entrapment efficiency (Figure 2A). The changes in the size and entrapment were minimal even after three weeks of storage, pointing to formulations' suitability for short and long term use. For maximal drug deposition in the deep lungs, aerodynamic diameter of the particles should be in the respirable range. The MMAD of and FPF of the formulations were 2.31±0.03 μm and 53.7±2.69%, suggesting that the formulations are likely to deposit in the deep lung as reported by us and others.8,10,17 The presence of PEG chains on the outer layer of the polymeric hybrid micelles induces a repulsive force around the particle surface that reduces the adhesive forces and allows a more efficient aerosolization of the micellar suspension in the air. Micelles collected from the cascade impactor were also evaluated for their stability and integrity: The micelles had an average size of 14.53±1.99 nm and entrapment efficiency of 49.66±2.71%, suggesting particles would be robust for inhalational delivery. The micelles were intact and retained their physical properties after nebulization by the microsprayer (Figure 2B). On the whole, the physics of the micellar formulation suggests that these particles can be delivered as aerosolized droplets for inhalational therapy of PAH.
Figure 2.
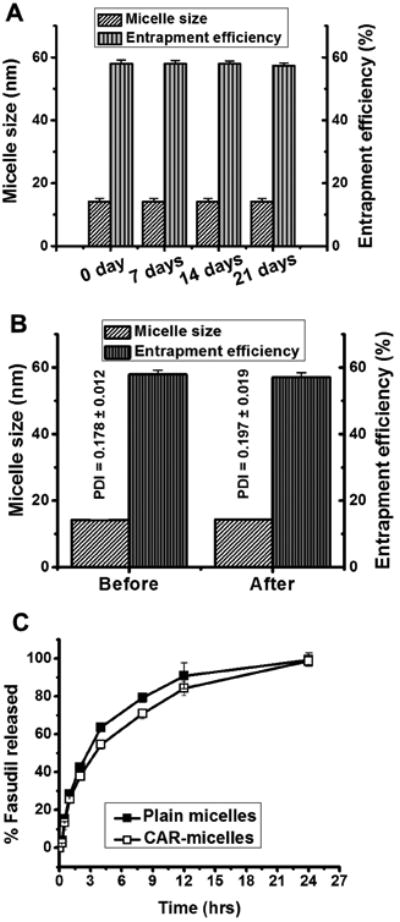
(A) The stability of CAR-micelles stored at 4°C for a period of 3 weeks. (B) Aerosolization stability of formulations. (C) In-vitro release profiles of plain and CAR conjugated micelles in SLF at 37°C. Data represent mean ± standard deviation (n=3).
NMR to confirm the encapsulation of fasudil in the core-shell structure of micelles
We confirmed the formation of fasudil containing micelles by 1H NMR spectroscopic studies. We ran the spectra of fasudil in CD3OD, DSPE-PEG5000 in D2O and fasudil-loaded micelles in D2O and CD3OD (Figure 1). NMR of fasudil showed peaks at 2.18, 3.33, 3.44, 7.89, 8.47, 8.57, 8.67 and 9.45 ppm. DSPE-PEG5000, when dissolved in D2O, forms empty micelles that gave rise to resonance signals for PEG blocks at 4.70 ppm. However, fasudil-loaded micelles showed only the peaks for DSPE-PEG5000 (4.70 ppm) but no peaks for fasudil. The absence of fasudil peak indicates that the drug was encapsulated in the micelles and no drug was present on the micelles' surface. To confirm that drug was in the micelle core, we lysed micelle containing fasudil in CD3OD and recorded the NMR spectra that showed peaks for both fasudil and DSPE-PEG5000. The peak for fasudil observed in this sample was because of the release of the drug from the micelles. NMR data suggest that fasudil was confined within the micelle core, which resulted in the insufficient motion of fasudil protons in the core and eventually disappearance of fasudil peaks.
Figure 1.
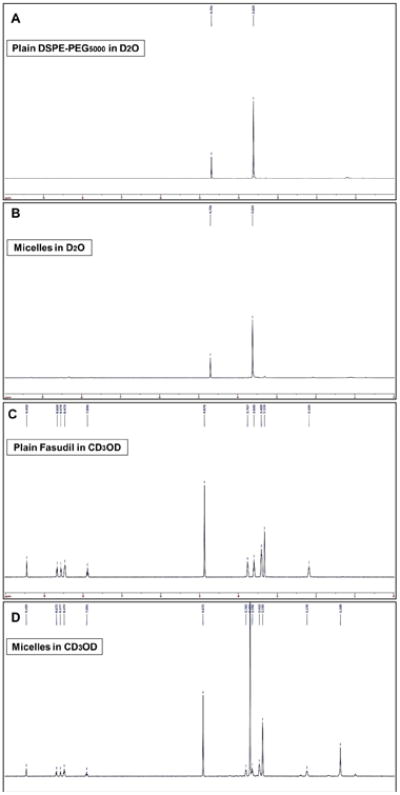
1H NMR spectra of (A) plain DSPE-PEG5000 in D2O, (B) plain micelles in D2O, (C) plain fasudil in CD3OD and (D) Micelles containing fasudil in CD3OD.
In-vitro fasudil release from micelles in SLF
SLF was used to study the release of fasudil from the micelles in the actual lung environment. We observed a slow and controlled release of drug from micellar core, with only 28.3±1.22% release in the first hour (Figure 2C). CAR-micelles showed a reduced drug release, 25.8±2.21%, which may be due to the presence of cyclic peptides on the surface. About 90% of the drug was released after 12 hrs in the same fashion. In contrast to other sustained release formulations, which show an initial burst release and leads to significant loss of drug reservoir, micelles showed initially a slower release, pointing to the assumption that fasudil was not present on the outer surface but rather confined within the micellar core. Lack of initial burst release can be explained by the glassy stiff core, originated from the two long-chain fatty acyl groups of the DSPE segment, of micelles that restricts mobility of the drug molecules.26. The retarded release may have resulted from diffusion of the drug from micellar core.9,27 Further, the solubility of drug/formulation often dictates the absorption from the lungs. Upon landing on the airway surface, particles are wetted and dissolved in the airway lining fluid. Particles with reduced solubility, such as budesonide in Pulmicort Respules®, take relatively long time to dissolve and partition between the phases of the airway lining, and are cleared from the airways by the mucociliary transport and phagocytosis. But inhaled drug particles with high solubility, such as budesonide in DSPE-PEG micelles, enter and dissolve in the airway lining fluid faster and are less susceptible to mucociliary clearance.8 Based on this evidence, we expect our formulation will be dissolved in the respiratory fluid and release the drug which, after crossing the air-blood barrier, may either directly enter the general circulation and traverse the pulmonary capillaries via the adventitial side of the endothelial cells.28
Cellular uptake and cytotoxicity of formulations
Cellular uptake studies were performed in rat PASM cells, the therapeutic target of fasudil. Fasudil prevents smooth muscle contraction in response to several vasoconstrictors, including endothelin, serotonin, and angiotensin, by inhibiting rho-kinase pathway. Compared to plain micelles, CAR-micelles were internalized at a greater extent by activated cells (Figure 3A). This could be explained by the interaction of CAR peptide and heparan sulfate overexpressed on cell surface. This observation agrees with previous data that TGF-β increases endogenous heparan sulfate proteoglycan expression29, and CAR peptide has an affinity to bind with a specific heparan sulfate15, and that CAR accumulates in activated cells that mimic PAH-like conditions.
Figure 3.
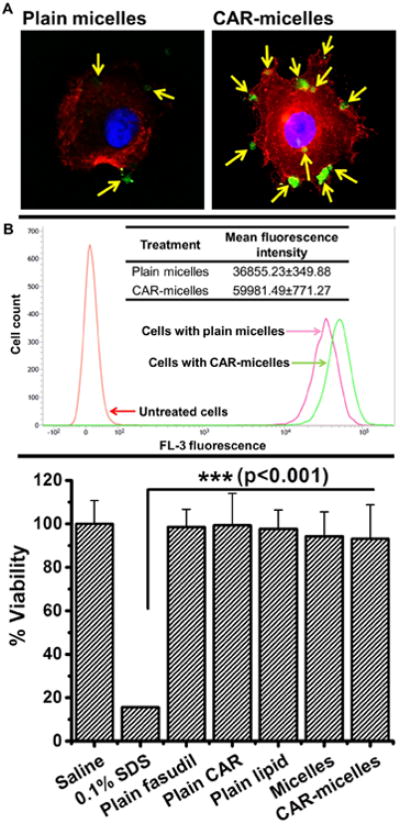
(A) Uptake of plain and CAR conjugated micelles (green color) by TGF-β activated rat PASM cells (actin stained red). (B) Flow cytometric analysis of cellular uptake of formulations by activated cells (data represent mean ± standard deviation, n=3). (C) Viability of rat PASM cells upon incubation with formulations or individual components of formulation (data represent mean ± standard deviation, n=12).
We further confirmed the cellular uptake results by flow cytometry studies. CAR-micelles showed a ∼1.7 fold increase in the uptake than plain micelles (Figure 3B), as demonstrated by the shift in peak of fluorescence intensity. Based on these results, we anticipate that CAR-micelles would provide more localized and prolonged action in-vivo.
As synthetic lipids were used for the formulation of micelles, we evaluated their effect on the viability of cells by an MTT assay. We considered % cell viability obtained with saline treatment as 100 and compared with other treatments (Figure 3C). SDS, a potent surfactant, is very cytotoxic and showed a mere ∼16% cell viability. Other treatments, either ingredients alone or formulations, showed not less than ∼90% cellular viability, which suggest their suitability as vehicles for drug delivery.
Isolated perfused rat lung studies
IPRL studies were performed to quantitate fraction-drug retained in the lung after pulmonary administration. The rationale for inhalation delivery of fasudil is to extend the residence time in the lungs and reduce its systemic exposure. The ex-vivo lung perfusion allows quantifying the amount of drug that leaves the lung and enters the pulmonary circulation. When plain fasudil was administered into the isolated lungs, fasudil concentration in the perfusate was detected within 5 minutes of the instillation which increased rapidly, suggesting fast absorption via the lung epithelium. But drug release from micelles was rather slow; the drug appeared in the perfusate in 15-20 minute (Figure 4A). The drug concentration in the perfusate, which mimics the systemic circulation, collected from lungs treated with micellar formulation were lower than that collected from lungs treated with the plain drug. The amount of drug retained in the lung treated with plain or micellar drug was also determined in the lung homogenates after the perfusion. The drug amount in micelle treated lungs was about two-fold greater than the amount in the homogenates of lungs treated with plain drugs (Figure 4B). Both experiments point to the extended residence time in the lung when the drug was administered as micellar formulations; extended lung-residence-time of the drug is likely to extend the pulmonary selective vasodilation in PAH. Increased residence time may have resulted from slower release of the drug from the micelles, evasion of macropahgeal uptake of nanosize particles, and reduced clearance.
Figure 4.
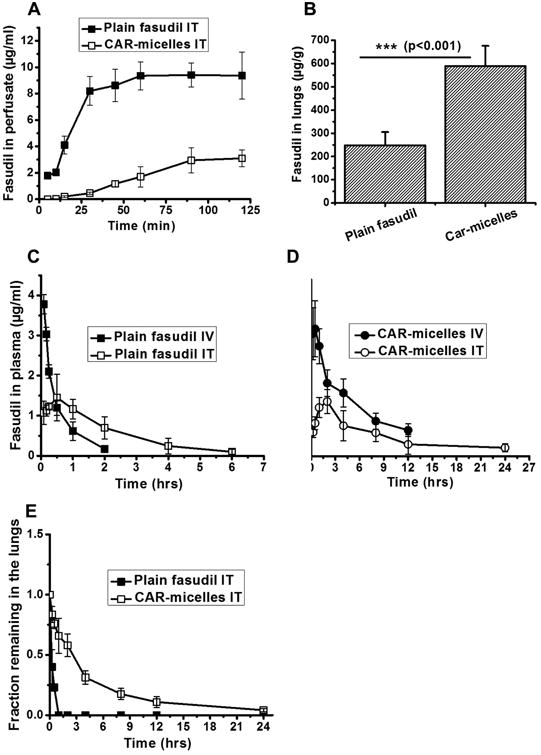
Isolated perfused rat lung studies showing the concentration of (A) plain and entrapped fasudil over a period of 2 hrs. (B) Concentration of fasudil (μg/g lung weight) in homogenized rat lungs which received either plain drug or CAR-micelles. In-vivo absorption profiles of (C) plain fasudil and (D) CAR-micelles containing fasudil, administered either intravenously or intratracheally. (E) Fraction of fasudil remaining in the lungs after intratracheal administration to rats calculated from plasma drug concentrations-time profile using Wagner-Nelson method. Dose of fasudil was 6 mg/kg in all of the above experiments. Data represent mean ± standard deviation (n=3).
Pharmacokinetics of fasudil loaded CAR-micelles
The pharmacokinetics of fasudil as plain drug or formulation after IV and IT administrations were evaluated in adult male SD rats. Plain fasudil administered intratracheally showed a ∼3-fold increase in the half-life as compared to the IV half-life, suggesting a reduction in the rate of absorption after pulmonary administration (Figure 4C). However, a significant extension in the half-life was observed (∼15-fold greater than plain fasudil IV) when the drug was administered in the form of micelles (7.93±1.51 hrs) via the intratracheal route (Figure 4D). The Cmax of CAR-micelles fell below the Cmax for plain drug administered IV or IT (Table 2). The reduction in Cmax may have resulted from the reduced availability of the drug from CAR-micelles as observed in the IPRL studies described above. But free fasudil underwent faster absorption and much of the drug was available in the peripheral vasculature. Plain IV and IT fasudil showed no plasma concentration after 2 and 6 hrs, respectively; CAR micelles showed detectable plasma levels of fasudil for 24 hrs, suggesting slower appearance of the drug in the plasma because of the retarded drug release from the micelles. True to the plasma concentration-time data, the fraction-dose remaining in the lung, calculated by Wagner-Nelson method30, was also higher in the lungs treated with micellar formulation than those treated with the plain drug (Figure. 4E). Based on the fasudil plasma concentration-time data, fraction-dose remaining in the lung and published IC50 for rho-kinase inhibition (0.158 μM)31, we expect that micellar fasudil will maintain the therapeutic concentrations of the drug for a prolonged time and reduce the dosing frequency.
Table 2.
Pharmacokinetic profiles of plain fasudil and CAR-micelles containing fasudil. Data represent mean ± standard deviation (n=3).
| Treatment | Cmax (μg/ml) | t1/2 (hr) | AUC0-inf (μg/ml*hr) |
|---|---|---|---|
| Fasudil IV | 3.78±0.23 | 0.53±0.05 | 27.64±3.91 |
| Fasudil IT | 1.45±0.58 | 1.55±0.08 | 21.72±4.08 |
| CAR-micelles IV | 1.80±0.64 | 3.32±0.59 | 29.62±4.77 |
| CAR-micelles IT | 0.77±0.17 | 7.93±1.51** | 20.21±2.34 |
Means are significantly different (p<0.01).
Acute hemodynamic efficacy of CAR-micelles
The hemodynamic efficacy of the optimized formulation was evaluated in a rodent model, MCT induced PAH. We and others have reported that MCT administered subcutaneously at a dose of 50 mg/kg produces PAH in rats in 28 days. MCT-induced PAH exhibits elevated MPAP, right ventricular hypertrophy and muscularization of small pulmonary arteries.18 The normal MPAP of healthy rats was ∼12-16 mm Hg, which increased to ∼45-50 mm Hg in PAH animals.32 IT fasudil produced more pronounced reduction in MPAP than IV fasudil. But a significant reduction in the MSAP was also observed (Figures 5A and 5B) because of the fast absorption of plain drug and immediate availability of drug in the circulation. Formulations, both plain and CAR conjugated micelles, given via the IT route showed a major reduction in MPAP (∼60%) compared with plain drug; but reduction in MSAP was not as great as that of MPAP (Figures 5C and 5D), pointing to the fact that the formulations reduce rate of rise of the drug level in the circulation. The vasodilatory duration, calculated from the time point when pressure returned to the initial value33, was longer for CAR-micelles than the duration for plain drug and unmodified micelles (Figure 5E). To evaluate the selectivity of the formulation toward pulmonary vasodilation, we calculated the lung targeting indices (LTI) from the ratio of area above the efficacy curves (AUEC) for MPAP and MSAP: LTI = AUEC (MPAP)/AUEC (MSAP). CAR-micelles showed a ∼2 and ∼3.5-fold increase in lung selectivity than plain micelles and plain fasudil (Figure 5F). These results support our hypothesis that CAR conjugation enables the formulations to accumulate more on the diseased site and show prolonged hemodynamic effect without affecting the systemic pressure. Compared with liposomal fasudil13, CAR-micelles were superior to liposomes in terms of pulmonary specificity and prolonged vasodilation. CAR-micelles produced ∼2- and ∼3-fold increase in vasodilatory duration and lung targeting, respectively, as compared to that produced by liposomal fasudil.
Figure 5.
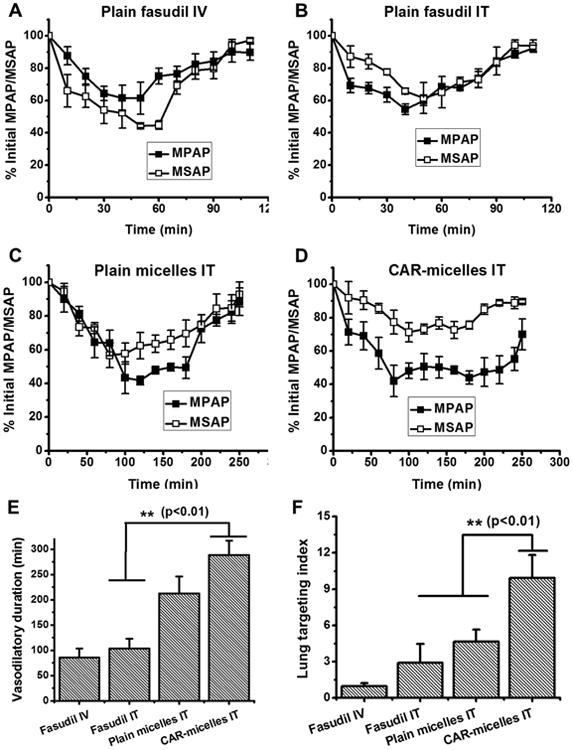
Acute hemodynamic efficacy of (A) plain fasudil IV, (B) plain fasudil IT, (C) plain micelles IT, (D) CAR-micelles IT, (E) duration of vasodilatory effects and (F) lung targeting indices of plain fasudil or formulations in MCT induced PAH rats. Data represent mean ± standard deviation (n=3).
Safety studies of formulation
Lastly, we have evaluated the short-term safety of the formulations after intratracheal administration. CAR-micelles and saline did not cause any significant increase in wet lung weight but SDS produced a two-fold increase in lung weight compared to saline treated rats (Figure 6A). In fact, increase in wet lung weight suggests formation of edema due to accumulation of extracellular fluid in the epithelial cells of the respiratory wall. Similar to wet lung weight, SDS treated rats showed elevated levels of LDH and ALP compared with rats that received CAR-micelles and plain drug (Figure 6B). The presence of ALP in BAL fluid is an indication of alveolar type II cell proliferation due to type I damage, and presence of LDH represents cell damage and lysis. Based on these data, we can rule out the concerns regarding the safety of DSPE and PEG. DSPE is a biodegradable lipid and degraded by endogenous phospholipase A2; PEG with molecular weight <20 kD is excreted through renal clearance.34,35 Increasing use of PEG modified delivery systems have raised concerns regarding its long-term safety. In fact, a number studies suggest generation of antibodies against PEG35 although many of the studies ignore the fate of PEG after the disintegration of lipids.36,37 However, molecular weight, branching of polymer, dose and dosing frequency, direct/indirect conjugation with drugs/delivery systems and the route of administration of PEG determine their fate and safety that demands further evaluation. Overall, these acute safety data point toward the assumption that CAR-micelles are safe to administer via intratracheal route, but long-term safety studies after multiple administrations would provide more useful information regarding the suitability of formulations.
Figure 6.
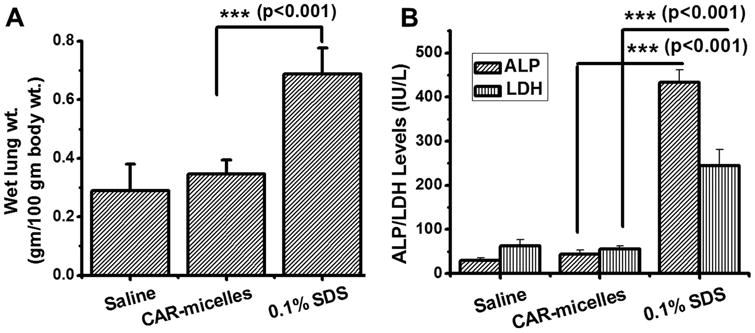
The safety of the formulations in the lung: (A) Effect of formulation on the wet lung weight and (B) levels of injury markers (APL, LDH) in BAL fluid. Data represent mean ± s.d. (n=4).
Conclusions
CAR conjugated DSPE-PEG micelles release fasudil at a continuous fashion in a simulated lung fluid. CAR-micelles were efficiently taken up by TGF-β activated rat PASM cells. Similar to continuous release patterns, fasudil showed prolonged circulation-time and prolonged retention after intratracheal administration of micellar formulations. Fasudil encapsulated in CAR-micelles produced reduction in MPAP for an extended period without affecting much MSAP in PAH rats. BAL studies indicated the safety of micelles as pulmonary delivery carriers. However, data concerning the chronic administration and drug's influence in ameliorating pathological changes in PAH can only dictate the clinical feasibility of this micelle based formulation.
Acknowledgments
We acknowledge Drs. Eva Nozik-Grayck and Kurt Stenmark at the University of Colorado, Denver for providing PASM cell lines. We thank Kshitij Verma who helped us in conducting the NMR studies and analysis. This work was supported in part by an American Recovery and Reinvestment Act Fund, NIH 1R15HL103431 to Dr. Fakhrul Ahsan.
References
- 1.Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharm Res. 2007;24(1):1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 2.Trivedi R, Kompella UB. Nanomicellar formulations for sustained drug delivery: strategies and underlying principles. Nanomedicine (Lond) 2010;5(3):485–505. doi: 10.2217/nnm.10.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baginski L, Gobbo OL, Tewes F, Salomon JJ, Healy AM, Bakowsky U, Ehrhardt C. In vitro and in vivo characterisation of PEG-lipid-based micellar complexes of salmon calcitonin for pulmonary delivery. Pharm Res. 2012;29(6):1425–1434. doi: 10.1007/s11095-012-0688-6. [DOI] [PubMed] [Google Scholar]
- 4.Haeri A, Sadeghian S, Rabbani S, Anvari MS, Lavasanifar A, Amini M, Dadashzadeh S. Sirolimus-loaded stealth colloidal systems attenuate neointimal hyperplasia after balloon injury: a comparison of phospholipid micelles and liposomes. Int J Pharm. 2013;455(1-2):320–330. doi: 10.1016/j.ijpharm.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Brandenburg KS, Rubinstein I, Sadikot RT, Onyuksel H. Polymyxin B self-associated with phospholipid nanomicelles. Pharm Dev Technol. 2012;17(6):654–660. doi: 10.3109/10837450.2011.572893. [DOI] [PubMed] [Google Scholar]
- 6.Cesur H, Rubinstein I, Pai A, Onyuksel H. Self-associated indisulam in phospholipid-based nanomicelles: a potential nanomedicine for cancer. Nanomedicine. 2009;5(2):178–183. doi: 10.1016/j.nano.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang R, Xiao R, Zeng Z, Xu L, Wang J. Application of poly(ethylene glycol)-distearoylphosphatidylethanolamine (PEG-DSPE) block copolymers and their derivatives as nanomaterials in drug delivery. Int J Nanomedicine. 2012;7:4185–4198. doi: 10.2147/IJN.S34489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sahib MN, Darwis Y, Peh KK, Abdulameer SA, Tan YT. Rehydrated sterically stabilized phospholipid nanomicelles of budesonide for nebulization: physicochemical characterization and in vitro, in vivo evaluations. Int J Nanomedicine. 2011;6:2351–2366. doi: 10.2147/IJN.S25363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gill KK, Nazzal S, Kaddoumi A. Paclitaxel loaded PEG(5000)-DSPE micelles as pulmonary delivery platform: formulation characterization, tissue distribution, plasma pharmacokinetics, and toxicological evaluation. Eur J Pharm Biopharm. 2011;79(2):276–284. doi: 10.1016/j.ejpb.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 10.Abdulla JM, Tan YT, Darwis Y. Rehydrated lyophilized rifampicin-loaded mPEG-DSPE formulations for nebulization. AAPS PharmSciTech. 2010;11(2):663–671. doi: 10.1208/s12249-010-9428-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Craparo EF, Teresi G, Bondi ML, Licciardi M, Cavallaro G. Phospholipid-polyaspartamide micelles for pulmonary delivery of corticosteroids. Int J Pharm. 2011;406(1-2):135–144. doi: 10.1016/j.ijpharm.2010.12.024. [DOI] [PubMed] [Google Scholar]
- 12.Gupta N, Patel B, Ahsan F. Nano-Engineered Erythrocyte Ghosts as Inhalational Carriers for Delivery of Fasudil: Preparation and Characterization. Pharm Res. 2014 doi: 10.1007/s11095-013-1261-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta V, Gupta N, Shaik IH, Mehvar R, McMurtry IF, Oka M, Nozik-Grayck E, Komatsu M, Ahsan F. Liposomal fasudil, a rho-kinase inhibitor, for prolonged pulmonary preferential vasodilation in pulmonary arterial hypertension. J Control Release. 2013;167(2):189–199. doi: 10.1016/j.jconrel.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukumoto Y, Yamada N, Matsubara H, Mizoguchi M, Uchino K, Yao A, Kihara Y, Kawano M, Watanabe H, Takeda Y, Adachi T, Osanai S, Tanabe N, Inoue T, Kubo A, Ota Y, Fukuda K, Nakano T, Shimokawa H. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ J. 2013;77(10):2619–2625. doi: 10.1253/circj.cj-13-0443. [DOI] [PubMed] [Google Scholar]
- 15.Urakami T, Jarvinen TA, Toba M, Sawada J, Ambalavanan N, Mann D, McMurtry I, Oka M, Ruoslahti E, Komatsu M. Peptide-directed highly selective targeting of pulmonary arterial hypertension. Am J Pathol. 2011;178(6):2489–2495. doi: 10.1016/j.ajpath.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dabholkar RD, Sawant RM, Mongayt DA, Devarajan PV, Torchilin VP. Polyethylene glycol-phosphatidylethanolamine conjugate (PEG-PE)-based mixed micelles: some properties, loading with paclitaxel, and modulation of P-glycoprotein-mediated efflux. Int J Pharm. 2006;315(1-2):148–157. doi: 10.1016/j.ijpharm.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 17.Patel B, Gupta V, Ahsan F. PEG-PLGA based large porous particles for pulmonary delivery of a highly soluble drug, low molecular weight heparin. J Control Release. 2012;162(2):310–320. doi: 10.1016/j.jconrel.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Gupta V, Gupta N, Shaik IH, Mehvar R, Nozik-Grayck E, McMurtry IF, Oka M, Komatsu M, Ahsan F. Inhaled PLGA particles of prostaglandin E(1) ameliorate symptoms and progression of pulmonary hypertension at a reduced dosing frequency. Mol Pharm. 2013;10(5):1655–1667. doi: 10.1021/mp300426u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, McMurtry IF, Oka M. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med. 2005;171(5):494–499. doi: 10.1164/rccm.200405-637OC. [DOI] [PubMed] [Google Scholar]
- 20.Fujita H, Fukumoto Y, Saji K, Sugimura K, Demachi J, Nawata J, Shimokawa H. Acute vasodilator effects of inhaled fasudil, a specific Rho-kinase inhibitor, in patients with pulmonary arterial hypertension. Heart Vessels. 2010;25(2):144–149. doi: 10.1007/s00380-009-1176-8. [DOI] [PubMed] [Google Scholar]
- 21.Sawant RR, Torchilin VP. Polymeric micelles: polyethylene glycol-phosphatidylethanolamine (PEG-PE)-based micelles as an example. Methods Mol Biol. 2010;624:131–149. doi: 10.1007/978-1-60761-609-2_9. [DOI] [PubMed] [Google Scholar]
- 22.Lee H, Soo PL, Liu J, Butler M, Allen C. Polymeric micelles for formulation of anticancer drugs. In: MM A, editor. Nanotechnology for Cancer Therapy, ed. Tyler & Francis Group LLC; 2006. pp. 317–356. [Google Scholar]
- 23.Davidsen J, Vermehren C, Frokjaer S, Mouritsen OG, Jorgensen K. Drug delivery by phospholipase A(2) degradable liposomes. Int J Pharm. 2001;214(1-2):67–69. doi: 10.1016/s0378-5173(00)00634-7. [DOI] [PubMed] [Google Scholar]
- 24.Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv Drug Deliv Rev. 2004;56(9):1273–1289. doi: 10.1016/j.addr.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 25.El-Sherbiny IM, Smyth HD. Biodegradable nano-micro carrier systems for sustained pulmonary drug delivery: (I) self-assembled nanoparticles encapsulated in respirable/swellable semi-IPN microspheres. Int J Pharm. 2010;395(1-2):132–141. doi: 10.1016/j.ijpharm.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torchilin VP. Lipid-core micelles for targeted drug delivery. Curr Drug Deliv. 2005;2(4):319–327. doi: 10.2174/156720105774370221. [DOI] [PubMed] [Google Scholar]
- 27.Sarisozen C, Vural I, Levchenko T, Hincal AA, Torchilin VP. Long-circulating PEG-PE micelles co-loaded with paclitaxel and elacridar (GG918) overcome multidrug resistance. Drug Deliv. 2012;19(8):363–370. doi: 10.3109/10717544.2012.724473. [DOI] [PubMed] [Google Scholar]
- 28.Gomberg-Maitland M, Olschewski H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur Respir J. 2008;31(4):891–901. doi: 10.1183/09031936.00097107. [DOI] [PubMed] [Google Scholar]
- 29.Dodge GR, Kovalszky I, Hassell JR, Iozzo RV. Transforming growth factor beta alters the expression of heparan sulfate proteoglycan in human colon carcinoma cells. J Biol Chem. 1990;265(29):18023–18029. [PubMed] [Google Scholar]
- 30.Sanaka M, Yamamoto T, Ishii T, Kuyama Y. The Wagner-Nelson method can generate an accurate gastric emptying flow curve from CO2 data obtained by a 13C-labeled substrate breath test. Digestion. 2004;69(2):71–78. doi: 10.1159/000077391. [DOI] [PubMed] [Google Scholar]
- 31.Oka M, Fagan KA, Jones PL, McMurtry IF. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br J Pharmacol. 2008;155(4):444–454. doi: 10.1038/bjp.2008.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta V, Ahsan F. Inhalational therapy for pulmonary arterial hypertension: current status and future prospects. Crit Rev Ther Drug Carrier Syst. 2010;27(4):313–370. doi: 10.1615/critrevtherdrugcarriersyst.v27.i4.20. [DOI] [PubMed] [Google Scholar]
- 33.Jiang BH, Tawara S, Abe K, Takaki A, Fukumoto Y, Shimokawa H. Acute vasodilator effect of fasudil, a Rho-kinase inhibitor, in monocrotaline-induced pulmonary hypertension in rats. J Cardiovasc Pharmacol. 2007;49(2):85–89. doi: 10.1097/FJC.0b013e31802df112. [DOI] [PubMed] [Google Scholar]
- 34.Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10(21):1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 35.Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed Engl. 2010;49(36):6288–6308. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- 36.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53(2):283–318. [PubMed] [Google Scholar]
- 37.Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42(6):463–478. doi: 10.1016/s0163-7827(03)00033-x. [DOI] [PubMed] [Google Scholar]