

Abstract
Patients with mutations in ITK are susceptible to viral infections, particularly Epstein Barr Virus, suggesting that these patients have defective function of CD8+ cytolytic T lymphocytes (CTLs). Here, we evaluated the effects of ITK-deficiency on cytolysis in murine CTLs deficient in ITK, and both human and murine cells treated with an ITK inhibitor. We find that ITK-deficiency leads to a global defect in the cytolysis of multiple targets. The absence of ITK both affected CTL expansion and delayed the expression of cytolytic effectors during activation. Furthermore, absence of ITK led to a previously unappreciated intrinsic defect in degranulation. Nonetheless, these defects could be overcome by early or prolonged exposure to IL-2, or by addition of IL-12 to cultures, revealing that cytokine signaling could restore the acquisition of effector function in ITK-deficient CD8+ T cells. Our results provide new insight into the effect of ITK and suboptimal TCR signaling on CD8+ T cell function, and how these may contribute to phenotypes associated with ITK-deficiency.
INTRODUCTION
CD8+ cytotoxic T lymphocytes (CTLs) are critical for combatting viral infections and tumors through the directed lysis of target cells. Accordingly, mutations in genes affecting CTL cytolytic function have been found in a number of primary immunodeficiencies associated with impaired viral clearance and tumor development.
Granule-dependent, contact-mediated killing of virally infected cells by CTLs is initiated upon T cell receptor (TCR) engagement, which causes a series of cellular changes resulting in the release of cytolytic effectors at the site of contact with target cells. These stages include the initial adhesion of CTLs to target cells and the rapid accumulation of a rich cortical actin network (1), which then clears to form a ring at the edge of immunological synapse, the special organization of membrane and signaling proteins that forms at the interface between a T cell and its target. Actin clearance is closely followed by reorientation of the centrosome (2) and the polarization of cytotoxic granules toward the target cell along a reorganized microtubule network (3, 4), leading to centrosome docking and granule fusion at the plasma membrane. The release of lytic granule contents at the secretory domain of the synapse, including the pore-forming molecule perforin, allows granzymes to enter the cytoplasm of target cells and initiate cell death (5–8). Through this ordered series of events, CTLs are able to rapidly and effectively eliminate virally infected targets during an immune response.
In order to trigger cytolysis, TCR engagement initiates signaling cascades associated with the formation of signaling complexes at the plasma membrane. Inducible T cell kinase (ITK) is a non-receptor tyrosine kinase that is a component of the LAT-SLP76 signaling complex, which is formed downstream of TCR activation. ITK phosphorylates PLCγ1, a key enzyme required for generation of critical second messengers during TCR signaling. Accordingly, the loss of ITK leads to reduced TCR-induced PLCγ1 phosphorylation and downstream impairments in Ca2+ flux and ERK signaling, as well as altered actin cytoskeletal regulation (9–11). Studies of CD4+ T cells from Itk−/− mice have shown that suboptimal TCR signaling in the absence of ITK leads to dramatic effects on CD4+ T cell differentiation, and altered CD4+ T cell function (12–14) (reviewed in (15)), including decreased IL-2 production and altered responses to IL-2 (9, 13, 16–19). Notably however, most of these studies have primarily focused on either total T cell or CD4+ T cell populations, leaving the role of ITK in CD8+ T cells relatively less well explored.
Recently, loss of function mutations in ITK were reported in a subset of patients with fulminant infectious mononucleosis triggered by Epstein Barr virus (EBV) infection (20–22). In addition, lymphomas, defective antibody responses, and a broader susceptibility to viral infection were also reported in these patients (reviewed in (23)), highlighting a potential requirement for ITK for proper CTL function. Intriguingly, this clinical phenotype resembles a number of other primary immunodeficiencies, including X-linked lymphoproliferative syndrome (XLP-1), a disease caused by mutations affecting the small adaptor molecule, signaling lymphocyte activation molecule (SLAM)-associated protein (SAP). We have previously shown that CTLs from SAP-deficient mice exhibit specific defects in killing B cells, despite normal cytolysis of other targets (24). Analogous observations have been made in cells from patients with XLP-1 (25), likely accounting for the inability of SAP-deficient CTLs to clear EBV-infected B cells. The similarities in clinical phenotypes between ITK-deficiency and XLP-1 raised the question of whether ITK-deficiency also similarly affects cytolytic effector function. Although Itk−/− mice can mount protective immune responses against vaccinia virus, vesicular stomatitis virus, and lymphocytic choriomeningitis virus (26, 27), viral clearance is delayed, likely reflecting poor activation of CD8+ T cells under conditions of suboptimal TCR signaling. However, whether or not there were defects in granule-mediated cytolysis of specific targets, or at specific stages of cytolysis, has not been well explored. A more complete examination of the role of ITK in CTL effector function would be useful for better understanding the human disease.
Here, we used the OT-I TCR transgenic system to examine the role of ITK in CD8+ T cell cytolytic effector function. We found that ITK was required for killing of multiple different target cells, suggesting global defects in cytolysis in the absence of ITK. Although ITK-deficient CD8+ T cells showed decreased expansion and expression of effector molecules after activation, treatment of differentiated WT CTLs with an ITK inhibitor still led to defects in cytolysis, suggesting direct effects of ITK-deficiency on the process of killing. Examination of discrete stages of CTL function revealed that ITK-deficiency did not affect the early stages of killing, including adhesion to targets and polarization of the centrosome and lytic granules, which were intact in Itk−/− CTLs. Instead, ITK-deficiency in CTLs was associated with defects in degranulation, a late stage of target killing, which could be recapitulated by treatment of WT mouse CTLs or activated CD8+ T cells from healthy human donors with an ITK-specific inhibitor. Nonetheless, we also found that early or prolonged incubation with IL-2 or IL-12 could rescue these defects, supporting a role for cytokines in rescuing T cell activation defects. Our results provide evidence for novel roles for ITK and TCR signaling in regulating both the early differentiation and expansion of CTLs, as well as the late stages of cytolytic activity that may contribute to reduced viral clearance in patients with mutations in ITK.
MATERIALS AND METHODS
Mice
Wild type (WT) OT-I (28), Itk−/− (29) OT-I TCR transgenic, and C57Bl/6 (Jackson Laboratories) mice were maintained in a Specific Pathogen Free facility and used between 6–10 weeks of age. For in vitro experiments, cells from either male or female mice were used. Animal husbandry and experiments were performed in accordance with approved protocols by the National Human Genome Research Institute Animal Use and Care Committee at the National Institutes of Health.
Cell culture
To generate in vitro activated mouse CTLs, splenocytes from WT or Itk−/− OT-I mice were harvested and stimulated at 0.5x106 cells/mL with 10nM OVA257-264 peptide (AnaSpec) for 3 days in 10% complete media (RPMI 1640 plus 10% FBS, 2mM L-glutamine, 50U/mL penicillin/streptomycin, and 50μM β-mercaptoethanol). In some experiments, 10 IU/mL recombinant human IL-2 (rHIL-2) and/or 20ng/mL IL-12 (Peprotech) were included, where indicated. On day 3, cells were washed once and seeded in fresh media plus rHIL-2 at 0.5x106 cells/mL every 48 hours. All experiments were performed with CTLs between 6 and 7 days after primary in vitro stimulation, unless otherwise indicated. Resting B cells were purified by negative selection with anti-CD43 microbeads (Miltenyi) and activated with 1μg/mL LPS from E. coli (Enzo Life Sciences) in 10% complete media for 2–3 days before use as targets in assays. EL4 and MC57 cell lines were maintained in complete Dulbecco’s modified Eagle’s medium (DMEM) plus 5% FBS. Blood from healthy donors was obtained at the NIH Clinical Center under NIH Clinical Center IRB-approved protocol 99-CC-0168 “Collection and Distribution of Blood Components from Healthy Donors for In Vitro Research Use.” Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by density-gradient centrifugation using Lymphocyte Separation Medium (MP Biomedical), washed twice in phosphate buffered saline (PBS), and resuspended at 1x106 cells/mL. One mL of cells was then added to each well of a 24-well plate and placed at 37°C. Mixed buffy coats for anti-allogeneic stimulation were irradiated and resuspended at 1x106 cells/mL, and phytohemagglutinin (PHA) was added to the buffy coats at 2μg/mL. To stimulate lymphocytes, 1mL of activated buffy coat was added to each well for a final ratio of 1:1 stimulators:responders in 1μg/mL PHA. PHA blasts were split as needed, and CD8+ T cells isolated using a CD8+ T cell isolation kit (MACS Miltenyi). Bulk CD8+ T cells were cultured for use in experiments. For inhibition experiments, previously activated WT OT-I CTLs or human CD8+ T cells were pre-treated for 10 minutes at 37°C with the ITK inhibitor, 10n (gift of Craig Thomas, NCATS, Bethesda, MD), at indicated concentrations and used directly in assays without washing.
Staining and flow cytometry
For staining surface markers, cells were washed and blocked in FACS buffer (PBS plus 1% FCS) in the presence of Fc block. Samples were stained with anti-CD8α (clone 53–6.7, BioLegend), anti-CD25 (clone 7D4, eBioscience), anti-CD62L (clone MEL-14, eBioscience), anti-CD69 (clone H1.2F3, eBioscience), anti-Vα2 (clone B20.1, BD Biosciences), anti-CD244 (clone C9.1, BD Biosciences), anti-Ly108 (clone 13G3, BD Biosciences), or anti-CD27 (clone LG.3A10, BioLegend), at 4 C for 30 minutes in FACS buffer, protected from light, followed by fixation with 4% paraformaldehyde (PFA, Electron Microscopy Sciences). For intracellular staining, cells were fixed and permeabilized with BD Cytofix/Cytoperm (BD Biosciences) at 4 C for one hour. Samples were then stained with anti-granzyme B (clone GB11, BD Biosciences) for one hour at 4 C, protected from light. For phospho-antibody staining, cells were fixed with 4% paraformaldehyde, methanol-permeabilized at −20 C, and stained for 60 minutes at 4 C with anti-phosphoS6 (clone D57.2.2E, Cell Signaling) in PBS plus 1% Triton X-100 and 0.5% bovine serum albumin (BSA). Data were acquired on either a Calibur1 or LSRII flow cytometer (BD) and analyzed using FlowJo software (Tree Star).
Proliferation assay
To evaluate the proliferative capacity of cells, splenocytes were stained with 1μM Cell Trace Violet (CTV, Life Technologies) in PBS at 37 C for 10 minutes. Stained cells were washed 3 times with complete media and then stimulated in the presence of OVA257-264. Cells were collected at indicated time points, stained with anti-CD8α (clone 53–6.7, BioLegend) and evaluated via flow cytometry.
Cytotoxicity assays
In vitro cytolytic activity was determined using either a lactate dehydrogenase (LDH) release or flow-based assay. For LDH release, CytoTox Non-radioactive Cytotoxicity Assays (Promega) were used according to the manufacturer’s instructions. Briefly, targets were pulsed with 1μM OVA257–264 peptide for 1 hour at 37°C, washed twice, and resuspended in phenol red-free RPMI with 2% FBS (assay buffer). Activated CTLs were washed and resuspended in assay buffer, added to 96 well plates and titrated in assay buffer. Targets or assay buffer were added to wells to achieve appropriate effector:target ratios and control groups, and plates were incubated for indicated times at 37°C. Supernatants were then transferred to unused plates containing assay substrate and the OD read at 490nm on a Thermomax plate reader to measure lactate dehydrogenase (LDH) release. Percent cytotoxicity was calculated per manufacturer’s suggestions. For the flow-based method, targets were stained with 1μM Cell Trace Violet (CTV, Life Technologies) as described above. Targets were either left unpulsed or pulsed with 1μM OVA257–264 peptide for 1 hour at 37°C, washed twice, and resuspended in assay buffer. Activated CTLs were washed and resuspended, added to 96 well plates and titrated in assay buffer. Non-pulsed, or pulsed targets were added to wells with CTLs to achieve appropriate effector:target ratios; additional wells set up with CTLs or target cells alone to control for spontaneous cell death. After indicated times at 37°C, plates were centrifuged, and supernatants discarded. Cells were then stained with anti-CD8α (clone 53–6.7, BioLegend) antibodies and LiveDead green (Life Technologies), washed with FACS buffer, and fixed with 4% PFA. Plates were read on a LSRII instrument using a high throughput sampler. For analysis, the CTV+ LiveDead+ population represents the target cells that have been killed, while the CTV+ LiveDead− population represents the remaining viable target cells in each well. Percent cytotoxicity was calculated as: 100 – [(viable CTV+ cells in sample)/(viable CTV+ cells in control)] x 100, where CTV+ cells in sample are cells in experimental wells, and viable CTV+ cells in control are cells in wells without T cells.
To examine cytolytic activity in vivo, indicated numbers of previously activated WT or ITK-deficient CTLs, or a PBS control were adoptively transferred via retro-orbital injection into naïve WT C57BL/6 hosts. LPS-activated B cell targets from WT GFP mice were labeled with either 0.2μM or 2μM Cell Proliferation Dye eFlour450 (eBioscience) and left unpulsed or pulsed with 1μM OVA257–264 peptide for 1 hour at 37°C, respectively. B cells were then mixed at a 1:1 ratio and transferred via retro-orbital injection into mice 24 hours after delivery of CTLs. Spleens were harvested at indicated time points and populations analyzed via flow cytometry. Transferred B cells were distinguished from recipient B cells by gating on the GFP positive population, and peptide pulsed versus non-pulsed targets based on the intensity of eFluor450 fluorescence. Percent cytotoxicity was calculated as follows: % cytotoxicity = 100 – ([(Tpulsed/Tnon-pulsed)/(Cpulsed/Cnon-pulsed)] x 100), where Tpulsed is the percentage of peptide-pulsed targets harvested from spleens of recipients, Tnon-pulsed is the percentage of non-pulsed targets harvested from spleens of recipients, Cpulsed is the percentage of peptide-pulsed targets harvested from spleens of PBS recipients, and Cnon-pulsed is the percentage of non-pulsed targets harvested from spleens of PBS recipients.
Conjugate assays
For FACS-based conjugate assays, LPS-activated primary B cells, EL4, or MC57 targets were stained with 0.1μM carboxyfluorescein diacetate succinimidyl ester (CFSE), and pulsed with peptide at indicated concentrations for 1 hour at 37°C or left non-pulsed as a control. After washing, targets were mixed with previously activated CTLs at a 2:1 T:target ratio in 96-well round bottom plates, spun down, and incubated for 20 minutes at 37°C. Cells were washed and stained with anti-CD8α-PerCPCy5.5 or –APC (clone 53–6.7, BioLegend) and conjugates enumerated via flow cytometry, where the CD8+ CFSE double positive population represented T cells forming conjugates with targets.
Degranulation assays
For degranulation assays (30), activated CTLs were stimulated in plates coated with anti-CD3ε (BioXCell) or mixed at a 1:1 ratio with peptide-pulsed or non-pulsed targets at 37°C in the presence of anti-CD107a-PE-labeled antibody (clone 1D4B, BioLegend). Upon degranulation, CD107a is exposed on the cell surface, allowing binding and subsequent internalization of the anti-CD107a-PE. At indicated time points, plates were placed on ice and cells transferred into cold PBS, stained with anti-CD8α (clone 53–6.7, BioLegend) antibody, and analyzed via flow cytometry. For analysis, the percent of PE+ cells in the CD8+ gate indicates cells that have degranulated, having been exposed to anti-CD107a during the assay.
RNA isolation and analysis
Total RNA was isolated from WT and Itk−/− CD8+ T cells at indicated time points using the RNeasy Mini Kit (Qiagen) and was reverse transcribed with random hexamer primers and the M-MLV Reverse Transcriptase (Applied Biosystems). Quantitative RT-PCR was performed on Step One Plus Real-time PCR System (Applied Biosystems) using TaqMan assays (Applied Biosystems) for indicated genes. Samples were normalized to 18S RNA and data are expressed as relative to WT levels using the 2−ΔΔCTmethod.
Immunofluorescence confocal microscopy
To prepare conjugates for immunofluorescence microscopy, targets were pulsed with 1μM OVA257–264 at 37°C for 1 hour, washed twice, and resuspended in pre-warmed phenol red-free RPMI (imaging media). Activated CTLs were washed and resuspended in imaging medium and mixed with peptide-pulsed targets at a 1:1 ratio. Cells were incubated at 37°C for 15 minutes to allow conjugate formation and then plated on glass multi-well slides previously coated with 0.01% poly-L-lysine for 5 minutes at 37°C. Cells were fixed and permeabilized with cold methanol on ice or fixed at room temperature with 2% paraformaldehyde for 5 minutes, followed by several washes in PBS. Methanol-fixed cells were blocked for 30 minutes at room temperature in 1% BSA in PBS plus Fc block (blocking buffer). PFA-fixed cells were quenched for 10 minutes with 5mM glycine, and permeabilized and blocked with 0.2% saponin in blocking buffer for 30 minutes at room temperature. Cells were incubated with primary antibodies (polyclonal actin and γ-tubulin, Sigma, and polyclonal granzyme B, Abcam) in blocking buffer for 1 hour at room temperature, washed in either blocking buffer or blocking buffer containing 0.2% saponin, followed by a 45-minute incubation with secondary antibodies at room temperature, and washed several times. Samples were preserved using ProLong Gold with DAPI (Life Technologies) and no. 1.5 cover glass (VWR), and imaged using an Axio Observer Z1 microscope (Carl Zeiss Inc.). For quantification of centrosome and granule polarization, structures in the uropod (or distal 1/3) of T cells were scored as “distal.” Structures in the main body (or middle third) of the T cell polarized toward the target cell were scored as “partial,” and structures in contact with the plasma membrane at the immunological synapse were scored as “polarized.” Granules were scored as “dispersed” when seen localized throughout more than one section of the body of the cell.
Transmission electron microscopy (TEM)
WT and Itk−/− OT-I splenocytes that were frozen after three days activation in the presence of OVA257–264 as described above, were thawed into medium plus IL-2 and incubated for 48 hours to allow for the recovery and expansion of CTLs. CTLs were then either used unlabeled or incubated overnight with horseradish peroxidase (HRP, Boehringer Ingelheim) to label granules, washed and used to generate CTL:target cell conjugates by incubating for 20–65 minutes with EL-4 targets previously pulsed with between 10nM and 1μM OVA257–264. Conjugated samples were fixed in 1.5% glutaraldehyde/2% paraformaldehyde and processed for TEM analysis, as previously described (31, 32). Samples were analyzed using a FEI Tecnai G2 Spirit BioTWIN transmission EM (Eindhoven, The Netherlands) and images captured with an Eagle 4K CCD camera using FEI TIA software. For quantitation of degranulation and target death, images of CTL conjugated to targets for 45 minute in the presence of 1μM OVA257–264 peptide were collected for both WT (n=21) and Itk−/− (n=11) CTLs populations, and scored for the presence or absence of material between the cells (indicative of degranulation) and target death (indicated by ER swelling).
Statistical analysis
All statistical analyses (Student’s t tests, paired sample tests, and two-way ANOVA) were performed using Microsoft Excel or GraphPad Prism software. P values less than 0.05 were considered statistically significant.
RESULTS
ITK-deficient CTLs have impaired cytolytic effector function against targets
Previous studies on the effects of ITK-deficiency on murine CD8+ T cell function revealed both decreased and delayed viral clearance, accompanied by decreased CTL expansion in vivo and in vitro (26, 27). However, whether there are defects against distinct targets, such as B cells, and whether there are specific defects in the distinct stages of cytolysis on an individual cell basis, have not been examined. To evaluate the effects of ITK on CD8+ T cell cytotoxicity, we used the OT-I TCR transgenic mouse model. T cells from OT-I mice express a clonal TCR that recognizes a peptide, OVA257-264, in the context of H2Kb (28). This system allowed us to evaluate killing of different targets presenting the same antigen in a controlled environment, using defined numbers of effectors and targets. Furthermore, although ITK-deficient mice show altered thymic development of CD8+ T cells, expression of the OT-I transgene largely rescues these phenotypes (33).
To generate effector CTLs, splenocytes from WT and ITK-deficient OT-I mice were stimulated in vitro with OVA257–264 peptide for three days, followed by culture in IL-2 to allow expression of cytolytic effectors and acquisition of cytolytic capabilities (Figure 1A). Consistent with their TCR signaling defects, Itk−/− OT-I CD8+ T cells initially exhibited delayed proliferation (Figure 1B), measured by the dilution of the amine reactive dye, Cell Trace Violet (CTV), as well as differences in the initial induction and down-regulation of TCR surface activation markers (Figure 1C). However, by day six, when WT CTLs are functional to kill, the mean fluorescence intensity of surface markers including Vα2, CD69, CD25, as well as the percentage of CD62L+ cells and cell viability (see below) were grossly similar between WT and Itk−/− OT-I CD8+ T cells (Figure 1C). Similarly, expression of SLAM family members and other costimulatory molecules such as CD27, which is mutated in another immunodeficiency associated with increased susceptibility to EBV, was similar between WT and ITK-deficient CTLs (data not shown). Therefore, we used day 6 CTLs to compare cytolysis between WT and Itk−/− cells.
Figure 1.

ITK-deficient CTLs express similar surface marker levels after activation. (A) Schematic of in vitro activation of OT-I splenocytes. (B) Proliferation of CD8+ T cells in total ex vivo splenocytes cultures from WT or ITK-deficient OT-I mice stimulated in the presence of 10nM OVA257-264 peptide, evaluated with Cell Trace Violet (CTV). Histogram overlay depicts CTV in WT (black) or Itk−/− (grey) CD8+ T cells examined directly ex vivo (dotted lines) or after 48 hours of culture (solid). (C) Representative histograms of surface marker staining on WT (black line) or Itk−/− (grey solid) OT-I CD8+ T cells at indicated time points during in vitro activation. IL-2 was added after day 3 and day 5 of culture. Data are representative of one of greater than three independent experiments.
CTLs from SAP-deficient patients, who have clinical phenotypes similar to ITK-deficient patients, show defects in cytolysis of EBV-infected B cells but normal killing of other targets (25, 34–36). To evaluate whether Itk−/− OT-I CTLs also showed specific defects in killing B cells, we used three target cell types that were pulsed with 1μM of OVA257-264 peptide: LPS-activated B cells from WT C57Bl/6 mice, the EL4 lymphoma T cell line, and the MC57 non-lymphocyte fibrosarcoma cell line. While in vitro activated WT OT-I CTLs could kill WT B cells effectively, CTLs from Itk−/− OT-I mice exhibited impaired cytotoxicity against B cell targets (Figure 2A). Defects were confirmed using a flow-based assay where death of targets and CTLs could be individually monitored and were observed at all time points tested, up to 8h of cytolysis (data not shown). Defects were also seen in an in vivo transfer model, where activated CTLs from Itk−/− OT-I mice had impaired cytotoxicity against co-transferred WT activated B cell targets pulsed with OVA257-264 peptide (Supplemental figure 1). However, unlike CTLs from SAP-deficient mice that show defective killing primarily of B cell targets (24), ITK-deficient CTLs also failed to efficiently kill both the peptide-pulsed EL4 T lymphocyte (Figure 2B) and MC57 fibrosarcoma cell lines (Figure 2C). Together these data suggest that unlike SAP-deficiency, ITK–deficiency leads to a global defect in cytolysis by CTLs.
Figure 2.
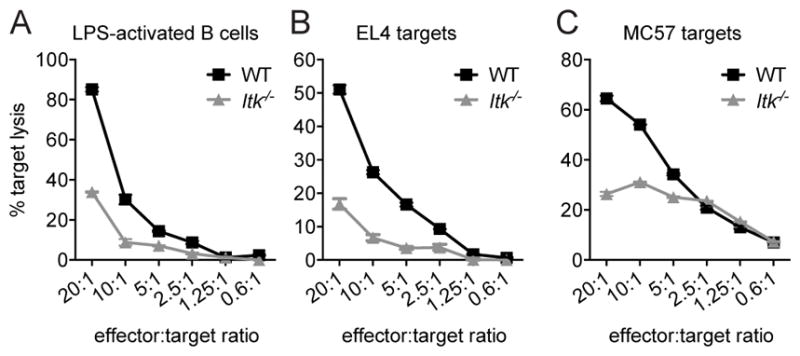
ITK-deficient CTLs have impaired cytolytic function against targets. In vitro cytolysis of (A) LPS-activated WT B cells, (B) EL4, or (C) MC57 targets pulsed with 1μM OVA257-264 peptide by WT OT-I (black) or Itk−/− OT-I (grey) day 6 CTLs at decreasing CTL:target ratios, as measured by LDH-release assays. Data for LPS-activated B cells are representative of one of greater than three independent experiments, EL4 targets are representative of greater than ten experiments, and MC57 targets are representative of one of two experiments. Graphs show mean of triplicate wells ± SD. Similar results were obtained with a flow-based cytolysis assay.
ITK-deficient cells show decreased expression of effector molecules
Following activation, CD8+ T cells differentiate into CTLs, which express cytolytic effector molecules critical for the cytolysis of target cells. Notably, expression of several of these effector molecules is dependent on mTOR and AKT-mediated pathways (37, 38). We have recently found that ITK-deficient CD4+ T cells show impaired activation of mTORC1, as evidenced by decreased phosphorylation of ribosomal protein S6, which is phosphorylated by S6 Kinase, a direct target of the mTORC1 complex (13). Similarly, we observed decreased phosphorylation of S6 during early activation of ITK-deficient CD8+ T cells (Figure 3A). Consistent with these observations, both granzyme B (Figure 3B) and perforin (Figure 3C) were reduced in in vitro activated Itk−/− OT-I CTLs when compared with WT cells. Thus, ITK-deficiency prevents full expression of cytolytic effectors on day 6 of CTL culture.
Figure 3.
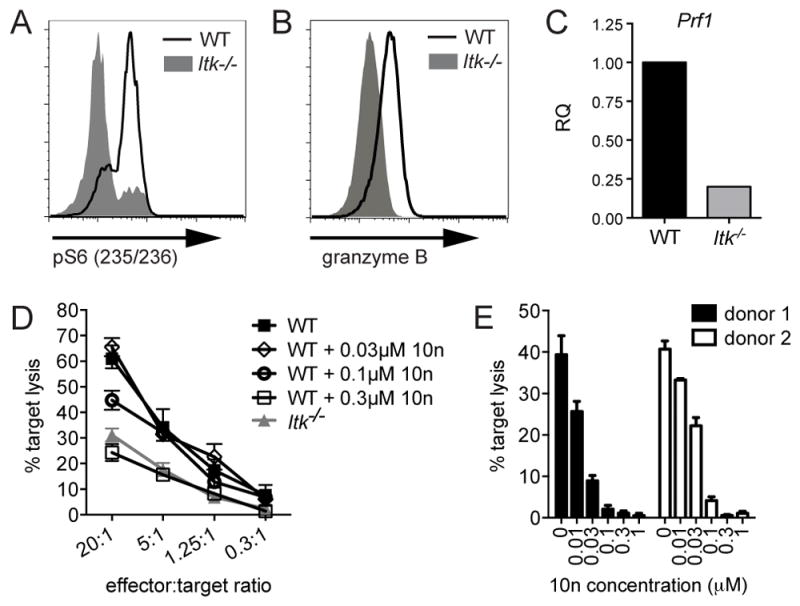
ITK-deficient CTLs show decreased expression of lytic effector molecules, and additional defects in cytolysis. (A) Whole splenocytes from WT (black line) or Itk−/− (grey solid) OT-I CD8+ T cells were activated for six hours in the presence of 10nM OVA257-264 peptide, stained and analyzed for pS6 (S235/236) using flow cytometry. Histogram is representative of three independent experiments. (B) Granzyme B expression levels measured by flow cytometry in day 7 in vitro activated WT (black) or Itk−/− (grey solid) CTLs. Histogram is representative of greater than three independent experiments. (C) Prf mRNA transcript levels in day 7 in vitro activated WT (black) or Itk−/− (grey), determined by qRT-PCR. Data are representative of two independent experiments. (D) In vitro cytolysis of EL4 targets pulsed with 1μM OVA257-264 peptide by previously activated ITK-deficient (grey triangles) or WT OTI CTLs treated with indicated concentrations of the ITK inhibitor, 10n (black open diamonds, circles, or squares) immediately before cytolysis assay, or left untreated as a control (black closed squares). Graph shows mean of triplicates ± SD, and is representative of three independent experiments. (E) In vitro cytolysis of P815 target cells pulsed with 2μg/mL OKT3 by allo-reactive human CD8+ T cells generated from two independent healthy donors and treated with 10n immediately before cytolysis assay. Graph shows mean of triplicates ± SD at 4-hour time points using 20:1 CD8:target cell ratio. Data representative of two experiments using cells from two healthy donors each.
ITK-deficiency also results in an intrinsic impairment in cytolysis
To determine whether reduced expression of effector molecules was the sole cause of the reduced cytotoxicity observed in day 6 ITK-deficient CTLs, we treated previously activated WT OT-I CTLs with 10n, an inhibitor of ITK, immediately prior to their use. This treatment allowed for short-term inhibition of ITK during cytolysis, while minimizing effects on differentiation. Notably, incubation of WT CTLs with 10n during the cytolysis assay reproduced the defects in killing seen in activated ITK-deficient OT-I CTLs (Figure 3D). Similarly, treatment with 10n of allo-activated human CD8+ T cell blasts generated from healthy peripheral blood mononuclear cell (PBMC) donors also led to impaired killing of P815 targets, a mouse mastocytoma cell line expressing high levels of Fc receptors that can present anti-CD3, making it a target for human CD8+ T cells (Figure 3E). These data suggest that while suboptimal TCR signaling in the absence of ITK affects the activation and the expression of cytolytic effectors, impaired killing is not fully attributed to altered activation and differentiation of Itk−/− CD8+ T cells.
Itk−/− CTLs show normal adhesion and actin ring formation
To better understand the roles of ITK and TCR signaling in regulating cytolytic activity, we examined how ITK-deficiency affects discrete stages of CTL function. Killing by CTLs is initiated when TCR engagement triggers adherence of CTLs to targets. Similar to defects observed in Itk−/− CD4+ T cells (39), freshly isolated Itk−/− OT-I CD8+ T cells showed decreased adhesion to B cell targets in a flow-based adhesion assay (Supplemental figure 2A). However, once activated, Itk−/− OT-I CTLs were able to effectively form conjugates with either peptide-pulsed LPS-activated WT B cell targets (Figure 4A) or EL4 targets (Supplemental figure 2B), when compared with WT CTLs. These results suggest that once CTLs are generated, impaired adhesion is unlikely to contribute to defects in killing by Itk−/− CTLs.
Figure 4.

Itk−/− CTLs have normal adhesion and actin ring formation during immunological synapse formation. (A) Adhesion after 20 minutes conjugation of previously activated WT (black) or Itk−/− (grey) CTLs to LPS-activated WT B cells pulsed with 1μM OVA257-264 or unpulsed B cells as a control. Graph represents mean ± SD of percent CD8+ target+ cells in total CD8+ events. Data are representative of one of greater than three experiments. (B) Representative images of maximum projections of WT or Itk−/− CTLs in conjugate pairs with LPS-activated WT B cell targets pulsed with 1μM OVA257-264 (first and third columns, respectively). 1μm slice of reconstructed z stacks rotated in the yz plane (second and fourth columns). Nuclei (blue), CD8 (red), actin (green). Scale bars = 5μm. (C) Quantification of actin ring formation at the immunological synapse between WT (black) and Itk−/− (grey) CTLs and LPS-activated WT B cell targets. Bars represent mean ± SEM from more than three independent experiments (WT total n=105, Itk−/− total n=82).
Adhesion is accompanied by the accumulation and subsequent centralized clearance of actin at the immunological synapse (3, 4, 31). Previous work had shown that ITK-deficient CD4+ T cells have defects in actin polarization, likely due to a kinase-independent scaffolding role for ITK in stabilizing VAV1-SLP76 interactions during signaling (11). To examine actin organization in Itk−/− OT-I CTLs, we evaluated actin localization in CTLs by immunofluorescence confocal microscopy (Figure 4B). Actin accumulation at the CTL:target interface appeared normal in Itk−/− OT-I CTLs. Furthermore, the ring-like organization of the actin cytoskeleton, as evaluated in 3-dimensional reconstruction of z-stacks turned en face, did not differ between WT and Itk−/− cells in conjugates with peptide-pulsed LPS-activated B cells (Figure 4B and C) or EL4 targets (Supplemental figure 2C). This, in combination with their normal adhesion, suggested that once activated, CTLs do not require ITK for the early stages of their interactions with target cells.
Itk−/− CTLs exhibit polarized centrosomes and lytic granules during target cell cytolysis
Following immunological synapse formation, the centrosome reorients toward the interface between T and target cells, thus directing lytic granules toward their target for effective killing (3, 4). Studies suggest that centrosome polarization in T cells requires PLCγ1 activation, but is a DAG signaling-dependent, calcium-independent process (40). Because ITK directly phosphorylates PLCγ1, which is responsible for DAG production during TCR signaling, we hypothesized that impaired centrosome reorientation could contribute to defects in cytolysis in Itk−/− CTLs. To evaluate this question, we co-stained for actin and γ-tubulin as a marker for the centrosome, and examined their localization using confocal immunofluorescence (IF) microscopy in WT and ITK-deficient CTLs (Figure 5A). We found that ITK-deficient CTLs polarized their centrosomes as efficiently as WT in response to peptide-pulsed EL4 targets (Figure 5B). Nonetheless, we found that Ca2+ mobilization in response to anti-CD3 stimulation was still markedly reduced in activated CTLs in the absence of ITK (Supplemental figure 2D), confirming that PLCγ1 activation was still impaired in ITK-deficient CTLs (10, 17, 41).
Figure 5.

Itk−/− CTLs show normal centrosome and lytic granule polarization during cytolysis of targets. (A) Centrosome reorientation shown as maximum projections. Nuclei (blue), EL4 targets (red), actin (green), γ-tubulin (white). Centrosome location highlighted by yellow arrowheads. Scale bars = 5μm. (B) Quantification of centrosome reorientation in WT (black, total n=129) and Itk−/− (grey, total n=128) CTLs. (C) Lytic granule polarization shown as maximum projections. Nuclei (blue), EL4 targets (red), actin (green), granzyme B (white). Lytic granule location highlighted by yellow arrowheads. Scale bars = 5μm. (D) Quantification of lytic granule polarization in WT (total n=127) and Itk−/− (total n=151) CTLs in conjugates with EL4 targets pulsed with 1μM OVA257-264 peptide. Bars in panels B and D represent mean ± SEM from greater than three independent experiments.
The polarization of lytic granules has also been linked to the strength of TCR signaling in the OT-I system, where weak signals generated by low avidity ligands induced centrosome polarization without triggering concomitant lytic granule polarization (32, 42). These data suggest that reorientation of the centrosome and polarization of lytic granules can be decoupled under suboptimal TCR-triggering conditions. Although the absence of ITK during TCR engagement also results in impaired TCR signaling, we found polarization of lytic granules in Itk−/− OT-I CTLs was equivalent to WT CTLs in response to peptide-pulsed EL4 targets, as evaluated by granzyme B staining (Figure 5C and 5D). Thus, once activated CTLs are generated, ITK is not required for centrosome or lytic granule polarization toward targets, despite defective activation of PLCγ1.
TCR-triggered degranulation is reduced in the absence of ITK
The final stage of granule-dependent killing of target cells by CTLs is degranulation of the cytotoxic granules, which leads to the killing of target cells. To evaluate degranulation, we used a flow-based secretion assay that measures cycling of lysosomal associated membrane protein 1 (LAMP1) to the cell surface in response to TCR stimulation (30). Although there was some degree of variability, we found that Itk−/− OT-I CTLs exhibited reduced degranulation, as measured by LAMP1 cycling, in response to either plate-bound anti-CD3 (Figure 6A), or activated targets, including both peptide-pulsed LPS-activated WT B cell (Figure 6B) and EL4 cell targets (Figure 6C). Importantly, intracellular staining confirmed that total LAMP1 content was equivalent between WT and ITK-deficient CTLs (Supplemental figure 3A), suggesting that reduced degranulation in the absence of ITK was not due to differences in total LAMP1 content between WT and ITK-deficient CTLs.
Figure 6.
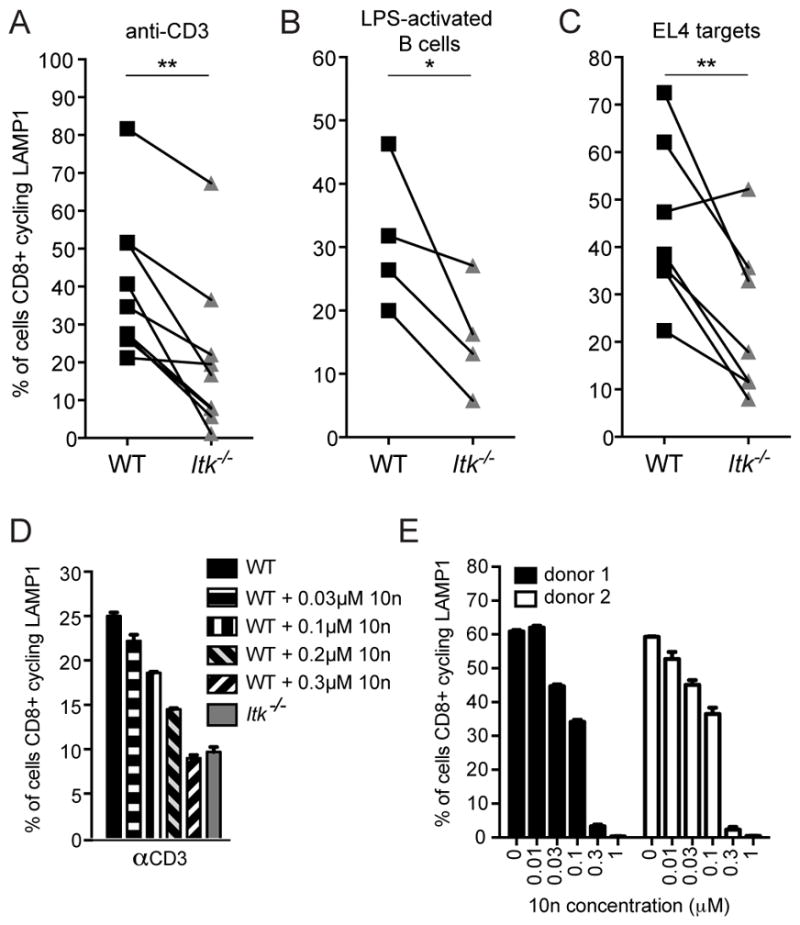
ITK-deficient CTLs exhibit reduced TCR-triggered degranulation. Degranulation measured in a flow-based LAMP1 cycling assay in WT (black) or Itk−/− (grey) CTLs in response to (A) plate-bound anti-CD3, and 1μM OVA257-264-pulsed (B) LPS-activated B cell or (C) EL4 targets. Each line represents data from paired mice from an independent LAMP1 cycling experiment, where CD107a-PE positive cells in the CD8+ gate were determined to be positive for degranulation. *P<0.05, **P<0.01 calculated by paired sample t-tests. (D) Degranulation in response to 5μg/mL plate-bound anti-CD3 in day 6 WT OT-I CTLs (black solid bar), WT OT-I CTLs pre-treated with the ITK inhibitor, 10n, at 0.03 μM (horizontal striped bar), 0.1 μM (vertical striped bar), 0.2 μM (black and grey diagonal bar) or 0.3 μM (black and white inverse diagonal bar), or Itk−/− OT-I CTLs (grey solid bar). Graph representative of two independent experiments. (E) Degranulation in allo-reactive human CD8+ T cells from two healthy donors treated with increasing concentrations of 10n in response to 5μg/mL of plate-bound OKT3. Graph represents data using the same donors as in Figure 3E and is representative of one of two independent experiments.
To confirm that impaired degranulation by Itk−/− CTLs was due to the loss of ITK activity, we treated previously activated WT OT-I CTLs with increasing concentrations of the ITK-inhibitor, 10n, during degranulation assays. Inhibitor treatment of WT CTLs led to a reduction in degranulation similar to that seen in Itk−/− OT-I CTLs (Figure 6D). In contrast, upstream processes such as adhesion were not affected by the ITK inhibitor (Supplemental figure 3B). Furthermore, treatment of allo-activated human CD8+ T cells with 10n also led to impaired degranulation (Figure 6E), again supporting a defect in degranulation that was independent of impaired differentiation and expression of lytic effectors. Together, these results suggest that ITK activity plays a previously unappreciated role in degranulation, the final stage of CTL killing, without affecting upstream events.
Transmission electron microscopy revealed centrosome docking in ITK-deficient CTLs
To help understand the cellular basis for the reduction in LAMP1 cycling observed in FACS-based secretion assays, we examined the immunological synapse in WT and ITK-deficient CTL:target conjugates using transmission electron microscopy (TEM), which provides more detailed structural information at a higher resolution than can be obtained using immunofluorescence techniques. TEM images revealed that ITK-deficient CTLs could establish contact sites with peptide-pulsed EL4 targets (Figure 7 and Supplemental figure 3C). Consistent with our observations by immunofluorescence microscopy, TEM images showed polarized centrosomes (Figure 7) and accumulation of granules (Supplemental figure 3C) at the synapse in both WT and ITK-deficient CTLs. Furthermore, the greater resolution allowed by TEM revealed that, as in WT cells, the polarized centrosomes of ITK-deficient cells moved right up to the cell surface (Figure 7), indicating centrosome docking was unaffected in these cells. Also consistent with our in vitro cytolysis assays, 95.5% (20/21) of WT CTL conjugates after 45 minutes incubation were associated with dead/dying targets (indicated by swollen target ER), and/or showed accumulation of material in the gaps between the two cells, indicative of lytic protein release and target cytolysis. While some dying targets (45.5%, 5/11 conjugates) were also present in preparations using ITK-deficient CTLs, 54.5% (6/11) of conjugates at 45 minutes showed no released material between the cells and were associated with healthy-looking targets. Thus, consistent with our observations in light microscopy, ITK-deficient cells show normal early stages of cytolysis, including those up to centrosome docking, but appear to have impaired degranulation.
Figure 7.

The centrosome polarizes to the synapse in Itk−/− CTLs conjugated to targets. Representative TEM images of the immune synapse region of WT and Itk−/− CTLs conjugated to targets for 20 minutes in the presence of 10nM OVA257-264 peptide, showing centrosomes (black asterisks) tightly polarized to the plasma membrane. Granules are indicated by white asterisks. The WT conjugate (left) has debris in the secretory cleft (SC) between the cells and swollen target cell endoplasmic reticulum (ER), indicative of granule exocytosis and target apoptosis, whereas the ITK−/− conjugate (right) shows an empty secretory cleft and healthy ER, suggesting no degranulation and no target death. Representative sample from two independent experiments. G = Golgi, N = nucleus. Scale bars = 500nm.
Cytokine signaling restores degranulation and cytotoxicity in ITK-deficient CTLs
Cytokines such as IL-2 have long been known to enhance lymphocyte cytotoxicity in culture, particularly for Natural Killer (NK) cells. For example, IL-2 restores cytotoxic capabilities in NK cells from patients with primary immunodeficiencies that exhibit defects in lymphocyte degranulation (43, 44). However, very little is understood about the contribution of IL-2 to the regulation of CTL degranulation, in part due to the requirement for IL-2 for generation of CTLs. Since ITK-deficient T cells have significant defects in IL-2 production (9, 10, 29), we evaluated the effects of IL-2 on the maturation and function of ITK-deficient CTLs.
We first asked whether prolonged exposure to IL-2 affected degranulation and cytolysis in ITK-deficient CTLs. To compare degranulation between cells cultured in IL-2 for different amounts of time, we activated independent CD8+ OT-I splenocyte cultures over the course of five days, first stimulating with OVA257-264 alone for 3 days and then resuspending the cells in fresh media plus IL-2 every 48 hours after day 3 of culture (Figure 8A). This provided us with CTLs at different time points after primary activation that could be assayed for degranulation on the same day. Under these conditions, cells that had been in culture longer were exposed to an additional round of IL-2. We observed that viability in WT OT-I CTLs began to decrease after eight days of culture, from an average of 83.3% on day 7 to 25.8% on day 9, as evaluated by a membrane permeable dye. In contrast, ITK-deficient CTLs were more resistant to cell death, with viability only decreasing from 76% on day 7 to 62.8% on day 9 (Supplemental figure 4A). Moreover, prolonged culture rescued degranulation in viable Itk−/− OT-I CTLs on day 9 of culture, reaching levels equivalent to those seen in viable WT OT-I CTLs that were stimulated for the same period of time (Figure 8B, left). This finding was not secondary to reduced degranulation in the viable WT cells, but rather to an increase in ITK-deficient cells. Prolonged incubation with IL-2 also restored the ability of ITK-deficient CTLs to kill targets by day 9, as evidenced by both LDH release and flow-based assays, which evaluate cell death specifically in targets (Figure 8B, right and data not shown). The expression of granzyme B also improved after 9 days of culture of ITK-deficient CTLs (Figure 8C). Thus, prolonged culture in the presense of IL-2 restored both degranulation and cytolysis, as well as the expression of cytolytic effector molecules in ITK-deficient OT-I CTLs. Of note, however, treatment of WT CTLs with 10n still inhibited degranulation and killing on day 9 (Figure 8D), whereas 10n did not affect cytolysis by ITK-deficient cells (Supplemental figure 4B).
Figure 8.

Degranulation and cytotoxicity are restored in ITK-deficient CTLs after prolonged or early culture in IL-2, or the addition of exogenous IL-12 to cultures. (A) Schematic of CD8+ T cell activation and cytokine addition time course. (B) Percent of WT (black) or Itk−/− (grey) CTLs cycling LAMP1 in response to 5μg/mL of plate-bound anti-CD3 evaluated at indicated days after the start of primary activation (left panel). CD107a-PE positive cells in the CD8+ gate were determined to be positive for degranulation. In vitro cytolysis of EL4 targets pulsed with 1μM OVA257-264 peptide by WT OT-I (black) or Itk−/− OT-I (grey) CTLs at a 20:1 effector:target ratio, evaluated at indicated days after primary activation (right panel). Graphs show percent cytotoxicity ± SD at 4-hour time points and are representative of three independent experiments. *P<0.05, **P<0.01, ***P<0.001 calculated by Student’s T-test. (C) Granzyme B MFIs ± SD at indicated time points. Data are representative of two independent experiments. (D) Left panel: degranulation in day 9 CTLs in response to 5μg/mL plate-bound anti-CD3 in WT OT-I (black solid bar), WT OT-I pre-treated with the ITK inhibitor, 10n, at 0.03 μM (horizontal striped bar), 0.1 μM (vertical striped bar), or 0.3 μM (diagonal striped bar), Itk−/− OT-I CTLs (grey solid bar), or day 6 Itk−/− OT-I CTLs (open bar). Graph is representative of two independent experiments. Right panel: in vitro cytolysis of EL4 targets pulsed with 1μM OVA257-264 peptide by day 9 ITK-deficient (grey closed triangles) or WT OT-I CTLs treated with indicated concentrations of the ITK inhibitor, 10n (black open diamonds, cirlces, or squares), immediately before cytolysis assays, or left untreated as a control (black closed squares). Day 6 Itk−/− OT-I CTLs are also shown (grey open triangles). Graph shows mean of triplicates ± SD, and is representative of two independent experiments. (E) Left panel: In vitro cytolysis of 1μM OVA257-264 peptide-pulsed EL4 target cells by WT (black) or Itk−/− OT-I CTLs (grey) activated with 10nM OVA257-264 peptide and then cultured in IL-2 for three days (OVA), or Itk−/− OT-I CTLs activated with 10nM OVA257-264 peptide plus 10 IU IL-2 before continued culture in IL-2 for an additional three days (OVA+IL-2, grey open triangles). Graphs show percent cytotoxicity ± SD on indicated days and are representative of two independent experiments. Right panel: Degranulation measured in a flow-based LAMP1 cycling assay in response to plate-bound anti-CD3 in WT (black) or Itk−/− (grey) OT-I CTLs activated with 10nM OVA257-264 peptide only and then cultured in IL-2 for three days (OVA) or Itk−/− OT-I CTLs activated with 10nM OVA257-264 peptide plus 10 IU IL-2 before continued culture in IL-2 for and additional three days (OVA+IL-2, grey open triangles). Graphs show percent CD8+ T cells cycling LAMP1 ± SD, where CD107a-PE positive cells in the CD8+ gate were determined to be positive for degranulation on indicated days and are representative of two independent experiments. (F) Granzyme B MFI ± SD in day 6 WT (black) or Itk−/− (grey) OT-I CTLs activated for 3 days in the presence of 10nM OVA257-264 peptide only and then cultured in IL-2 for three days (OVA), or 10nM OVA257-264 peptide plus 10 IU IL-2 (OVA+IL-2) before continued culture in IL-2 for three days. Data are representative of two independent experiments. (G) CD25 MFIs ± SD in day 3 WT (black) or Itk−/− (grey) OT-I CD8+ T cells activated for three days in the presence of 10nM OVA257-264 peptide only (OVA) or 10nM OVA257-264 peptide plus 10 IU IL-2 (OVA+IL-2). Data are representative of two independent experiments. (H) In vitro cytolysis of EL4 targets pulsed with 1μM OVA257-264 peptide by WT OT-I (black) or Itk−/− OT-I (grey) CTLs at a 20:1 effector:target ratio, evaluated on day 6 after primary activation with 10nM OVA257-264 peptide ± exogenous IL-2 or IL-12 at indicated time points. Graphs show percent cytotoxicity ± SD at 4-hour time points and are representative of three independent experiments. *P<0.05, ***P<0.001, calculated by Student’s T-test, ns = not significant. (I) CD25 MFI ± SD in WT (black) or Itk−/− OT-I (grey) CD8+ T cells on day three after primary activation with 10nM OVA257-264 peptide only (OVA) or 10nM OVA257-264 peptide plus IL-12 (OVA+IL-12). Data are representative of two independent experiments.
Given these observations, we then asked whether the addition of IL-2 to cultures at the beginning of in vitro activation could alter the acquisition of effector function in CTLs. To test this, WT and Itk−/− CD8+ T cells were activated in the presence of OVA257-264 +/− IL-2 during days 0–3, followed by culture in IL-2 alone, and then degranulation and cytolysis assays were performed on days 4–9 (Figure 8A). The addition of IL-2 beginning on day 0 of in vitro culture boosted killing by WT CD8+ T cells at an earlier time point (increasing from 18.1±3.5% to 34.9± 0.1%, on day 4), but had a minimal effect on target cell lysis after day 4 (Supplemental figure 4C, left panel). The addition of IL-2 to ITK-deficient CD8+ T cell cultures beginning on day 0 also increased target killing on day 4 (15.9±2.3% to 24.6±3.2%), but further augmented target lysis throughout the duration of culture, shifting the acquisition of lytic function to an earlier time points (Figure 8E, left panel and Supplemental figure 4C, right panel). Similar results were obtained in degranulation assays (Figure 8E, right panel and Supplemental figure 4D). While granzyme B expression was also restored by day 6 of culture in ITK-deficient CTLs supplemented on day 0 with IL-2 (Figure 8F), full rescue of cytolysis to WT CTL levels was still not observed until day 8–9 (Figure 8E). Of note, the addition of IL-2 earlier to our cultures also increased surface levels of the high-affinity IL-2Rα, CD25, on day 3 in both WT and Itk−/− CD8+ T cells (Figure 8G). Taken together, these data suggest that IL-2 can provide a synergizing signal that enhances the kinetics of complete activation of CTLs in the absence of optimal TCR signaling.
In addition to IL-2, other cytokines, such as IL-12, have been shown to promote the development of effector CTLs in vivo (38, 45, 46). To evaluate whether IL-12 enhanced activation of CTL function under conditions of suboptimal TCR signaling in the absence of ITK, we activated CD8+ T cells from WT and Itk−/− OT-1 mice as above, but in the presence or absence of IL-12 (Figure 8A). The addition of both IL-12 and IL-2 to cultures starting on day 3 after OVA257-264 stimulation significantly increased target lysis by WT CTLs on day 6 compared with cultures that did not receive exogenous IL-12 on day 3 (Figure 8H). Similarly, killing by ITK-deficient CTLs was also significantly increased in the presence of IL-12 after OVA257-264 stimulation, and achieved equivalent target lysis to WT CTLs activated under the same conditions (Figure 8H). Interestingly, the addition of IL-12 on day 0 did not significantly increase target killing by WT CTLs on day 6 when compared with conditions in which no IL-12 was added at the beginning of cultures. However, addition of IL-12 on day 0 did increase cytolysis by ITK-deficient CTLs (Figure 8H). This rescue was more complete than that seen with IL-2 alone (Figure 8E). We also noted that when added at the beginning of cultures, IL-12 improved the expression of CD25 on ITK-deficient CD8+ cells on day 3, suggesting that IL-12 also enhanced the ability of ITK-deficient cells to respond to IL-2. Taken together these data suggest that cytokines such as IL-12 can promote the acquisition of CTL function under conditions of suboptimal TCR signaling in the absence of ITK, and that this may occur in part by increasing responses to IL-2.
DISCUSSION
ITK is an important modulator of TCR signaling, required for maximum PLCγ1 activation and calcium signaling in T cells. Previous work has suggested that although Itk−/− mice can mount a protective immune response against viral infection, the kinetics of viral clearance were delayed in absence of ITK (26, 27). This information, coupled with reports that patients with mutations in ITK are particularly susceptible to EBV and other viral infections, led us to ask how ITK-deficiency directly affects the process of killing by CTLs. We found that, unlike cells deficient in SAP that exhibit specific defects in cytolysis of B cells, ITK-deficient cells exhibit global defects in cytolysis. We further found that ITK-deficiency both affects CTL expansion and delays expression of cytolytic effectors during activation, and moreover, leads to an intrinsic defect in degranulation. Nonetheless, these defects could be overcome by early or prolonged culture in IL-2, or the addition of exogenous IL-12, suggesting that cytokine signaling can restore the acquisition of effector function in ITK-deficient CD8+ T cells. Our results provide new insight into the effects of ITK and suboptimal TCR signaling on CD8+ T cell function, and how these may contribute to phenotypes associated with ITK-deficiency in humans.
Given altered actin accumulation in ITK-deficient CD4+ T cells (11), we were surprised to find that the early stages of CTL killing were normal in the absence of ITK. Indeed, ex vivo CD8+ T cells from Itk−/− OTI mice exhibited reduced adhesion to target cells, similar to ITK-deficient CD4+ T cells. However, once CTLs were fully activated, ITK-deficiency did not affect adherence or actin recruitment to target cells. This suggests that the cells that expanded during in vitro activation were now more functional. It is intriguing to speculate that this rescue may be at least partially attributable to the presence of IL-2, which can rescue adhesion and other defects in NK cells from patients with other primary immunodeficiencies (see below) (44, 47, 48).
Downstream of TCR engagement, the activation of PLCγ1 leads to the hydrolysis of PIP2 to generate two major second messengers: DAG and IP3. Localized DAG gradients generated by TCR activation serve as a polarizing signal, regulating centrosome reorientation toward target cells by recruiting PKC isozymes (41, 49). Although ITK is important for the full activation of PLCγ1 in T cells, we found that centrosome docking at the synapse, microtubule reorganization, and lytic granule polarization toward target cells appeared normal in Itk−/− CTLs in conjugates with either dying or live targets. Thus, the DAG gradient and downstream activation of ERK in ITK-deficient CTLs appears to be sufficient to drive cell polarization. Whether ITK-deficiency alters expression or activity of other compensatory regulators of DAG levels, such as DAG kinases, remains an intriguing question.
The other major product of PLCγ1, IP3, triggers store-operated calcium entry into the cell through the action of ER calcium sensors, STIM1 and 2, and the calcium release-activated channel (CRAC), ORAI1, at the plasma membrane. Consistent with impaired PLCγ1 activation, ITK-deficient CTLs show defective Ca2+ mobilization. It is therefore of interest that there is an absolute dependence on calcium for lytic granule secretion. Although the precise signals that couple surface receptor signaling to degranulation machinery in CTLs are still not fully understood, CTLs do not degranulate in the presence of EGTA, and cytotoxic lymphocytes from patients with mutations in STIM1 or ORAI1 have defects in secretion, while polarization in these cells remains intact (50–52) (and data not shown). Similarly, Itk−/− CD8+ T cells exhibit defects in degranulation, while not affecting earlier stages of CTL polarization. Nonetheless, we note that in our hands, treatment with ionomycin, a calcium ionophore that bypasses TCR signaling to induce calcium flux, could not rescue degranulation in Itk−/− CTLs in the presence of TCR stimulation, despite inducing strong Ca2+ influx (data not shown). Indeed, although both human CTLs and T cell clones can degranulate in response to ionomycin alone (50, 53), this is less clear for primary murine CTLs. Thus calcium flux may be necessary, but not sufficient, for degranulation in primary murine CTLs, at least under the conditions we have examined.
Once granules reach the plasma membrane, microscopy has revealed them to be highly dynamic structures that must dock before fusion and exocytosis can occur (4). Previous work has suggested that efficient granule docking, defined as the tethering of vesicles to the plasma membrane (reviewed in (54)), is dependent on strength of TCR signaling (32, 42, 55). Other studies have described distinct morphological phenotypes in CTLs that are unable to kill targets but lack different components of the secretory pathway. For example, cells lacking MUNC13-4 accumulate polarized granules trapped at docking sites along the synapse membrane, indicating these cells were blocked at the exocytic event (56, 57). In contrast, cells lacking RAB27a accumulate granules around the centrosome and along microtubules, suggesting an earlier failure to dissociate from the microtubule network and/or dock at the plasma membrane (58). Our TEM studies show that unlike MUNC13-4-deficient cells, there was no evidence of accumulation of granules trapped along the synapse at docking sites at the membrane. This suggests that killing in ITK-deficient CTLs may blocked at a post-polarization, pre-granule docking step, more similar to RAB27a-deficient cells. While expression of Rab27a, as well as Unc13d, Stx11, Syt7, Snap23, which all encode regulators of docking during degranulation, was equivalent in WT and ITK-deficient CTLs (data not shown), we cannot rule out impaired levels or localization of these proteins, nor defective secondary modifications, due to the limited availability of good antibodies. Moreover, it should be noted that since exocytosis and docking occur rapidly in WT CTLs, cells blocked at the pre-docking step can appear morphologically similar to normal cells and may only be identified by lack of target death. Thus, it is not possible to distinguish definitively whether the apparent lack of target death in individual conjugates with ITK-deficient CTLs is because killing is indeed blocked or just taking place more slowly. Nonetheless, together with the decrease in target killing observed, these high-resolution images provide clues as to how the absence of ITK may affect the process of degranulation, and raise the possibility that there is a delay or block in the transfer of granules from the microtubule network to the membrane and/or in granule docking itself.
Although our inhibitor data suggests that ITK plays a role in TCR-triggered degranulation, intrinsic defects in degranulation are not the only problem in Itk−/− CTLs. Indeed, we find that ITK-deficient cells do not proliferate as well as WT CD8+ cells, consistent with previous studies (33), and show reduced expression of the downstream effectors, granzyme B and perforin. Previous work has shown that through the HIF1α pathway, mTORC1 signaling controls a diverse transcriptional program including the expression of cytolytic effectors in T cells (37). Consistent with these findings, we show here that ITK-deficient CD8+ T cells have altered mTORC1 signaling at early time points during activation, as indicated by reduced pS6. Together, these results suggest that ITK-deficiency in CD8+ T cells has two consequences. First, when naïve CD8+ T cells in the periphery encounter antigen, a suboptimal TCR signal in the absence of ITK generates a reduced population of CTLs that express less granzyme B, perforin and perhaps other effectors. Upon second antigen encounter, this population of sub-optimally activated CTLs is less capable of degranulating efficiently in the absence of ITK. In this scenario, the lytic granules that do undergo fusion may contain less granzyme B and perforin, thus making ITK-deficient CTLs poor cytotoxic lymphocytes that are less effective at clearing virally infected targets at early time points during infection.
However, we further show that prolonged culture or early exposure of ITK-deficient CTLs to IL-2 rescued degranulation and cytolysis of target cells. The loss of ITK in CTLs is reminiscent of many phenotypes associated with altered peptide ligands (APLs) or reduced TCR signaling. It is therefore possible that in our system, IL-2 provides a synergizing signal that enables or accelerates complete activation of CTLs in the absence of optimal TCR signaling, similar to the rescue of activation that has been seen for cells stimulated with APLs (59). How IL-2 exerts these effects remains an important question. Expression of surface markers in the absence of ITK improved after addition of IL-2 on day 3 of culture. Notably, the addition of exogenous IL-2 to cultures during the initial in vitro activation further rescued the kinetics of CD25 surface expression, suggesting direct effects on transcription that permit increased IL-2 responsiveness in ITK-deficient CD8+ T cells. Alternatively, there may also be other non-transcriptional effects of IL-2. For example, IL-2 stimulation of Wiskott-Aldrich Syndrome protein (WASp)-deficient NK cells in vitro led to increased phosphorylation of the WASp homolog WAVE2 and improved cytotoxicity (44), suggesting that IL-2 directly affects activation of signaling intermediates. It is interesting to speculate that such IL-2-mediated effects, which may promote or accelerate cytolysis, could contribute to the eventual clearance of viral infections in ITK-deficient mice. Furthermore, although not the focus of this work, prolonged culture of ITK-deficient CTLs in IL-2 also revealed that they are less susceptible to death. This is consistent with previous reports that ITK-deficient CD4+ cells are resistant to apoptosis under certain conditions (60, 61). Whether impaired cell death contributes to the lymphoproliferation associated with ITK-deficiency in patients is unknown, but it is interesting to note that cells from XLP-1 patients show a defect in restimulation-induced cell death that is thought to contribute to the lymphoproliferation in SAP-deficiency (62). Whether there are other defects in human CD8+ T cells deficient in ITK that make patients particularly susceptible to EBV, particularly those that may affect expansion of EBV-specific CTLs, remains an important question.
In addition to the importance of IL-2 signaling for the generation of CTLs (63, 64), other cytokines such as IL-12 have been shown to be critical for the development of both effector and memory CTLs, particularly under inflammatory conditions (38, 45, 46). Our data show that while IL-2 alone can rescue the kinetics of acquisition of granzyme B expression and cytolytic activity in the absence of ITK, the addition of IL-12 to cultures further boosts WT CTL effector function and rescues killing of targets by ITK-deficient CTLs at early time points. Indeed the addition of IL-12 appeared to accelerate the acquisition of cytolytic capability in both WT and ITK-deficient cells as early as 4 days post-stimulation (data not shown). Notably, the kinetics of CD25 expression are also rescued in Itk−/− CD8+ T cells by IL-12 addition to cultures. Our findings are consistent with reports that IL-12 increases IL-2 responsiveness by activating transcriptional programs in T cells (38, 65, 66). It is therefore interesting to speculate that the susceptibility of ITK-deficient patients to some infections, but not others, could be influenced by the presence or absence of inflammatory signals such as IL-12, which could augment IL-2 responses necessary to generate fully functional CTLs under suboptimal TCR signaling conditions.
Overall, this work demonstrates two significant effects of ITK-deficiency on CTL function: first, decreased expansion associated with impaired/delayed expression of effectors and second, a potentially novel role for ITK in regulating degranulation in CTLs without affecting upstream processes such as adhesion or cell polarization. Importantly, we also offer additional evidence for the role of IL-2 and IL-12 in integrating TCR and costimulatory signaling pathways for the generation of fully functional CTL responses. Together, this work provides insight into the defects that may account in part for the particular susceptibility to viral infections observed in patients with mutations in ITK and other TCR signaling components.
Supplementary Material
Acknowledgments
The authors would like to thank J. Reilley and R. Handon for assistance with mouse husbandry, S. Wincovitch for advice on microscopy, and C. Thomas for the 10n inhibitor.
Footnotes
This work was supported in part by intramural funds of the National Human Genome Research Institute (PLS and SMK) and by funding from the Wellcome Trust (103930 and 100140) (JCS and GMG).
References
- 1.Kupfer A, Dennert G. Reorientation of the microtubule-organizing center and the Golgi apparatus in cloned cytotoxic lymphocytes triggered by binding to lysable target cells. J Immunol. 1984;133:2762–2766. [PubMed] [Google Scholar]
- 2.Cannon JL, Burkhardt JK. The regulation of actin remodeling during T6-cell-APC conjugate formation. Immunol Rev. 2002;186:90–99. doi: 10.1034/j.1600-065x.2002.18609.x. [DOI] [PubMed] [Google Scholar]
- 3.Stinchcombe JC, Majorovits E, Bossi G, Fuller S, Griffiths GM. Centrosome polarization delivers secretory granules to the immunological synapse. Nature. 2006;443:462–465. doi: 10.1038/nature05071. [DOI] [PubMed] [Google Scholar]
- 4.Ritter AT, Asano Y, Stinchcombe JC, Dieckmann NM, Chen BC, Gawden-Bone C, van Engelenburg S, Legant W, Gao L, Davidson MW, Betzig E, Lippincott-Schwartz J, Griffiths GM. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity. 2015;42:864–876. doi: 10.1016/j.immuni.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennert G, Podack ER. Cytolysis by H-2-specific T killer cells. Assembly of tubular complexes on target membranes. J Exp Med. 1983;157:1483–1495. doi: 10.1084/jem.157.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masson D, Tschopp J. A family of serine esterases in lytic granules of cytolytic T lymphocytes. Cell. 1987;49:679–685. doi: 10.1016/0092-8674(87)90544-7. [DOI] [PubMed] [Google Scholar]
- 7.Masson D, Tschopp J. Isolation of a lytic, pore-forming protein (perforin) from cytolytic T-lymphocytes. J Biol Chem. 1985;260:9069–9072. [PubMed] [Google Scholar]
- 8.Henkart P, Henkart M, Millard P, Frederikse P, Bluestone J, Blumenthal R, Yue C, Reynolds C. The role of cytoplasmic granules in cytotoxicity by large granular lymphocytes and cytotoxic T lymphocytes. Adv Exp Med Biol. 1985;184:121–138. doi: 10.1007/978-1-4684-8326-0_9. [DOI] [PubMed] [Google Scholar]
- 9.Liu KQ, Bunnell SC, Gurniak CB, Berg LJ. T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. J Exp Med. 1998;187:1721–1727. doi: 10.1084/jem.187.10.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schaeffer EM, Debnath J, Yap G, McVicar D, Liao XC, Littman DR, Sher A, Varmus HE, Lenardo MJ, Schwartzberg PL. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science. 1999;284:638–641. doi: 10.1126/science.284.5414.638. [DOI] [PubMed] [Google Scholar]
- 11.Dombroski D, Houghtling RA, Labno CM, Precht P, Takesono A, Caplen NJ, Billadeau DD, Wange RL, Burkhardt JK, Schwartzberg PL. Kinase-independent functions for Itk in TCR-induced regulation of Vav and the actin cytoskeleton. J Immunol. 2005;174:1385–1392. doi: 10.4049/jimmunol.174.3.1385. [DOI] [PubMed] [Google Scholar]
- 12.Gomez-Rodriguez J, Wohlfert EA, Handon R, Meylan F, Wu JZ, Anderson SM, Kirby MR, Belkaid Y, Schwartzberg PL. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J Exp Med. 2014;211:529–543. doi: 10.1084/jem.20131459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomez-Rodriguez J, Meylan F, Handon R, Hayes ET, Anderson SM, Kirby MR, Siegel RM, Schwartzberg PL. Itk is required for Th9 differentiation via TCR-mediated induction of IL-2 and IRF4. Nat Commun. 2016;7:10857. doi: 10.1038/ncomms10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sahu N, Venegas AM, Jankovic D, Mitzner W, Gomez-Rodriguez J, Cannons JL, Sommers C, Love P, Sher A, Schwartzberg PL, August A. Selective expression rather than specific function of Txk and Itk regulate Th1 and Th2 responses. J Immunol. 2008;181:6125–6131. doi: 10.4049/jimmunol.181.9.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect Biol. 2010;2:a002287. doi: 10.1101/cshperspect.a002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gomez-Rodriguez J, Sahu N, Handon R, Davidson TS, Anderson SM, Kirby MR, August A, Schwartzberg PL. Differential expression of interleukin-17A and -17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity. 2009;31:587–597. doi: 10.1016/j.immuni.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fowell DJ, Shinkai K, Liao XC, Beebe AM, Coffman RL, Littman DR, Locksley RM. Impaired NFATc translocation and failure of Th2 development in Itk-deficient CD4+ T cells. Immunity. 1999;11:399–409. doi: 10.1016/s1074-7613(00)80115-6. [DOI] [PubMed] [Google Scholar]
- 18.Singleton KL, Gosh M, Dandekar RD, Au-Yeung BB, Ksionda O, Tybulewicz VL, Altman A, Fowell DJ, Wulfing C. Itk controls the spatiotemporal organization of T cell activation. Sci Signal. 2011;4:ra66. doi: 10.1126/scisignal.2001821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sahu N, Venegas AM, Jankovic D, Mitzner W, Gomez-Rodriguez J, Cannons JL, Sommers C, Love P, Sher A, Schwartzberg PL, August A. Selective expression rather than specific function of Txk and Itk regulate Th1 and Th2 responses. J Immunol. 2008;181:6125–6131. doi: 10.4049/jimmunol.181.9.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mansouri D, Mahdaviani SA, Khalilzadeh S, Mohajerani SA, Hasanzad M, Sadr S, Nadji SA, Karimi S, Droodinia A, Rezaei N, Linka RM, Bienemann K, Borkhardt A, Masjedi MR, Velayati AA. IL-2-inducible T-cell kinase deficiency with pulmonary manifestations due to disseminated Epstein-Barr virus infection. Int Arch Allergy Immunol. 2012;158:418–422. doi: 10.1159/000333472. [DOI] [PubMed] [Google Scholar]
- 21.Linka RM, Risse SL, Bienemann K, Werner M, Linka Y, Krux F, Synaeve C, Deenen R, Ginzel S, Dvorsky R, Gombert M, Halenius A, Hartig R, Helminen M, Fischer A, Stepensky P, Vettenranta K, Kohrer K, Ahmadian MR, Laws HJ, Fleckenstein B, Jumaa H, Latour, Schraven B, Borkhardt A. Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia. 2012 doi: 10.1038/leu.2011.371. [DOI] [PubMed] [Google Scholar]
- 22.Huck K, Feyen O, Niehues T, Ruschendorf F, Hubner N, Laws HJ, Telieps T, Knapp S, Wacker HH, Meindl A, Jumaa H, Borkhardt A. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest. 2009;119:1350–1358. doi: 10.1172/JCI37901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh S, Bienemann K, Boztug K, Borkhardt A. Interleukin-2-inducible T-cell kinase (ITK) deficiency - clinical and molecular aspects. J Clin Immunol. 2014;34:892–899. doi: 10.1007/s10875-014-0110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao F, Cannons JL, Dutta M, Griffiths GM, Schwartzberg PL. Positive and Negative Signaling through SLAM Receptors Regulate Synapse Organization and Thresholds of Cytolysis. Immunity. 2012;36:1003–1016. doi: 10.1016/j.immuni.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palendira U, Low C, Chan A, Hislop AD, Ho E, Phan TG, Deenick E, Cook MC, Riminton DS, Choo S, Loh R, Alvaro F, Booth C, Gaspar HB, Moretta A, Khanna R, Rickinson AB, Tangye SG. Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biol. 2011;9:e1001187. doi: 10.1371/journal.pbio.1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bachmann MF, Littman DR, Liao XC. Antiviral immune responses in Itk-deficient mice. J Virol. 1997;71:7253–7257. doi: 10.1128/jvi.71.10.7253-7257.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atherly LO, Brehm MA, Welsh RM, Berg LJ. Tec kinases Itk and Rlk are required for CD8+ T cell responses to virus infection independent of their role in CD4+ T cell help. J Immunol. 2006;176:1571–1581. doi: 10.4049/jimmunol.176.3.1571. [DOI] [PubMed] [Google Scholar]
- 28.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 29.Liao XC, Littman DR. Altered T cell receptor signaling and disrupted T cell development in mice lacking Itk. Immunity. 1995;3:757–769. doi: 10.1016/1074-7613(95)90065-9. [DOI] [PubMed] [Google Scholar]
- 30.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 31.Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 2001;15:751–761. doi: 10.1016/s1074-7613(01)00234-5. [DOI] [PubMed] [Google Scholar]
- 32.Jenkins MR, Tsun A, Stinchcombe JC, Griffiths GM. The strength of T cell receptor signal controls the polarization of cytotoxic machinery to the immunological synapse. Immunity. 2009;31:621–631. doi: 10.1016/j.immuni.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atherly LO, Lucas JA, Felices M, Yin CC, Reiner SL, Berg LJ. The Tec family tyrosine kinases Itk and Rlk regulate the development of conventional CD8+ T cells. Immunity. 2006;25:79–91. doi: 10.1016/j.immuni.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 34.Sharifi R, Sinclair JC, Gilmour KC, Arkwright PD, Kinnon C, Thrasher AJ, Gaspar HB. SAP mediates specific cytotoxic T-cell functions in X-linked lymphoproliferative disease. Blood. 2004;103:3821–3827. doi: 10.1182/blood-2003-09-3359. [DOI] [PubMed] [Google Scholar]
- 35.Dupre L, Andolfi G, Tangye SG, Clementi R, Locatelli F, Arico M, Aiuti A, Roncarolo MG. SAP controls the cytolytic activity of CD8+ T cells against EBV-infected cells. Blood. 2005;105:4383–4389. doi: 10.1182/blood-2004-08-3269. [DOI] [PubMed] [Google Scholar]
- 36.Hislop AD, Palendira U, Leese AM, Arkwright PD, Rohrlich PS, Tangye SG, Gaspar HB, Lankester AC, Moretta A, Rickinson AB. Impaired Epstein- Barr virus-specific CD8+ T-cell function in X-linked lymphoproliferative disease is restricted to SLAM family-positive B-cell targets. Blood. 2010;116:3249–3257. doi: 10.1182/blood-2009-09-238832. [DOI] [PubMed] [Google Scholar]
- 37.Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, Panteleyev AA, Okkenhaug K, Cantrell DA. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209:2441–2453. doi: 10.1084/jem.20112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Labno CM, Lewis CM, You D, Leung DW, Takesono A, Kamberos N, Seth A, Finkelstein LD, Rosen MK, Schwartzberg PL, Burkhardt JK. Itk functions to control actin polymerization at the immune synapse through localized activation of Cdc42 and WASP. Curr Biol. 2003;13:1619–1624. doi: 10.1016/j.cub.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quann EJ, Merino E, Furuta T, Huse M. Localized diacylglycerol drives the polarization of the microtubule-organizing center in T cells. Nat Immunol. 2009;10:627–635. doi: 10.1038/ni.1734. [DOI] [PubMed] [Google Scholar]
- 41.Liu X, Kapoor TM, Chen JK, Huse M. Diacylglycerol promotes centrosome polarization in T cells via reciprocal localization of dynein and myosin II. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1306180110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anikeeva N, Somersalo K, Sims TN, Thomas VK, Dustin ML, Sykulev Y. Distinct role of lymphocyte function-associated antigen-1 in mediating effective cytolytic activity by cytotoxic T lymphocytes. Proc Natl Acad Sci U S A. 2005;102:6437–6442. doi: 10.1073/pnas.0502467102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trinchieri G, Matsumoto-Kobayashi M, Clark SC, Seehra J, London L, Perussia B. Response of resting human peripheral blood natural killer cells to interleukin 2. J Exp Med. 1984;160:1147–1169. doi: 10.1084/jem.160.4.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orange JS, Roy-Ghanta S, Mace EM, Maru S, Rak GD, Sanborn KB, Fasth A, Saltzman R, Paisley A, Monaco-Shawver L, Banerjee PP, Pandey R. IL-2 induces a WAVE2-dependent pathway for actin reorganization that enables WASp-independent human NK cell function. J Clin Invest. 2011;121:1535–1548. doi: 10.1172/JCI44862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–1151. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, Popescu F, Xiao Z. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. 2006;211:81–92. doi: 10.1111/j.0105-2896.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 47.Fontana S, Parolini S, Vermi W, Booth S, Gallo F, Donini M, Benassi M, Gentili F, Ferrari D, Notarangelo LD, Cavadini P, Marcenaro E, Dusi S, Cassatella M, Facchetti F, Griffiths GM, Moretta A, Notarangelo LD, Badolato R. Innate immunity defects in Hermansky-Pudlak type 2 syndrome. Blood. 2006;107:4857–4864. doi: 10.1182/blood-2005-11-4398. [DOI] [PubMed] [Google Scholar]
- 48.Rohr J, Beutel K, Maul-Pavicic A, Vraetz T, Thiel J, Warnatz K, Bondzio I, Gross-Wieltsch U, Schundeln M, Schutz B, Woessmann W, Groll AH, Strahm B, Pagel J, Speckmann C, Janka G, Griffiths G, Schwarz K, zur Stadt U, Ehl S. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica. 2010;95:2080–2087. doi: 10.3324/haematol.2010.029389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quann EJ, Liu X, Altan-Bonnet G, Huse M. A cascade of protein kinase C isozymes promotes cytoskeletal polarization in T cells. Nat Immunol. 2011;12:647–654. doi: 10.1038/ni.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maul-Pavicic A, Chiang SC, Rensing-Ehl A, Jessen B, Fauriat C, Wood SM, Sjoqvist S, Hufnagel M, Schulze I, Bass T, Schamel WW, Fuchs S, Pircher H, McCarl CA, Mikoshiba K, Schwarz K, Feske S, Bryceson YT, Ehl S. ORAI1- mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc Natl Acad Sci U S A. 2011;108:3324–3329. doi: 10.1073/pnas.1013285108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 52.Shaw PJ, Feske S. Regulation of lymphocyte function by ORAI and STIM proteins in infection and autoimmunity. J Physiol. 2012;590:4157–4167. doi: 10.1113/jphysiol.2012.233221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu D, Martina JA, Wu XS, Hammer JA, 3rd, Long EO. Two modes of lytic granule fusion during degranulation by natural killer cells. Immunol Cell Biol. 2011;89:728–738. doi: 10.1038/icb.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pattu V, Halimani M, Ming M, Schirra C, Hahn U, Bzeih H, Chang HF, Weins L, Krause E, Rettig J. In the crosshairs: investigating lytic granules by high-resolution microscopy and electrophysiology. Front Immunol. 2013;4:411. doi: 10.3389/fimmu.2013.00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jenkins MR, Griffiths GM. The synapse and cytolytic machinery of cytotoxic T cells. Curr Opin Immunol. 2010;22:308–313. doi: 10.1016/j.coi.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santoro A, Cannella S, Bossi G, Gallo F, Trizzino A, Pende D, Dieli F, Bruno G, Stinchcombe JC, Micalizzi C, De Fusco C, Danesino C, Moretta L, Notarangelo LD, Griffiths GM, Arico M. Novel Munc13-4 mutations in children and young adult patients with haemophagocytic lymphohistiocytosis. J Med Genet. 2006;43:953–960. doi: 10.1136/jmg.2006.041863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, Lambert N, Ouachee-Chardin M, Chedeville G, Tamary H, Minard-Colin V, Vilmer E, Blanche S, Le Deist F, Fischer A, de Saint Basile G. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115:461–473. doi: 10.1016/s0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- 58.Stinchcombe JC, Barral DC, Mules EH, Booth S, Hume AN, Machesky LM, Seabra MC, Griffiths GM. Rab27a is required for regulated secretion in cytotoxic T lymphocytes. J Cell Biol. 2001;152:825–834. doi: 10.1083/jcb.152.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Voisinne G, Nixon BG, Melbinger A, Gasteiger G, Vergassola M, Altan-Bonnet G. T Cells Integrate Local and Global Cues to Discriminate between Structurally Similar Antigens. Cell reports. 2015;11:1208–1219. doi: 10.1016/j.celrep.2015.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller AT, Berg LJ. Defective Fas ligand expression and activation-induced cell death in the absence of IL-2-inducible T cell kinase. J Immunol. 2002;168:2163–2172. doi: 10.4049/jimmunol.168.5.2163. [DOI] [PubMed] [Google Scholar]
- 61.Sun Y, Peng I, Webster JD, Suto E, Lesch J, Wu X, Senger K, Francis G, Barrett K, Collier JL, Burch JD, Zhou M, Chen Y, Chan C, Eastham-Anderson J, Ngu H, Li O, Staton T, Havnar C, Jaochico A, Jackman J, Jeet S, Riol-Blanco L, Wu LC, Choy DF, Arron JR, McKenzie BS, Ghilardi N, Ismaili MH, Pei Z, DeVoss J, Austin CD, Lee WP, Zarrin AA. Inhibition of the kinase ITK in a mouse model of asthma reduces cell death and fails to inhibit the inflammatory response. Sci Signal. 2015;8:ra122. doi: 10.1126/scisignal.aab0949. [DOI] [PubMed] [Google Scholar]
- 62.Snow AL, Marsh RA, Krummey SM, Roehrs P, Young LR, Zhang K, van Hoff J, Dhar D, Nichols KE, Filipovich AH, Su HC, Bleesing JJ, Lenardo MJ. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J Clin Invest. 2009;119:2976–2989. doi: 10.1172/JCI39518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW, Leonard WJ. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med. 1998;188:2067–2074. doi: 10.1084/jem.188.11.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malek TR, Yu A, Scibelli P, Lichtenheld MG, Codias EK. Broad programming by IL-2 receptor signaling for extended growth to multiple cytokines and functional maturation of antigen-activated T cells. J Immunol. 2001;166:1675–1683. doi: 10.4049/jimmunol.166.3.1675. [DOI] [PubMed] [Google Scholar]
- 65.O’Sullivan A, Chang HC, Yu Q, Kaplan MH. STAT4 is required for interleukin-12-induced chromatin remodeling of the CD25 locus. J Biol Chem. 2004;279:7339–7345. doi: 10.1074/jbc.M309979200. [DOI] [PubMed] [Google Scholar]
- 66.Yanagida T, Kato T, Igarashi O, Inoue T, Nariuchi H. Second signal activity of IL-12 on the proliferation and IL-2R expression of T helper cell-1 clone. J Immunol. 1994;152:4919–4928. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.