

Abstract
Integration of signaling and metabolic pathways enables and sustains lymphocyte function. While metabolic changes occurring during T cell activation are well-characterized, the metabolic demands of differentiated T lymphocytes are largely unexplored. Here we defined the bioenergetics of TH17 effector cells generated in vivo. These cells depend on OXPHOS2 for energy and cytokine production. Mechanistically, the essential role of OXPHOS in TH17 cells results from their limited capacity to increase glycolysis in response to metabolic stresses. This metabolic program is observed in mouse and human TH17 cells, including those isolated from Crohn’s disease patients, and is linked to disease, as inhibiting OXPHOS reduces the severity of murine colitis and psoriasis. These studies highlight the importance of analyzing metabolism in effector lymphocytes within in vivo inflammatory contexts and suggest a therapeutic role for manipulating OXPHOS in TH17 driven diseases.
Keywords: TH17, inflammation, immune metabolism
Introduction
During activation and differentiation, T cells adapt their metabolism in highly coordinated ways to provide energy and substrates for growth, trafficking, and the synthesis and secretion of immune-active molecules (1, 2). Regulating metabolic pathways has thus emerged as an attractive approach to developing new therapeutic strategies to control immune function (3, 4).
TH17 cells are CD4+, αβ T cell receptor-expressing cells that secrete the pro-inflammatory cytokines IL-17A, IL-17F, IL-21, and GM-CSF, which makes these cells critical in host defense against infections and cancer (5, 6). By contrast, abnormally increased TH17 cell activity can result in autoimmune and inflammatory disease (5, 7). TH17 cells can be generated in vitro from naïve CD4+ T cells in the presence of activating stimuli and appropriate polarizing cytokines (8–10). Under these conditions, HIF-1α3, which upregulates the expression of glycolysis genes (11), and Pdk14, which inhibits the flux of pyruvate into the TCA cycle (12), promote TH17 differentiation (13–16). Consistent with these findings, mice deficient in genes that control glycolysis have impaired TH17 differentiation in vivo (13, 15–17).
In contrast to the detailed understanding of the metabolic requirements for differentiation of naïve CD4+ T cells into TH17, the metabolic requirements of differentiated TH17 effector cells have not been defined. At least two studies suggest that the metabolism of TH17 cells post-differentiation differs from the glycolytic metabolism used during differentiation (16, 18). In particular, blocking glycolysis and/or glycolysis-linked biosynthesis is ineffective at treating TH17-driven diseases once TH17 cells are present (16, 18). Hence, metabolic targeting of TH17-driven disease processes requires analysis of the metabolism and bioenergetics of differentiated TH17 cells within in vivo inflammatory contexts.
To develop a metabolically-targeted approach to control TH17-mediated inflammation, we analyzed the bioenergetics of differentiated TH17 cells and their metabolic requirements for the secretion of pro-inflammatory cytokines and the induction of colitis. We paid particular attention to two key parameters that influence T cell metabolism and function (19, 20). First, we compared the metabolic profiles of TH17 effector cells differentiated in vitro to those differentiated in vivo, as we and others have shown that T cells activated in vitro adapt a different metabolic phenotype than cells similarly activated in vivo (21, 22). Secondly, we took particular note of the inflammatory environment, comparing for the first time the metabolic requirements of cells isolated from normal lymphoid tissues with those from inflammatory lesions.
METHODS
Mice
C57BL/6 mice were obtained from Charles River. OT-II mice (B6.Cg-Tg (TcraTcrb)425Cbn/J), SJL mice (B6.SJL-PtprcaPep3b/BoyJ), and IL-17GFP knockin mice (C57BL/6-Il17atm1Bcgen/J), were purchased from Jackson Laboratories. Mice were kept under specific pathogen-free conditions and provided with food and water ad libitum. The animal studies were conducted under protocols approved by the University of Michigan Committee on Use and Care of Animals.
PBMC and biopsy specimens
PBMC from healthy subjects and patients with IBD5 were isolated via Ficoll gradient fractionation and treated overnight with indicated compounds. All experiments using human PBMC were collected in accordance with the University of Michigan Institutional Review Board and written informed consent was obtained.
Ileum intestinal biopsy samples taken from two patients with CD6 undergoing intestinal resection due to disease severity and inadequate responses to medical treatment. Biopsy specimens were obtained from an inflamed area of the large intestine of a patient with active UC7, were used to isolate LPMC8. One CD patient and the UC patient were receiving corticosteroids, and the remaining CD patient was treated with mesalazine. Each patient who took part in the study gave written informed consent and the study protocol was approved by the local Ethics Committees (Tor Vergata University Hospital, Rome).
In vitro TH17 differentiation
Naïve cells were isolated from the spleens of 8–12 week-old mice using CD4+ CD62L+ T Cell Isolation Kit II (Miltenyi Biotec) or EasySep Mouse Naïve CD4+ T Cell Isolation Kit (StemCell Technologies) following manufacturer protocols. Cells (100,000 to 200,000) were plated in RPMI-1640 (Corning Cellgro) and supplemented with 10% heat-inactivated FBS (Hyclone), 1% Glutamax (Gibco), 1% Penicillin/Streptomycin (Sigma), and 0.1% 2-mercaptoethanol (Gibco) on anti-CD3-coated (2.5 μg/mL, BD Biosciences) 96-well plates with anti-CD28 (10 μg/mL, BD Biosciences) and TH17 differentiation cocktail (see below) for four days in a 37 °C incubator with 5% CO2. Alternatively, splenocytes from OT-I and OT-II mice were cultured with up to 0.5 μg/mL of OVA peptide 257–264 for OT-1 and OVA 323–339 peptide for OT-2 (RS Synthesis) and supplemented with a TH17 differentiation cocktail. Unless otherwise stated, TH17 differentiation cocktail was prepared with IL-1β (10 ng/mL), IL-6 (10 ng/mL), IL-23 (10 ng/mL), and human TGF-β (2.5 ng/mL). All cytokines were purchased from R&D Systems. In vitro-differentiated TH17 cells were >90% CD45.1+CD4+ at the end of differentiation.
In vivo TH17 differentiation
Naïve cells (approximately 100,000) isolated from OT-I or OT-II mice were transferred into B6.SJL mice by tail vein injection. Six to 16 hours later, mice were immunized subcutaneously, two to four sites per mouse, with 50 μL of 2:1:1 mixture of M. Tuberculosis H37 Ra (Difco), 100 mg dissolved in 10 mL of CFA (Sigma): OVA 323–339 peptide (4 μg/mL water): PBS. Cells from lymph nodes and spleens were isolated seven to nine days post-immunization, subjected to red blood cell lysis (Sigma), filtered through 70 μm cell strainer (Max Scientific), and purified by surface CD4 microbeads (Miltenyi 130-049-201). In experiments analyzing cytokine expression, donor cells (CD4+CD45.2+) and recipient cells (CD4+CD45.1+) were gated by FACS. When higher-purity donor cells were required, CD4+ cells were sorted by flow cytometry using either IL-17-GFP or PE-CD45.2 surface marker (1:500, eBioscience 12-0454-82). Sorted in vivo-differentiated TH17 were >90% CD4+CD45.2. Alternatively, naïve cells (approximately 100,000) isolated from OT-II (Thy1.1+Thy1.2+) mice were transferred into B6 (Thy1.1-Thy1.2+) by tail vein injection. After immunization as above, lymph node and splenic CD4+ T cells were enriched by negative selection using Mylteni CD4+ T Cell Isolation Kit followed by positive selection with anti-CD90.1-PE (BD Biosciences) and anti-PE microbeads. Sorted in vivo-differentiated TH17 cells were typically >80% CD4+CD90.1.
Lamina propria isolation
Dissected mucosa were freed of mucus and epithelial cells in sequential with DTT and EDTA, and then digested with 2 mg/mL of Liberase (Roche) as previously decribed (23). After digestion, the medium containing the mononuclear cells was collected and centrifuged at 400g for 10 minutes. After two washes in calcium- and magnesium-free HBSS, the pellet was resuspended in 40% Percoll solution. 2 mL each of 100%, 60%, 40%, and 30% Percoll were layered in a 15 mL tube. The tube was centrifuged at 400g for 25 minutes, and LPMCs were collected from the interface at the 60%–40% Percoll layers.
Cell sorting
Cells were harvested at the end of the differentiation protocol, counted, and resuspended to 5x106/mL of 1% FBS/PBS prior to passing through a 50 μm filter (Partec) and sorted on MoFlo Astrios or MoFlo XDP (Beckman Coulter) at the University of Michigan Flow Cytometry Core. After sorting, cells were washed once with full media and stored as pellet at −80°C for RNA or protein extraction.
Intracellular staining
Cells were processed for intracellular staining following the manufacturer’s protocol (eBiosciences 88-8823-88). Mouse antibodies used were anti-IL-17a (clone eBio17B7, eBiosciences), anti-CD4 (clone eBioRM4-5, eBiosciences), anti-IFNγ (clone XMG1.2, eBiosciences), anti-FoxP3 (clone FJK-16S, eBiosciences), anti-CD62L (clone MEL-14, eBiosciences), anti-CD45.2 (clone 104, eBiosciences), anti-CD44 (clone IM7, eBiosciences), RORγt (clone AFKJS-9, eBiosciences), and anti-CD90.1 (clone OX-7, BD Biosciences). Murine LPMC were stained with the following antibodies: anti–CD45-APC-Cy7 (1:50, final dilution, clone 30f11 BD Biosciences), anti–CD3-Pacific Blue (1:50, final dilution, clone 500a2, BD Biosciences); anti-CD8-FITC (1:50, final dilution; clone 53–6.7, BD Biosciences) and intracellularly with anti-IL-17A-APC (1:50 final dilution; clone Ebio17b7, eBioscience); and anti-IFNγ-PECy7 (1:50 final dilution; clone XMG1.2 BD Bioscience). Human antibodies used were anti-human CD3 from eBiosciences (clone OKT3) and anti-human IL-17a antibody (clone eBio64CAP17). Human LPMC were stained with the following Abs: anti–CD45-APC-H7 (1:50, final dilution, clone 2D1 BD Biosciences) and anti–CD3-Pacific Blue (1:50, final dilution, clone UCHT1, BD Biosciences). To assess the intracellular expression of IFNγ and IL-17A, cells were stained with the following Abs: anti-IFNγ-APC (1:50 final dilution, clone 25723.11, BD Biosciences) and anti-IL-17A-PE (1:50, final dilution, clone 033-782, BD Biosciences). Where indicated, cells were stimulated with PMA, phorbol 12-myristate 13 acetate, (Sigma), ionomycin calcium salt (Sigma), and Brefeldin A (eBiosciences) for four to five hours prior to fixation in 2% formaldehyde/PBS (Thermo Scientific). Cells were analyzed using a FACSVerse cytometer and FACSuite software (BD Biosciences).
Anti-CD3 activation in vivo
Female C57BL/6 mice received compound at specified dose or vehicle (40% DMSO/PBS) via intraperitoneal injection (i.p.) in 200 μL. One hour later, anti-CD3e (5 μg/mouse) (BD Biosciences) was injected i.p. in PBS. Four hours after anti-CD3e injection, serum was collected by cardiac puncture and cytokines were analyzed by ELISA.
Seahorse measurements
Cells (7x105) were plated per well on an XF24 plate coated with 0.5mg/mL poly-D lysine (Sigma), centrifuged in 100 μL of extracelluar flux (XF) media supplemented with 1 mM sodium pyruvate (Sigma), 11 mM glucose (Sigma), 1% FBS, pH 7.4, filtered through a 0.22 μm filter. Extracellular flux (XF) media (530 μL) was then added to each well and incubated for 30 minutes in a CO2-free incubator at 37 °C, then run per the manufacturer’s protocol with oligomycin, carbonylcyanide-4-(trifluoromethoxy)phenylhydrazone, and antimycin A injections in ports A, B, and C respectively.
Cell proliferation analysis
Naïve cells were labeled with 10 μM CFSE (Life Technologies) in 1 ml of RPMI + 2% FBS for 10 minutes at 37 °C, then washed with RPMI and resuspended into full medium culture. For in vivo measurements, 2x106 labeled cells were transferred through tail vein injection into B6SJL mice.
Lactate measurement
Sorted in vivo TH17 cells and in vitro TH17 cells were plated at 100,000 cells per well in a 96-well plate and treated with indicated compounds. After the indicated time, cells were pelleted and supernatants from triplicate cultures were analyzed by the colorimetric method using Glycolysis Cell-Based Assay Kit (Cayman) according to the manufacturer’s protocol.
ATP measurement
Sorted in vivo TH17 cells and in vitro TH17 cells were plated at 100,000 cells per well in a 96-well plate and treated with indicated compounds. After the indicated time, cells were pelleted and lysed with a lysis buffer from ATP Bioluminescence Assay Kit CLS II (Roche) supplemented with Halt Protease Inhibitor Cocktail (Thermo Scientific) and Halt Phosphatase Inhibitor Cocktail (Thermo Scientific) prior to heat extraction. To determine the intracellular ATP level, the lysate was processed according to the manufacturer’s protocol. Analysis was performed in triplicate cultures.
Real-time PCR
RNA was isolated with RNeasy (Qiagen) according to the manufacturer’s instructions. RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and cDNA was then used for quantitative PCR by SYBR Green Gene Expression Assay on an ABI 7900HT analyzer (Applied Biosystems). Primer sequences are as follows: hif1a forward 5′-CGGCGAGAACGAGAAGAA-3′, reverse 5′-AAACTTCAGACTCTTTGCTTCG-3′; pdk1 forward 5′-GGACTTCGGGTCAGTGAATGC-3′, reverse 5′-TCCTGAGAAGATTGTCGGGGA-3′; rorc2 forward 5′-CCACAGATCTTGCAAGGGATC-3′, reverse 5′-CCGCTGAGAGGGCTTCAC-3′; foxp3 forward 5′-TGGCAGAGAGGTATTGAGGG-3′, reverse 5′-CTCGTCTGAAGGCAGAGTCA-3′, Ldha forward 5′-CTGGGTCCTGGGAGAACAT-3′, reverse 5′-GTGCCCAGTTCTGGGTTAAG-3′; aldoa forward 5′-TCAGTGCTGGGTATGGGTG-3′, reverse 5′-GCTCCTTAGTCCTTTCGCCT-3′; hk2 forward 5′-TGATCGCCTGCTTATTCACGG-3′, reverse 5′-AACCGCCTAGAAATCTCCAGA-3′; β-actin forward 5′-ATGGAGGGGAATACAGCCC-3′, reverse 5′-TTCTTTGCAGCTCCTTCGTT-3′.
For hypoxia pathway activity, RT2 Profiler Mouse Hypoxia Signaling Pathway PCR Array (Qiagen) was used following the manufacturer’s protocol.
Gene expression profiling
Microarray analysis was performed on sorted CD4+IL-17GFP+ isolated from in vitro- or in vivo-differentiated TH17. RNA was extracted with RNeasy Plus Mini Kit (Qiagen) and samples were processed and analyzed at the University of Michigan Microarray Core Facility with GeneChip Mouse Gene 1.0 ST Array (Affymetrix). RNA was analyzed on Bioanalyzer (Agilent Technologies). All samples had >9 RNA integrity number (RIN). Microarray quality control was performed with R (Bioconductor). The data were processed with Robust Multi-chip Average (RMA) normalization. Technical replicates were averaged for fold-change. Genes were considered differentially expressed if they were > log2-fold different. The microarray data are available in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/gds) under the accession number GSE68017.
Gene Set Enrichment Analysis
All of the genes differentially expressed by >log2-fold difference between in vivo (CD45.2+CD4+)- or in vitro (CD4+)-differentiated TH17 were selected and analyzed using GSEA.9 Three GSEA tests were employed using default program settings, testing the complete 2log-fold gene set.
Transfection of siRNA10
Splenocytes from OT-II mice were cultured with up to 0.5 μg/mL of OVA 323–339 peptide (RS Synthesis) and supplemented with a TH17 differentiation cocktail as described above. After three to four days, cells were nucleofected with 4D-Nucleofector according to the Amaxa protocol (Lonza). Briefly, 5x106 cells were resuspended in 98 μL nucleofector solution V4XP with 300 pM ON-TARGETplus siRNA SMARTpool (GE Healthcare Dharmacon), transferred to a cuvette, and then electroporated using the DN-100 pulsing parameter. Cells were immediately transferred into wells containing a 37°C pre-warmed culture medium and used one day after electroporation.
Animal Disease models
For induction of acute colitis, 2.5 mg of TNBS11 in 50% ethanol was administered to lightly anesthetized mice through a 3.5Fr catheter inserted into the rectum on day 0. The catheter tip was inserted 4 cm proximal to the anal verge, and 150 μL of fluid was slowly instilled into the colon, after which the mouse was held in a vertical position for 30 seconds. Starting at day 1 mice received an i.p. dose of vehicle (0.01% DMSO in PBS) or oligomycin (0.25 mg/kg) once a day, or ursolic acid orally (0.75 mg/kg in 1% gum acadia) twice a day. On day 2, colon tissue was collected for histology. Alternatively, colitis was induced by giving mice water containing 3% dextran sodium sulfate (DSS) (MP Biomedicals, MW=36,000–50,000) for 3 days.
For the induction of psoriasis, Aldara (a 5% cream of imiquimod; 3M Pharmaceuticals) was applied to the ear of mice daily for 5 days. Starting at day 1 mice received an i.p. dose of vehicle (0.01% DMSO in PBS) or oligomycin (0.25 mg/kg) once a day, or ursolic acid orally (0.75 mg/kg in 1% gum acadia) twice a day.
Histology
From the colitis animal models, upon necropsy, colon tissue was separated and Swiss rolls were prepared. Tissues were fixed in 10% buffered formalin and paraffin-embedded. The sections (5 μm) were stained with H&E. A pathology score (0–10) was determined based on analysis of the following: goblet cell hyperplasia, crypt pyolymphocytic infiltrate, surface mucosa ulceration, mucosa pyolymphocytic infiltrate, lamina propria fibrosis, lamina propria crypt abscess, crypt dilatation, crypt hyperplasia, and crypt branching. An experienced veterinary pathologist without knowledge of the tissues’ origin performed histologic scoring.
Measurements of cytokines
Cytokines were measured in cell free culture supernatants of triplicate cultures by ELISA kits (R&D Systems) at the University of Michigan Cancer Center Immunology Core.
Metabolomics
Naïve (CD4+) from non-immunized OT-II mice, and in vivo TH17 (CD4+CD90.1+), and in vitro TH17 (CD4+CD90.1+) cells generated as described above were purified by magnetic sorting. Immediately after isolation, cells were cultured in RPMI in the presence of an equimolar concentration of unlabeled and isotope-labeled and U-13C(6)-glucose (Sigma# 660663) or U-13C(5)-glutamine (Sigma# 605166). After three hours, T cell pellets were extracted in 80% methanol at dry ice temperatures, and metabolite fractions corresponding to input cell numbers were dried using a speed-vac in preparation for liquid chromatography-based targeted tandem mass spectrometry (LC-MS/MS). Metabolomics analyses were performed and the data was processed and analyzed as described in the literature (24, 25).
Statistical analysis
Statistical analysis was conducted using two-tailed unpaired or paired Student’s t-test using Prism (GraphPad Software). A value of p ≤ 0.05 was considered significant. Results were considered reproducible if all independent experiments had comparable p values that were significant.
Results
To determine if TH17 cells differentiated in vivo depend on the same metabolic processes as those previously described for cells differentiated in vitro (14, 17, 26), protocols were optimized to produce populations of TH17 cells in vivo and in vitro that had nearly equivalent expression of IL-17 and RORγt with limited levels of IFNv and FOXP3 (Supplemental Figure 1). To generate cells in vivo, OT-II spleen cells were transferred to syngeneic wild-type host animals and then activated by immunization with ovalbumin peptide in CFA (Figure 1A). In vitro-differentiated TH17 cells were generated by placing naïve T cells in culture and activating them with agonistic anti-CD3 and -CD28 antibodies or by activating OT-II T cells with OVA-pulsed antigen presenting cells in the presence of IL-1β, IL-6, IL-23, and TGF-β (Figure 1A).
Figure 1. Differential expression of genes controlling glycolysis in in vivo- and in vitro-differentiated TH17.
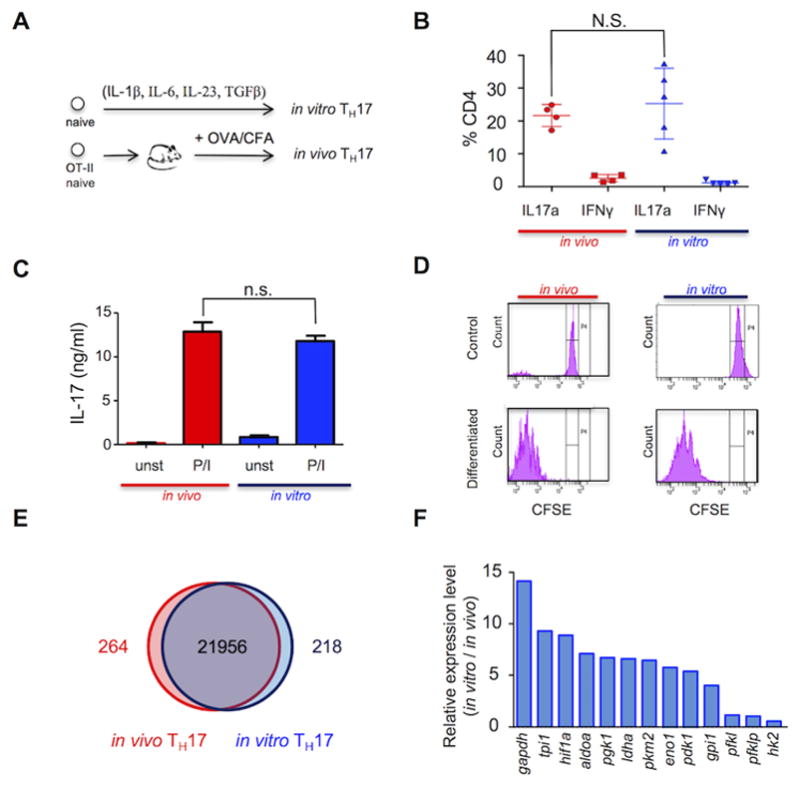
(A) Systems used to generate in vitro and in vivo TH17 cells. (B and C) In vivo (red) and in vitro (blue)-differentiated TH17 cells stimulated with PMA/ionomycin. The percentage of CD4+IL17a+ and CD4+IFNγ+ cells was determined by FACS (B) and the production of IL-17 in cell-free supernatant from triplicate cultures by ELISA (C). (D) Cellular replication profile of in vivo (red) and in vitro (blue)-differentiated TH17 cells assessed using CFSE dilution and FACS in cells before (“control”) and after differentiation (“differentiated”). (E) Gene expression of in vivo-differentiated TH17 cells (CD45.2+ CD4+IL-17GFP+) (red circle) and in vitro-differentiated TH17 cells (CD45.1+CD4+IL-17GFP+) (blue circle) analyzed by microarray. Common and differential expression of genes (>2log) is shown in a Venn diagram. (F) Expression of glycolytic enzymes graphed as a ratio of in vitro-/in vivo-differentiated TH17 cells analyzed by qPCR. (B) Plot of 4 (in vivo) and 5 (in vitro) independent experiments. (C,D,F) Representative of three independent experiments. N.S.: non-significant (unpaired Student’s t-test) (error bars, SEM).
The proportion of CD4+ cells with detectable intracellular IL-17 was maximal after differentiation for four days in vitro and for seven days in vivo (Supplemental Figure 1). Notably, the in vivo-differentiated CD4+ T cells, generated after transfer of 105 donor cells, expressed IL-17, RORγt, but not IFNγ or FOXP3 (Figure 1B and Supplemental Figure 1 C and D), at levels that were comparable to those of T cells generated in vitro with IL-1β, IL-6, IL-23, and TGF-β. Importantly, they secreted equivalent amounts of IL-17 in response to stimulation with PMA and ionomycin (Figure 1C). Analysis by CFSE labeling showed that a similar number of cell divisions was achieved in cells differentiated for four days in vitro and seven days in vivo (Figure 1D).
Having optimized the differentiation conditions, we compared the gene expression profiles of in vitro- and in vivo-derived TH17 cells. To isolate purified TH17 cells, OT-II mice were crossed with syngeneic IL-17GFP knock-in mice to generate F1 animals that expressed the IL-17GFP reporter and ovalbumin-specific TCR. These mice were used to isolate and characterize gene expression selectively in CD4+IL-17GFP+ T cells, which represented approximately 2% of the T cells after in vitro or in vivo differentiation. Importantly, the purified TH17 cells were not re-stimulated with PMA and ionomycin prior to RNA isolation and gene expression analysis, so as to provide a more biologically informative comparison of the transcriptomes of the in vitro versus in vivo TH17-differentiated states. More than 98% of all genes were expressed equivalently in the in vitro- and in vivo-derived cells (Figure 1E). GSEA was applied to identify pre-defined gene sets with statistically significant concordant differences between in vitro and in vivo conditions (Table 1). Gene sets defined as RORγt-regulated transcripts or linked to differentiated TH17 cells were, as expected, expressed equivalently in the in vitro- and in vivo-differentiated TH17 cells (Table 1). Conversely, multiple metabolism-related gene sets defined by the KEGG pathway database, including glycolysis-related genes, had significantly lower concordant expression in in vivo-differentiated cells. Lower expression of glycolytic genes in in vivo-differentiated TH17 cells was confirmed using quantitative RT-PCR by measuring transcript levels of 13 selected glycolytic enzymes, all of which were lower in in vivo cells (Figure 1F). Remarkably, both HIF1α and Pdk1, two genes that have been previously shown to regulate TH17 differentiation via controlling glycolysis, were substantially more highly expressed in the in vitro-generated effector TH17 cells. These data suggested that in vivo-differentiated IL-17+ cells function with significantly reduced utilization of glycolysis.
Table 1.
| Pathway | NES | p-value | FDR (q-value) |
|---|---|---|---|
| Ox Phos | −1.79 | 0 | 0.006 |
|
|
|||
| FA Met | −1.95 | 0.002 | 0.001 |
|
|
|||
| Glycolysis/Gluconeogenesis | −1.64 | 0.004 | 0.02 |
|
|
|||
| Citrate Cycle | −1.75 | 0.002 | 0.009 |
|
|
|||
| PPP | −1.66 | 0.012 | 0.017 |
|
|
|||
| TH17 signature | −1.37 | 0.094488 | 0.094 |
|
|
|||
| TFs associted to RORγ | −0.73 | 0.883019 | 0.883 |
|
|
|||
Gene set enrichment analysis (GSEA) of the running enrichment score (ES) based on fold change of in vitro TH17 (CD45.1+ CD4+IL-17GFP+) versus in vivo TH17 (CD45.2+ CD4+IL-17GFP+). NES (normalized enrichment score); FDR (false discovery rate).
In vivo TH17 exhibit low glycolysis
To determine if glycolytic activity differed between TH17 cells differentiated in vivo and in vitro as predicted by the gene expression profiles, we measured the rate of lactate production. The in vivo-differentiated TH17 cells exhibited a two-fold lower rate of lactate production than the in vitro-generated counterparts (Figure 2A). To further interrogate and compare glucose metabolism in these cell types, we analyzed the abundance of metabolites in glycolysis and the PPP12 using targeted mass spectrometry-based metabolomics (25). Indeed, when normalized to the levels in in vitro-differentiated cells, the in vivo-differentiated TH17 cells had dramatically smaller metabolite pool sizes across both pathways (Figure 2 C and D).
Figure 2. In vivo-differentiated TH17 cells are oxidative.
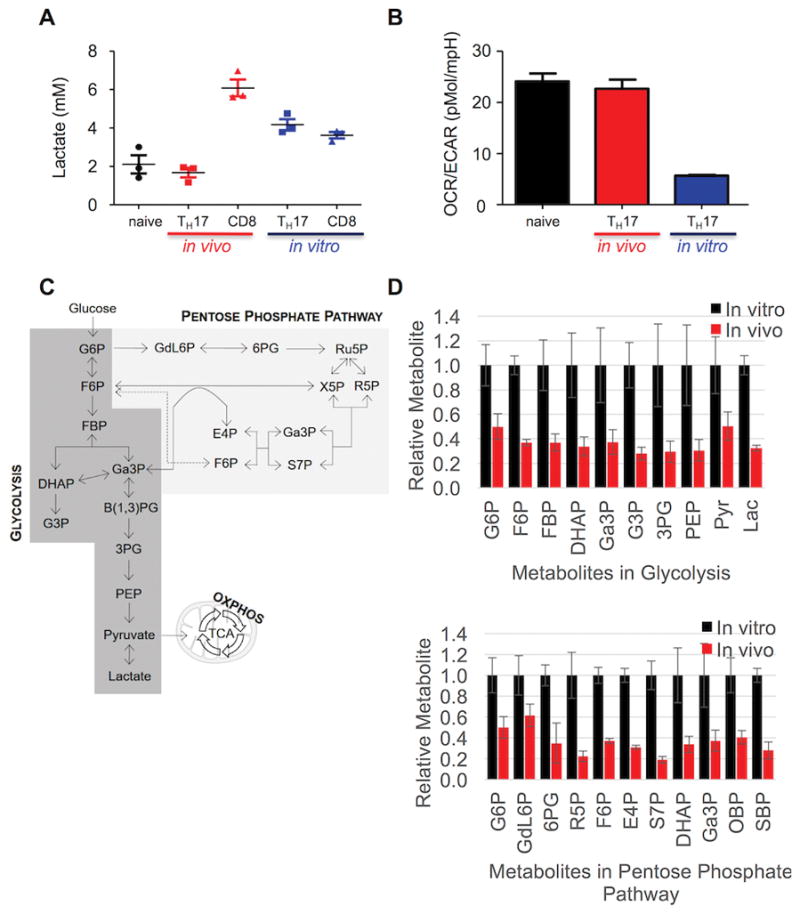
(A) Lactate concentration was determined in cell-free supernatant from triplicate cultures of naïve CD4 cells, in vivo-differentiated TH17 cells (CD4+CD90.1+), in vivo CD8 cells (CD8+CD90.1+), in vitro TH17 cells, or in vitro CD8 cells stimulated with PMA/ionomycin. (B) OXPHOS and glycolysis of naïve, in vivo (red)- and in vitro (blue)-differentiated TH17 cells was analyzed via OCR and ECAR on a Seahorse Bioanalyzer and expressed as OCR/ECAR ratio. (C) Schematic depiction of glucose metabolism in glycolysis, the PPP, and the TCA cycle. (D) Relative metabolite pool sizes normalized to those in the in vitro-derived TH17 cells for each of the metabolites in glycolysis (top) and the PPP (bottom). Metabolic profiles for in vitro-derived TH17 cells are depicted in black and in vivo TH17 profiles in red. Error bars represent the SEM for n=3, and data are representative of four independent experiments. G6P, glucose 6-phosphate; F6P, fructose 6-phosphate; FBP, fructose-bisphosphate; DHAP, dihydroxyacetone phosphate; Ga3P, glyceraldehyde 3-phosphate; G3P, glycerol 3-phosphate; 3PG, 3-phosphoglycerate; PEP, phosphoenol pyruvate; Pyr, pyruvate; Lac, lactate; GdL6P, glucono-delta-lactone 6-phosphate; 6PG, 6-phosphogluconate; R5P, ribose 5-phosphate; Ru5P, ribulose 5-phosphate; X5P, xylulose 5-phosphate; E4P, erythrose 4-phosphate; S7P, sedoheptulose 7-phosphate; OBP, octulose bis-phosphate; SBP, sedoheptulose bis-phosphate.
To exclude the possibility that the reduced glycolytic metabolism of in vivo differentiated TH17 cells was simply due to the isolation procedure and brief ex vivo manipulation, we activated OVA-specific, OT-I CD8+ T cells in vivo by injection of OVA peptide, similarly isolated them, and measured their rate of lactate production. As seen in Figure 2A, CD8+ T cells activated in vivo demonstrated much higher rates of lactate production than did CD4+ TH17 cells, thereby suggesting the metabolic program in the latter is not simply attributable as a procedure-related effect.
In vivo TH17 are oxidative
The balance between ATP synthesis from glycolysis and OXPHOS can be compared among different cell populations by calculating the ratio of the OCR13 to the ECAR14, the latter is primarily attributed to lactic acid production from glucose through glycolysis (21, 27). Since in vivo-differentiated TH17 effectors use less glycolysis, we reasoned their balance of OXPHOS and glycolysis might be shifted toward oxidative metabolism compared to in vitro-differentiated TH17 cells. OCR and ECAR measurements showed that in vivo-differentiated TH17 cells possess a significantly higher OCR/ECAR ratio than in vitro cells when measured immediately after isolation (Figure 2B). This difference is primarily accounted for by reduced glycolysis in the in vivo-generated cells (Figure 2D and Supplemental Figure 2A) and has also been observed in T cells activated in vivo by alloantigen (21).
To exclude the possibility that differences in the OCR/ECAR ratios could be explained by the different stimuli used to activate T cells in vitro than those used in vivo, we compared OCR/ECAR values of T cells activated in vitro by anti-CD3/anti-CD28 stimulation under polarizing conditions to OT-II T cells activated by OVA-pulsed antigen-presenting cells. TH17 cell populations differentiated in response to either stimulation protocol in vitro had lower OCR/ECAR values compared with TH17 cells differentiated in vivo (Supplemental Figure 2B). In addition, stimulation of differentiated TH17 cells with PMA and ionomycin, which is commonly used to increase IL-17 production, did not affect the OCR/ECAR balance (Supplemental Figure 2B).
Since there is often a reciprocal relationship between levels of ATP generated by glycolysis and OXPHOS, the differences in OCR/ECAR values suggest ATP levels in in vivo-differentiated TH17 cells should be more dependent on OXPHOS than in in vitro cells. To test this hypothesis, cells were treated for four hours with oligoymcin, a selective inhibitor of mitochondrial ATP synthase. Oligomycin reduced OCR in both the in vitro and in vivo differentiated TH17 effector cells to roughly comparable levels (Figure 3A). By contrast, after three hours, oligomycin reduced the intracellular ATP concentration by 70% in the in vivo TH17 cells, whereas ATP levels in the in vitro cells were unchanged from control (Figure 3B). When intracellular ATP levels were assessed over the time of oligomycin treatment, there was an initial drop in ATP in both the in vitro and in vivo cells; ATP levels in the in vitro TH17 effector cells returned to baseline, whereas ATP levels remained low in the in vivo effector TH17 cells. The transient drop in ATP in the in vitro effector TH17 cells (Figure 3C), followed by the recovery of ATP production in the presence of oligomycin, demonstrate the cells’ metabolic adaptability and suggests that in vitro-differentiated cells increase glycolysis to compensate for energy stress (28).
Figure 3. In vivo TH17 are metabolically inflexible.
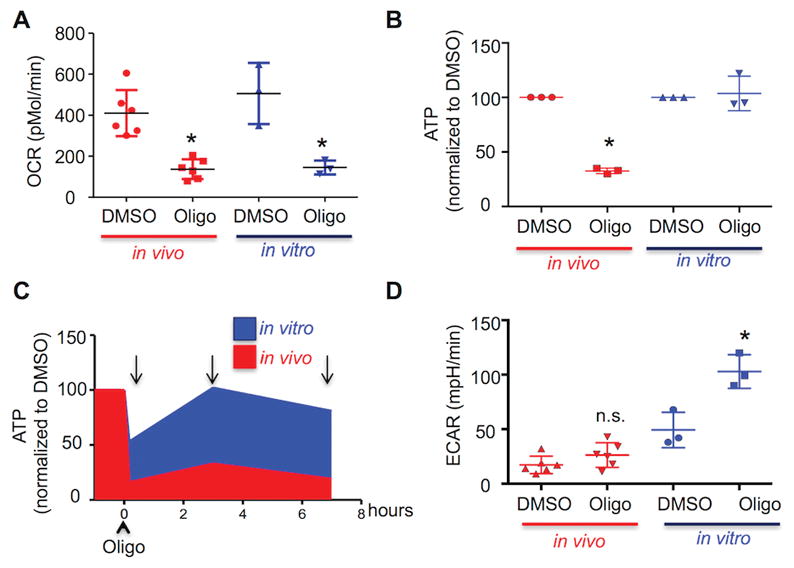
(A–D) In vivo (red)- and in vitro (blue)-differentiated TH17 cells were treated for one hour with DMSO or oligomycin (1μM) and then stimulated with PMA/ionomycin. OCR was analyzed on a Seahorse Bioanalyzer (A); intracellular ATP was evaluated by bioluminescence (B,C); ECAR was analyzed on a Seahorse Bioanalyzer (D). (B,C) Results are expressed as percentage of DMSO-treated cells. (C) Arrows indicate time in which intracellular ATP was evaluated. (A–D) Representative of three independent experiments; *p < 0.001; N.S., not significant (unpaired Student’s t-test); error bars, SEM.
In vitro TH17 cells activate glycolysis when OXPHOS is blocked
The ability to adapt metabolism by increasing glycolytic flux to meet energy demands in the face of oxygen deprivation is well-established in many tissue types. However, some cell types lack this capacity (29), which presents a putative liability that could be exploited therapeutically. Therefore, we tested if in vivo-generated TH17 cells lack metabolic flexibility; namely, the ability to increase glycolysis to preserve effector function when OXPHOS is blocked.
Consistent with this possibility, oligomycin doubled the ECAR in in vitro TH17 cells but had no effect on the ECAR of in in vivo-differentiated TH17 cells (Figure 3D). Next, we modulated the increase in glycolysis caused by oligomycin to the extent necessary to maintain lactate production at basal levels by partially blocking glycolysis (by inhibiting hexokinase 2 with 2-deoxyglucose; 2-DG) concurrent with oligomycin treatment. Lactate production increased ≥two-fold when in vitro effector TH17 cells were treated with oligomycin in the absence of 2-DG, but did not increase in the in vivo-differentiated TH17 cells (Figure 4A). By comparison, treatment with 2-DG inhibited the oligomycin-induced increase in lactate production from in vitro effector TH17 cells (Figure 4A) to the level observed in the in vivo effector TH17 cells. Importantly, co-treatment with 2-DG sensitizes in vitro-differentiated TH17 cells to oligomycin, causing both intracellular ATP and IL-17 to fall by more than 90% (Figure 4B and C). From these results, we conclude that the ability to increase glycolysis to generate ATP in response to energy restriction allows in vitro-differentiated TH17 cells to function independently of OXPHOS. This property is not observed with in vivo TH17 cells, thus rendering them exquisitely susceptible to OXPHOS inhibition.
Figure 4. In vivo-differentiated TH17 cells have restricted glycolytic capacity.

(A–C) In vivo-differentiated TH17 (red), in vitro TH17 (blue), and in vitro TH17 treated with 2-deoxyglucose (red stripes) were treated for one hour with DMSO or oligomycin (1μM) and then stimulated with PMA/ionomycin. Lactate concentration in cell-free supernatant from triplicate cultures was evaluated by a colorimetric assay (A), intracellular ATP triplicate cultures by bioluminescence (B), and IL-17 production in cell-free supernatant from triplicate cultures was analyzed by ELISA (C). (A–C) Results are expressed as percentage of DMSO-treated cells. (A–C) Representative of three independent experiments; *p < 0.001 (unpaired Student’s t-test); (error bars, mean ± SEM).
To further confirm that TH17 cells in vivo but not those in vitro rely on OXPHOS for IL-17 production, oligomycin was applied to (a) in vivo differentiated TH17 cells, (b) naïve CD4 T cells differentiated in vitro using anti-CD3 + anti-CD28 coated plates, (c) OT-II splenocytes differentiated in vitro to TH17 cells by OVA antigen in the presence or absence of muramyldipepdide, the active component of adjuvant CFA (30, 31) that was used for in vivo TH17 differentiation (Supplemental Figure 3B,C,E). Oligomycin inhibited IL-17 production in the in vivo differentiated TH17 cells whereas in vitro differentiated TH17 cells were insensitive, irrespective of the differentiation protocol used.
Since IL-1β and IL-2 can regulate glycolysis in T cells (32, 33), we differentiated naïve CD4 cells with a polarization cocktail in the presence or absence of IL-1β, with or without IL-2 blocking antibodies. Consistent with the literature (34, 35), the absence of IL-1β reduced the number of TH17 cells from ~25% to ~5% (Supplemental Figure 3B) while IL-2 blocking antibodies increased the number of TH17 from ~25% to ~80% (Supplmental Figure 3D), confirming their biological activity. Moreover, in vitro differentiated TH17 cells were insensitive to oligomycin, irrespective of whether of IL-1β and IL-2 blocking antibodies were present during the differentiation (Supplemental Figures 3B and 3D). As TH17 differentiated in vitro with several different protocols are all insensitive to oligomycin, the reliance on OXPHOS for cytokine production appears to be a distinctive feature of in vivo derived TH17 cells.
Glutamine metabolism is not affected by TH17 differentiation conditions
Glucose and glutamine are two of the primary carbon substrates across many aspects of T cell biology (3). These nutrients are able to displace one another in important metabolic pathways, including mitochondrial metabolism in the TCA cycle (3). Given the increased utilization of glycolysis in in vitro cells, and the increased reliance of in vivo cells on mitochondrial metabolism, we predicted that the in vivo cells would more avidly metabolize glutamine carbon in the mitochondria. To test this hypothesis, we traced glucose and glutamine carbon into intermediary metabolism using the non-radioactive carbon-13 isotope and targeted mass spectrometry-based metabolomics (24). The data from these studies confirmed the glycolytic phenotype of the in vitro TH17 cells, and surprisingly revealed that the in vitro cells were generally more metabolic— consuming and metabolizing both more glucose and more glutamine carbon (data not shown). However, when glucose and glutamine entry into the TCA cycle was analyzed as a function of total carbon, major global differences were not observed (data not shown). These results suggest that in vivo-differentiated TH17 cells do not rely on glutamine metabolism to fuel the TCA cycle, and that the major difference between in vivo and in vitro TH17 cells is at the level of glycolytic flux.
Pdk1 controls the metabolic flexibility of TH17 cells in vitro
We next sought to identify molecular differences between in vitro- and in vivo-differentiated TH17 cells to define the mechanism of metabolic flexibility. Established glycolytic activators during TH17 differentiation include HIF1α and Pdk1. HIF1α is transiently upregulated in normoxic conditions during TH17 cell differentiation (13, 17) (Supplemental Figure 3F). There is evidence that HIF1α-driven glycolysis skews differentiation of T cells toward a TH17 phenotype (13, 17); however, its role in the bioenergetics of already differentiated TH17 has not been defined. When HIF1α in in vitro-differentiated TH17 cells was reduced by siRNA transfection there was a slight reduction in the expression of glycolytic enzymes (Figure 5A), however, the expression of RORγt remained constant and production of IL-17 was comparable to cells transfected with scramble siRNA. These data suggest that HIF1α is important during TH17 differentiation (17, 36) but is dispensable for IL-17 production in differentiated TH17 cells (Figure 5B). Moreover, in cells transfected with either control siRNA or with HIF1α, siRNA, treatment with oligomycin did not reduce lactate or IL-17 production (Figure 5B and Supplemental Figure 3G), indicating that HIF1α expression does not control sensitivity to oligomycin and dependence on OXPHOS.
Figure 5. Pdk1 regulates in vitro TH17 metabolic flexibility.
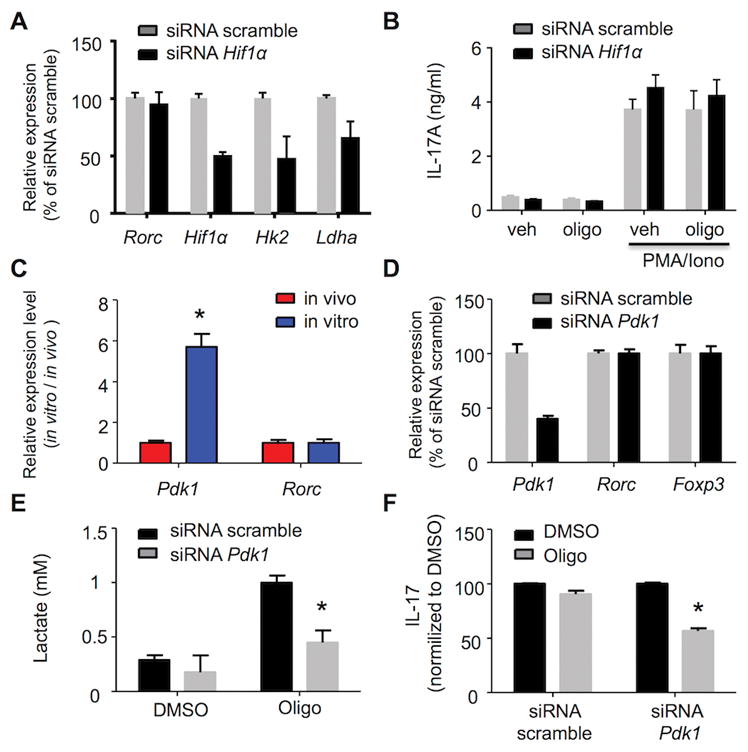
(A,B,D,E,F) In vitro-differentiated TH17 cells were nucleofected with siRNA scramble or siRNA targeting HIF1α (A and B) or Pdk1 (D–F) were treated for one hour with DMSO or oligomycin (1 μM) and then stimulated with PMA/ionomycin. Gene expression level was analyzed by RT-PCR (A and D), lactate concentration in cell-free supernatant from triplicate cultures was determined by a colorimetric assay (E), and IL-17 production in cell-free supernatant from triplicate cultures was analyzed by ELISA (B and F). (C) Gene expression in vivo-differentiated TH17 (red) and in vitro-differentiated TH17 (blue) cells was analyzed by RT-PCR. Representative of two (C) and three (A,B,D,E,F) independent experiments; *p < 0.05 (unpaired Student’s t-test); error bars, mean ± SEM.
Pdk1 inhibits the pyruvate dehydrogenase complex (PDC), which leads to an increase in pyruvate to lactate conversion and a reduction of mitochondrial pyruvate oxidation. Indeed, Pdk1 expression was nearly six-fold higher in the cells derived in vitro than in those derived in vivo (Figure 5C). As expected, siRNA knock-down of Pdk1 expression in differentiated TH17 cells did not affect expression of either RORγt or FOXP3 (Figure 5D). In contrast, lowering Pdk1 expression significantly reduced the amount of lactate produced in response to oligomycin (Figure 5E) and made cells dependent on OXPHOS for IL-17 production (Figure 5F). Thus we deduce that Pdk1 provides metabolic flexibility to in vitro TH17 effectors and that low Pdk1 expression by in vivo cells can account for their OXPHOS-dependent phenotype.
TH17 cells employ OXPHOS for cytokine production in vivo
A primary function of effector TH17 cells is the secretion of cytokines such as IL-17 (37). Synthesis and secretion of cytokines are energy-intensive processes that cease when intracellular ATP is reduced (38). Hence, we sought to determine if the metabolic differences between the in vitro- and in vivo-differentiated cells are important for this TH17 effector function by examining IL-17 expression following OXPHOS inhibition.
Treating in vivo-derived effector TH17 cells with oligomycin for one hour ex vivo reduced IL-17+ cells by 40–60% (Figure 6A), along with the amounts of IL-17 (Figure 6B) and other TH17 signature cytokines like GM-CSF, TNF, and IL-21 that were secreted (Figure 6C–F). In contrast, oligomycin had a much smaller effect on IL-17 production by in vitro effector TH17 cells (10–30% decrease in CD4+IL-17+ cells and 30–40% decrease in IL-17 titer) (Figure 6C–F). The differential sensitivity of IL-17 production to oligomycin is independent of when oligomycin is added relative to the time at which cells are analyzed and the specific polarization cocktail employed (Supplemental Figure 3). Together, these results show that vivo-differentiated TH17 cells rely on OXPHOS for cytokine production and suggest that TH17 function in vivo can be modulated by inhibition of oxidative metabolism.
Figure 6. OXPHOS inhibition reduces TH17 cytokine production.
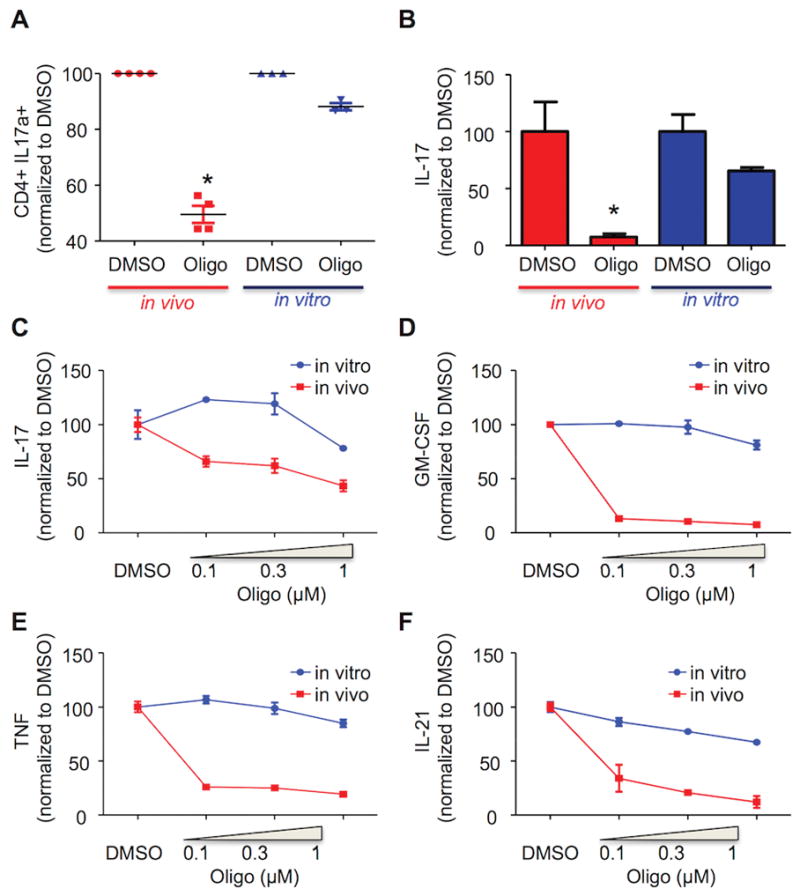
(A–F) In vivo (red)- or in vitro (blue)-differentiated TH17 cells were treated for one hour with DMSO or the indicated concentration of oligomycin and then stimulated with PMA/ionomycin. IL-17 expression was analyzed by FACS (A). The production of IL-17a, GM-CSF, TNF, and IL-21 in cell-free supernatant from triplicate cultures was evaluated by ELISA (B–F). (A–F) Results are expressed as percentage of DMSO-treated cells. (A) For in vivo TH17, each dot represents an individual mouse; for in vitro TH17, an independent cell culture. (A–F) Representative of three independent experiments; *p < 0.001; N.S., not significant (A: paired Student’s t-test; B–F: unpaired Student’s t-test); error bars, mean ± SEM.
Modulating OXPHOS regulates TH17 effector function in vivo
We aimed to determine if the metabolic sensitivity of IL-17 production to oligomycin identified ex vivo is also observed in vivo. For these experiments, mice were injected with anti-CD3 antibody to induce IL-17 production in the presence or absence of oligomycin, and plasma levels of IL-17 were measured. To confirm that the IL-17 produced in this model is RORγt-dependent, a control group of animals received ursolic acid (an RORγt inhibitor) together with the anti-CD3 antibody (39). Both oligomycin and ursolic acid reduced IL-17 serum to similar levels (Figure 7A). Notably, this effect was specific for TH17 cells, as production of other cytokines (e.g. TNF; Figure 7B) was not affected. By contrast, echinomycin, which was effective in blocking the transcription of HIF1α target genes in vivo (Figure 7C), had no effect on anti-CD3-induced IL-17, indicating that HIF1α does not regulate TH17 effector function in vivo (Figure 7A, right).
Figure 7. OXPHOS inhibition reduces IL-17 production in vivo.

(A–C) Mice were treated via i.p. injection with vehicle, ursolic acid (A), oligomycin (A,B), and echinomycin for one hour and then stimulated i.p. with agonistic anti-CD3e antibody for three hours. (A and B) IL17a (A) and TNF (B) plasma concentrations were determined by ELISA. (C) Expression of HIF1α target genes was analyzed using RNA extracted from at least three spleens per treatment group by qPCR and expressed as a ratio between vehicle and 0.25 mg/kg echinomycin-treated animals. Dashed line depicts the cutoff of >two-fold difference. Dark gray bars show higher expression in vehicle-treated samples. (D,E) B6 mice were dosed with oligomycin or ursolic acid and then challenged intrarectally with TNBS. (D) Graph shows histology score of individual mice two days after TNBS challenge. (E) Representative images (F,G) from B6 mice dosed with oligomycin or ursolic acid followed by daily application of Aldara cream (F) Graph shows ear thickness of individual mice five days after Aldara treatment (G) Representative histology (A,B,D,F) Each dot represents an individual mouse. (A,C,F) Representative of three independent experiments. (D) Results from two independent experiments are plotted together; *p < 0.001; **p < 0.05; N.S., not significant (unpaired Student’s t-test); error bars, mean ± SEM.
To test if inhibiting OXPHOS controls inflammatory cytokine production in vivo, we assessed the effect of oligomycin in a murine TNBS colitis model in which development of disease is IL-17-dependent (40). In this model, oligomycin was comparable to ursolic acid in reducing disease (Figure 7D and E). To further evaluate the role of OXPHOS in IL-17 production in vivo, we next tested the effect of oligomycin and ursolic acid in a murine IL-17-driven model of psoriasis (41, 42). In this model, oligomycin reduced ear thickness, keratinocyte proliferation, and immune cell infiltration (Figure 7G and F). The efficacy of oligomycin in this model was comparable of ursolic acid.
To determine if oligomycin reduces IL-17 production directly on cells isolated from sites of inflammation, LPMCs were isolated from mice with colitis and healthy controls and treated ex vivo with oligomycin before stimulation with PMA and ionomycin. Since the CD4 antigen is cleaved during isolation of LPMCs, we compared the effect of oligomycin on total CD3+ cells relative to CD3+CD8+ cells. Animals with colitis had almost three-fold more CD3+IL-17+IFNγ-cells relative to controls (Figure 8A and Supplemental Figure 4A,B,E) and oligomycin reduced the number of CD3+IL-17+IFNγ-cells in LPMCs from mice with colitis by ~70% (Figure 8B and Supplemental Figures 4B and 4E). By contrast, oligomycin had no effect on the production of IL-17 in CD3+CD8+ cells, and only slightly increased the number of CD3+CD8+IFNγ+ cells (Figure 8C), consistent with the glycolytic phenotype of CD8+ effector cells (Figure 2A). From these results we conclude that, similar to the bioenergetic requirements of effector TH17 cells generated by adjuvant challenge in vivo, OXPHOS is required for IL-17 production by TH17 cells at sites of colonic inflammation.
Figure 8. OXPHOS controls IL-17 production in IBD.

(A–D) LPMCs that were isolated from mice treated with ethanol (A) or TNBS (B and C), or from human IBD patients (D) were treated for one hour with DMSO or oligomycin (1 μM) and then stimulated with PMA/ionomycin. IL-17 and IFNγ production in CD3+ and CD8+ was evaluated by FACS analysis. Each dot represents an individual mouse (A–C) or subject (D). (A–C) Representative of three independent experiments; *p < 0.05; n.s., not significant (paired Student’s t-test) (error bars, mean ± SEM). (E) Schematic representation of the metabolic pathways used to fuel effector function in TH17 cells. TH17 cells derived in vitro predominantly utilize glycolysis to generate ATP to sustain effector function (left). In addition, these cells exhibit metabolic flexibility and can engage OXPHOS-derived ATP when glycolysis is inhibited. TH17 cells derived in vivo (right) generate ATP used to sustain effector function through mitochondrial OXPHOS and are unable to engage glycolysis to sustain ATP levels following OXPHOS inhibition.
We next asked if OXPHOS supports the IL-17 production by differentiated human TH17 cells. To address this question, purified PBMCs from healthy subjects and patients with IBD were stimulated with PMA and ionomycin. Oligomycin reduced IL-17 expression and secretion in CD4+ lymphocytes from both healthy subjects and patients with IBD, whereas echinomycin, an inhibitor of HIF1α, had no effect (data not shown). Other TH17 cytokines, including TNF and GM-CSF, were also sensitive to oligomycin and unaffected by echinomycin (data not shown). Thus, the secretion of cytokines by human TH17 cells isolated from blood depends on OXPHOS, similar to murine TH17 effector cells generated in vivo.
Lastly, LPMCs were purified from human tissue biopsied at sites of inflammation identified by endoscopy in subjects with active IBD. After isolation, cells were stimulated with PMA and ionomycin in the absence or presence of oligomycin. Oligomycin significantly decreased the percentage of CD3+IL-17+IFNγ- and T cells, producing both IFNγ and IL-17, whereas the proportion of CD3+IFNγ+IL-17- was not changed (Figure 8D). These data provide evidence that human TH17 cells at sites of colonic inflammation selectively depend on OXPHOS for their function, similar to murine TH17 cells generated in vivo thereby rendering them susceptible to OXPHOS inhibitors. These results also underscore the importance of using in vivo models to understand metabolic control of T cell function (43).
DISCUSSION
Here we show that the environment in which naïve T lymphocytes differentiate into effector TH17 cells has a profound influence on their metabolism post-differentiation. TH17 cells differentiated in vitro are metabolically flexible and meet the bioenergetic demands of cytokine production (which can account for up to 50% of total cellular ATP requirements in lymphocytes (38)) using either OXPHOS or glycolysis (Figure 8E). These results are consistent with previous studies showing that lymphocytes are metabolically flexible in vitro and can upregulate glycolysis when OXPHOS is inhibited or when certain nutrients are limiting (44), a feature thought to promote lymphocyte fitness (45). By contrast, TH17 cells derived in vivo rely on OXPHOS to supply ATP needed for cytokine production (Figure 8E). The finding that in vivo-derived effector TH17 cells are metabolically inflexible and cannot upregulate glycolysis when bioenergetically challenged was unexpected. This result has significant implications for the discovery and development of therapeutics to control immune responses, in that the inability to compensate to metabolic stress render TH17 cells vulnerable to drugs that target their metabolism (4).
The dependence of effector TH17 cells on oxidative metabolism in vivo contrasts with the glycolytic phenotype described for other pro-inflammatory lineages of differentiated CD4 cells (14, 26) and for CD8 effector cells (46, 47) and is comparable to the metabolic phenotype of memory CD8 cells that require OXPHOS for survival and function (48, 49). Both memory CD8 T cells and TH17 cells can survive in vivo for extended periods and produce more differentiated progeny (50, 51); hence, it is possible that dependence of TH17 cells on OXPHOS is linked to this enhanced survival advantage in vivo. In support of this hypothesis, increased oxidative metabolism promotes stemness and survival in CD8 cells (43, 52). Similarly, Kawalekar et al. showed that 4-1BBζ CAR T cells are characterized by increased central memory differentiation, along with enhanced mitochondrial biogenesis, and greater oxidative metabolism compared to antigen stimulated CD28ζ CAR T cells (53). These data further support the hypothesis that increased oxidative phosphorylation with limited glycolysis is necessary for central memory differentiation and long-term survival (43, 54).
A key outstanding question regarding the metabolism of immune cells is how environmental cues influence the metabolic pathways that shape immune responses (3, 20). Environmental signals in vitro, in the form of nutrient availability, oxygen tension, cytokine concentration, and length and type of cellular stimulation, are all known to affect cellular bioenergetics (reviewed in (3)). To probe the possibility that the metabolism of effector TH17 cells varies as a result of the context in which they are found, we assessed the bioenergetics of TH17 cells isolated from different anatomical loci and at sites of inflammation, which differ with respect to nutrient availability and cytokine levels (20). This point is quite relevant in the intestine, where the microenvironment contains substrates derived from food, as well as unique metabolites from the intestinal microbiome that can impact immune responses and which are not present in lymph nodes (55). Notwithstanding, we found that TH17 cells isolated from the lymph nodes, spleen, and thymus (data not shown) of immunized mice; human peripheral blood; and cells isolated from inflamed lamina propria all employ OXPHOS to generate the energy required for production of IL-17. These data indicate that the bioenergetics of TH17 cells are independent of the specific compartment from which cells are isolated and suggests that the observed oxidative phenotype is an intrinsic property of in vivo differentiated TH17 cells. Given that limited oxygen availability in the inflamed intestine can promote glycolysis (20), the finding that TH17 derived from oxygen-poor sites of inflammation retain an oxidative phenotype suggests that the environment in which TH17 cells differentiate instructs their metabolic fate, as opposed to the environments in which they function. However, we cannot exclude the possibility that TH17 effector cells isolated from other tissues and microenvironments, such as tumors, have alternate metabolic programs that more closely match that of in vitro-derived TH17 cells (20). Collectively, our data are consistent with other studies showing that immune cells adapt specific metabolic phenotypes in vivo (21, 22, 56); hence, conclusions about metabolism and energy production by immune cells should not be based entirely on results from in vitro models.
We explored the basis for the difference in metabolic flexibility between in vitro and in vivo TH17 cells. HIF1α is activated by reduced O2 tension and in normoxic conditions by inflammatory cytokines, reactive oxygen species, growth factors, and bacterial products (57). Even though HIF1α is necessary for TH17 cell differentiation (13, 17) and promotes glycolysis in immune cells (20), reducing its expression in differentiated TH17 cells had no effect on IL-17 production, even when mitochondrial ATP production was inhibited. These findings suggest that differences in the metabolic programs of in vitro and in vivo TH17 cells do not result from differential expression of HIF1α and are consistent with transient expression of HIF1α during TH17 cell differentiation (Supplemental Figure 3F) (17, 36).
Pdk1 is another gene that regulates glucose metabolism. By inhibiting the activity of the pyruvate dehydrogenase complex (PDHc), Pdk1 increases the rate that glucose-derived pyruvate is converted into lactate (58). The expression of Pkd1 is regulated by nutrient levels and several signaling pathways (59, 60), and is expressed in in vitro-differentiated TH17 cells at more than a six-fold excess of in vivo-differentiated cells. Rathmell et al. recently found that by increasing glycolysis, Pdk1 drives TH17 differentiation in vitro (16). In addition to functioning during TH17 differentiation, our data show that Pdk1 activity has a role in IL-17 production in in vitro-differentiated TH17 cells under metabolic stress, but that Pdk1 does not affect in vivo-derived TH17 cell metabolism. Furthermore, low Pdk1 expression in TH17 cells generated in vivo likely explains their particular dependence on OXPHOS for IL-17 production.
The Crabtree effect is a phenomenon whereby exposure to a high concentration of glucose limits OXPHOS (61). Unlike in the Warburg effect, cells retain the ability to switch to mitochondrial respiration when glycolysis is inhibited (61). The Crabtree effect has been observed in lymphocytes (62) and cancer cells (61), but the underlying molecular mechanism remains poorly characterized. In vitro, TH17 cells are exposed to a high concentration of glucose (≥11 mM versus 4 mM in vivo) and glucose concentration can drive the expression of Pdk1 (60). An intriguing possibility is that the metabolic phenotype of in vitro TH17 cells can be explained by the Crabtree effect and that that the Crabtree effect is regulated at a molecular level by the expression of Pdk1. Additional studies will be required to test this hypothesis.
TH17 cells are a central component of immune host defense. There is evidence that these cells also contribute to pathological inflammation associated with autoimmune diseases, including IBD (6, 63) and psoriasis (41, 42). The critical role of IL-17 in psoriasis is documented by the efficacy of anti-IL-17A antibodies (64), but blocking IL-17A alone is not sufficient to limit colitis (65) suggesting that therapeutic interventions in IBD need to target a more complete spectrum of TH17 effector functions. In agreement with this hypothesis, adoptive transfer experiments have shown that TH17 cells are more effective than TH1 in inducing murine colitis (65); TH17 cells accumulate in intestinal inflammatory lesions in IBD (66, 67); genetic polymorphism of cytokines controlling TH17 generation and function is associated with an increased risk of IBD (68). The metabolic dependence of in vivo-derived TH17 cells on OXPHOS provides the possibility of controlling TH17 cell-driven disease by restricting OXPHOS. Here we showed that oligomycin, a specific and potent inhibitor of OXPHOS, reduced key TH17 cytokines produced by human and murine cells, is effective in blocking TH17 effector functions in cells from sites of colonic inflammation, and controls disease in murine TNBS-induced colitis and psoriasis. The differences in the metabolic pathways available to TH17 effector cells generated in vitro and in vivo highlight the importance of studying metabolism in vivo, particularly with respect to designing interventions that exploit cellular bioenergetics and support the concept that modulation of OXPHOS has therapeutic potential for diseases driven by TH17 cells.
Supplementary Material
Acknowledgments
We thank Andrea Viale (MD Anderson Cancer Center) for experimental advice.
Footnotes
This work was supported in part by a Dale F. Frey Breakthrough Scientist Award from the Damon Runyon Cancer Research Foundation (DFS-09-14 to C.A.L.) and the NIH (AI-47450 to G.D.G.).
Oxidative phosphorylation
hypoxia-inducible factor 1-alpha
pyruvate dehydrogenase kinase, isozyme 1
Inflammatory bowel disease
Crohn’s disease
Ulcerative colitis
Lamina Propria Mononuclear Cells
Gene Set Enrichment Analysis
Small interfering RNA
Trinitrobenzenesulfonic acid
Pentose phosphate pathway
Oxygen consumption rate
Extracellular acidification rate
Conflict of interest: LF, LYH, MS, XL, KD, CL, BS, XH, AWO, and GDG have ownership interests in Lycera Corp.
References
- 1.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wahl DR, Byersdorfer CA, Ferrara JL, Opipari AW, Jr, Glick GD. Distinct metabolic programs in activated T cells: opportunities for selective immunomodulation. Immunol Rev. 2012;249:104–115. doi: 10.1111/j.1600-065X.2012.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol. 2015 doi: 10.1016/j.it.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters A, Lee Y, Kuchroo VK. The many faces of Th17 cells. Curr Opin Immunol. 2011;23:702–706. doi: 10.1016/j.coi.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber S, Gagliani N, Flavell RA. Life, death, and miracles: Th17 cells in the intestine. Eur J Immunol. 2012;42:2238–2245. doi: 10.1002/eji.201242619. [DOI] [PubMed] [Google Scholar]
- 7.O’Connor W, Jr, Zenewicz LA, Flavell RA. The dual nature of T(H)17 cells: shifting the focus to function. Nat Immunol. 2010;11:471–476. doi: 10.1038/ni.1882. [DOI] [PubMed] [Google Scholar]
- 8.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, Monticelli S, Lanzavecchia A, Sallusto F. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature. 2012;484:514–518. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]
- 10.Hu X, Wang Y, Hao LY, Liu X, Lesch CA, Sanchez BM, Wendling JM, Morgan RW, Aicher TD, Carter LL, Toogood PL, Glick GD. Sterol metabolism controls TH17 differentiation by generating endogenous RORgamma agonists. Nat Chem Biol. 2015;11:141–147. doi: 10.1038/nchembio.1714. [DOI] [PubMed] [Google Scholar]
- 11.Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol. 2011;76:347–353. doi: 10.1101/sqb.2011.76.010678. [DOI] [PubMed] [Google Scholar]
- 12.Saunier E, Benelli C, Bortoli S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int J Cancer. 2016;138:809–817. doi: 10.1002/ijc.29564. [DOI] [PubMed] [Google Scholar]
- 13.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michalek RD, V, Gerriets A, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, Haeberli L, Huck C, Turka LA, Wood KC, Hale LP, Smith PA, Schneider MA, MacIver NJ, Locasale JW, Newgard CB, Shinohara ML, Rathmell JC. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. The Journal of clinical investigation. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bahre H, Tschirner SK, Gorinski N, Gohmert M, Mayer CT, Huehn J, Ponimaskin E, Abraham WR, Muller R, Lochner M, Sparwasser T. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. 2014;20:1327–1333. doi: 10.1038/nm.3704. [DOI] [PubMed] [Google Scholar]
- 19.Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol. 2014;14:435–446. doi: 10.1038/nri3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C, Glick GD, Ferrara JL. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Science translational medicine. 2011;3:67ra68. doi: 10.1126/scitranslmed.3001975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, Faicchia D, Marone G, Tramontano D, Corona M, Alviggi C, Porcellini A, La Cava A, Mauri P, Matarese G. The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity. 2016;44:406–421. doi: 10.1016/j.immuni.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monteleone G, Biancone L, Marasco R, Morrone G, Marasco O, Luzza F, Pallone F. Interleukin 12 is expressed and actively released by Crohn’s disease intestinal lamina propria mononuclear cells. Gastroenterology. 1997;112:1169–1178. doi: 10.1016/s0016-5085(97)70128-8. [DOI] [PubMed] [Google Scholar]
- 24.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc. 2012;7:872–881. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glick GD, Rossignol R, Lyssiotis CA, Wahl D, Lesch C, Sanchez B, Liu X, Hao LY, Taylor C, Hurd A, Ferrara JL, Tkachev V, Byersdorfer CA, Boros L, Opipari AW. Anaplerotic metabolism of alloreactive T cells provides a metabolic approach to treat graft-versus-host disease. J Pharmacol Exp Ther. 2014;351:298–307. doi: 10.1124/jpet.114.218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berridge MV, Herst PM, Tan AS. Metabolic flexibility and cell hierarchy in metastatic cancer. Mitochondrion. 2010;10:584–588. doi: 10.1016/j.mito.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, Kapoor A, Yao W, Brunetto E, Kang Y, Yuan M, Asara JM, Wang YA, Heffernan TP, Kimmelman AC, Wang H, Fleming JB, Cantley LC, DePinho RA, Draetta GF. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellouz F, Adam A, Ciorbaru R, Lederer E. Minimal structural requirements for adjuvant activity of bacterial peptidoglycan derivatives. Biochem Biophys Res Commun. 1974;59:1317–1325. doi: 10.1016/0006-291x(74)90458-6. [DOI] [PubMed] [Google Scholar]
- 31.Adam A, Ciorbaru R, Ellouz F, Petit JF, Lederer E. Adjuvant activity of monomeric bacterial cell wall peptidoglycans. Biochem Biophys Res Commun. 1974;56:561–567. doi: 10.1016/0006-291x(74)90640-8. [DOI] [PubMed] [Google Scholar]
- 32.Chatterjee S, Thyagarajan K, Kesarwani P, Song JH, Soloshchenko M, Fu J, Bailey SR, Vasu C, Kraft AS, Paulos CM, Yu XZ, Mehrotra S. Reducing CD73 expression by IL1beta-Programmed Th17 cells improves immunotherapeutic control of tumors. Cancer Res. 2014;74:6048–6059. doi: 10.1158/0008-5472.CAN-14-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM, Laidlaw BJ, Araki K, Ahmed R, Kaech SM, Craft J. The Interleukin-2-mTORc1 Kinase Axis Defines the Signaling, Differentiation, and Metabolism of T Helper 1 and Follicular B Helper T Cells. Immunity. 2015;43:690–702. doi: 10.1016/j.immuni.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O’Shea JJ. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Flach H, Onizawa M, Wei L, McManus MT, Weiss A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat Immunol. 2014;15:393–401. doi: 10.1038/ni.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuniga LA, Jain R, Haines C, Cua DJ. Th17 cell development: from the cradle to the grave. Immunol Rev. 2013;252:78–88. doi: 10.1111/imr.12036. [DOI] [PubMed] [Google Scholar]
- 38.Krauss S, Brand MD, Buttgereit F. Signaling takes a breath--new quantitative perspectives on bioenergetics and signal transduction. Immunity. 2001;15:497–502. doi: 10.1016/s1074-7613(01)00205-9. [DOI] [PubMed] [Google Scholar]
- 39.Xu T, Wang X, Zhong B, Nurieva RI, Ding S, Dong C. Ursolic acid suppresses interleukin-17 (IL-17) production by selectively antagonizing the function of RORgamma t protein. J Biol Chem. 2011;286:22707–22710. doi: 10.1074/jbc.C111.250407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin Y, Lin Y, Lin L, Zheng C. IL-17/IFN-gamma interactions regulate intestinal inflammation in TNBS-induced acute colitis. J Interferon Cytokine Res. 2012;32:548–556. doi: 10.1089/jir.2012.0030. [DOI] [PubMed] [Google Scholar]
- 41.Cai Y, Shen X, Ding C, Qi C, Li K, Li X, Jala VR, Zhang HG, Wang T, Zheng J, Yan J. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity. 2011;35:596–610. doi: 10.1016/j.immuni.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, Becher B. Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. The Journal of clinical investigation. 2012;122:2252–2256. doi: 10.1172/JCI61862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. 2014;15:1104–1115. doi: 10.1038/ni.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tripmacher R, Gaber T, Dziurla R, Haupl T, Erekul K, Grutzkau A, Tschirschmann M, Scheffold A, Radbruch A, Burmester GR, Buttgereit F. Human CD4(+) T cells maintain specific functions even under conditions of extremely restricted ATP production. Eur J Immunol. 2008;38:1631–1642. doi: 10.1002/eji.200738047. [DOI] [PubMed] [Google Scholar]
- 45.Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015 doi: 10.1016/j.it.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, Johnson RS, Goldrath AW. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. 2013;14:1173–1182. doi: 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, Durovic B, Jauch A, Hess C. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14:1064–1072. doi: 10.1038/ni.2687. [DOI] [PubMed] [Google Scholar]
- 48.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kryczek I, Zhao E, Liu Y, Wang Y, Vatan L, Szeliga W, Moyer J, Klimczak A, Lange A, Zou W. Human TH17 cells are long-lived effector memory cells. Science translational medicine. 2011;3:104ra100. doi: 10.1126/scitranslmed.3002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, Sukumar M, Reger RN, Yu Z, Kern SJ, Roychoudhuri R, Ferreyra GA, Shen W, Durum SK, Feigenbaum L, Palmer DC, Antony PA, Chan CC, Laurence A, Danner RL, Gattinoni L, Restifo NP. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011;35:972–985. doi: 10.1016/j.immuni.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, Mohney RP, Klebanoff CA, Lal A, Finkel T, Restifo NP, Gattinoni L. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. The Journal of clinical investigation. 2013;123:4479–4488. doi: 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Jr, Patel PR, Guedan S, Scholler J, Keith B, Snyder N, Blair I, Milone MC, June CH. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 54.Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–684. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, Mazmanian SK. Specialized metabolites from the microbiome in health and disease. Cell Metab. 2014;20:719–730. doi: 10.1016/j.cmet.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, Romano A, De Simone S, Procaccini C, La Rocca C, Carrieri PB, Maniscalco GT, Salvetti M, Buscarinu MC, Franzese A, Mozzillo E, La Cava A, Matarese G. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol. 2015;16:1174–1184. doi: 10.1038/ni.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. The Journal of clinical investigation. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel MS, Nemeria NS, Furey W, Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem. 2014;289:16615–16623. doi: 10.1074/jbc.R114.563148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498:109–112. doi: 10.1038/nature12154. [DOI] [PubMed] [Google Scholar]
- 60.Xu J, Han J, Epstein PN, Liu YQ. Regulation of PDK mRNA by high fatty acid and glucose in pancreatic islets. Biochem Biophys Res Commun. 2006;344:827–833. doi: 10.1016/j.bbrc.2006.03.211. [DOI] [PubMed] [Google Scholar]
- 61.Diaz-Ruiz R, Rigoulet M, Devin A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim Biophys Acta. 2011;1807:568–576. doi: 10.1016/j.bbabio.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 62.Guppy M, Greiner E, Brand K. The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes. Eur J Biochem. 1993;212:95–99. doi: 10.1111/j.1432-1033.1993.tb17637.x. [DOI] [PubMed] [Google Scholar]
- 63.Monteleone I, Sarra M, Pallone F, Monteleone G. Th17-related cytokines in inflammatory bowel diseases: friends or foes? Curr Mol Med. 2012;12:592–597. doi: 10.2174/156652412800620066. [DOI] [PubMed] [Google Scholar]
- 64.Mease PJ, I, McInnes B, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, Landewe R, Nash P, Pricop L, Yuan J, Richards HB, Mpofu S, Group FS. Secukinumab Inhibition of Interleukin-17A in Patients with Psoriatic Arthritis. N Engl J Med. 2015;373:1329–1339. doi: 10.1056/NEJMoa1412679. [DOI] [PubMed] [Google Scholar]
- 65.Leppkes M, Becker C, Ivanov, Hirth S, Wirtz S, Neufert C, Pouly S, Murphy AJ, Valenzuela DM, Yancopoulos GD, Becher B, Littman DR, Neurath MF. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology. 2009;136:257–267. doi: 10.1053/j.gastro.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 66.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rovedatti L, Kudo T, Biancheri P, Sarra M, Knowles CH, Rampton DS, Corazza GR, Monteleone G, Di Sabatino A, Macdonald TT. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut. 2009;58:1629–1636. doi: 10.1136/gut.2009.182170. [DOI] [PubMed] [Google Scholar]
- 68.Graham DB, Xavier RJ. From genetics of inflammatory bowel disease towards mechanistic insights. Trends Immunol. 2013;34:371–378. doi: 10.1016/j.it.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.