

Abstract
Crosstalk between B and T cells in transplantation is increasingly recognized as being important in the alloimmune response. T cell activation of B cells occurs by a 3-stage pathway, culminating with costimulation signals. We review the distinct T cell subtypes required for B-cell activation and discuss the formation of the germinal center (GC) after transplantation, with particular reference to the repopulation of the GC after depletional induction, and the subsequent effect of immunosuppressive manipulation of T cell-B cell interactions. In addition, ectopic GCs are seen in transplantation, but their role is not fully understood. Therapeutic options to target T cell-B cell interactions are of considerable interest, both as immunosuppressive tools, and to aid in the further understanding of these important alloimmune mechanisms.
The 2 major cellular components that constitute the alloimmune response in transplantation, T and B cells, play major roles in graft rejection. In the absence of immunosuppression, organ transplantation elicits intense responses from T and B cells, resulting in cell-mediated rejection and antibody-mediated rejection (AMR), respectively. Unsurprisingly, “pure” alloimmune responses, limited exclusively to either type of rejection are infrequent in clinical settings. Increasingly, it is recognized that the role of B cells in transplantation is not restricted to their effector function, the humoral response, alone-antigen presentation of B cells also contributes to the optimal immune response.1 Similarly, although graft rejection had been considered largely mediated by T cell effector function, there is growing evidence that B cells and their immunoglobulin products (alloantibody) may play a role in the process.2 In this review, we wish to focus on the crosstalk between T cells and B cells in AMR after transplantation.
B CELL IN TRANSPLANTATION
T Cell–Dependent B-Cell Activation in Transplantation
After transplantation, there are 3 signaling pathways required for T cell–dependent activation of B cells. Initial B-cell activation is driven by alloantigen (Figure 1). Alloantigen is delivered to the mature B cell (IgM+IgD+)–rich area known as the B-cell follicle (cortex) within the secondary lymphoid organ.
FIGURE 1.
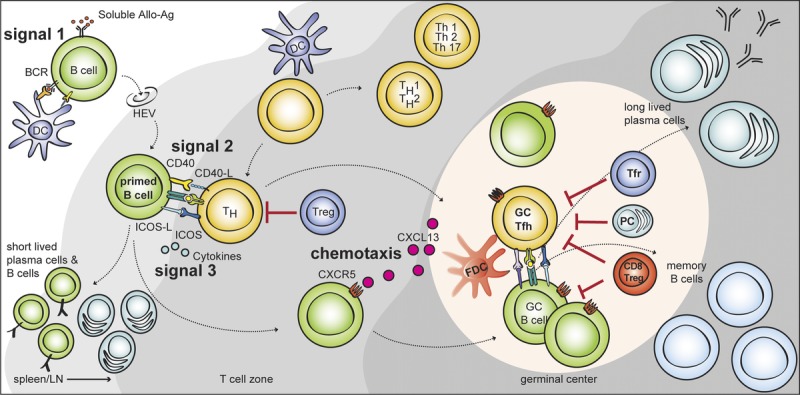
T cell–dependent B cell activation via multiple T-B interactions after allostimulation. Naive mature B cells are activated through BCR recognition (signal 1) and migrate to the T-B border via HEVs. Primed B cells receive further signals from costimulation (signal 2), and cytokines (signal 3) in the T-B border. Some activated CD4 T cells can acquire characteristics of Tfh cell lineage and migrate into the B cell follicle via CXCR5. These Tfh cells provide IL-21 and costimulation and induce the proliferation of cognate B cells, isotype switching, and somatic mutation. This massive B cell expansion and differentiation leads to the formation of hyperplastic GC in the B cell follicle. Tfr cells and CD8 Treg cells are thought to suppress this GC response either directly, by depleting B cells or indirectly by modulating Tfh cells. The GC response induces the differentiation of isotype-switched affinity mature B cells into memory B cells or into long-lived plasma cells. HEVs, high endothelial venules.
These naive mature B cells are able to recognize both membrane-bound and soluble alloantigens as a function of the B-cell receptor (BCR). BCR recognition (signal 1) of the cognate alloantigen provides an activation signal via CD19 complex (which comprises CD19, CD21, CD81, and CD225).3–5 The BCR is also responsible for internalization (endocytosis) of alloantigens derived from the allogeneic cells, which are degraded and presented via Major Histocompatibility Complex II molecules.6,7 Primed B cells are translocated into the T cell–rich area (T cell zone, paracortex) and only B cells which interact with cognate follicular T helper (Tfh) cells receive further activation signals.
In addition to antigen recognition through the BCR, the second signal for B cell activation is costimulation (signal 2). A cognate interaction between helper T (Th) cells and B cells provides multiple costimulation signals for B-cell activation. CD40L from T cells has been extensively studied; signaling via CD40 on B cells drives B-cell proliferation, antibody affinity maturation, and class switch recombination (via activation of NF-κB).8 In mice, deficiency of CD40 or CD40L results in the absence of IgG production and Ig class switch defects.9,10 Corroborating this, targeting CD40 or CD40L in organ transplantation results in a reduction of alloantibody production.11,12 After alloantigen recognition and costimulation, B-cell activation requires cytokines (signal 3), produced by various Th cells including Th1, Th2, and Th17. In support of this various cytokines are known to affect antibody production. Furthermore, numerous cytokines (including IL-6, IL-21, IL-12, IL-23, and IL-27) appear to be capable of inducing enhancing or sustaining Tfh cell–like phenotypes, which become important in the germinal center (GC) response. These cytokines act through phosphorylation of STAT1, STAT3, or STAT4 to regulate the Tfh cell–associated gene expression.13–15 However, it is now known that BCL-6 is the required transcription factor for Tfh-cell differentiation,16,17 which implies that the Tfh cell is a separate lineage of Th cell. This is supported by the finding that the expression of BCL-6 is suppressed by IL-2 acting through STAT 5,18 and that BCL-6 expression represses of T-bet (Th1 differentiation) and RORγc (Th17 differentiation).15 More GC-associated cytokines will be discussed later.
T Cell–Independent Signals for B-Cell Activation
Although B-cell activation is predominantly through T cell–dependent mechanisms, there is interest in T cell–independent mechanisms by which B-cell activation might occur in organ transplantation.
Innate Immunity
Complement split products are bound by complement receptors CD21 (C3b, C3dg, and C3d) and CD35 (C3b and C4b), which then inactivate them to iC3b and iC4b, respectively.19 CD21 expression is limited to B cells and follicular dendritic cells, whereas CD35 is expressed more widely. In crosstalk between complement and the adaptive immune system, opsonized alloantigen activates the classical complement pathway and may engage the complement receptor, CD21/CD35, to form a coreceptor with the BCR, thereby lowering the threshold for B-cell activation.20,21
Cytokine Stimulation
Part of the TNF family of cytokines, the B-lymphocyte stimulation (BLyS) family of ligands (BLyS, B cell activating factor [BAFF]-B cell–activating factor) and receptors (BAFFr-B-cell activating factor receptor; present on each B cell, TACI transmembrane activator and calcium modulator and cyclophilin ligand interactor; present on memory B cells and BCMA-B cell maturation antigen; present on plasma cells) are costimulators of B cells and necessary for the survival and maturation of B cells to plasma cells.22 A proliferation-inducing ligand (APRIL), like BAFF, is produced as a transmembrane protein, and then proteolytically cleaved to be released in soluble form.23 In mice, BAFF has been shown to independently promote TLR7/9 expression and autoantibody production, in the absence of T cells.24
Other mechanisms for T cell–independent B-cell activation include BCR recognition of polysaccharides and costimulation with TLRs.25 Although this is predominantly of importance in bacterial immunity, it may be of relevance in ABO-incompatible or xenotransplantation.
GC DEVELOPMENT
The GC is a specialized structure containing proliferating antigen-specific B cells that promote antigen-dependent affinity maturation, which is located within B-cell follicles of secondary lymphoid organs. The GC response arises from the interaction between Tfh and B cells. It is now known that Tfh cells and their interaction with B cells are required for GC formation, the generation of long-lasting antibody production,26–28 and high-affinity antibody generation.
Tfh and B Cell Interaction
Tfh cells upregulate the chemokine receptor 5 (CXCR5) while downregulating chemokine receptor 7. This facilitates migration toward B-cell follicle.29,30 Additional surface molecules including PD-1, inducible T cell costimulator, and SLAM family of receptors are highly expressed by Tfh cells (Figure 2). These molecules are now used for the identification of Tfh cells. It is now known that BCL-6 is the key transcription factor for Tfh cell differentiation.15–17 CXCR5+PD-1hiICOS+BCL-6+ CD4 T cells also have been shown to localize to the GCs. Taken together, Tfh cells are a terminally differentiated effector T cell lineage.31
FIGURE 2.
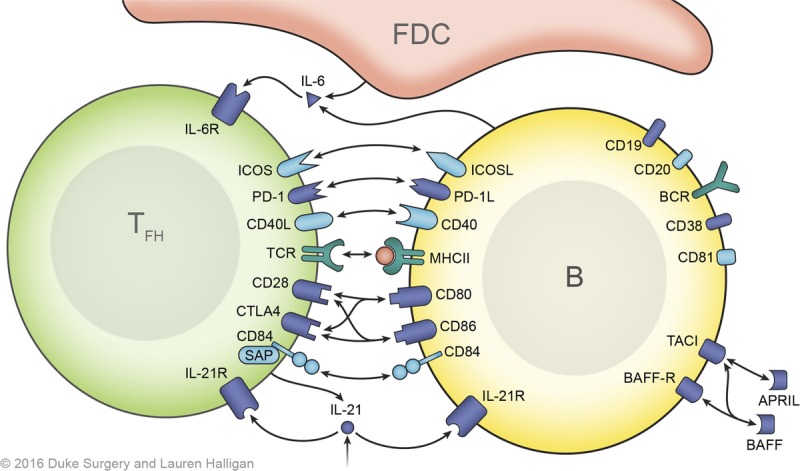
T-B cell interactions and signals. The initial B cell activation signal is provided by cognate BCR-alloantigen interaction and requires the CD19, CD21, CD81 complex for an optimal signaling. Cognate Tfh cells provide costimulation after presentation of antigenic peptides in MHC class II by the B cells in the GC. T-B interactions, especially ICOS-ICOSL, CD40-CD40L, and CD28-B7 interactions, are central to this process. Members of the SLAM (Signaling lymphocytic activation molecule) family of receptors, particularly CD84, are also important for stable T-B cell interactions, which also contribute to Tfh cell differentiation through SAP (SLAM associated protein)-mediated signaling. Cytokines, IL-6 and IL-21 contribute to Tfh cell differentiation via upregulation of BCL-6. The crosstalk between T and B cells in the GC is bidirectional mediated by ligand-receptor interactions, soluble mediators (cytokines). Biologics targeting these signals can modulate posttransplant humoral response.
As shown in Figure 1, the T cells located at the T-B border are considered to be precursors of Tfh cells, which briefly express some of the Tfh cell–associated markers. It is believed that these cells initiate primed B-cell proliferation and differentiation. Only once B cells receive the described activation signals, will they proliferate and differentiate along the both follicular and extrafollicular pathway.32 Although some fully differentiate into short-lived plasma cells and memory B cells (via the extrafollicular pathway), others move through the dark zone (centroblasts; CXCR4+CD83+CD86+) into the light zone of the GC via chemokine receptor CXCR5 (binding its ligand CXCL13; Figure 1). These B cells (known as centrocytes; CXCR4−CD83+CD86+) have already experienced somatic hypermutation and interact with follicular dendritic cells and Tfh cells. Selected B cell clones, via the cognate interaction with Tfh cells, differentiate into plasma cells (short-lived and long-lived) and memory B cells or, alternatively, some migrate back to the dark zone for further proliferation.8,33 It is notable that memory B cells emerge earlier than plasmablast with less Tfh cell requirement.34,35 Clonal expansion of these cells, to become the hyperplastic GC, occurs as result of their interaction with cognate-Tfh (Tfh:CXCR5+ PD-1hiICOS+ CD4+) cells, via CD40 signaling and IL-21.36 Further costimulation signaling, via ICOS and PD-1 interactions with ICOS-L and PD-L1/L2 on the B cell provides optimal signals for hyperplastic GC formation and antibody production.37–39 Defects of these surface molecules in human have been shown to result in impaired GC formation and an improper antibody response.37,38,40,41
CD28 and CTLA4 interactions to CD80 or CD86 on B cells are involved in both positive and negative regulation of B cells. Signaling of CD28 via CD86 promotes B-cell proliferation and Ig class switching, whereas interaction of CD28 to CD80 inhibits this response.42,43 CTLA4 counteracts the CD28 signal by binding to CD80/86 with higher affinity.44 In transplantation, the efficacy of CTLA4-Ig (Belatacept) to suppress the antidonor humoral response has been demonstrated.45–48 However, like to ICOS and PD-1, it is still unclear if this costimulation signal is critical for T cell-B cell cooperation directly, or whether the mode of action is predominantly by limiting Tfh cell or B cell function/proliferation.
The costimulation interactions described between T cells and B cells require additional binding to provide physical stabilization. Initially, this is by adhesion molecules, LFA-1 and ICAM; however, subsequent maintenance of the T-B conjugation is dependent on SLAM-associated protein interactions, including CD84.49 Targeting these interactions with an anti-LFA-1mAb also significantly reduces both alloantibody production as well as the GC formation (manuscript in preparation). Additionally, CD84 deficiency has been shown to result in an impaired humoral response in mice.49
Within the GC, cytokines (IL-6 and IL-21) remain crucial for further B-cell differentiation.50 Tfh cells are the major source of IL-21. Both Tfh cells and GC-B cells express IL-21 receptors. IL-21 signaling in GC-B cells promotes proliferation, plasma cell differentiation, and antibody production.51–53 IL-21 signaling also stimulates Tfh formation.54,55 IL-6 provided by dendritic cells (DC, including follicular DC) induces IL-21 production from Tfh cells.56 Interestingly, IL-6 production from DC is enhanced after IFN-γ activation.57 The role of IFN-γ may be very limited because the BCL-6 represses IFN-γ expression. It also has been shown that Tfh cells produce less IFN-γ.15,58 From the autoimmune literature, it would appear that IFN-γ promotes BAFF production from myeloid cells that may be indirectly involved in the GC response.59 Goenka et al60,61 elegantly showed that within the GC, a further role of BAFF produced by Tfh cells for GC B cells. In transplantation, the role of BAFF is a developing field and its effect on Tfh within the GC remains to be fully elucidated.
Distinct T Cell Subtypes in the GC
Tfh cells are known to be the key Th cell phenotype; however, the mechanism of Tfh cells in GC homeostasis is incompletely understood. Here, we briefly outline the other known T cell phenotypes involved in the process:
T Follicular Regulatory Cell
The selection of GC-B cells expressing high-affinity variants of BCR via cognate Tfh cells prevents an irrelevant GC response. However, this does not explain the control of possible cross-reactive GC-B cells which can recognize both target and self-antigen. More recently, CXCR5-expressing regulatory T (Treg) cells (FoxP3+) (T follicular regulatory cells [Tfr]) have been identified in the GC.62–64 Like Tfh cells, the high surface expression of CXCR5 is a useful marker to identify the subset of Tfr cells involved in regulation of GC in secondary lymphoid organ.65 The Tfr cells share a similar genetic profile to Tfh cells and Treg cells including BCL-6, FoxP3, CXCR5, PD-1, ICOS and CTLA-4. An important distinguishing feature is that the Tfr cell does not express certain molecules necessary for Bc-ell help, such as CD40L, IL-4, and IL-21.63 A lack of Tfr cell promotes increased Tfh, GC-B cells, and antibody-secreting cells.62 However, the relevance of these cells in transplantation is not fully elucidated, and it is still unclear how these Tfr cells affect the GC response.
CD8 T Cells in Humoral Response
The other notable cell population involved in the GC response is CD8 T cells. It has been shown that a fraction of the CD8 T cell population suppresses B cell response.66 More recently, CD8+ Treg cells which are crucial for the maintenance of self-tolerance in the GC were identified in autoimmune disease models.67 These suppressive CD8 T cells do not share common developmental pathway with CD4 Treg cells. Unlike Tfr cells, CD8 regulatory cells in the GC express ICOS ligand (ICOS-L) but they do not express ICOS or PD-1.68 However, they are both dependent on the transcription factor Helios to maintain suppressive phenotype.69 In transplantation, Bumgardner and colleagues70 also observed the role of CD8 T cells in alloantibody reduction. In their model, allo-primed CD8 T cells targeted allo-primed B cells via perforin and FasL in an antigen-specific manner.71
KINETICS OF GC DEVELOPMENT AFTER ORGAN TRANSPLANTATION
The GC response after organ transplantation has not been fully described, this is mostly due to scarcity of posttransplant samples of secondary lymphoid samples in human patients. The GC is, however, an important site of interaction between T cells and B cells, which has been investigated more thoroughly in animal models. In nonhuman primate model, we have traced the in vivo GC response with immunohistochemical analysis. The GC-associated Tfh cell has been also visualized via PD-1 and their functional cytokine, IL-21 in rhesus macque (Figure 3).72,73 Interestingly, we did not see an impressive development of GC after skin transplantation despite DSA elevation (data not shown). It is partially because the baseline level of GC response is high in monkeys because they are not germ-free animals. The robust Th1 immune response, especially IL-2, induced by skin transplantation (without immunosuppression) may also limit the Tfh cell differentiation.18 IL-2 represses Tfh cell differentiation by upregulating BLIMP-1, a transcription factor that is mutually antagonistic to BCL-6, which is critical for Tfh cell differentiation.
FIGURE 3.
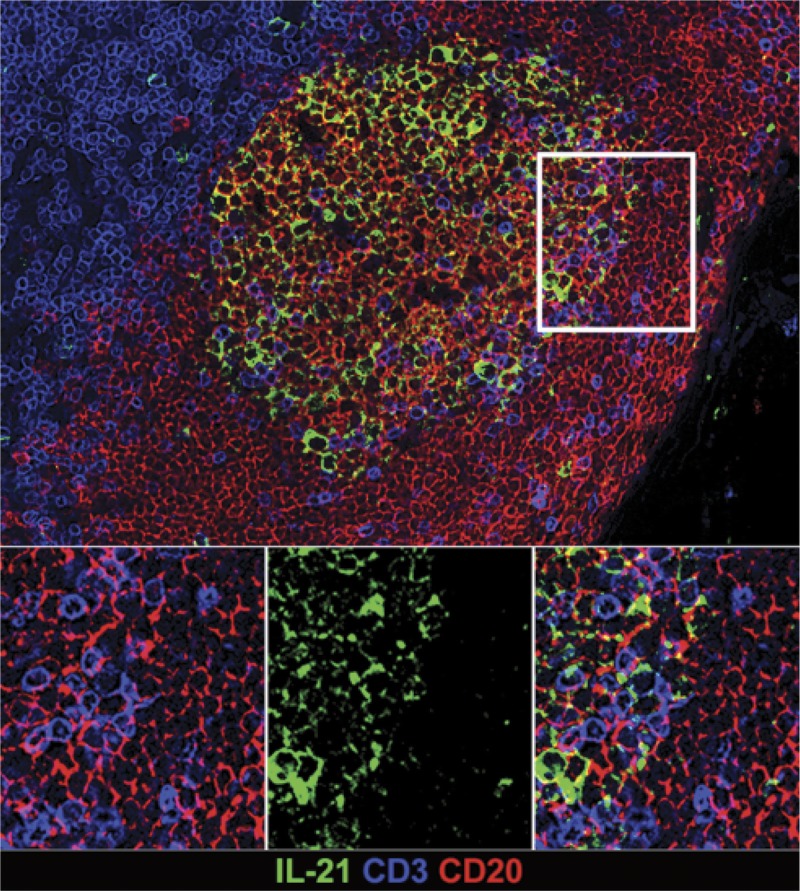
In situ IL-21 staining in the GC. IL-21 expression is increased in hyperplastic follicles during AMR. Representative immunofluorescence image of GC staining with CD3 (blue), CD20 (red), IL-21 (green) in lymph node of rhesus macaque.
We have developed an NHP AMR model which mimics clinical observations seen with alemtuzumab. Cai et al74 found that 42% of their alemtuzumab-treated and sirolimus-treated renal transplant recipients developed HLA antibodies, 60% of which were donor-specific. Other studies have observed more frequent occurrences of acute AMR after renal transplantation with alemtuzumab induction.75–78 We treated our monkeys with CD3-Immunotoxin, Alefacept (LFA-3-Ig;anti-CD2 mAb), and tacrolimus.79 Comparing this combination therapy with those monkeys treated with CD3-Immunotoxin and tacrolimus alone, we found that Alefacept treatment resulted in significant production of de novo antibody. Because the Kirk group80,81 showed that higher CD2 expression on activated (or memory) CD8 T cells, our findings,45,79,82 support Bumgardner’s work on the role of CD8 T cells in GC regulation, as previously described.
We have also assessed T cell depletion in different immune-compartments and GC of the secondary lymphoid organs after CD3 immunotoxin. We found that GC size and frequency are highly correlated to both DSA production and AMR score.45,82 In the AMR-inducing model, T cell depletion allowed us to study the reconstruction of GC after eradication due to the depletional therapy. GC areas containing proliferating B cells (Ki67+) were measured using Hoechst staining of nuclei, demonstrating markedly less focal staining than the smaller marginal B cells.72,73 In kidney transplantation, we found that T cell depletion results in elimination of GC in the first week after transplantation (Figure 4). There was also an absence of visible GC in the spleen and Peyer patches within the gut (Figure 5). In addition, systemic T cell depletion resulted in a reduction in the circulating number of B cells, despite the B-cell follicle remaining intact. This suggests that, in the nondepleted steady state, B cells from the GC contribute to circulating B cell numbers.45,82
FIGURE 4.
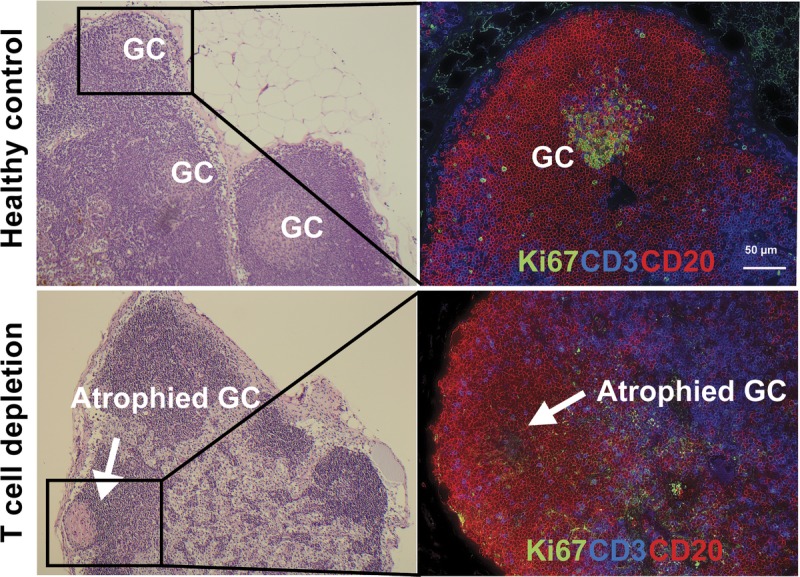
Eradication of GC in the lymph node after T cell depletion. Representative haematoxylin & eosin (left) and immunofluorescence image (right) of GC staining with CD3 (blue), CD20 (Red), Ki67 (green) in lymph node of rhesus macaque. Upper panels show sections of LN from healthy control with GCs, and lower panels show atrophied GC (white arrow) after T cell depletion.
FIGURE 5.
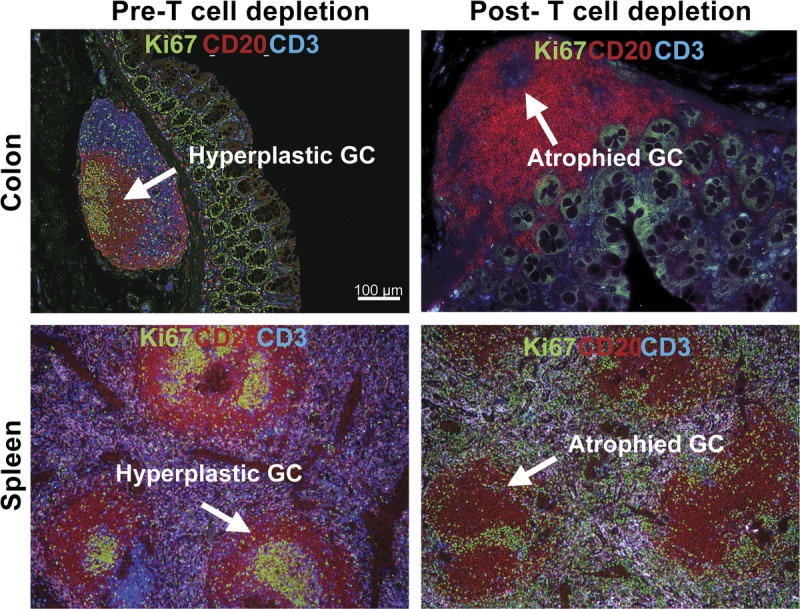
Disrupted GC after T cell depletion in spleen and colon. Representative IHC panels show sections of colon (upper panel) and spleen (lower panel) from before and after T cell depletion with large lymphoid aggregation (Peyer patch) stained with Ki67, CD20, and CD3 antibodies. A, Similar to the lymph node, Peyer patch and spleen comprises T cell area and B-cell follicle with GC containing large proliferating B cells. B, GC in Peyer patch in the descending colon and spleen of rhesus macaques were disrupted after T cell depletion.
Immunohistochemistry within the B-cell follicles this early after transplantation with T cell depletion showed no evidence of T cell or proliferating B cells (Ki67). The GC-like structures without cell components may represent the effect of rapid T cell depletion on the GC (Figures 4 and 5). However, at this stage, although very few T cells were identified in the B-cell follicle, residual T cells in the medulla appear more evident.
By 2 to 4 weeks posttransplantation, the GC shows evidence of rapid reconstruction. Within the medulla, there is clear evidence of T cell presence, with small numbers of T cells detectable in the B-cell follicle and GC. It would appear that, after T cell depletion, repopulation of the T cell occurs in the LN before the circulation. Supporting this finding, Kirk et al83 also showed that any remaining T cells after Alemtuzumab treatment are found in the lymph node, specifically the mantle zone. Of note in the NHP model, T cells are not fully removed by T cell depletion.79
In a mouse model of chronic AMR, using human CD52 transgenic mice,84 after alemtuzumab treatment, we also demonstrated eradication of the GC with residual T cells in the LN (MS in preparation). This mouse model also showed that T cells in the LN were more resistant to those in spleen (data not shown). Like our NHP observations, it is notable that there was more remaining T cells and faster repopulation in the lymph node compartments compared with spleen. We currently do not know what the driving force of GC reconstruction is, under the current immunosuppressive regimen. We speculate that CNI-based regimen does not efficiently suppress Tfh cell during homeostatic repopulation. It might be due to their great efficacy on suppressing Th1 response. It is also possible that Tfh cells are relatively spared by depletional induction due to their physical location (surrounded by densely populated B cells in GC). These remnant Tfh cells (or memory Tfh cells) could repopulate relatively faster under the lack of Th1 immune response. Currently, we are further analyzing these T cells in the GC to discover what type of T cell subpopulation they represent.
By 30 days after transplantation, recipients treated with CD3-IT with either tacrolimus or rapamycin showed GCs which are similar to the naïve animals. In the spleen and gut, GC reconstruction was negligible. This suggests that it is the residual T cells, after depletion, which contribute to GC reconstruction. In our AMR model, with considerable immunosuppression, we found that in conjunction with elevated DSA, the rebound increase in GC size and frequency at later time points exceeded baseline. This finding suggests that T-B interactions in the GC can be completely eradicated by current cytolytic induction. However, repopulating Tfh cells within the GC provide B cell help, despite potent immunosuppression to control T cell–mediated rejection.
ECTOPIC GC (TERTIARY STRUCTURE) IN TRANSPLANTATION
In addition to the changes within hyperplastic GC, it is known that GC-like structures develop in transplanted organs (kidney, lung transplantation). These ectopic intragraft GCs (also known as tertiary lymphoid organs) appear during chronic rejection; however, their role in graft rejection is debated. Originally, it was believed that they were related to rejection response by providing both a humoral response85 and/or allo-T cell priming86 directly at the site of alloantigens. Clinically, it is has also been suggested that ectopic GCs correlate with poor survival.87 However, these ectopic GCs have also been found in transplanted organs with no evidence of dysfunction.88
In nonhuman primate kidney transplantation model, we have made similar demonstrations of ectopic GCs in kidney transplant recipients in whom immunosuppressive drug therapy has been discontinued after initial long-term treatment. We observed that the tertiary GC structure developed rapidly in conjunction with graft rejection. However, even with well-developed T cell and B cell zones, there was no evidence of a B-cell clonal expansion (data not shown).
We also found evidence of ectopic GC within a well-functioning graft after drug discontinuation. These ectopic GCs are composed of T and B cells with often hyperplastic GC (showing B cell clonal expansion), although there was no evidence of circulating DSA. As found in other lymphoid organs including lymph nodes, spleen and Peyer patch (Figures 4 and 5), GCs in the tertiary lymphoid aggregates are clearly identifiable based on their typical architecture. Staining of ectopic GC within the graft showed Ki67 + B cells (Figure 6). We also observed an inverse BAFF staining pattern which suggests a consumption of BAFF in the GC-B cells in the ectopic GC—a finding which was also demonstrated in the hyperplastic GCs (data not shown). It remains unclear which factors mediate the differential GC morphologies in the graft after discontinuation of immunosuppression. Further analysis of T and B cells in the ectopic and hyperplastic GCs in well functioning (tolerant) nonhuman primate kidney recipient is likely to provide an insight on how these structures regulate the alloimmune response in recipients with long-term graft function and survival.
FIGURE 6.
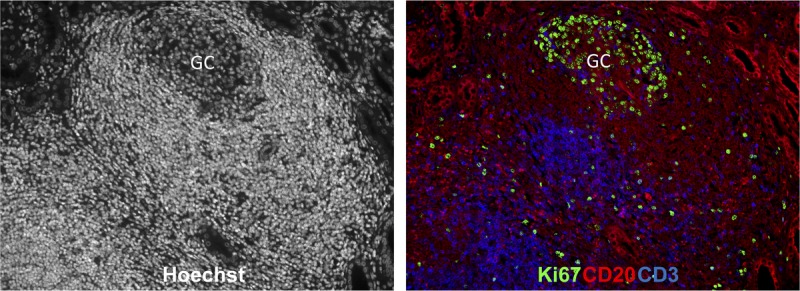
Ectopic GC in well-functioning graft. Representative GC response in the well-functioning kidney graft. Immunological profiles of Hoechst (left), CD20/CD3/Ki67 positive cells (right) within kidney graft section from renal transplant rhesus recipients. The sections were stained with Hoechst dye (white), CD3 (blue), CD20 (red), and Ki67 (green) antibodies.
POSSIBLE DRUG INTERVENTIONS ON T-B CROSSTALK IN TRANSPLANTATION
There are a number of drug therapies which have the ability to influence this crosstalk between T and B cells.
Costimulation Blockade
Costimulation pathway blockade has been in use in preclinical and clinical transplantation for some time.89 Belatacept is a second-generation fusion protein (CTLA4-Ig) which blocks the CD28/CTL4:CD80/86 costimulation axis and has been licensed for use in transplantation as maintenance therapy since 2011. Clinically, a Cochrane review has found that there is no difference in effectiveness of belatacept compared with CNI-based maintenance, with respect to graft rejection or survival, although it is less nephrotoxic than CNI.90 This review however acknowledges that the evidence so far has not fully evaluated the role of belatacept in transplantation, particularly with respect to costimulation blockade resistant rejection. Of pertinent interest, in a recent article based on an alemtuzumab, belatacept, rapamycin-based regimen in human kidney transplantation, it has been shown that, after depletional induction, the rapidly repopulating B-cell phenotype is predominantly naive and regulatory in nature.47
CD40 ligand pathway inhibitors are also currently of interest in preclinical models of transplantation.91 In a case report of liver transplantation in a patient with CD40L deficiency, it was been noted that although B-cell immunity is severely compromised due to CD40L being required for B-cell activation, GC formation and class switching, the lack of CD40L on T cells and NK cells did not result in any differences in T cell and NK-cell responses early after transplantation.92 In NHP models of transplantation, using CD40L blockade (Chi220) resulted in prolonged graft function, and peripheral B-cell depletion, although this was associated with severe systemic CMV infection.93 We have investigated CD40L (2C10) blockade, in conjunction with belatacept and, as described above in our AMR inducing model, and noted decreased DSA production, and suppression of the GC response.45 To date, no human studies of CD40 inhibition in transplantation are available.
Although anti-LFA1 blockade is appealing, Efalizumab (Anti LFA-1 blocking monoclonal antibody) has been used in a phase I/II trial human kidney transplantation, where although the graft rejection rates were low and mild, higher-dose treatment was associated with PTLD.94 Anti-LFA-1 has been also used in combination with belatacept in an NHP model of renal transplantation where it failed to significantly prolong graft survival.95 Efalizumab is also no longer commercially available.
Modulation of B Cell Development Via B-Lymphocyte Stimulator/BAFF or APRIL Pathways
Belimumab (Benlysta) is a fully human IgG1 monoclonal antibody directed against circulating BLyS factor or BAFF which binds soluble human BLyS is currently in clinical use in autoimmune disease.
Atacicept is a fusion protein which targets not only BLyS, but also APRIL pathways. Again, clinical use to date has been reserved to autoimmune indications, and outcomes have been somewhat controversial.96 However, as discussed in our NHP models, in a T cell depletion model, using Taci-Ig, successfully reduced the DSA response.82
SUMMARY
The role of crosstalk between B cells and T cells is increasingly recognized as being an important in the activation of the B cell and development of the humoral response in transplantation. In particular, this crosstalk is vital for the development of the GC. There are multiple pharmaceutical possibilities for intervention, although further studies in preclinical transplant models are necessary to fully evaluate the mechanisms described.
ACKNOWLEDGMENTS
The authors would like to thank Lauren Halligan (Department of Surgery, Duke University) for her contribution creating the illustration in the article.
Footnotes
This work is supported in part by grants: NIH U01 AI074635 (S.J.K.), NIH U19 AI051731 (S.J.K.), and AHA Enduring Hearts Foundation Research Award 15SDG25710165 (J.K.).
The authors declare no conflicts of interest.
S.J.K. and J.K. share the corresponding authorship for this work.
J.K. participated in the concept, article writing, figures drawing. M.M. participated in the concept, article cowriting. E.P, C.B., and J.H. participated in article cowriting. S.J.K. participated in the concept and article writing.
Correspondence: Stuart J Knechtle, MD, Duke Transplant Center, 207 Research Dr., Jones 365, Durham, NC 27710. (stuart.knechtle@dm.duke.edu); Jean Kwun, PhD, Duke Transplant Center, 207 Research Dr., Jones 365, Durham, NC 27710. (jean.kwun@duke.edu).
Kwun et al provide an in-depth review on the T cell subtypes, cytokines and molecules regulating B cell activation and germinal center development, and therapeutic options to control donor-specific B cell responses and antibody production.
REFERENCES
- 1.Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity Nat Rev Immunol 2010. 10236–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwun J, Knechtle SJ. Overcoming chronic rejection—can it B? Transplantation 2009. 88955–961 [DOI] [PubMed] [Google Scholar]
- 3.van Zelm MC, Reisli I, van der Burg M. An antibody-deficiency syndrome due to mutations in the CD19 gene N Engl J Med 2006. 3541901–1912 [DOI] [PubMed] [Google Scholar]
- 4.van Zelm MC, Smet J, Adams B. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency J Clin Invest 2010. 1201265–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thiel J, Kimmig L, Salzer U. Genetic CD21 deficiency is associated with hypogammaglobulinemia J Allergy Clin Immunol 2012. 129801–810.e6 [DOI] [PubMed] [Google Scholar]
- 6.Mitchison A. Latent help to and from H-2 antigens Eur J Immunol 1992. 22123–127 [DOI] [PubMed] [Google Scholar]
- 7.Mitchison NA. T cell–B-cell cooperation Nat Rev Immunol 2004. 4308–312 [DOI] [PubMed] [Google Scholar]
- 8.MacLennan IC. Germinal centers Annu Rev Immunol 1994. 12117–139 [DOI] [PubMed] [Google Scholar]
- 9.Durandy A, Kracker S, Fischer A. Primary antibody deficiencies Nat Rev Immunol 2013. 13519–533 [DOI] [PubMed] [Google Scholar]
- 10.van Zelm MC, Bartol SJ, Driessen GJ. Human CD19 and CD40L deficiencies impair antibody selection and differentially affect somatic hypermutation J Allergy Clin Immunol 2014. 134135–144 [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Yin H, Xu J. Reversing endogenous alloreactive B cell GC responses with anti-CD154 or CTLA-4Ig Am J Transplant 2013. 132280–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haanstra KG, Ringers J, Sick EA. Prevention of kidney allograft rejection using anti-CD40 and anti-CD86 in primates Transplantation 2003. 75637–643 [DOI] [PubMed] [Google Scholar]
- 13.Schmitt N, Morita R, Bourdery L. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12 Immunity 2009. 31158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonard WJ. Cytokines and immunodeficiency diseases Nat Rev Immunol 2001. 1200–208 [DOI] [PubMed] [Google Scholar]
- 15.Yu D, Rao S, Tsai LM. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment Immunity 2009. 31457–468 [DOI] [PubMed] [Google Scholar]
- 16.Johnston RJ, Poholek AC, DiToro D. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation Science 2009. 3251006–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nurieva RI, Chung Y, Martinez GJ. Bcl6 mediates the development of T follicular helper cells Science 2009. 3251001–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ballesteros-Tato A, León B, Graf BA. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation Immunity 2012. 36847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roozendaal R, Carroll MC. Complement receptors CD21 and CD35 in humoral immunity Immunol Rev 2007. 219157–166 [DOI] [PubMed] [Google Scholar]
- 20.Fearon DT, Locksley RM. The instructive role of innate immunity in the acquired immune response Science 1996. 27250–53 [DOI] [PubMed] [Google Scholar]
- 21.Dempsey PW, Allison ME, Akkaraju S. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity Science 1996. 271348–350 [DOI] [PubMed] [Google Scholar]
- 22.Treml JF, Hao Y, Stadanlick JE. The BLyS family: toward a molecular understanding of B cell homeostasis Cell Biochem Biophys 2009. 531–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vincent FB, Saulep-Easton D, Figgett WA. The BAFF/APRIL system: emerging functions beyond B cell biology and autoimmunity Cytokine Growth Factor Rev 2013. 24203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groom JR, Fletcher CA, Walters SN. BAFF and MyD88 signals promote a lupus-like disease independent of T cells J Exp Med 2007. 2041959–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vinuesa CGMICM. Antibody Responses to Polysaccharides. New York: Kluwer Academic; 2003. [Google Scholar]
- 26.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol 2011. 29621–663 [DOI] [PubMed] [Google Scholar]
- 27.Tangye SG, Ma CS, Brink R. The good, the bad and the ugly—TFH cells in human health and disease Nat Rev Immunol 2013. 13412–426 [DOI] [PubMed] [Google Scholar]
- 28.Haynes NM, Allen CD, Lesley R. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1 high germinal center-associated subpopulation J Immunol 2007. 1795099–5108 [DOI] [PubMed] [Google Scholar]
- 29.Kim CH, Rott LS, Clark-Lewis I. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells J Exp Med 2001. 1931373–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ansel KM, McHeyzer-Williams LJ, Ngo VN. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines J Exp Med 1999. 1901123–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linterman MA, Rigby RJ, Wong R. Roquin differentiates the specialized functions of duplicated T cell costimulatory receptor genes CD28 and ICOS Immunity 2009. 30228–241 [DOI] [PubMed] [Google Scholar]
- 32.Lee SK, Rigby RJ, Zotos D. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells J Exp Med 2011. 2081377–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Victora GD, Schwickert TA, Fooksman DR. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter Cell 2010. 143592–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shinnakasu R, Inoue T, Kometani K. Regulated selection of germinal-center cells into the memory B cell compartment Nat Immunol 2016. 17861–869 [DOI] [PubMed] [Google Scholar]
- 35.Weisel FJ, Zuccarino-Catania GV, Chikina M. A temporal switch in the germinal center determines differential output of memory B and plasma cells Immunity 2016. 44116–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chtanova T, Tangye SG, Newton R. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells J Immunol 2004. 17368–78 [DOI] [PubMed] [Google Scholar]
- 37.Good-Jacobson KL, Szumilas CG, Chen L. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells Nat Immunol 2010. 11535–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawamoto S. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut Science 2012. 336485–489 [DOI] [PubMed] [Google Scholar]
- 39.Bauquet AT, Jin H, Paterson AM. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells Nat Immunol 2009. 10167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grimbacher B, Hutloff A, Schlesier M. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency Nat Immunol 2003. 4261–268 [DOI] [PubMed] [Google Scholar]
- 41.Warnatz K, Bossaller L, Salzer U. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency Blood 2006. 1073045–3052 [DOI] [PubMed] [Google Scholar]
- 42.Jeannin P, Delneste Y, Lecoanet-Henchoz S. CD86 (B7-2) on human B cells. A functional role in proliferation and selective differentiation into IgE- and IgG4-producing cells J Biol Chem 1997. 27215613–15619 [DOI] [PubMed] [Google Scholar]
- 43.Suvas S, Singh V, Sahdev S. Distinct role of CD80 and CD86 in the regulation of the activation of B cell and B cell lymphoma J Biol Chem 2002. 2777766–7775 [DOI] [PubMed] [Google Scholar]
- 44.Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses Nat Rev Immunol 2011. 11852–863 [DOI] [PubMed] [Google Scholar]
- 45.Kim EJ, Kwun J, Gibby AC. Costimulation blockade alters germinal center responses and prevents antibody-mediated rejection Am J Transplant 2014. 1459–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Badell IR, Russell MC, Cardona K. CTLA4Ig prevents alloantibody formation following nonhuman primate islet transplantation using the CD40-specific antibody 3A8 Am J Transplant 2012. 121918–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu H, Samy KP, Guasch A. Postdepletion lymphocyte reconstitution during belatacept and rapamycin treatment in kidney transplant recipients Am J Transplant 2016. 16550–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Young JS, Chen J, Miller ML. Delayed cytotoxic T lymphocyte-associated protein 4-immunoglobulin treatment reverses ongoing alloantibody responses and rescues allografts from acute rejection Am J Transplant 2016. 162312–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cannons JL, Qi H, Lu KT. Optimal germinal center responses require a multistage T cell: B cell adhesion process involving integrins, SLAM-associated protein, and CD84 Immunity 2010. 32253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eto D, Lao C, DiToro D, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One. 2011:e17739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Recher M, Berglund LJ, Avery DT. IL-21 is the primary common γ chain-binding cytokine required for human B-cell differentiation in vivo Blood 2011. 1186824–6835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Linterman MA, Beaton L, Yu D. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses J Exp Med 2010. 207353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zotos D, Coquet JM, Zhang Y. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism J Exp Med 2010. 207365–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nurieva RI, Chung Y, Hwang D. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages Immunity 2008. 29138–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vogelzang A, McGuire HM, Yu D. A fundamental role for interleukin-21 in the generation of T follicular helper cells Immunity 2008. 29127–137 [DOI] [PubMed] [Google Scholar]
- 56.Karnowski A, Chevrier S, Belz GT. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1 J Exp Med 2012. 2092049–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clark EA, Grabstein KH, Shu GL. Cultured human follicular dendritic cells. Growth characteristics and interactions with B lymphocytes J Immunol 1992. 1483327–3335 [PubMed] [Google Scholar]
- 58.Oestreich KJ, Huang AC, Weinmann AS. The lineage-defining factors T-bet and Bcl-6 collaborate to regulate Th1 gene expression patterns J Exp Med 2011. 2081001–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harigai M, Kawamoto M, Hara M. Excessive production of IFN-gamma in patients with systemic lupus erythematosus and its contribution to induction of B lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13B J Immunol 2008. 1812211–2219 [DOI] [PubMed] [Google Scholar]
- 60.Goenka R, Scholz JL, Sindhava VJ. New roles for the BLyS/BAFF family in antigen-experienced B cell niches Cytokine Growth Factor Rev 2014. 25107–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goenka R, Matthews AH, Zhang B. Local BLyS production by T follicular cells mediates retention of high affinity B cells during affinity maturation J Exp Med 2014. 21145–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chung Y, Tanaka S, Chu F. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions Nat Med 2011. 17983–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Linterman MA, Pierson W, Lee SK. Foxp3+ follicular regulatory T cells control the germinal center response Nat Med 2011. 17975–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wollenberg I, Agua-Doce A, Hernández A. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells J Immunol 2011. 1874553–4560 [DOI] [PubMed] [Google Scholar]
- 65.Yu D, Vinuesa CG. The elusive identity of T follicular helper cells Trends Immunol 2010. 31377–383 [DOI] [PubMed] [Google Scholar]
- 66.Noble A, Zhao ZS, Cantor H. Suppression of immune responses by CD8 cells. II. Qa-1 on activated B cells stimulates CD8 cell suppression of T helper 2 responses J Immunol 1998. 160566–571 [PubMed] [Google Scholar]
- 67.Kim HJ, Verbinnen B, Tang X. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance Nature 2010. 467328–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim HJ, Wang X, Radfar S. CD8+ T regulatory cells express the Ly49 Class I MHC receptor and are defective in autoimmune prone B6-Yaa mice Proc Natl Acad Sci U S A 2011. 1082010–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim HJ, Barnitz RA, Kreslavsky T. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios Science 2015. 350334–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zimmerer JM, Pham TA, Sanders VM. CD8+ T cells negatively regulate IL-4-dependent, IgG1-dominant posttransplant alloantibody production J Immunol 2010. 1857285–7292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zimmerer JM, Horne PH, Fiessinger LA. Inhibition of recall responses through complementary therapies targeting CD8+ T cell- and alloantibody-dependent allocytotoxicity in sensitized transplant recipients Cell Transplant 2013. 221157–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hong JJ, Amancha PK, Rogers K. Spatial alterations between CD4(+) T follicular helper, B, and CD8(+) T cells during simian immunodeficiency virus infection: T/B cell homeostasis, activation, and potential mechanism for viral escape J Immunol 2012. 1883247–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hong JJ, Amancha PK, Rogers KA. Early lymphoid responses and germinal center formation correlate with lower viral load set points and better prognosis of simian immunodeficiency virus infection J Immunol 2014. 193797–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cai J, Terasaki PI, Bloom DD. Correlation between human leukocyte antigen antibody production and serum creatinine in patients receiving sirolimus monotherapy after Campath-1H induction Transplantation 2004. 78919–924 [DOI] [PubMed] [Google Scholar]
- 75.Knechtle SJ, Pirsch JD, Fechner H. Campath-1H induction plus rapamycin monotherapy for renal transplantation: results of a pilot study Am J Transplant 2003. 3722–730 [DOI] [PubMed] [Google Scholar]
- 76.Willicombe M, Roufosse C, Brookes P. Antibody-mediated rejection after alemtuzumab induction: incidence, risk factors, and predictors of poor outcome Transplantation 2011. 92176–182 [DOI] [PubMed] [Google Scholar]
- 77.Willicombe M, Brookes P, Santos-Nunez E. Outcome of patients with preformed donor-specific antibodies following alemtuzumab induction and tacrolimus monotherapy Am J Transplant 2011. 11470–477 [DOI] [PubMed] [Google Scholar]
- 78.LaMattina JC, Mezrich JD, Hofmann RM. Alemtuzumab as compared to alternative contemporary induction regimens Transpl Int 2012. 25518–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Page EK, Page AJ, Kwun J. Enhanced de novo alloantibody and antibody-mediated injury in rhesus macaques Am J Transplant 2012. 122395–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weaver TA, Charafeddine AH, Agarwal A. Alefacept promotes co-stimulation blockade based allograft survival in nonhuman primates Nat Med 2009. 15746–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lo DJ, Weaver TA, Stempora L. Selective targeting of human alloresponsive CD8+ effector memory T cells based on CD2 expression Am J Transplant 2011. 1122–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kwun J, Page E, Hong JJ. Neutralizing BAFF/APRIL with atacicept prevents early DSA formation and AMR development in T cell depletion induced nonhuman primate AMR model Am J Transplant 2015. 15815–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kirk AD, Hale DA, Mannon RB. Results from a human renal allograft tolerance trial evaluating the humanized CD52-specific monoclonal antibody alemtuzumab (CAMPATH-1H) Transplantation 2003. 76120–129 [DOI] [PubMed] [Google Scholar]
- 84.Kwun J, Oh BC, Gibby AC. Patterns of de novo allo B cells and antibody formation in chronic cardiac allograft rejection after alemtuzumab treatment Am J Transplant 2012. 122641–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thaunat O, Field AC, Dai J. Lymphoid neogenesis in chronic rejection: evidence for a local humoral alloimmune response Proc Natl Acad Sci U S A 2005. 10214723–14728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nasr IW, Reel M, Oberbarnscheidt MH. Tertiary lymphoid tissues generate effector and memory T cells that lead to allograft rejection Am J Transplant 2007. 71071–1079 [DOI] [PubMed] [Google Scholar]
- 87.Moreso F, Seron D, O’Valle F. Immunephenotype of glomerular and interstitial infiltrating cells in protocol renal allograft biopsies and histological diagnosis Am J Transplant 2007. 72739–2747 [DOI] [PubMed] [Google Scholar]
- 88.Brown K, Sacks SH, Wong W. Tertiary lymphoid organs in renal allografts can be associated with donor-specific tolerance rather than rejection Eur J Immunol 2011. 4189–96 [DOI] [PubMed] [Google Scholar]
- 89.Snanoudj R, Zuber J, Legendre C. Co-stimulation blockade as a new strategy in kidney transplantation: benefits and limits Drugs 2010. 702121–2131 [DOI] [PubMed] [Google Scholar]
- 90.Masson P, Henderson L, Chapman JR. Belatacept for kidney transplant recipients. Cochrane Database Syst Rev. 2014;11:CD010699. doi: 10.1002/14651858.CD010699.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Angin M, Poirier N, Dilek N. Gene transfer of human CD40Ig does not prevent rejection in a non-human primate kidney allotransplantation model Transpl Immunol 2012. 27139–145 [DOI] [PubMed] [Google Scholar]
- 92.Tseng M, Ge S, Roberts R. Liver transplantation in a patient with CD40 ligand deficiency and hyper-IgM syndrome: clinical and immunological assessments Am J Transplant 2016. 161626–1632 [DOI] [PubMed] [Google Scholar]
- 93.Pearson TC, Trambley J, Odom K. Anti-CD40 therapy extends renal allograft survival in rhesus macaques Transplantation 2002. 74933–940 [DOI] [PubMed] [Google Scholar]
- 94.Vincenti F, Mendez R, Pescovitz M. A phase I/II randomized open-label multicenter trial of efalizumab, a humanized anti-CD11a, anti-LFA-1 in renal transplantation Am J Transplant 2007. 71770–1777 [DOI] [PubMed] [Google Scholar]
- 95.Anderson DJ, Lo DJ, Leopardi F. Anti-leukocyte function-associated antigen 1 therapy in a nonhuman primate renal transplant model of costimulation blockade-resistant rejection Am J Tranplant 2016. 161456–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cogollo E, Silva MA, Isenberg D. Profile of atacicept and its potential in the treatment of systemic lupus erythematosus Drug Des Devel Ther 2015. 91331–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]