

Abstract
Oxygen is supplied as supportive treatment for patients suffering from acute respiratory distress syndrome. Unfortunately, high oxygen concentration increases reactive oxygen species (ROS) generation, which causes DNA damage and ultimately cell death in the lung. Although 8-oxoguanine-DNA glycosylase (OGG-1) is involved in repairing hyperoxia-mediated DNA damage, the underlying molecular mechanism remains elusive. Here, we report that ogg-1 deficient mice exhibited a significant increase of pro-inflammatory cytokines (TNF-α, IL-6 and IFN-γ) in the lung after being exposed to 95% oxygen. In addition, we found that ogg-1 deficiency downregulated (macro)autophagy when exposed to hyperoxia both in vitro and in vivo, which was evident by decreased conversion of LC3-I to LC3-II, reduced LC3 punctate staining, and lower Atg7 expression compared to controls. Using chromatin immunoprecipitation assay, we found that OGG-1 associated with the promoter of Atg7, suggesting a role for OGG1 in regulation of Atg7 activity. Knocking down OGG-1 decreased the luciferase reporter activity of Atg7. Further, inflammatory cytokine levels in MLE-12 cells were downregulated following autophagy induction by starvation and rapamycin treatment and upregulated when autophagy was blocked using 3-methyladenine and chloroquine. atg7 KO mice and Atg7 siRNA-treated cells exhibited elevated levels of phospho-NF-κB and intensified inflammatory cytokines, suggesting that Atg7 impacts inflammatory responses to hyperoxia. These findings demonstrate that OGG-1 negatively regulates inflammatory cytokine release by coordinating molecular interaction with autophagic pathway in the hyperoxia-induced lung injury.
Keywords: DNA repair, lung injury, NF-κB, pathway, reactive oxygen species (ROS), oxygen toxicity
Introduction
Treatment of acute respiratory distress syndrome (ARDS), a critical clinical condition, is focused on treating the underlying inciting trigger, such as infection (e.g., pneumonia or sepsis). In addition, supportive care includes lung-protective ventilation with lower tidal volumes and conservative fluid management (1). However, prolonged exposure to high oxygen concentration dramatically increases the risk of lung injury by inducing reactive oxygen species (ROS). ROS accumulation can mediate acute and chronic lung injury by causing DNA damage (2) and inflammation (3, 4). To date, two distinct forms of DNA damage, base adduction and the phosphodiester backbone disruption, are reported to be induced by excessive oxygen exposure both in vitro and in vivo (5).
As the initiation protein in base excision repair (BER), 8-oxoguanine DNA glycosylase (OGG-1) plays an indispensable role in directly recognizing DNA damage sites and in recruiting other DNA repair partners to the damage sites (5, 6). OGG-1 has been shown to prevent the accumulation of oxidative DNA damage (6). In addition, OGG-1 is implicated in the regulation of inflammation, and ogg-1 knockout (KO) mice exhibited vulnerability to Pseudomonas aeruginosa-induced organ dysfunction, neutrophil infiltration and oxidative stress (7, 8). However, the role and mechanism that OGG-1 plays in hyperoxia-induced inflammatory responses remain to be addressed in animal models.
Autophagy is essential for various cellular events, such as cell survival, differentiation, development and homeostasis. While impaired autophagy is implicated in a number of diseases, such as cancer, Crohn’s disease (9, 10), and inflammatory conditions (11). To date, more than 30 autophagy-related (Atg) genes have been reported in yeast. It is generally believed that all of the core Atg proteins are important for autophagosome formation (12). For instance, loss of Atg-7 (an E-1 like activating enzyme) fails to influence autophagy during Drosophila intestine cell death (13). In addition, the enhanced apoptosis and inflammation in the liver tissue of beclin1-mutant mice facilitate carcinoma progression (14). These reports suggest an inhibitory role of autophagy in inflammatory responses. Furthermore, it is reported that OGG-1 plays a role in the regulation of the inflammatory response. However, whether or not a relationship exists between OGG-1 and autophagy in inflammatory response is unknown. Nuclear factor-κB (NF-κB), a critical transcription factor involved in a spectrum of inflammatory responses (15, 16), is implicated in the negative regulation of inflammation by autophagy. IκB kinase could be degraded through an autophagy pathway (16). Further, beclin1 ablated mice exhibited apoptosis, NF-κB activation and increased TNF-α production (14). These findings suggest that autophagy inhibits inflammatory responses by regulating NF-κB signaling pathways.
Although studies indicate that the turnover of OGG-1 may be regulated by autophagy (17), a direct link between OGG-1 and autophagy in hyperoxia-induced toxicity remains unknown. Here we propose that OGG-1 may alleviate hyperoxic toxicity by decreasing inflammation, which is dependent on autophagy. Our data demonstrate that OGG-1 may inhibit inflammatory responses by influencing Atg7 activity. The current study elucidated the interaction between OGG-1 and Atg-7, and the regulatory role in inflammatory responses during hyperoxia-induced injury.
Materials and Methods
Animals
ogg-1 KO mice were generated by Dr. D. Barnes (18) and were provided by Dr. Susan Ackerman at the Jackson laboratory (18, 19). atg7 KO mice (in a C57BL/6J background) were kindly provided by Dr. Youwen He (Duke University). Mice were bred in the animal facility at the University of North Dakota, and the animal experiments were approved by the institutional animal care and use committee (IACUC) at UND. Mice were placed in cages inside an airtight Plexiglas chamber (55×40×50 cm) and exposed to 95% O2 (throughout this manuscript) at different time points (room air for controls). Oxygen concentrations were monitored with a mini Oxygen Ted 60T meter (Teledyne Analytical Instruments, City of industry, CA). O2 tension in cell culture medium below was 647 ± 9 mmHg determined as described (20). After bronchoalveolar lavage (BAL), the trachea and lung were obtained for cell biology assays or fixed in 10% formalin for histological analysis (21, 22).
Cells
Mouse alveolar macrophages (AM) were isolated by bronchial alveolar lavage (BAL) as previously described (23, 24). Murine lung epithelial cell line (MLE-12) and murine alveolar macrophage cell line (MH-S) were obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained as previously reported (24).
Cell transfection
Cells were transfected with OGG-1 siRNA (Santa Cruz Biotechnology, Santa Cruz, CA), Atg7 siRNA (Invitrogen, Carlsbad, CA) using LipofectAmine 2000 reagent (Invitrogen) in serum-free DMEM medium following the manufacturer’s instructions. The cells were collected after 24 h of transient transfection to evaluate the expression of respective genes (23).
Chromatin immunoprecipitation (ChIP)
Briefly, 2×107 MLE-12 cells were cross-linked in 20 mL DMEM medium containing 1% final HCHO at room temperature for 5 min. The reaction was stopped by adding 2 ml glycine (2M) and incubated for 5 min at room temperature. The cells were washed two times with ice-cold 1×PBS and collected in 1 mL 1×PBS containing fresh protease inhibitor cocktail. Cell pellets were precipitated by centrifugation at 700 g for 5 min at 4°C, resuspended in 1 mL lysis buffer (0.5% SDS; 20 mM Tris-HCl, pH 8; 2 mM EDTA; 0.5 mM EGTA; 1 mM PMSF and protease inhibitor cocktail). About 1 mL aliquot of lysate was sonicated using the Covaris S220 Focused-ultrasonicator (Low cell program: Output=105, Duty cycle=2) for 10 min. To preclear the chromatin, the sonicated cell suspension was diluted 10-fold with dilution buffer and incubated with 60 µL salmon sperm DNA/protein G Agarose 50% Slurry for 1 h with rotation at 4°C. The precleared chromatin was incubated with 2 µg of polyclonal anti-OGG-1 (Thermo) or normal IgG (controls) overnight. The samples were left incubated with 60 µL protein G Agarose for 1 h at 4°C. The unbound protein was sequentially washed using standard low salt, high salt, lithium chloride and TE washes, and DNA was eluted and crosslinks reversed overnight at 65°C. DNA was isolated following treatment with Proteinase K and phenol extraction, and diluted in 150 µL of water. Quantitative real-time PCR (qRT-PCR) was performed and amount of protein bound to DNA calculated as percent input using a standard curve of the input DNA. The primer sequences used in qRT-PCR for Atg7 (for both promoter region or coding region) are presented in supplementary table 1. The samples were analyzed by qRT-PCR as described previously (25).
Luciferase reporter assay
Transient transfections were performed with 70% confluent MLE-12 cells plated in 12-well plates by using an Atg7-reporter-luc plasmid (Promega, Madison WI) following the manufacturer’s instruction. 24 h after transfection, the cells were exposed to hypeorxia for 24 h. Cell lysates were subjected to luciferase activity analysis using the Luciferase Reporter Assay System (Promega) (25).
Inflammatory cytokine profiling
After infection, lungs were lavaged five times with 1.0 ml volume of lavage fluid to obtain BAL fluid. The supernatant of the first 0.5 mL of BAL fluid was collected to measure cytokine concentrations using an ELISA kit (eBioscience Co., San Diego, CA). For ELISA assay, 96-well plates (Corning Costar 9018, Corning, NY) were first coated with 100 µL/well capturing antibodies in coating buffer overnight at 4 °C (23). Then the aliquots of BAL fluid were added to the coated wells. After incubating with corresponding detection-HRP-conjugated antibodies, the plate was read at 450 nm and analyzed to determine the cytokine concentrations according to the standards of cytokines.
Western blotting and co-immunoprecipitation assay
Cells or lung homogenates were collected and lysed with RIPA lysis buffer (Thermofisher, Rockford, IL) containing a protease inhibitor cocktail. Protein concentrations were determined by Bio-Rad Protein Assays (Bio-Rad, USA). The individual protein sample was resolved by SDS-PAGE and then electro-transferred onto the nitrocellulose membrane (10). Membranes were blocked for 30 min with 5% skim milk in TBST buffer composed of 50 mM Tris (pH 7.6), 150 mM NaCl and 0.1% Tween-20 and incubated with the primary antibody overnight at 4 °C. Antibodies against Atg7 was purchased from Invitrogen; Anti-IL-6, NF-κB, phospho-NF-κB (ser536, sc-33020), OGG-1, GAPDH, p38, p-p38 (D-8, SC-7973), and β-actin were purchased from Santa Cruz Biotechnology, Inc. GAPDH or β-actin was used as the loading control. After incubation with secondary antibodies, ECL detection reagents (Santa Cruz Biotechnology, Inc.) were used to detect signals (26–30).
For co-immunoprecipitation, the supernatants were incubated with bare protein A/G-Sepharose beads (Invitrogen, Carlsbad, CA) for 2 h at 4 °C and then incubated with anti-Atg7 or OGG-1 antibodies (Santa Cruz Biotechnology) bound to protein A/G-Sepharose beads overnight at 4 °C. The beads were then washed three times with lysis buffer and boiled for 5 min after resuspended in SDS sample loading buffer. Protein expressions were analyzed by Western blotting (31).
Confocal microscopy and indirect immunofluorescence staining
Cells were grown in 3 cm glass-bottomed dishes (MatTek, Ashland, MA). After hyperoxia exposure, the cells were fixed in 4% paraformaldehyde and then permeabilized with 0.2% Triton X-100 in PBS. After incubated with the blocking buffer for 30 min, primary Abs were added to the dishes at a 1:500 dilution in blocking buffer and continued incubation overnight. The next day, cells were washed three times with washing buffer (27, 32). After incubation with appropriate fluorophore-conjugated secondary Abs, the cells were kept in mounting medium before taking images (33). DAPI (Sigma-Aldrich, St. Louis, MO) was used to stain the nucleus briefly before taking images. To obtain high resolution results, the images were taken by a CLSM 510 Meta confocal microscope (Carl Zeiss MicroImaging, Thornwood, NY).
OGG-1 activity assay
A 24-mer oligonucleotide containing 8-oxoguanine at the 10th position (Trevigen) was radiolabeled with γ-32P-ATP. An identical 24-mer without 8-oxoguanine was also provided in parallel reactions. The 10 µl labeling reaction contained 20 pmol of single-strand 8-oxoguanine oligonucleotide, 5 pmol of γ-32P-ATP, T4 polynucleotide kinase, DTT and appropriate kinase buffer (37 °C for 1 h), then diluted in 90 µl TE buffer. 20 pmol of complementary oligonucleotide was then added to form duplex DNA. Activity assays composed of 0.2 pmol of labeled duplex oligonucleotide, 2 µl of 10× REC buffer (100 mM HEPES, pH 7.4, 1 M KCl, 100 mM EDTA, and 1 mg/ml BSA), and 10 µl of protein extracts isolated from both cell types in a total volume of 20 µl (37 °C for 1 h). Bromphenol blue dye was added, and the reaction contents were resolved on 20% denaturing polyacrylamide gel electrophorese.
Comet assay
The Comet assay was performed according to Trevigen manufacturer’s instruction. Briefly, after exposure of cells to hyperoxia, cell suspension were harvested by centrifugation and then resuspended in ice cold PBS (Ca++ and Mg++ free) at a concentration of 1×105 cells/mL. The cell suspension was mixed with molten low melting agarose at a ratio of 1: 10 (vol/vol). 75 µl of the mixture was immediately transferred onto the comet assay slide. After cell lysis at 4° using prechilled lysis solution, slides were treated with alkaline solution (0.3 M NaOH, 1 mM EDTA) for 30 min to unwind the double-stranded DNA. Slides were electrophoresed at 1 vol/cm for 20 min. The images were captured with high resolution by LSM 510 Meta confocal microscope (Carl Zeiss MicroImaging, Inc, Thornwood, NY) after staining with SYBR green dye and the tail lengths indicating DNA damage extent were assessed by the CometScore software (Trevigen).
Nitroblue tetrazolium (NBT) assay
This assay was applied to detect released superoxide. The color of NBT dye changes upon reduction by released superoxide. The dye was yellow in color and after reduction by superoxide forms a blue formazan product. After hyperoxia exposure, the dye was added as previously described (23).
Dihydrodichlorofluorescein diacetate (H2DCF) assay
H2DCF dye (Molecular Probes, Carlsbad, CA) only emit green fluorescence upon reaction with superoxide inside cells. Cells were seeded in the 96 well plates and treated as above. After 1 h incubation with the dye, fluorescence was measured using a fluorescence plate reader (BioTek, Winooski, VT) (7, 34).
Lipid peroxidation assay
After hyperoxia exposure, lungs were homogenized and lysed in 62.5 mM Tris-HCl (pH=6.8) supplemented with a protease inhibitor cocktail (Thermo, Rockford, IL). Malondialdehyde could be measured in a colorimetric assay (Calbiochem, San Diego, CA) according to the manufacturer’s instructions. Then the protein concentration was measured and adjusted to a uniform level for the assay (19).
Myeloperoxidase (MPO) assay
Lung tissue samples were homogenized in 50 mM hexadecyltrimethylammonium bromide, 50 mM KH2PO4, pH 6.0, 0.5 mM EDTA at 1 mL/100 mg of tissue and centrifuged for 15 min at 12,000 rpm at 4°C. Precipitate was collected and 100 mL of reaction buffer (0.167 mg/ml O-dianisidine, 50 mM KH2PO4, pH 6.0, 0.0005% mM H2O2) were added to 100 ml of the sample. Absorbance of each sample in triplicate was read at 460 nm at 2 min intervals (23, 35).
Histopathology analysis
Lung tissues were fixed in 10% formalin in PBS using a routine histologic procedure. The fixed tissue samples were processed for obtaining standard hematoxylin and eosin (H&E) staining and examined for differences in morphology post infection (21, 22).
Immunohistochemistry
Lung tissues were fixed in 4% paraformaldehyde overnight at 4°C, processed, and cut into 4-µm sections. Masked antigens were uncovered by microwaving the sections for 20 min in EDTA (pH = 8) buffer. After the sections were cooled at room temperature, nonspecific binding was blocked by incubating in PBS containing 5% bovine serum albumin for 30 minutes. The sections were incubated with rabbit polyclonal antibody against Atg7 (Santa Cruz Biotechnology) overnight (1:1000). The secondary antibody (Santa Cruz Biotechnology) was applied (1:500) and incubated at 37°C for 1 h. Finally, the slides were visualized using DAB immunostaining under a light microscope (32).
Statistical Analysis
All experiments were performed in triplicate and repeated at least three times. Data were presented as percentage changes compared to controls ± S.D. from the three independent experiments. Group means were compared by student t-test using prism software, and a difference was accepted at p<0.05 (7).
Results
Hyperoxia induces cellular damage, inflammatory responses and increased expression of OGG-1 in vitro and in vivo
To study the role of OGG-1 in hyperoxia-mediated inflammatory processes, we employed in vitro models to evaluate hyperoxia-induced cellular damage and inflammatory response. We chose murine lung epithelial cell line (MLE-12) to examine hyperoxia-induced DNA damage, as it has been widely used as an in vitro model. DNA damage was quantified by a comet assay that detects DNA strand breaks as part of the general migration of nuclear components. The lengths of comet tails in this assay are proportional to the abundance of damaged DNA fragments. We showed that hyperoxia exposure markedly increased DNA strand breakage compared to controls evaluated using confocal laser scanning microscopy (CLSM) (Figure 1A). After exposure to 95% O2 for 24 h, the average tail length was about 40 µm in hyperoxic-exposed cells versus 10 µm in control cells (p<0.01) (Figure 1B). As DNA damage induces expression and activation of DNA repair proteins, we next assessed base excision repair (BER) pathway initiating enzyme OGG-1 after hyperoxia exposure, and found that OGG-1 activity was increased by threefold compared to controls (Figure 1C). Since damaged cells may be associated with inflammatory responses, we then examined levels of pro-inflammatory cytokines (TNF-α, IL-6, and IFN-γ) in MLE-12 cells and found that these cytokines are significantly increased under hyperoxia both at 6 h and 24 h (Figure 1D). To demonstrate the physiological relevance of the inflammatory response, we exposed C57BL/6J mice to 95% oxygen using a Plaxis chamber with controlled oxygen pressure. Our results demonstrate that the levels of TNF-α, IL-6, IFN-γ, as well as OGG-1, were also increased in the lungs of mice in a time-dependent manner under hyperoxia (Figure 1E).
FIGURE 1. OGG-1 responds to hyperoxic DNA damage and inflammation in lung cells.
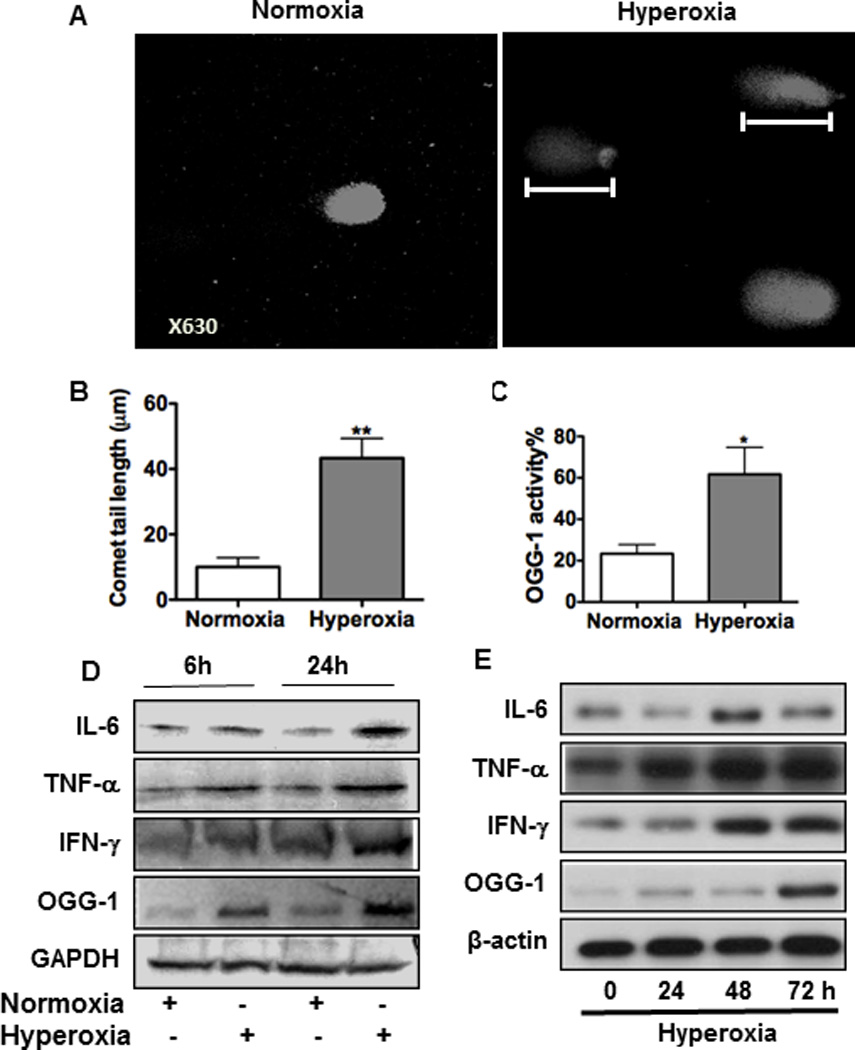
MLE-12 cells were incubated in room air or 95% O2. (A) DNA strand breaks were detected by a comet assay through measuring tail length (indicated by rulers) with confocal laser scanning fluorescence microscopy (CLSM). (B) Tail lengths were markedly increased in lung cells by hyperoxia compared to the control (P<0.001). (C) OGG-1 activity under 24 h hyperoxia was determined by incision enzymatic assay. (D) Increased inflammatory responses in MLE-12 cells after 6 h or 24 h exposure to hyperoxia by immunoblotting analysis. (E) Inflammatory responses in mice increased with exposure time (24, 48 and 72 h) by immunoblotting analysis. Data were representative of three experiments with similar results (student t-test, *p< 0.05, **p< 0.01).
ogg-1 KO mice exhibit heightened inflammatory responses
We previously showed that OGG-1 was increased in hyperoxia-induced inflammatory responses in vitro. We then used ogg-1 KO mice to study the role of ogg-1 in inflammation. We first assessed lung histology and found increased polymorphonuclear neutrophil (PMN) infiltration in the lung of ogg-1 KO mice after oxygen exposure, indicating severe lung injury in these animals (Figure 2A). We further demonstrated that PMN’s in bronchoalveolar lavage (BAL) fluid and blood was higher in ogg-1 KO mice than WT mice (Figure 2B and C). These data suggest that lung injury was increased in ogg-1 KO mice, correlating with the progression of lung injury by oxidation. BAL fluid was collected to measure cytokine concentrations after 48 h exposure to 95% O2 to assess the extent of inflammatory responses. The BAL fluid of ogg-1 KO mice contained significantly increased secretion of inflammatory cytokines including TNF-α, IL-6, and IFN-γ compared to that of WT mice (about 2-fold) as measured by ELISA (Figure 2D–F). Consistent with the secreted levels, these cytokines (TNF-α, IL-6, and IFN-γ) were also found to be increased in ogg-1 KO lung tissues detected by immunoblotting analysis (Figure 2G). Collectively, these data indicate that ogg-1 KO mice manifested intensified inflammatory response under hyperoxia as compared to WT mice.
FIGURE 2. Increased PMN, oxidation injury and inflammatory responses in lungs of ogg-1 KO mice.
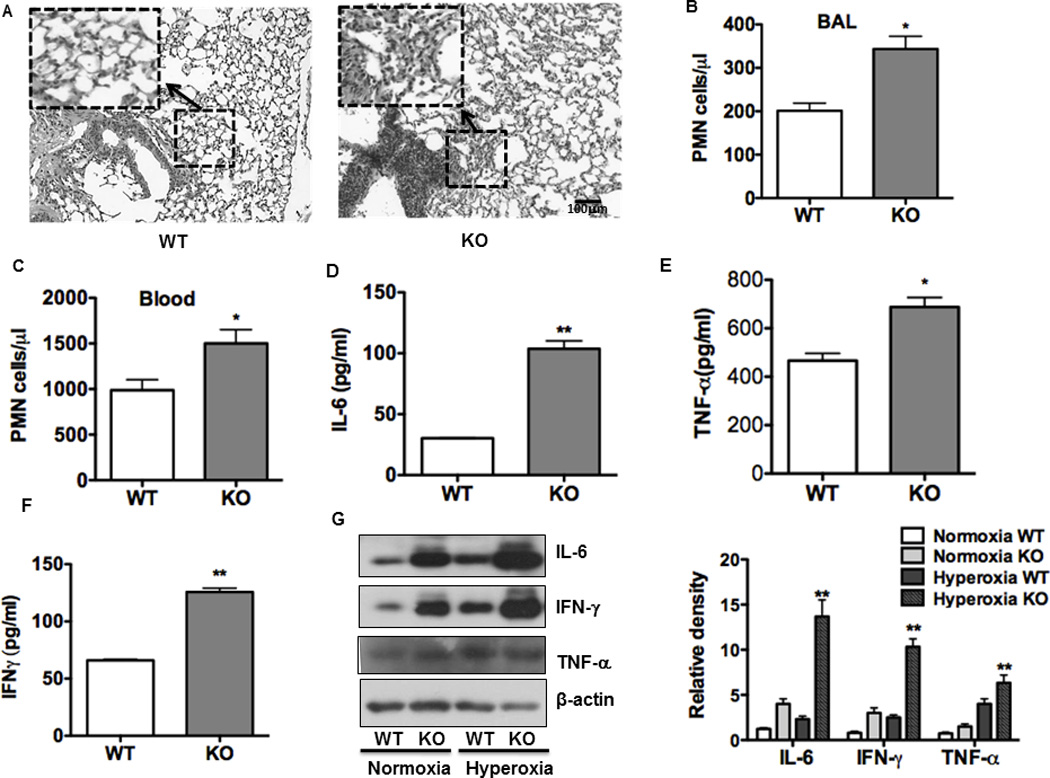
(A) Hyperoxia increased lung injury and inflammation as assessed by H&E staining. (B) and (C) Increased PMN infiltration and an acute inflammatory response were observed in the lung (B) and blood (C) of ogg-1 KO mice compared to WT mice (n=6) following hyperoxia for 48 h. (D)–(F), Increased inflammatory cytokines in BAL fluid of ogg-1 KO mice compared to those of WT mice by ELISA. (G) Increased expression of inflammatory cytokines in lungs of ogg-1 KO mice compared to WT mice by immunoblotting analysis. ogg-1 KO mice and WT mice were exposed to hyperoxia (95%) for 48 h. Gel data were quantified using ImageJ densitometry. Data were representative of three experiments with similar results (student t-test, *p< 0.05, **p< 0.01).
ogg-1 KO mice exhibit decreased autophagy under hyperoxia
Since recent studies indicate that autophagy is involved in lung injury (37), we set out to evaluate whether autophagy is altered in ogg-1 KO mice after hyperoxic exposure. We observed a reduction in both Atg7 and LC3-II (conversion of LC3-I to LC3-II) after exposing ogg-1 KO mice to hyperoxia (Figure 3A). Furthermore, we observed that phosphorylation of NF-κB (p65 subunit), which is a master transcription factor in various cellular processes including inflammation, was increased in ogg-1 KO mouse lungs under hyperoxia (Figure 3A). Importantly, immunohistochemistry analysis based on Atg7 antibody staining showed decreased expression of Atg7 in ogg-1 KO mouse lungs under hyperoxia (Figure 3B), which is a commonly-used method for evaluating antigen expression in lung tissue (36–39). To further confirm these observations in mice, we transfected MLE-12 cells with OGG-1 siRNA and then exposed them to 95% O2 for 48 h. Consistent with the animal data, we found that ogg-1 silencing reduced the expression of OGG-1 protein and resulted in down-regulation of Atg7 expression in lung epithelial cells (Figure 3C). To further differentiate the cell population in mice during ogg-1 deficiency, we next transfected a tandem RFP-GFP-LC3 plasmid as well as OGG-1 siRNA into alveolar macrophage cells (MH-S) and found that the punctate foci in cells were significantly decreased in OGG-1 siRNA silenced cells as detected by CLSM (Figure 3D), indicating that inhibiting OGG-1 also impeded autophagy in macrophages. However, the exposure to hyperoxia still induced significant autophagy (but smaller punctate foci) even in OGG-1 knockdown conditions as quantified at least 30 cells. These data were further validated in alveolar epithelial MLE-12 Cells (Figure 3E). These findings suggest that both macrophages and alveolar epithelia may also be involved in lung pathophysiology due to autophagy suppression.
FIGURE 3. ogg-1 KO mice exhibit impaired autophagy under hyperoxia.
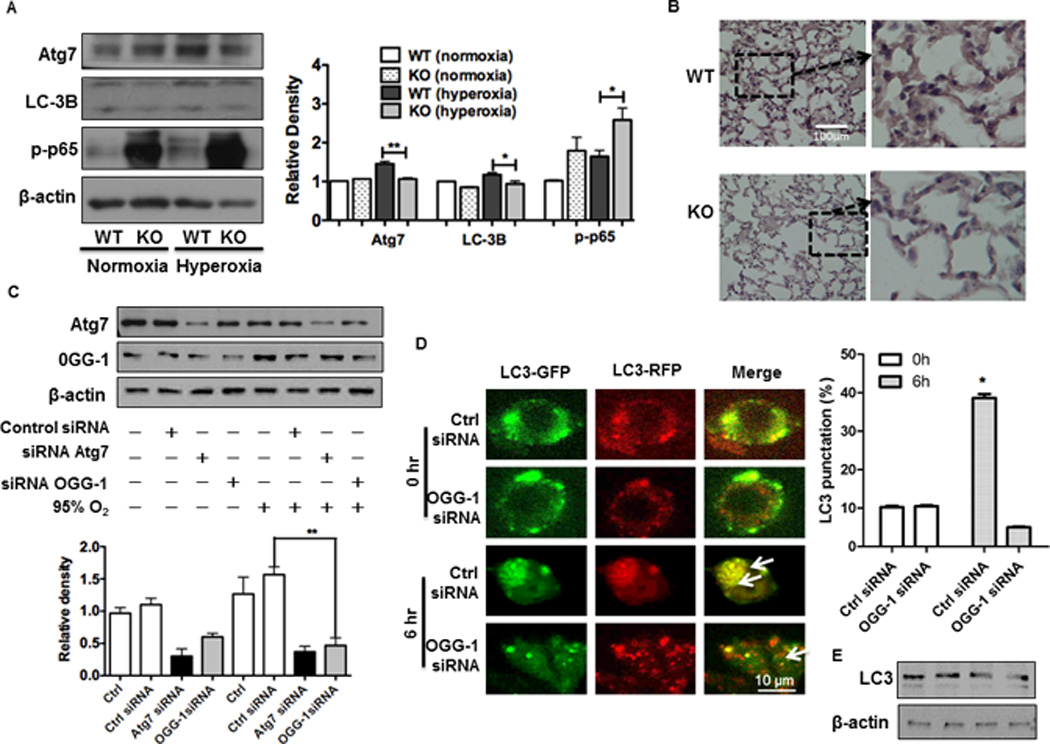
(A) Immunoblotting analysis of Atg7, p-NF-κB and LC3 in lungs of ogg-1 KO and WT mice (n=6) exposed to hyperoxia for 48 h. (B) Decreased expression of Atg7 in ogg-1 KO mice by immunohistochemistry. (C) Decreased Atg7 in MLE-12 cells after 48 h exposure to hyperoxia by immunoblotting analysis. Gel data were quantified using densitometry with Image J. (D) Tandem GFP-RFP-LC3 plasmids and OGG-1 siRNA were transfected to MH-S cells and then cells were exposed to hyperoxia for 6 h. Arrows indicate LC3 puncta. Data were representative of three experiments with similar results (student t-test, *p< 0.05, **p< 0.01). (E) Immunoblot analysis of LC3 with RFP-GFP-LC3/siOGG-1 transfected to MLE-12 cells that were exposed to hyperoxia for 6 h (lane 1: Ctrl siRNA 0 h; lane 2:OGG-1 siRNA 0 h; lane 3: Ctrl siRNA 6 h; lane 4: OGG-1 siRNA 6 h).
Interaction between OGG-1 and Atg7
To further elucidate the relationship between OGG-1 and autophagy, we attempted to probe whether there is an interaction between OGG-1 and Atg7. To our pleasant surprise, we found an interaction between OGG-1 and Atg7 as analyzed using fluorescent microscopy (Figure 4A) and Co-IP assay (Figure 4B), and this interaction was significantly increased under hyperoxia (Figure 4B, panels 3 and 4 [normoxia vs. hyperoxia, OGG-1 IP]). To further substantiate this observation, we used ChIP assay to identify the epigenetic regulation mechanism, and showed that OGG-1 bound to the DNA of Atg7 in MLE-12 cells stimulated with hyperoxia. Quantitative real-time PCR analysis demonstrated that OGG-1 binding to Atg7 DNA was increased compared to controls after exposure to hyperoxia for 24 h, while Atg7 binding to OGG-1 DNA was not so significant (data not shown). We further examined whether there is a direct impact for OGG-1 on Atg7 promoter, and our data suggest that OGG-1 was associated with the promoter of Atg7 (Figure 4C), while showing much less association with promoter upstream or downstream sequences. Furthermore, we may not exclude the possibility that GAPDH (as a functional protein in this case) may bind to OGG-1, but this binding was not influenced by hyperoxia nor as extensively as atg7 in the same condition (Figure 4C). In addition, knocking down OGG-1 with specific siRNA significantly decreased the luciferase reporter activity of Atg7 (Figure 4D). These data strongly indicate that OGG-1 interacts with Atg7 gene promoter to regulate its expression, hence impacting autophagy during hyperoxic lung toxicity.
FIGURE 4. OGG-1 interacts with Atg7.
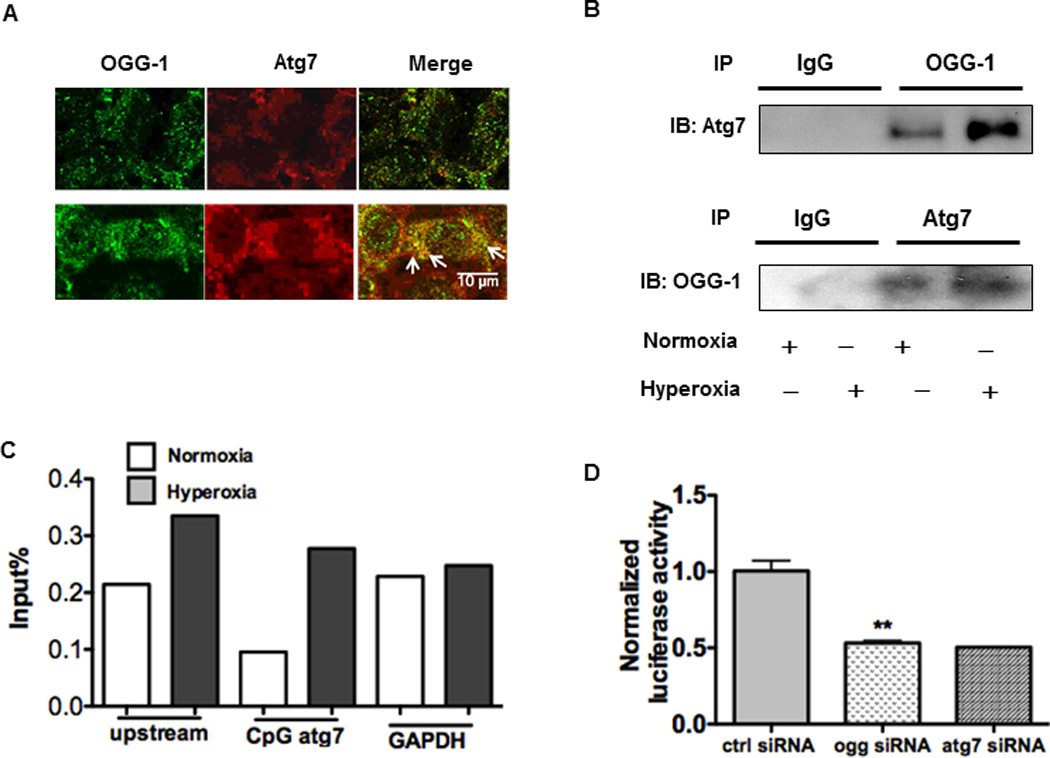
(A) Co-localization of OGG-1 and Atg7 was observed by microscopy (see arrows). (B) Interaction between OGG-1 and Atg7 detected using co-immunoprecipitation (CoIP) assay. IB, immunoblotting. (C) ChIP and real-time PCR analysis of OGG-1 for DNA binding of Atg7 or GAPDH in MLE-12 cells ed under hyperoxia for 24 h. (D) Normalized luciferase activity of a reporter containing the promoter constructs of Atg7 in MLE-12 cells. Cells were transfected with siRNA of OGG-1. 24 h later, the cells were transfected with Atg7-reporter plasmid and then followed by hyperoxia exposure for 24 h. Data were representative of three experiments with similar results (student t-test, *p< 0.05, **p< 0.01).
Atg7 deficiency contributes to intensified inflammatory responses under hyperoxia
To further characterize the effect of autophagy on inflammatory responses under hyperoxia, we next blocked autophagy using Atg7 siRNA or autophagy inhibitors and evaluated the levels of inflammatory cytokines (IL-6, TNF-α) in MLE-12 cells (Figure 5A). Cells were pre-treated with autophagy blockers (3-MA and chloroquine for 4 h) or inducers (4 h starvation or rapamycin) before hyperoxia exposure. We found that autophagy blockers significantly inhibited hyperoxia-induced autophagy, leading to stronger inflammatory responses. However, autophagy inducers significantly dampened inflammatory responses (Figure S1). These finding indicate that autophagy negatively regulates inflammatory responses during hyperoxia.
FIGURE 5. Atg7 deficiency contributes to intensified inflammatory responses under hyperoxia in vitro and in vivo.
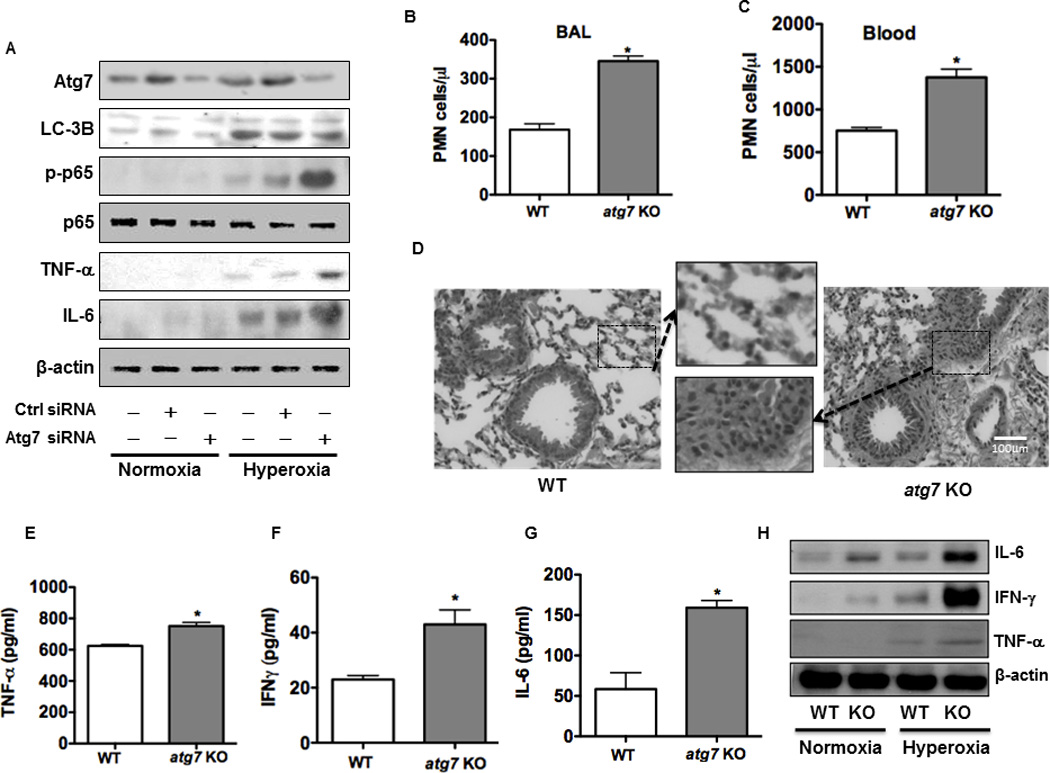
(A) Increased expression of cytokines (IL-6, TNF-α) and p-NF-κB after knocking down Atg7 with siRNA in MLE-12 cells. (B) and (C) PMN infiltration and inflammatory response were increased in the lung (B) and blood (C) of atg7 KO mice compared to WT mice. (D) Increased lung injury and inflammation as assessed by H&E staining. (E)-(G) Increased inflammatory cytokines in BAL fluid of atg7 KO mice compared to WT mice by ELISA. (H) Increased expression of inflammatory cytokines in the lungs of atg7 KO mice (n=6) compared to WT mice after 48 h hyperoxic exposure by immunoblotting analysis. Data were representative of three experiments with similar results (student t-test, *p< 0.05).
atg7 KO mice manifest increased inflammatory responses and lung injury
To delve into the role of autophagy in inflammatory responses, we evaluated the alteration of inflammatory response and lung injury in atg7 KO mice following hyperoxic exposure. Our data demonstrated that PMN’s in BAL fluid and blood was higher in atg7 KO mice than that in WT mice (Figures 5B and C). As a direct indicator of lung injury, we also assessed lung histology and found increased PMN infiltration in the lung of atg7 KO mice, indicating that PMN’s may be involved in the exacerbated lung injury in these animals compared to WT mice (Figure 5D). The levels of increased lung injury in atg7 KO mice reflect the role of autophagy in alleviating lung inflammatory responses and preventing subsequent tissue damage.
To identify additional critical factors in inflammatory responses due to the loss of Atg7, we measured cytokine concentrations in atg7 KO mouse lungs after 48 h exposure to 95% O2. BAL fluid of atg7 KO mice exhibited significantly increased inflammatory cytokine secretion (TNF-α, IL-6, and IFN-γ) by approximately 2-fold, compared to those of WT mice determined by ELISA (Figures 5E–G). Moreover, these cytokines were also increased with hyperoxia in atg7 KO lung tissues vs. those in WT mice as assessed by immunoblotting analysis (Figure 5H). Collectively, these data indicate that atg7 KO mice displayed intensified pro-inflammatory responses under hyperoxia compared to WT mice.
To further elucidate the mechanism of the autophagy-induced downregulation of inflammation, we blocked autophagy using Atg7 specific siRNA or inhibitors to investigate whether autophagy has effects on NF-κB. We noticed that autophagy blockers promoted the activation of NF-κB as evidenced by increased phosphorylation levels of NF-κB p65 subunit (ser536) and increased inflammatory responses, while autophagy inducers significantly reduced NF-κB phosphorylation and dampened inflammatory responses (Figure S1).
ROS is downstream of autophagy-regulated inflammatory responses
We speculate that ROS, acting as a downstream factor of autophagy, is a critical contributor to inflammatory responses due to effects on inflammation and tissue injury. As expected, we observed that ROS production was significantly increased in both ogg-1 KO mice (Figure S2) and atg7 KO mice (data not shown) stimulated with hyperoxia. We also observed similar results in OGG-1 or Atg7 siRNA silenced cells with hyperoxia, as shown in vivo (Figure S3A). To validate our data and confirm the role of ROS, we pretreated cells with two NADPH inhibitors diphenylene iodonium (DPI) (10 µM) and N-acetylcysteine (NAC) (5 µM) for 16 h, respectively. We then exposed the cells to hyperoxia for 6 h, and found that cytokine expression and NF-κB phosphorylation were significantly blunted by ROS inhibitors, suggesting that ROS may be involved in regulating inflammatory responses under hyperoxia (Figure S3B).
OGG-1 plays a role in regulating nuclear translocation of NF-κB
NF-κB is a ubiquitous transcription factor that modulates the production of various pro-inflammatory cytokines, such as TNF-α and IL-6. We observed heightened phosphorylation of NF-κB in ogg-1 KO lungs under hyperoxia compared to that in WT lungs (Figure 6A). In vitro silencing OGG-1 positively regulated NF-κB expression under hyperoxia using immunoblotting analysis (Figure 6B). Resting NF-κB bound to IκB located in the cytoplasm, while activated NF-κB was translocated to nuclei. Morphologically, we tracked the localization of NF-κB by CLSM. After knocking down OGG-1 with siRNA in MLE-12 cells, significantly increased translocation of NF-κB from cytoplasm to nuclei was observed compared to wild-type cells as quantified with different cells (Figure 6C).
FIGURE 6. OGG-1 plays a role in regulating the translocation of NF-κB.
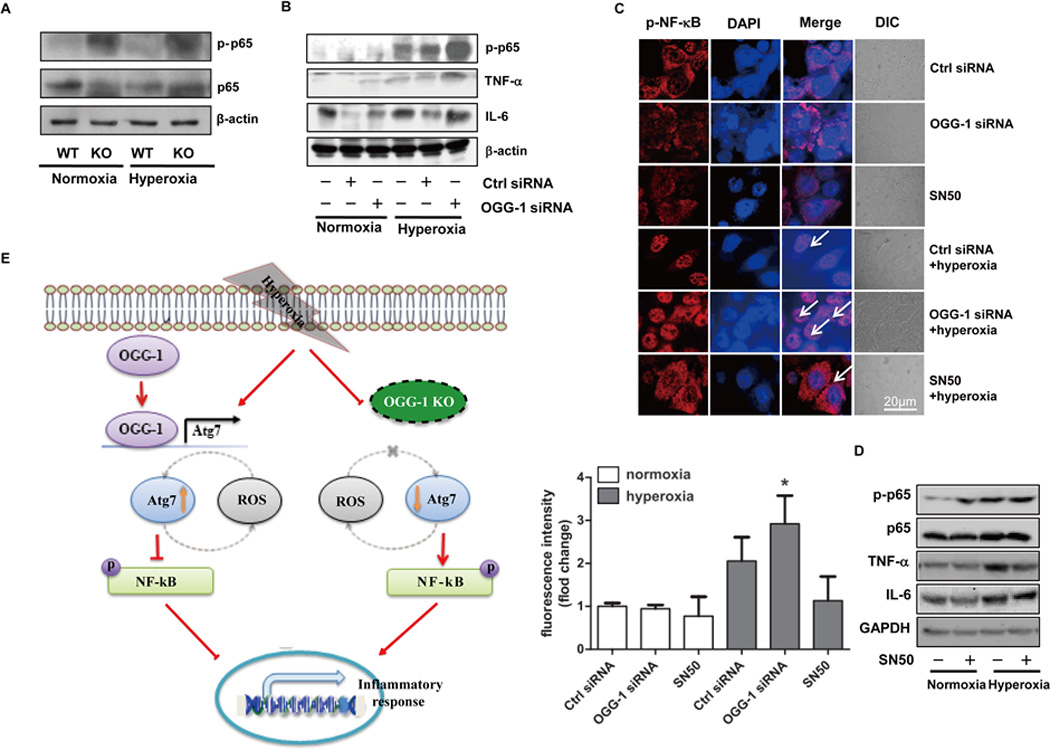
(A) Increased expression of NF-κB in ogg-1 KO mice after 48 h hyperoxia by immunoblotting analysis. Six mice were in each goup. (B) Knocking down OGG-1 with siRNA in MLE-12 cells increased NF-κB expression under hyperoxia using immunoblotting analysis. (C) Translocation of NF-κB was observed by microscopy (arrows show the nuclear translocation). (D) Hyperoxia-induced cytokine production was inhibited by NF-κB inhibitor (SN50). Cells were pretreated with SN50 (1.8 µM) for 1 h before hyperoxia exposure. Data were representative of three experiments with similar results. (E) Schematic illustration of the signaling pathways for cell viability regulated by OGG-1 under hyperoxia.
To affirm this result, we next employed an NF-κB inhibitor (SN50 1.8 µM) to investigate the role of NF-κB in regulating the expression of various pro-inflammatory cytokines. Cells were pretreated with SN50 for 1 h before hyperoxia exposure, which resulted in significantly decreased pro-inflammatory cytokines, such as TNF-α and IL-6, compared to mock-treated control cells after hyperoxia exposure (Figure 6D). Taken together, these findings strongly indicate that the OGG-1/Atg7 axis is involved in pro-inflammatory cytokine production through the activation of NF-κB and subsequent nuclear translocation (Figure 6E).
Discussion
In the present study, we demonstrated a phenotype of high concentration oxygen-mediated toxicity in ogg-1 KO mice, including intensified lung injury, increased inflammatory response and impaired macroautophagy compared to WT mice. These findings are similar to those observed in ogg-1 KO mice in other disease models, where ogg-1 KO mice showed an increased vulnerability to P. aeruginosa-induced organ dysfunction, neutrophil infiltration and oxidative stress (7, 8). However, recent reports showed a decreased inflammatory response in RNAi depelted ogg-1 mice (40). This may be explained by several possible factors including differences in mouse models (ogg-1 KO mice vs. RNAi depletion) and different inflammatory inducers (hyperoxia vs. 8-oxoG ). Secondly, the disease models are different as we also found that OGG-1 may be detrimental to allergic conditions, which are driven by Th2-mediated inflammatory responses (43). In the present study, we revealed a role of OGG-1 in inflammatory responses with hyperoxia in mouse models.
High concentration oxygen, a supportive therapy measure for ARDS patients, may further compromise the physiological condition as oxygenation may impair other anti-inflammatory mechanism offered by adenosine A2A receptor (A2AR), HIF1a, and autophagic proteins (41–44). We and others have also reported that hyperoxia induced DNA damage response (45) and other response mechanisms (46), which is tightly regulated by a variety of signaling system (47–49). Additionally, the DNA repair response is also associated with oxidative cytotoxicity and tissue injury resulted from various triggering factors, including infection, chronic inflammation, trauma, and chemotherapeutic application (50–56). Despite a variety of hyperoxic-mediated pathogenic factors contributing to tissue damage, we propose that increased generation and release of superoxide with hyperoxia contribute to the heightened pathophysiology in the lung. Previous studies have shown that OGG-1’s involvement in tetrandrine-induced autophagy is ROS-dependent during human hepatocellular carcinogenesis (57). We also demonstrated significantly increased ROS levels in AM cells of ogg-1 KO mice with hyperoxia (Figure S2A and B). Thus, ROS may induce DNA damage, which in turn initiates the response by the DNA repair system including OGG-1. Loss of ogg-1 may impair the ability of cells to repair damaged DNA and further potentiate ROS generation, resulting in over-zealous inflammation and tissue damage. Our findings demonstrate that both ogg-1 KO mice and OGG-1 siRNA-transfected cells exhibited elevated ROS generation (Figure S2A and B, Figure S3A). To further elucidate ROS’s role in cytokine production under hyperoxia, we pre-treated cells with two NADPH blockers (diphenylene iodonium and N-aceltylcysteine) and found that inhibiting ROS production by these blockers could significantly decrease the levels of cytokines, suggesting that ROS, as a downstream effector of autophagy, contributed to augmented cytokine expression (Figure S3B). Our data support a hypothesis that absence of ogg-1 failed to repair DNA damage caused by hyperoxia, leading to further ROS damage to the cells. Because OGG-1 resides both in the mitochondria and nuclei, whether the nuclear or mitochondrial form of OGG-1 is predominantly involved in this mechanism remains to be defined.
It has been shown that a spectrum of DNA repair proteins plays important roles in autophagy signaling. For instance, SWI2/SNF2 DNA-dependent ATPase Domino could regulate cell growth, proliferation and autophagy (58). In addition, ATM, as a DNA damage sensor, regulates mTORC1 in response to ROS, which can be released after hyperoxia exposure (59). Interestingly, it has been reported that autophagy, though not sufficient, is important for OGG-1 turnover in cardiomyopathy and autophagy balanced the positive effects of ROS inhibition against the negative effect of ogg-1 loss (17). However, the effect of OGG-1 on autophagy has not been previously elucidated. In the current study, we found that ogg-1 KO mice exhibited an impaired autophagy under hyperoxia. Furthermore, autophagy regulators such as Atg7 and LC3II appear to be decreased after knocking down OGG-1 with siRNA in MLE-12 cells. Further, we demonstrate that OGG-1 interacted with Atg7, which was further augmented by hyperoxia. One possibility is that ogg-1 KO mice display aggravated hyperoxia-mediated lung injury via an impaired autophagy since altered autophagy loses the ability to restore damaged DNA. Instead, OGG-1 binds to the damaged DNA to form complex, which may accumulate in autophagosomes. Using ChIP, we showed that OGG-1 binds to the promoter of Atg7 under hyperoxia, indicating that damaged DNA on Atg7 under hyperoxia recruits OGG-1. It is also possible that OGG-1 interacts with Atg7 by other mechanisms. Thus, further investigation of this relationship is warranted. Understanding the role of OGG-1 in inflammatory responses is important for elucidating the mechanisms underlying the pathogenesis of hyperoxia-induced lung injury.
Autophagy is critical for oxygen-dependent cellular stress and an altered autophagic pathway has been reported in cell culture models of oxidative stress, including hyperoxia (60). Our findings indicate that hyperoxia activates autophagy and cell injury occurs both in vitro and in vivo. Using atg7 KO mice, we demonstrated that an impaired autophagy leads to severe lung injury as well as increased inflammatory response after exposure to hyperoxia, consistent with previous data of impaired autophagy in various disease models, such as pathogenic microbe infection (61–63).
To dissect the mechanism for dysregulated inflammatory responses in ogg-1 KO mice, we assessed potential cell-signaling pathways in the lung tissue under hyperoxia. We observed marked activation of NF-κB, which has been widely recognized as a major transcription factor for cytokine production in alveolar epithelial cells. We also analyzed the role of other transcriptional factors like STAT-5, whose contribution is not significant (data not shown). In addition, we also found that p38 was greatly activated in ogg-1 KO under hyperoxia (data not shown), in line with our previous in vitro observations (33). Atg7 has been shown to interact with NF-κB directly through molecular binding and Atg7 deficiency may impact NF-κB activity (64). Thus, we identified a link between OGG-1 and Atg7/NF-κB that drives host homeostasis during stress, warranting further investigation of the underlying mechanism.
In conclusion, we proposes a previously undescribed model for explaining an inflammatory phenotype of ogg-1 KO mice stimulated with hyperoxia: our data suggest an important role for OGG-1 in innate immunity in mice. OGG-1 deficiency impaired immune functions, resulting in high infiltration of PMN cells into the lung, and an intense inflammatory response. Mechanistically, we revealed a novel link between OGG-1 and Atg7-mediated autophagy directly contributing to a dysregulated cytokine profile in ogg-1 KO mice as well as in OGG-1-silenced MLE-12 cells. Importantly, OGG-1 interacts with Atg7 by molecular binding, which controls NF-kB nuclear translocation, leading to accumulated ROS and elevated pro-inflammatory cytokines. Collectively, these observations provide new insight into the role of OGG-1 in innate immunity against hyperoxia and might indicate novel targets for clinical interventions.
Supplementary Material
Acknowledgments
We thank Sarah Abrahamson and Steven Adkins of university of North Dakota imaging core for using microscopy and Flow Cytometry Core for measuring lymphocyte populations, respectively.
Funding: This project was supported by Flight Attendant Medical Research Institute (FAMRI, Grant #103007), NIH AI101973-01, AI109317-01A1, and AI097532-01A1 to MW and the University of North Dakota Core Facilities were supported by NIH grants INBRE P20GM103442, COBRE P30GM103329, and COBRE P20GM113123)
Footnotes
AUTHOR CONTRIBUTIONS
Y. Y., P. L., and M. W. designed research and wrote the manuscript. Y. Y., P. L., W. Z., S. T., X. Z., R. L., and Q. P. performed experiments. J. K., A. D., F. M., X. D., and J. J. provided regents and suggestions.
Conflict of interest: none declared.
Reference
- 1.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiu WH, Luo SJ, Chen CL, Cheng JH, Hsieh CY, Wang CY, Huang WC, Su WC, Lin CF. Vinca alkaloids cause aberrant ROS-mediated JNK activation, Mcl-1 downregulation, DNA damage, mitochondrial dysfunction, and apoptosis in lung adenocarcinoma cells. Biochem Pharmacol. 2012;83:1159–1171. doi: 10.1016/j.bcp.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 3.Carvalho CR, Schettino G, de Paula Pinto Maranhao B, Bethlem EP. Hyperoxia and lung disease. Curr Opin Pulm Med. 1998;4:300–304. doi: 10.1097/00063198-199809000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Barazzone C, White CW. Mechanisms of cell injury and death in hyperoxia: role of cytokines and Bcl-2 family proteins. Am J Respir Cell Mol Biol. 2000;22:517–519. doi: 10.1165/ajrcmb.22.5.f180. [DOI] [PubMed] [Google Scholar]
- 5.Barker GF, Manzo ND, Cotich KL, Shone RK, Waxman AB. DNA damage induced by hyperoxia: quantitation and correlation with lung injury. Am J Respir Cell Mol Biol. 2006;35:277–288. doi: 10.1165/rcmb.2005-0340OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youn CK, Song PI, Kim MH, Kim JS, Hyun JW, Choi SJ, Yoon SP, Chung MH, Chang IY, You HJ. Human 8-oxoguanine DNA glycosylase suppresses the oxidative stress induced apoptosis through a p53-mediated signaling pathway in human fibroblasts. Mol Cancer Res. 2007;5:1083–1098. doi: 10.1158/1541-7786.MCR-06-0432. [DOI] [PubMed] [Google Scholar]
- 7.Wu M, Huang H, Zhang W, Kannan S, Weaver A, McKibben M, Herington D, Zeng H, Gao H. Host DNA repair proteins in response to Pseudomonas aeruginosa in lung epithelial cells and in mice. Infect Immun. 2011;79:75–87. doi: 10.1128/IAI.00815-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mabley JG, Pacher P, Deb A, Wallace R, Elder RH, Szabo C. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 2005;19:290–292. doi: 10.1096/fj.04-2278fje. [DOI] [PubMed] [Google Scholar]
- 9.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 11.de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, Begun J, Plantinga TS, Joosten LA, van der Meer JW, Chamilos G, Netea MG, Xavier RJ, Dinarello CA, Romani L, van de Veerdonk FL. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A. 2014;111:3526–3531. doi: 10.1073/pnas.1322831111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin L, Baehrecke EH. Autophagy, cell death, and cancer. Mol & cell oncol. 2015;2:e985913. doi: 10.4161/23723556.2014.985913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang TK, Shravage BV, Hayes SD, Powers CM, Simin RT, Wade Harper J, Baehrecke EH. Uba1 functions in Atg7- and Atg3-independent autophagy. Nat cell biol. 2013;15:1067–1078. doi: 10.1038/ncb2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beutler B, Cerami A. The biology of cachectin/TNF--a primary mediator of the host response. Annu Rev Immunol. 1989;7:625–655. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- 16.Sha WC. Regulation of immune responses by NF-kappa B/Rel transcription factor. J Exp Med. 1998;187:143–146. doi: 10.1084/jem.187.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siggens L, Figg N, Bennett M, Foo R. Nutrient deprivation regulates DNA damage repair in cardiomyocytes via loss of the base-excision repair enzyme OGG1. FASEB J. 26:2117–2124. doi: 10.1096/fj.11-197525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci U S A. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu M, Audet A, Cusic J, Seeger D, Cochran R, Ghribi O. Broad DNA repair responses in neural injury are associated with activation of the IL-6 pathway in cholesterol-fed rabbits. J Neurochem. 2009;111:1011–1021. doi: 10.1111/j.1471-4159.2009.06390.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Martin WJ, 2nd, Kachel DL. Oxygen-mediated impairment of human pulmonary endothelial cell growth: evidence for a specific threshold of toxicity. J Lab Clin Med. 1989;113:413–421. [PubMed] [Google Scholar]
- 21.Wu M, Hussain S, He YH, Pasula R, Smith PA, Martin WJ., 2nd Genetically engineered macrophages expressing IFN-gamma restore alveolar immune function in scid mice. Proc Natl Acad Sci U S A. 2001;98:14589–14594. doi: 10.1073/pnas.251451498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, Zhou X, Ye Y, Li Y, Li J, Privratsky B, Wu E, Gao H, Huang C, Wu M. Lyn regulates inflammatory responses in Klebsiella pneumoniae infection via the p38/NF-kappaB pathway. Eur J Immunol. 2014;44:763–773. doi: 10.1002/eji.201343972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan K, Huang C, Fox J, Gaid M, Weaver A, Li G, Singh BB, Gao H, Wu M. Elevated inflammatory response in caveolin-1-deficient mice with Pseudomonas aeruginosa infection is mediated by STAT3 protein and nuclear factor kappaB (NF-kappaB) J Biol Chem. 2011;286:21814–21825. doi: 10.1074/jbc.M111.237628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye Y, Li X, Wang W, Ouedraogo KC, Li Y, Gan C, Tan S, Zhou X, Wu M. Atg7 deficiency impaires host defense against Klebsiella pneumoniae by impacting bacterial clearance, survival and inflammatory responses in mice. Am J Physiol Lung Cell Mol Physiol. 2014;307:L355–L363. doi: 10.1152/ajplung.00046.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou X, Li X, Ye Y, Zhao K, Zhuang Y, Li Y, Wei Y, Wu M. MicroRNA-302b augments host defense to bacteria by regulating inflammatory responses via feedback to TLR/IRAK4 circuits. Nat Commun. 2014;5:3619. doi: 10.1038/ncomms4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He YH, 2nd, Wu M, Kobune M, Xu Y, Kelley MR, Martin WJ. Expression of yeast apurinic/apyrimidinic endonuclease (APN1) protects lung epithelial cells from bleomycin toxicity. Am J Respir Cell Mol Biol. 2001;25:692–698. doi: 10.1165/ajrcmb.25.6.4550. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Hui L, Geiger NH, Haughey NJ, Geiger JD. Endolysosome involvement in HIV-1 transactivator protein-induced neuronal amyloid beta production. Neurobiol Aging. 2013;34:2370–2378. doi: 10.1016/j.neurobiolaging.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hui L, Chen X, Geiger JD. Endolysosome involvement in LDL cholesterol-induced Alzheimer’s disease-like pathology in primary cultured neurons. Life Sci. 2012;91:1159–1168. doi: 10.1016/j.lfs.2012.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan J, Pei DS, Yin XH, Hui L, Zhang GY. Involvement of oxidative stress in the rapid Akt1 regulating a JNK scaffold during ischemia in rat hippocampus. Neurosci Lett. 2006;392:47–51. doi: 10.1016/j.neulet.2005.08.057. [DOI] [PubMed] [Google Scholar]
- 30.Hui L, Chen X, Haughey NJ, Geiger JD. Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN Neuro. 2012;4:243–252. doi: 10.1042/AN20120017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Zou L, Liu Q, Li J, Zhou J, Wang Y, Li N, Liu T, Wei H, Wu M, Wan Y, Wu Y. Rapid generation of dendritic cell specific transgenic mice by lentiviral vectors. Transgenic Res. 2009;18:921–931. doi: 10.1007/s11248-009-9282-z. [DOI] [PubMed] [Google Scholar]
- 32.Wu M, Pasula R, Smith PA, Martin WJ., 2nd Mapping alveolar binding sites in vivo using phage peptide libraries. Gene Ther. 2003;10:1429–1436. doi: 10.1038/sj.gt.3302009. [DOI] [PubMed] [Google Scholar]
- 33.Kannan S, Pang H, Foster DC, Rao Z, Wu M. Human 8-oxoguanine DNA glycosylase increases resistance to hyperoxic cytotoxicity in lung epithelial cells and involvement with altered MAPK activity. Cell Death Differ. 2006;13:311–323. doi: 10.1038/sj.cdd.4401736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hui L, Chen X, Bhatt D, Geiger NH, Rosenberger TA, Haughey NJ, Masino SA, Geiger JD. Ketone bodies protection against HIV-1 Tat-induced neurotoxicity. J Neurochem. 2012;122:382–391. doi: 10.1111/j.1471-4159.2012.07764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kannan S, Audet A, Huang H, Chen LJ, Wu M. Cholesterol-rich membrane rafts and Lyn are involved in phagocytosis during Pseudomonas aeruginosa infection. J Immunol. 2008;180:2396–2408. doi: 10.4049/jimmunol.180.4.2396. [DOI] [PubMed] [Google Scholar]
- 36.Li X, He S, Li R, Zhou X, Zhang S, Yu M, Ye Y, Wang Y, Huang C, Wu M. Pseudomonas aeruginosa infection augments inflammation through miR-301b repression of c-Myb-mediated immune activation and infiltration. Nat Microbiol. 2016;1:16132. doi: 10.1038/nmicrobiol.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, He S, Zhou X, Ye Y, Tan S, Zhang S, Li R, Yu M, Jundt MC, Hidebrand A. Lyn delivers bacteria to lysosomes for eradication through TLR2-initiated autophagy related phagocytosis. PLoS Pathog. 2016;12:e1005363. doi: 10.1371/journal.ppat.1005363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He S, Li X, Li R, Fang L, Sun L, Wang Y, Wu M. Annexin A2 Modulates ROS and Impacts Inflammatory Response via IL-17 Signaling in Polymicrobial Sepsis Mice. PLoS Pathog. 2016;12:e1005743. doi: 10.1371/journal.ppat.1005743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li R, Fang L, Tan S, Yu M, Li X, He S, Wei Y, Li G, Jiang J, Wu M. Type I CRISPR-Cas targets endogenous genes and regulates virulence to evade mammalian host immunity. Cell Res. 2016;26:1273–128. doi: 10.1038/cr.2016.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aguilera-Aguirre L, Bacsi A, Radak Z, Hazra TK, Mitra S, Sur S, Brasier AR, Ba X, Boldogh I. Innate Inflammation Induced by the 8-Oxoguanine DNA Glycosylase-1-KRAS-NF-kappaB Pathway. J Immunol. 2014;193:4643–4653. doi: 10.4049/jimmunol.1401625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eckle T, Brodsky K, Bonney M, Packard T, Han J, Borchers CH, Mariani TJ, Kominsky DJ, Mittelbronn M, Eltzschig HK. HIF1A reduces acute lung injury by optimizing carbohydrate metabolism in the alveolar epithelium. PLoS Biol. 2013;11:e1001665. doi: 10.1371/journal.pbio.1001665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thiel M, Chouker A, Ohta A, Jackson E, Caldwell C, Smith P, Lukashev D, Bittmann I, Sitkovsky MV. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 2005;3:e174. doi: 10.1371/journal.pbio.0030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka A, Jin Y, Lee S-J, Zhang M, Kim HP, Stolz DB, Ryter SW, Choi AM. Hyperoxia-induced LC3B interacts with the Fas apoptotic pathway in epithelial cell death. Am J Respir Cell Mol Biol. 2012;46:507–514. doi: 10.1165/rcmb.2009-0415OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Sauler M, Shinn AS, Gong H, Haslip M, Shan P, Mannam P, Lee PJ. Endothelial PINK1 mediates the protective effects of NLRP3 deficiency during lethal oxidant injury. J Immunol. 2014;192:5296–5304. doi: 10.4049/jimmunol.1400653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu M, He Y-h, Kobune M, Xu Y, Kelley MR, Martin WJ. Protection of human lung cells against hyperoxia using the DNA base excision repair genes hOgg1 and Fpg. Am J Respir Crit Care Med. 2002;166:192–199. doi: 10.1164/rccm.200112-130OC. [DOI] [PubMed] [Google Scholar]
- 46.Zhang M, Lee S-J, An C, Xu J-f, Joshi B, Nabi IR, Choi AM, Jin Y. Caveolin-1 mediates Fas-BID signaling in hyperoxia-induced apoptosis. Free Radic Biol Med. 2011;50:1252–1262. doi: 10.1016/j.freeradbiomed.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fukumoto J, Cox R, Jr, Fukumoto I, Cho Y, Parthasarathy PT, Galam L, Lockey RF, Kolliputi N. Deletion of ASK1 Protects against Hyperoxia-Induced Acute Lung Injury. PloS one. 2016;11:e0147652. doi: 10.1371/journal.pone.0147652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kannan S, Pang H, Foster D, Rao Z, Wu M. Human 8-oxoguanine DNA glycosylase increases resistance to hyperoxic cytotoxicity in lung epithelial cells and involvement with altered MAPK activity. Cell Death Differ. 2006;13:311–323. doi: 10.1038/sj.cdd.4401736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gray JM, Karow DS, Lu H, Chang AJ, Chang JS, Ellis RE, Marletta MA, Bargmann CI. Oxygen sensation and social feeding mediated by a C. elegans guanylate cyclase homologue. Nature. 2004;430:317–322. doi: 10.1038/nature02714. [DOI] [PubMed] [Google Scholar]
- 50.Huang H, Weaver A, Wu E, Li Y, Gao H, Fan W, Wu M. Lipid-Based Signaling Modulates DNA Repair Response and Survival against Klebsiella pneumoniae Infection in Host Cells and in Mice. Am J Respir Cell Mol Biol. 2013;49:798–807. doi: 10.1165/rcmb.2013-0069OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu M, Huang H, Zhang W, Kannan S, Weaver A, Mckibben M, Herington D, Zeng H, Gao H. Host DNA repair proteins in response to Pseudomonas aeruginosa in lung epithelial cells and in mice. Infect Immun. 2011;79:75–87. doi: 10.1128/IAI.00815-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu M, Audet A, Cusic J, Seeger D, Cochran R, Ghribi O. Broad DNA repair responses in neural injury are associated with activation of the IL-6 pathway in cholesterol-fed rabbits. J Neurochem. 2009;111:1011–1021. doi: 10.1111/j.1471-4159.2009.06390.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.He Y-H, Wu M, Kobune M, Xu Y, Kelley MR, Martin WJ. Expression of yeast apurinic/apyrimidinic endonuclease (APN1) protects lung epithelial cells from bleomycin toxicity. Am J Respir Cell Mol Biol. 2001;25:692–698. doi: 10.1165/ajrcmb.25.6.4550. [DOI] [PubMed] [Google Scholar]
- 54.Li G, Yuan K, Yan C, Fox J, Gaid M, Breitwieser W, Bansal AK, Zeng H, Gao H, Wu M. 8-Oxoguanine-DNA glycosylase 1 deficiency modifies allergic airway inflammation by regulating STAT6 and IL-4 in cells and in mice. Free Radic Biol Med. 2012;52:392–401. doi: 10.1016/j.freeradbiomed.2011.10.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu M. DNA repair proteins as molecular therapeutics for oxidative and alkylating lung injury. Curr Gene Ther. 2005;5:225–236. doi: 10.2174/1566523053544245. [DOI] [PubMed] [Google Scholar]
- 56.O’Reilly MA. DNA damage and cell cycle checkpoints in hyperoxic lung injury: braking to facilitate repair. Am J Physiol Lung Cell Mol Physiol. 2001;281:L291–L305. doi: 10.1152/ajplung.2001.281.2.L291. [DOI] [PubMed] [Google Scholar]
- 57.Gong K, Chen C, Zhan Y, Chen Y, Huang Z, Li W. Autophagy-related gene 7 (ATG7) and reactive oxygen species/extracellular signal-regulated kinase regulate tetrandrine-induced autophagy in human hepatocellular carcinoma. J Biol Chem. 2012;287:35576–35588. doi: 10.1074/jbc.M112.370585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kwon MH, Callaway H, Zhong J, Yedvobnick B. A targeted genetic modifier screen links the SWI2/SNF2 protein domino to growth and autophagy genes in Drosophila melanogaster. G3 (Bethesda) 2013;3:815–825. doi: 10.1534/g3.112.005496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, Walker CL. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107:4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ryter SW, Choi AM. Regulation of autophagy in oxygen-dependent cellular stress. Curr Pharm Des. 2013;19:2747–2756. doi: 10.2174/1381612811319150010. [DOI] [PubMed] [Google Scholar]
- 61.Jia K, Thomas C, Akbar M, Sun Q, Adams-Huet B, Gilpin C, Levine B. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc Natl Acad Sci U S A. 2009;106:14564–14569. doi: 10.1073/pnas.0813319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy. 2009;5:937–945. doi: 10.4161/auto.5.7.9243. [DOI] [PubMed] [Google Scholar]
- 63.Amer AO, Byrne BG, Swanson MS. Macrophages rapidly transfer pathogens from lipid raft vacuoles to autophagosomes. Autophagy. 2005;1:53–58. doi: 10.4161/auto.1.1.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fujishima Y, Nishiumi S, Masuda A, Inoue J, Nguyen NM, Irino Y, Komatsu M, Tanaka K, Kutsumi H, Azuma T, Yoshida M. Autophagy in the intestinal epithelium reduces endotoxin-induced inflammatory responses by inhibiting NF-kappaB activation. Arch Biochem Biophys. 2011;506:223–235. doi: 10.1016/j.abb.2010.12.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.