

Abstract
While the majority of host cell protein (HCP) impurities are effectively removed in typical downstream purification processes, a small population of HCPs are particularly challenging. Previous studies have identified HCPs that are challenging for a variety of reasons. Lipoprotein lipase (LPL) – a Chinese hamster ovary (CHO) HCP that functions to hydrolyze esters in triglycerides – was one of ten HCPs identified in previous studies as being susceptible to retention in downstream processing. LPL may degrade polysorbate 80 (PS-80) and polysorbate 20 (PS-20) in final product formulations due to the structural similarity between polysorbates and triglycerides. In this work, recombinant LPL was found to have enzymatic activity against PS-80 and PS-20 in a range of solution conditions that are typical of mAb formulations. LPL knockout CHO cells were created with CRISPR and TALEN technologies and resulting cell culture harvest fluid demonstrated a significantly reduced polysorbate degradation without significant impact on cell viability when compared to wild type samples.
Keywords: polysorbate, CRISPR, CHO cell, host cell protein, lipase
Introduction
Host cell proteins (HCPs) are a class of impurities that must be removed from all cell-derived protein therapeutics. The FDA does not specify a maximum acceptable level of HCP, but HCP concentrations in final drug product must be controlled and reproducible from batch to batch (FDA, 1999). CHO cell culture supernatant contains a complex population of HCPs, the majority of which are slightly acidic (Jin et al., 2010). This complex mixture of HCP impurities enters the downstream purification process and must be reduced to low levels. Typically HCP concentrations are reduced to 1-100 ppm for final mAb formulations (Champion et al., 2005). All products are expected to have trace HCP levels, although the levels may be below the limit of detection.
A primary safety concern relates to the possibility that HCP impurities cause antigenic effects in human patients (Singh et al., 2012). It is impossible to predict the human response to trace HCP impurities, but it is hypothesized that the more dissimilar an impurity is to human proteins, the more likely the impurity is to elicit an immune response in humans (Wang et al., 2009). While it is uncommon, two clinical trials were recently withdrawn due to anti-CHO responses in human patients (Gutierrez et al., 2012; Hanania et al., 2015). In addition to adverse health consequences for the patient, enzymatically-active HCP impurities can potentially impact product quality during processing or long-term storage (Gao et al., 2011; Robert et al., 2009).
Prior works explored three routes by which HCP impurities can challenge downstream purification: product-association through strong attractive interactions with mAb products under protein A loading conditions (Levy et al., 2014), variable expression during extended cell culture (Valente et al., 2015), or co-elution with mAbs on polishing chromatographic media (Levy et al., 2015). Of the hundreds of extracellular HCPs expressed by CHO cells, 116 HCPs were identified as difficult to remove by at least one mechanism, with 10 HCPs evading clearance by all three methods (Figure 1). Such HCPs may present the greatest risk for persisting through purification operations into the final drug product and consequently warrant further investigation to facilitate their clearance during biopharmaceutical manufacturing. We focus this work on lipoprotein lipase.
Figure 1.
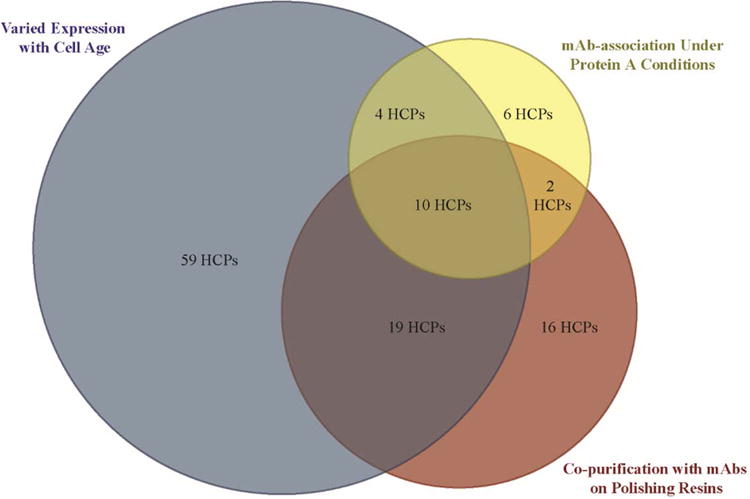
Venn diagram of difficult-to-remove HCP impurities and the mechanism by which they challenge clearance in biopharmaceutical manufacturing. Diagram not drawn to scale.
During long-term storage, the critical quality attributes of the product molecule must be maintained and degradation of excipients in the final product formulation must be minimized. Polysorbates are nonionic surfactants that are common additives in drug products. The majority of FDA-approved mAbs contain either polysorbate 80 (PS-80) or polysorbate 20 (PS-20, Marichal-Gallardo and Alvarez, 2012). Polysorbates protect mAbs from degradation during purification, filtration, freeze-drying, storage and final delivery (Kerwin, 2008). They are thought to stabilize high-concentration mAb solutions by competing with mAbs for surface adsorption (Mahler et al., 2009) or binding to the product molecules (Lee et al., 2011). Several different routes of polysorbate degradation in formulations have previously been identified (Dixit et al., 2016; Ha et al., 2002; Khossravi et al., 2002; Kishore et al., 2011; Hall et al., 2016), with degradation leading to accelerated product degradation due to increased aggregation (Khossravi et al., 2002) or oxidation due to peroxide formation (Ha et al., 2002).
We hypothesize that a specific difficult-to-remove CHO HCP impurity may contribute to polysorbate degradation and contribute to reduced mAb stability. Specifically, one of the difficult-to-remove HCPs noted above, lipoprotein lipase (LPL), which was also identified in other studies of HCP persistence (Aboulaich et al., 2014; Doneanu et al., 2012), hydrolyzes ester bonds within triglycerides to form alcohol and fatty acid molecules (Nilsson-Ehle et al., 1980). Given the structural similarities between polysorbates and triglycerides, it is hypothesized that LPL may enzymatically degrade polysorbates and consequently negatively impact mAb stability. A similar mechanism has recently been proposed to be associated with the degradation of PS-20 in a non-mAb product formulation by putative phospholipase B-like 2 (PLBL2) (Dixit et al., 2016). PLBL2 is an HCP impurity that was previously identified to have variable expression during extended culture (Valente at al., 2015).
Targeted gene disruption or knockout can be achieved using zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPRs) technologies. Several groups have demonstrated the application of gene disruption technologies in CHO cells (Grav et al., 2015; Ronda et al., 2014; Sun et al., 2015) for targeted gene deletion. Functional knockouts of FUT8 yield CHO cell lines that produce defucosylated antibodies (Grav et al., 2015; Ronda et al., 2014; Sun et al., 2015), while BAX and BAK knockouts yield CHO cell lines with high viability under long culture times (Grav et al., 2015). Recent advances in the sequencing of the CHO-K1 and the Chinese hamster genome (Brinkrolf et al., 2013; Xu et al., 2011) have aided the rational design of engineered CHO cell lines with desired properties.
In this study, we applied targeted gene disruption technologies to reduce expression of lipoprotein lipase and test if the enzyme is associated with the degradation of polysorbates including through the use of a mass spectrometry-based assay. We also explored the quantification of LPL expression also using a multiple selected ion reaction monitoring (MRM) assay.
Methods
E. coli expression of CHO LPL
The Chinese hamster LPL gene sequence (UniProKB entry G3H6V7) was synthesized by Life Technologies (Carlsbad, CA, USA). The synthesized sequence included NdeI and BamHI restriction enzyme sites at the 5′ and 3′ ends respectively and a six-His tag sequence was also added between the last codon of lpl and the BamHI site. The lpl sequence was amplified, purified and ligated into the pET11a vector; the lpl-containing pET11a vector was transformed into NEB5α competent cells (New England BioLabs, Ipswich, MA, USA) and plated on Amp-LB agar plates. Twelve positive colonies were selected and cultured overnight. The pET11a vector was then purified and digested and positive clones were confirmed via sequencing.
The lpl-containing pET11a plasmid was then transformed into BL21 competent cells in SOC broth (New England BioLabs) and plated on ampicillin. Colonies were selected and cultures were grown overnight to seed a 750 mL production culture. Induction was commenced at 0.4 OD with the addition of 400 μM IPTG (Sigma-Aldrich Chemical Co., St. Louis, MO, USA), followed by LPL expression for 3 hours.
LPL purification and refolding
The E. coli inoculum was harvested and centrifuged at 1000 g for 10 minutes to pellet the cells using an Eppendorf 5810R centrifuge (Hamburg, Germany). The supernatant was discarded and the cell pellets were frozen for future use. Cell pellets were thawed in lysis buffer – 75 mM tris, 120 mM NaCl, 5 mM EDTA, pH 7.7 – and cells were lysed in an M-110L Pneumatic Microfluidizer from Microfluidics (Westwood, MA, USA) at 9000 psi for at least 6 full cycles at 5 °C. Cell lysate, containing LPL inclusion bodies, was then ultracentrifuged in a Beckman Coulter Optima™ L-100 XP Ultracentrifuge (Brea, CA, USA) at 40,000 g for 1 hour to pellet the inclusion bodies. The inclusion bodies were solubilized in 6 M guanidine HCl, 300 mM NaCl, 10 mM imidazole, 20 mM sodium phosphate at pH 7.4. The solubilized LPL was loaded onto a HisPur™ Ni-NTA column from Thermo Scientific (Waltham, MA, USA), washed with 10 CV of 6 M guanidine HCl and eluted with 16 CVs of 20 mM sodium phosphate, 300 mM NaCl, 6 M guanidine HCl, 250 mM imidazole at pH 7.4. The elution pool was diluted in 6 M guanidine HCl to a final OD of 0.4. The solubilized protein was then reduced with the addition of dithiothreitol (DTT) at a final concentration of 15 mM.
A solution of refolding buffer was prepared containing 50 mM tris, 600 mM L-arginine, 2.5 mM calcium chloride and 5 mM cysteine at pH 8.5. The arginine is intended to prevent aggregation and there is evidence from previous work that calcium chloride can assist in proper folding of LPL into active dimers (Zhang et al., 2005). A volume of refolding buffer 50 times the volume of solubilized inclusion bodies was stirred gently at 5 °C while the LPL inclusion body solution was added at ∼0.2 mL/min using a peristaltic pump. After the addition of LPL was complete, gentle stirring was continued for 12 hours at constant temperature.
To confirm folding, reverse phase-HPLC was run with unfolded LPL (LPL solubilized in 6 M guanidine) and refolded LPL. The LPL was injected into a C18 column at 1 mL/min with a linear gradient from 0-100% acetonitrile in water over 45 minutes. The unfolded LPL eluted after 7 minutes and the majority of refolded LPL eluted after 4 minutes (data not shown).
CHO cell culture
A null CHO-K1 cell line (ATCC, Manassas, VA, USA) was adapted to serum-free, suspension culture in 125 mL shake flasks containing 20–30 mL SFM4CHO medium (GE Healthcare Life Sciences, Little Chalfont, UK). Following adaptation, the cells were subjected to extended culture with routine passaging at 3–5 day intervals in a 37 °C cell culture incubator at 5% CO2 and 80% relative humidity.
Single guide RNA (sgRNA) target design and plasmid construction for CRISPR and TALEN
The Cas9 expression vector was obtained from Addgene (Addgene #41815) and selected on 100 μg/mL ampicillin LB plates. The sgRNA target selections (Supplementary Table I) for lpl were filtered using a bioinformatics tool, CRISPy (Ronda et al., 2014). The target sgRNA expression vectors were constructed by cloning 455 bp gBlocks (Supplementary Table II) synthesized by Integrated DNA Technologies (Coralville, IA, USA) into pCR-Blunt-II TOPO vector (Life Technologies) according to the manufacturer's recommendations. The sgRNA expression vectors were transformed into E. coli One Shot TOP10 competent cells (Life Technologies) according to the manufacturer's recommendations and selected on 50 μg/mL kanamycin LB plates. Cas9 and sgRNA plasmids were purified using the QIAprep Spin Miniprep Kit (Qiagen, Hilden, Germany) and the construct sequences were verified by Sanger sequencing (DBI sequencing center, Newark, DE, USA). The TALENs were designed against lpl exon 4 (Supplementary Figure 1) and provided by the Gene Editing Institute at Christiana Care Hospital.
CRISPR- and TALEN-mediated DNA modification and subclone selection
1×106 cells were simultaneously transfected with Cas9 and sgRNA targeting lpl expression vectors, for CRISPR-mediated DNA modification, and TAL1 and TAL2 expression vectors, for TALEN-mediated DNA modification, using Amaza Cell Line Nucleofector Kit V for CHO-K1 cells (Lonza, Basel, Switzerland) according to the manufacturer's protocol. A transfection with pmaxGFP vector (Lonza) was applied to evaluate the transfection efficiency. Transfected cells were cultured for 48 hours to allow gene knockout. Single cell clones from cells treated with CRISPR or TALEN plasmids were generated by limiting dilution into a 96-well plate with a target density of 0.5 cells/well. Single clones were grown at 37 °C in a cell culture incubator at 5% CO2 and 80% relative humidity for three weeks and then further expanded in 6-well plates before testing for mutations. Genomic DNA was extracted from the CRISPR-treated cell population using the QIAamp DNA Mini Kit (Qiagen) for a T7 endonuclease I (T7EI) assay. The cells were counted using either a Fuchs-Rosenthal hemocytometer or Countess II cell counter (Life Technologies). Cell viability was determined by the Trypan blue exclusion method. CHO harvested cell culture fluid (HCCF) from wild-type and lpl knockout cell lines was separated from the cells by centrifugation (180 g, 10 min) and stored at -20 °C.
The genomic regions covering the lpl sgRNA target sites were PCR-amplified from genomic extracts. PCR products were purified using the QIAquick PCR Purification Kit (Qiagen) and then TOPO-cloned into the pCR-Blunt II-TOPO vector using the Zero Blunt TOPO PCR Cloning Kit (Life Technologies). The constructed plasmids were transformed into E. coli One Shot TOP10 competent cells (Life Technologies) according to the manufacturer's recommendations and selected on 50 μg/mL kanamycin LB plates. Single colonies were picked and grown in LB medium with 50 ug/mL kanamycin. Plasmids from single colonies were extracted using QIAprep Spin Miniprep Kit (Qiagen). Sequences of the plasmids were verified by Sanger sequencing (DBI sequencing center, Newark, DE, USA) using an M13 reverse primer.
T7 Endonuclease (T7EI) assay for targeting efficiency of CRISPR
The overall targeting efficiency of CRISPR can be assessed using a T7EI assay, which measures insertions and deletions (indels) derived from CRISPR activity. Genomic regions around the CRISPR target site were amplified from the genomic DNA extracts using Phusion high-fidelity polymerase (New England Biolabs, Ipswich, MA, USA) by touchdown PCR (Supplementary Table III). The PCR products were subjected to a reannealing process to enable heteroduplex formation (95 °C for 5 min; 95-85 °C ramped at -2 °C/s; 85-25 °C ramped at -0.1 °C/s; and held at 4 °C). Annealed PCR products were subsequently digested with T7EI (New England Biolabs) at 37 °C for 20 minutes and analyzed on a 2% TAE gel. The percentage of indels was quantified from band intensity analysis of the cut and uncut gel bands using ImageJ.
LPL expression analysis by LC-Multiple Reaction Monitoring (MRM) assay
Extracellular CHO HCPs were precipitated from HCCF with methanol as described previously (Valente et al., 2014) and residual detergent was removed by DetergentOUT™ GBS10–800 detergent removal kit (G-Biosciences, St. Louis, MO, USA) according to the manufacturer's protocol. Trypsin digestion was performed as described previously (Valente et al., 2014). Peptide pellets were resolubilized in 0.1% trifluoroacetic acid (TFA, Fisher Scientific), loaded onto C18 ZipTips (EMD Millipore, Billerica, MA, USA) and eluted in 50% acetonitrile with 0.1% TFA (Fisher Scientific).
MRM is a mass spectrometry-based technique that can precisely quantify small molecules, peptides, and proteins within complex matrices with high sensitivity, specificity and a wide dynamic range (Picotti et al., 2010). MRM is typically performed with triple quadrupole mass spectrometers wherein a precursor ion corresponding to the selected small molecules/peptides is selected in the first quadrupole and a fragment ion of the precursor ion was selected for monitoring in the third quadrupole (Choi et al., 2013). The LPL LC-MRM assay was performed on a QTrap 4000 (AB Sciex, Foster City, CA, USA) equipped with an UltiMate 3000 nLC system (Dionex, Sunnyvale, CA, USA). Digested CHO HCPs were injected onto a C18 trap column (Dionex), and washed with 2% acetonitrile with 0.1% formic acid (Mallinckrodt Chemicals, Phillipsburg, NJ, USA) for 5 min with a flow rate of 30 μl/min, then eluted onto a C18 column (Acclaim PepMap100, 75 μm × 150 mm, 3 μm, 100 Å, Dionex) by a program of 2–49% acetonitrile with 0.1% formic acid in 50 min, followed by 49% acetonitrile with 0.1% formic acid for 20 min. Column eluate was directly injected into a QTrap 4000 through a nanoSpray II source (AB Sciex) with an uncoated fused-silica Pico tip (New Objective, Woburn, MA, USA). The instrument was operated in positive ESI ion mode, with spray voltage at 2400 V and source temperature of 150 °C, with MRM triggered enhanced resolution scan and enhanced product ion scans. MRM transitions were generated with Skyline v2.5 (MacLean et al., 2010) and monitored through Analyst 1.6.2 (AB Sciex) with parameters specified in Table I. All raw MRM data were integrated for peak area with Skyline. Skyline is an open source software for method development and data analysis in target proteomics, and freely available (MacLean et al., 2010). Raw data from CRISPR and TALEN knockout cell lines were normalized to peptide NVLVTLYER, and raw data for the final selected five clones were normalized to ITGLDPAGPNFEYAEAPSR with C-terminal 13C15N-labeled R. The analysis was performed with three technical replicates.
Table I.
MRM assay parameters.
| Target peptide sequence | Precursor (m/z) | Product (m/z) | ID | Scan time (ms) | CE (V) | |
|---|---|---|---|---|---|---|
| LPL | ITGLDPAGPNFEYAEAPSR | 1002.987 | 793.424 | +2y7 | 20 | 72.9 |
| ITGLDPAGPNFEYAEAPSR | 1002.987 | 753.355 | +2y14+2 | 20 | 67.9 | |
| ITGLDPAGPNFEYAEAPSR | 1002.987 | 359.204 | +2y3 | 20 | 52.9 | |
| ITGLDPAGPNFEYAEAPSR | 1007.991 | 803.392 | +2y7 | 20 | 72.9 | |
| ITGLDPAGPNFEYAEAPSR | 1007.991 | 758.359 | +2y14+2 | 20 | 67.9 | |
| ITGLDPAGPNFEYAEAPSR | 1007.991 | 369.212 | +2y3 | 20 | 52.9 | |
| GLGDVDQLVK | 522.29 | 873.478 | +2y8 | 20 | 30.5 | |
| GLGDVDQLVK | 522.29 | 701.429 | +2y6 | 20 | 30.5 | |
| GLGDVDQLVK | 522.29 | 602.361 | +2y5 | 20 | 30.5 | |
|
| ||||||
| SPARC | NVLVTLYER | 553.814 | 780.425 | +2y6 | 20 | 35.4 |
| NVLVTLYER | 553.814 | 681.357 | +2y5 | 20 | 35.4 | |
| NVLVTLYER | 553.814 | 467.225 | +2y3 | 20 | 35.4 | |
CHO LPL protein expression analysis by western analysis
Extracellular CHO HCPs were precipitated with methanol as described previously (Valente et al., 2014) and resolubilized in phosphate buffered saline. Concentrations of resolubilized HCPs were measured using a Bradford assay. Two sets of 30 μg CHO HCPs were reduced by sodium dodecyl sulfate (SDS) and DTT before being separated on 4-20% Mini-PROTEAN TGX precast gel (Biorad Laboratories, Hercules, CA, USA). Proteins were then transferred to a 0.45 μm polyvinylidene difluoride membrane (Life Technologies). Membranes were blocked in 3% non-fat dry milk in Tris-buffered saline with TWEEN 20 (TBST, Sigma-Aldrich Chemical Co.) for 1 hour and probed overnight at 4 °C with LPL N-terminus primary antibody (1:500 dilution in 3% milk block), which recognizes LPL residues 27-79 (Santa Cruz Biotechnology, Dallas, TX, USA). A second membrane was probed overnight at 4 °C with LPL C-terminus primary antibody (1:250 dilution in 3% milk block), which recognizes LPL residues 297-326 (Abcam, Cambridge, UK). The membranes were then probed with alkaline phosphatase-conjugated mouse anti-rabbit IgG (1:5,000 dilution, Sigma-Aldrich Chemical Co.) for 45 minutes, detected using enhanced chemifluorescence (ECF, GE Healthcare Life Sciences) substrate following the manufacturer's instructions and imaged using a Typhoon FLA-7000 scanner (GE Healthcare Life Sciences).
LPL activity assay
Polysorbate degradation is measured indirectly by measuring the free fatty acid concentration because enzymatic hydrolysis of PS-80 yields oleic acid and an alcohol component and enzymatic hydrolysis of PS-20 yields lauric acid and an alcohol component. Measurements of LPL activity against PS-80 were carried out under various solution conditions. Refolded recombinant LPL was buffer-exchanged into the appropriate buffer prior to the activity assay. The conditions investigated were 10 mM sodium acetate at pH 5.0, 10 mM L-histidine at pH 6.0, and 50 mM bis-tris at pH 6.8. PS-80 was added to the buffer-exchanged LPL to a final concentration of 0.23 mM. Some samples also included either 10 mM calcium chloride or 10 mM sodium chloride. The polysorbate and LPL solutions were then incubated at 37 °C with constant mixing for 24 hours.
The polysorbate degradation assay was adapted from previous work (Khossravi et al., 2002) that was designed to measure degraded PS-20 from pancreatic lipase. 270 μM 9-(diazomethyl)anthracene (ADAM) (Sigma-Aldrich Chemical Co.) in methanol was added to each sample in a 3:1 volumetric ratio of ADAM solution to sample. The ADAM conjugation was carried out at room temperature using opaque 1.6 mL Eppendorf tubes with constant mixing for at least 6 hours. Following conjugation the samples were centrifuged at 13,000 g for 6 minutes (Eppendorf miniSpin centrifuge, Hamburg, Germany) and the supernatant was added to HPLC sample vials. A Viva C18 150 × 4.6 mm column from Restek (Bellefonte, PA) was used with a Shimadzu Prominence UFLC (Kyoto, Japan). The mobile phase was 97% acetonitrile and 3% methanol. Samples were all run in triplicate on the HPLC with injection volumes of 10 μL and a flow rate of 1 mL/min for 13 minutes per sample. The absorbance at 254 nm was analyzed for the characteristic peaks of degraded PS-80. A sample chromatogram comparing degraded and non-degraded PS-80 is shown in Supplementary Figure 2. The ADAM-labeled polysorbate degradation product has a characteristic peak at 7 minutes. The EnzyChrom™ Free Fatty Acid Kit (Fisher Scientific, Fair Lawn, NJ, USA) was used as a secondary method to confirm the results of the HPLC assay described by directly measuring the release of fatty acid by lipase.
CHO HCCF prepared from CRISPR transfected cells, TALEN transfected cells, and control cultures were independently buffer-exchanged into pH 6.8 buffer containing 10 mM CaCl2 (Sigma-Aldrich Chemical Co). PS-80 and PS-20 were then added to the buffer-exchanged HCPs to final concentrations of 0.23 mM (0.03% w/w) and 0.27 mM (0.03% w/w), respectively. The mixture was incubated at 37 °C for 12 hours with mixing. Enzymatic degradation of PS-80 and PS-20 were measured using the EnzyChrom™ Free Fatty Acid Kit to measure the concentration of fatty acid released during polysorbate hydrolysis. The LPL inhibitor study also included 2 μM apolipoprotein C-III (apoC-III, Sigma-Aldrich Chemical Co.) in the reaction mixture.
Quantification of oleic acid by LC-Multiple Reaction Monitoring (MRM) assay
LC-MRM analysis was performed on a QTrap 2000 (AB Sciex, Foster City, CA) equipped with an Agilent 1100 HPLC (Agilent, Wilmington, DE). The reaction product was diluted 50 times with 90% methanol (Avantor Performance Materials, Center Valley, PA), 0.1% acetic acid (Avantor Performance Materials). For normalization, U-13C oleic acid was spiked in as an internal standard at a concentration of 0.5 μM. Five μL of the diluted reaction mixture was injected onto a C18 reverse phase column (3 μm, 100 Å, 100 mm × 2 mm, Phenomenex, Torrance, CA), and isocratically eluted with 90% methanol, 0.1% acetic acid for 25 min at a flow rate of 200 μL/min. The eluate was introduced to the QTrap 2000 through a Turbo V source from 6 min to 15 min. The data were acquired in negative ion mode with pseudo MRM with the following settings: 20 psi curtain gas, -4500 V ion spray voltage, 450 °C temperature, 45 psi ion source gas 1, 50 psi ion source gas 2, and -16 V collision energy. Oleic acid and the internal standard were monitored at 281.2/281.2 and 299.2/299.2 respectively. Peak integration was performed with Analyst 1.6.2 (AB Sciex) in quantitation mode. Oleic acid concentrations were calculated based on a calibration curve. All samples were measured using biological triplicates in technical duplicates.
Results and discussion
E. coli-expressed CHO LPL impacts PS degradation
The experimentally measured degradation rates of PS-80 by CHO LPL produced in E. coli are shown in Figure 2. Activity was measured at 37 °C at pH 5.0, 6.0 and 6.8 in the presence of either NaCl, CaCl2 or no additional salt. These conditions were chosen to mimic FDA-approved mAb formulation conditions (Daugherty and Mrsny, 2006; Wang et al., 2009) and Ca2+ was previously found to promote the formation of active LPL dimers (Kobayashi et al., 2002). Overall, there is measurable PS-80 degradation in almost all of the conditions tested (and more than in a buffer-only control). Degradation rates were found to increase from 0.2±0.08 to 3.9±0.7 μmol PS-80/μmol LPL/hour in no salt and from 0.3±0.08 to 10.1±0.8 μmol PS-80/μmol LPL/hour in 10 mM CaCl2 with increasing pH (pH 5.0-6.8 in no salt and pH 6.0-6.8 in 10 mM CaCl2), consistent with prior work on lipase catalysis that showed maximum rates at higher pH (Pereira et al., 2003; Shirai et al., 1983). The addition of the two salts had only a small effect, contrary to previous findings (Kobayashi et al., 2002). The highest rate of degradation, at 10 μmol PS-80/μmol LPL/hour, was found at pH 6.8 with 10 mM CaCl2, but similar rates were found with NaCl and no additional salt; neither salt appears necessary for active LPL degradation of PS-80. The PS-80 degradation rates measured here were similar to those in previous findings with pancreatin, which contains lipases (Christiansen et al., 2010), and are consistent with the possibility that CHO LPL may influence PS stability under certain conditions. At the same time, E. coli is unable to perform N-linked and so the E. coli-expressed LPL may not have the same, full activity as a natively processed LPL (Semb and Olivecrona, 1989) thus underscoring the need to test the impact of CHO LPL.
Figure 2.
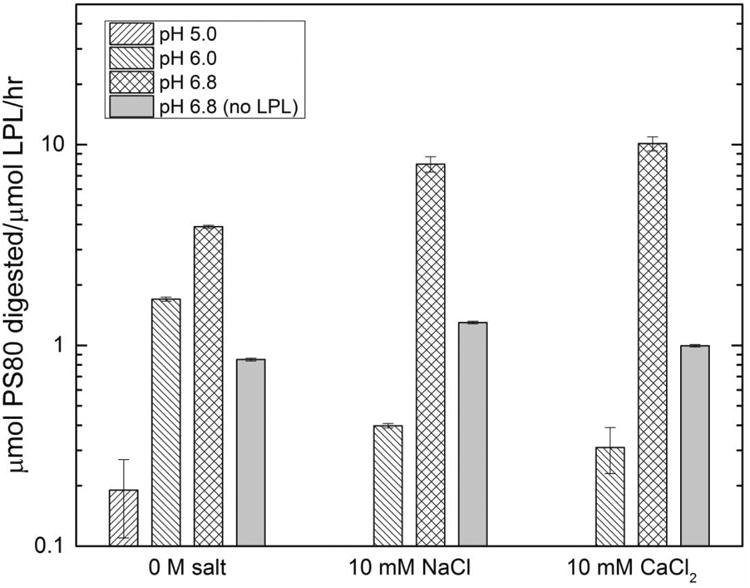
Average degradation rate of PS-80 in solutions containing recombinant CHO LPL (produced in E. coli) in different solution conditions at 37 °C for 24 hours.
Design of sgRNA target site for CRISPR knockout of LPL
A rational design of the sgRNA target site is necessary to knock out LPL function while minimizing off-target genome-editing on other CHO genes (Fu et al., 2014; Kuscu et al., 2014; Ran et al., 2013). The exon and its corresponding function were taken into consideration when selecting target sequences. The lpl gene is composed of 10 exons and 9 introns (Braun & Severson, 1992), with six exons encoding functional sites. Exons 5, 6, and 8 of lpl were the three target domains chosen for CRISPR genome editing. The Gly-Xaa-Ser-Xaa-Gly active site of LPL, encoded by exon 5, is essential for the hydrolysis of lipids. The heparin-binding site, encoded by exon 6, serves as a bridge between the protein and lipoprotein. A study has shown that an inactive heparin-binding site diminishes LPL activity (Lutz et al., 2001). N-linked glycosylation, with sites encoded by exons 2 and 8, are required for LPL to be catalytically active. The selected target sequences for functional lpl knockout (Supplementary Table I) are present only in the lpl gene in the CHO-K1 genome, contain the lowest number of off-target mismatches, and are predicted to disrupt at least one important functional site.
Three sgRNA expression vectors, each specific for their respective lpl exon target, were constructed and individually co-transfected with a Cas9 expression vector in CHO-K1 cells. The efficacy of CRISPR-mediated genome-editing was determined three days post-transfection, with transfection efficiency of 91% and cells at 63% viability (Supplementary Figure 3). The indel percentages observed in CRISPR-treated cell populations were 7%, 1%, and 0% for cells transfected with sg1, sg2, and sg3, respectively (Supplementary Figure 4). Indels not observed in cells treated with sg3 may be contributed by the instability of sgRNA formation, which depends on folding energy and G-quadruplex formation (Moreno-Mateos et al., 2015).
Characterization of CHO-K1 lpl knockout cell lines by sequencing, western analysis and LPL-specific MRM assay
In an initial experiment, 96 wells of TALEN and 96 wells of CRISPR clones were expanded by dilution cloning. 26 TALEN and 15 CRISPR clones (41 total) were subsequently expanded into 6 well plates. The LPL expression levels of the 41 isolated populations and a wild-type control in CHO HCCF were analyzed by an MRM assay, which measures protein expression at the peptide level. Treatment of CHO-K1 cells with TALEN and CRISPR reduced LPL expression by 80-99% (Figure 3). Here, we also highlight the establishment of an MRM assay that is specific to LPL to detect LPL peptides in CHO HCCF, a mixture with hundreds of HCPs.
Figure 3.
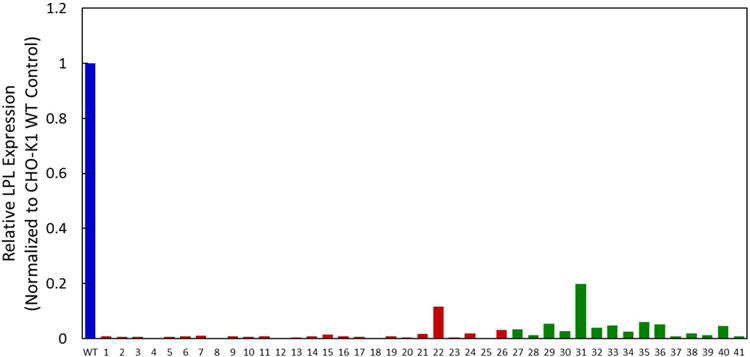
Relative LPL protein expression measured with a LPL-specific MRM assay for initial 26 TALEN-treated and 15 CRISPR-treated CHO-K1 cells. LPL expressions are normalized to the wildtype control, n = 1. Wildtype is shown in blue, TALEN in red, and CRISPR in green.
In an additional experiment, five separate CRISPR-based cell lines (42, 43, 44, 45, 46), were isolated and selected for further characterization, with three cell lines (42, 43, 44) targeting exon 5 and two cell lines (45, 46) targeting exon 6 showed indels in lpl (Supplementary Figure 5A). Sequencing results confirmed that there are two different mutations in cell lines 42, 43, and 44, and one mutation in cell lines 45 and 46, where cell lines 42, 43, and 44 each may contain two subpopulations with changes in lpl, while cell lines 45 and 46 each contain one subpopulation. Four of the five cell lines (43, 44, 45, and 46) contains subpopulations with frameshift mutations, which would result to the expression of a truncated LPL protein, if expressed (Supplementary Figure 5B). Cell line 42 contains one subpopulation with a frameshift mutation and another subpopulation that leads to a 6-amino acid deletion that occurs at the active site, which would result to the expression of a catalytically inactive LPL.
The five CHO-K1 lpl knockout cell lines, for which lpl knockout was confirmed by sequencing, were subjected to western analysis for their relative expression of LPL. Two LPL antibodies were selected to probe for the expression of N-terminal and C-terminal regions of the LPL protein. Western analysis shows reduced expression of LPL near the expected molecular weight of 53 kDa in the lpl knockout cell lines (Figure 4A and Supplementary Information Figure 6). Cell line 42 exhibited reduced expression of the modified 52.2 kDa LPL, but not the modified 24.7 kDa LPL protein. Cell lines 43, 44, and 46 did not exhibit expression of either native LPL or modified LPL. Native LPL expression was not detected in cell line 45. However, cell line 45 exhibited expression of a truncated LPL protein was detected by the N-terminus LPL antibody, as predicted by the change made in the lpl gene. The faint band in each lane below the molecular weight of LPL is a non-specific binding of the antibody and is seen in the western images provided by the manufacturer.
Figure 4.
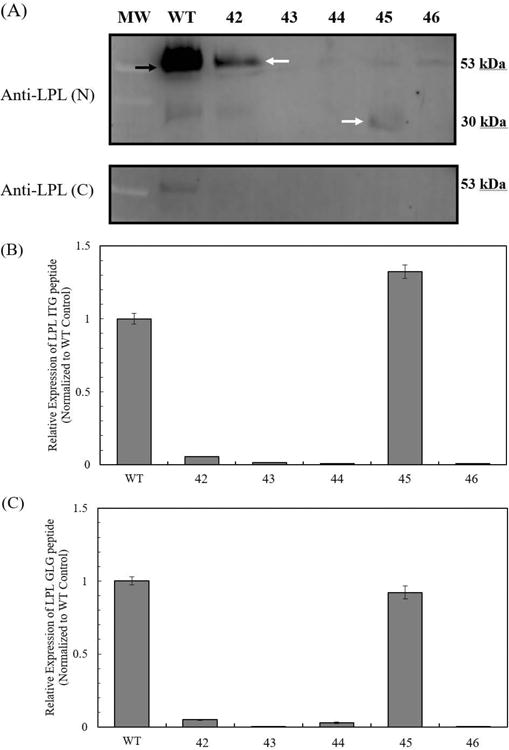
Characterization of LPL protein expression from CHO lpl knockout cell lines by western analysis and MRM. (A) Expression of LPL protein probed by the N-terminus LPL antibody (Anti-LPL(N)) and C-terminus LPL antibody (Anti-LPL (C)). The predicted molecular weight of LPL is 53 kDa. The red arrow indicates the native LPL band. The yellow arrow indicates the altered LPL protein expressed by cell line 42, with predicted molecular weight of 52.2 kDa. The white arrow indicates the altered LPL protein expressed by cell line 45, with predicted molecular weight of 29.9 kDa. (B) Relative expression of LPL ITG peptide by MRM. (C) Relative expression of LPL GLG peptide by MRM. LPL expressions are normalized to the wildtype control. Error bars represent the standard error of the mean from three technical replicates.
LPL expression from lpl knockout cells and wild-type cells were analyzed by an MRM assay. The peptide fragments used for the detection of LPL expression are ITGLDPAGPNFEYAEAPSR (ITG) and GLGDVDQLVKC (GLG). The positions of the peptide fragments and the starting regions affected by CRISPR in LPL are shown in Supplementary Figure 7. Lpl knockout by CRISPR reduced LPL expression by 95-99% in cell lines 42, 43, 44 and 46 (Figure 4B, C). CHO HCP from cell line 45 contains amounts of ITG and GLG peptides similar to those of the wild-type control, which may be representative of degraded LPL not detected previously by western rather than the presence of native LPL. Western and MRM results both support the reduction of native LPL expression in all five CHO-K1 lpl knockout cell lines. The successful knockout of LPL at the protein level occurred through frameshift mutations in the exons (cell lines 43, 44, 45, 46) that yielded either no expression or truncated expression of LPL proteins.
Culture performance of LPL knockout CHO-K1 cell lines
The five CHO-K1 lpl knockout cell lines (42, 43, 44, 45, 46) were cultivated in parallel to the wild-type CHO-K1 control to evaluate the effect of a functional lpl knockout on culture performance. All five lpl knockout cell lines showed similar integrated viable cell density (IVCD) to that of the wild-type control during the first six days of cultivation and by day 10 the IVCD of lpl knockouts differs from that of the wild-type control by -16% to +5% (Figure 5A). High levels of cell viability were maintained for 8 days across all cultures, with three lpl knockout cell lines (A, C, D) able to maintain longer cell culture durations with over 50% viability (Figure 5B). Changes to total extracellular protein in the HCCF on day 4 of cultivation were not statistically significant (p > 0.05), with the exception of cell lines 43 (p = 0.013) and 46 (p = 0.046), which produced 29% and 17% more protein than the wild-type control, respectively (Figure 5C).
Figure 5.
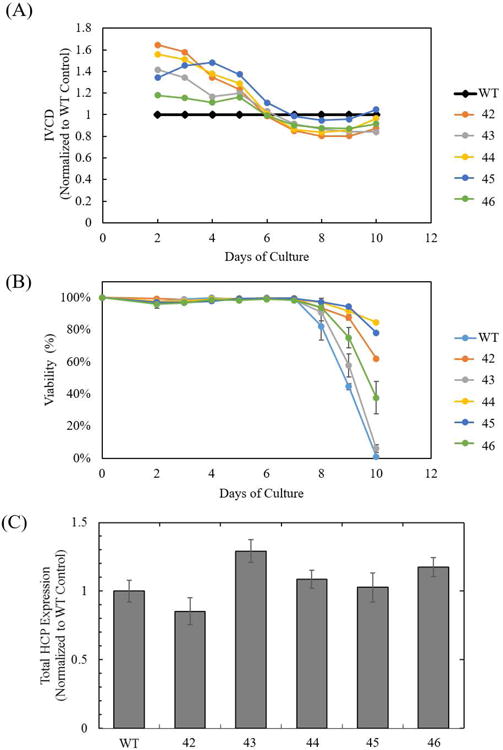
Cell culture performance of CHO-K1 lpl knockout cell lines. (A) Integrated viable cell density (IVCD), relative to the wild-type control, (B) viability cell culture profile of CHO-K1 wild-type, and cell lines 42, 43, 44, 45, and 46. Error bars represent the standard error of the mean from two biological replicates. (C) Total extracellular protein expression at day 4 of cell culture. Error bars represent the standard error of the mean from three biological replicates.
Reduction of IVCD in lpl knockout cell lines suggests that LPL activity, or free fatty acid production from LPL activity, affects cell growth. Previous studies have shown that the addition of free fatty acids and phospholipids in mammalian cell culture promotes cell growth (Wicha et al, 1979; Prasad, 1980; Schmid et al., 1991). Minimal changes to IVCD across the cell lines indicate that the absence of LPL does not appreciably impair cell growth while variable total protein production between the cell lines does not give a clear indication of LPL's role in HCP productivity. These observations demonstrate that LPL does not have a significant negative impact on cell performance.
Polysorbate degradation is reduced in lpl knockout cell lines
PS-80 or PS-20 with a final concentration of 0.03% w/w was incubated with concentrated CHO HCCF obtained from wild-type CHO-K1 cells or CHO-K1 lpl knockout cells. Samples used in an LPL inhibitor study were incubated with either PS-80 or PS-20 and 2 μM apoC-III, which has been shown to specifically inhibit LPL activity (Bobik, 2008; Larsson et al., 2013; McConathy et al., 1992). Wild-type CHO HCCF degraded 34% (85 μM) of the total PS-80 after 12 hours. Incubation of CHO wild-type HCCF with apoC-III reduced PS-80 degradation by 28% (Figure 6A) as measured by an MRM assay, which is consistent with prior observations regarding a CHO HCP impact on PS-80 stability. HCCF from the various lpl knockout lines degrade anywhere from 18% (45 μM) to 20% (50 μM) of total PS-80, representing a 41% to 47% improvement in PS-80 stability compared to the CHO-K1 wild-type control (Figure 6A). Moreover, addition of apoC-III did not alter the ability of lpl knockout HCCF to degrade PS-80.
Figure 6.
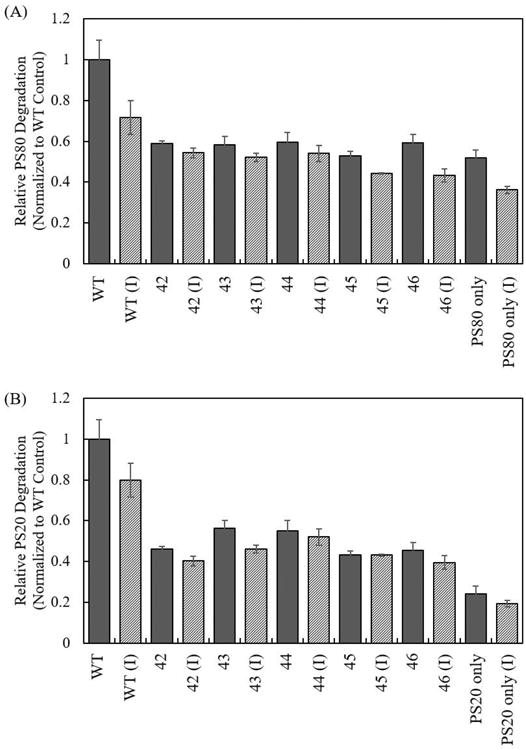
Effect of LPL on the degradation of (A) PS-80 and (B) PS-20 by CHO HCCF from CHO-K1 wild-type control (WT), CHO-K1 lpl knockout cell lines (42, 43, 44, 45, and 46), and apoC-III (I). Error bars represent the standard error of the mean from three biological replicates.
A similar set of experiments was carried out to study the effects of LPL on PS-20 degradation. HCCF from CHO wild-type control degraded 38% (102 μM) of total PS-20. Incubation of CHO wild-type HCCF with apoC-III reduced PS-20 degradation by 20% (Figure 6B). HCCF from CHO-K1 lpl knockouts degrade 16% (44 μM) to 21% (58 μM) of total PS-20, representing a 44% to 57% improvement in PS-20 stability, compared to the CHO-K1 wild-type control (Figure 6B) measured using the fatty acid assay. The addition of apoC-III did not alter the ability of lpl knockout HCCF to degrade PS-20, which suggests that LPL is not catalytically active in knockout cell lines, if present at all.
Joucla et al. identified LPL as composing 0.1% of the total CHO HCCF concentration (2013). Although the exact protein composition of HCCF used in each polysorbate degradation assay is unknown, the reduction in polysorbate degradation (41% to 57%) by CHO-K1 lpl knockouts HCCF suggests that LPL, though not abundantly present in CHO HCCF, plays an outsized role in polysorbate hydrolysis.
Conclusions
LPL is an HCP impurity that is expressed and secreted by CHO cells and is difficult to remove during downstream purification because it exhibits product association and similar retention characteristics to mAbs on polishing chromatographic resins. This study shows that persistence of LPL through downstream purification operations and into the final drug product can degrade polysorbates. CHO-K1 lpl knockout cell lines (generated using CRISPR) reduced LPL expression by greater than 95% while maintaining properties important for biopharmaceutical processing and reducing polysorbate degradation by 41% to 57%.
The gene-silencing and gene-disruption techniques used in this work can be applied to study LPL during biopharmaceutical process development or to reduce LPL expression during therapeutic protein manufacturing. Additionally, the techniques shown here are not specific to LPL and can be applied to study the impact of reduced expression of other difficult-to-remove HCP impurity.
Supplementary Material
Acknowledgments
We are grateful for the support from the National Science Foundation under grant no. CBET-0966644, CBET-1144726, and MCB-1412365, and the National Institute of Health IDeA Networks of Biomedical Research Excellence (NIH-NIGMS P20 GM103446) for support to the Gene Editing Institute at Christiana Care Hospital that provided the TALENs. JC was supported in part by NIH T32GM008550.
References
- Aboulaich N, Chung WK, Thompson JH, Larking C, Robbins D. A novel approach to monitor clearance of host cell proteins associated with monoclonal antibodies. Biotechnol Prog. 2014;30:1114–1124. doi: 10.1002/btpr.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobik A. Apolipoprotein CIII and atherosclerosis: beyond effects on lipid metabolism. Circulation. 2008;118:702–704. doi: 10.1161/CIRCULATIONAHA.108.794081. [DOI] [PubMed] [Google Scholar]
- Braun JEA, Severson DL. Regulation of the synthesis, processing and translocation of lipoprotein lipase. Biochem J. 1992;287:337–347. doi: 10.1042/bj2870337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkrolf K, Rupp O, Laux H, Kollin F, Ernst W, Linke B, Kofler R, Romand S, Hesse F, Budach WE, Galosy S, Muller D, Noll T, Wienberg J, Jostock T, Leonard M, Grillari J, Tauch A, Goesmann A, Helk B, Mott JE, Puhler A, Borth N. Chinese hamster genome sequenced from sorted chromosomes. Nat Biotechnol. 2013;31:694–695. doi: 10.1038/nbt.2645. [DOI] [PubMed] [Google Scholar]
- Champion KM, Madden H, Dougherty J, Shacter E. Defining your product profile and maintaining control over it, part 2. Bioprocess Int. 2005;3:52–57. [Google Scholar]
- Choi YS, Hou S, Choe LH, Lee KH. Targeted human cerebrospinal fluid proteomics for the validation of multiple Alzheimer's disease biomarker candidates. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;930:129–135. doi: 10.1016/j.jchromb.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen A, Backensfeld T, Weitschies W. Effects of non-ionic surfactants on in vitro triglyceride digestion and their susceptibility to digestion by pancreatic enzymes. Eur J Pharm Sci. 2010;41:376–382. doi: 10.1016/j.ejps.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Daugherty AL, Mrsny RJ. Formulation and delivery issues for monoclonal antibody therapeutics. Adv Drug Deliv Rev. 2006;58:686–706. doi: 10.1016/j.addr.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Dixit N, Salamat-Miller N, Salinas PA, Taylor KD, Basu SK. Residual host cell protein promotes polysorbate 20 degradation in a sulfatase drug product leading to free fatty acid particles. J Pharm Sci. 2016;105:1657–1666. doi: 10.1016/j.xphs.2016.02.029. [DOI] [PubMed] [Google Scholar]
- Doneanu C, Xenopoulos A, Fadgen K, Murphy J, Skilton S, Prentice H, Stapels M, Chen W. Analysis of host-cell proteins in biotherapeutic proteins by comprehensive online two-dimensional liquid chromatography/mass spectrometry. MAbs. 2012;4:24–44. doi: 10.4161/mabs.4.1.18748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA. Guidance for Industry. Q6B Specifications: Test procedures and acceptance criteria for biotechnological/biological products 1999 [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR–Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao SX, Zhang Y, Stansberry-Perkins K, Buko A, Bai S, Nguyen V, Brader ML. Fragmentation of a highly purified monoclonal antibody attributed to residual CHO cell protease activity. Biotechnol Bioeng. 2011;108:977–982. doi: 10.1002/bit.22982. [DOI] [PubMed] [Google Scholar]
- Grav LM, Lee JS, Gerling S, Kallehauge TB, Hansen AH, Kol S, Lee GM, Pedersen LE, Kildegaard HF. One-step generation of triple knockout CHO cell lines using CRISPR/Cas9 and fluorescent enrichment. Biotechnol J. 2015;10:1446–1456. doi: 10.1002/biot.201500027. [DOI] [PubMed] [Google Scholar]
- Gutierrez AH, Moise L, De Groot AS. Of [hamsters] and men. Hum vaccines Immunother. 2012;8:1172–1174. doi: 10.4161/hv.22378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha E, Wang W, Wang YJ. Peroxide formation in polysorbate 80 and protein stability. J Pharm Sci. 2002;91:2252–2264. doi: 10.1002/jps.10216. [DOI] [PubMed] [Google Scholar]
- Hall T, Sandefur SL, Frye CC, Tuley TL, Huang L. Polysorbates 20 and 80 degradation by group xv lysosomal phospholipase A2 isomer X1 in monoclonal antibody formulations. J Pharma Sci. 2016;105:1633–1642. doi: 10.1016/j.xphs.2016.02.022. [DOI] [PubMed] [Google Scholar]
- Hanania N, Noonan M, Corren J, Korenblat P, Zheng Y, Fischer S, Cheu M, Putnam W, Murray E, Scheerens H, Holweg C, Maciuca R, Gray S, Doyle R, McClintock D, Olsson J, Matthews J, Yen K. Lebrikizumab in moderate-to-severe asthma: pooled data from two randomised placebo-controlled studies. Thorax. 2015;0:1–9. doi: 10.1136/thoraxjnl-2014-206719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Szapiel N, Zhang J, Hickey J, Ghose S. Profiling of host cell proteins by two-dimensional difference gel electrophoresis (2D-DIGE): Implications for downstream process development. Biotechnol Bioeng. 2010;105:306–316. doi: 10.1002/bit.22532. [DOI] [PubMed] [Google Scholar]
- Joucla G, Le Senechal C, Begorre M, Garbay B, Santarelli X, Cabanne C. Cation exchange versus multimodal cation exchange resins for antibody capture from CHO supernatants: identification of contaminating host cell proteins by mass spectrometry. J Chromatogr B. 2013:942–943. 126–133. doi: 10.1016/j.jchromb.2013.10.033. [DOI] [PubMed] [Google Scholar]
- Kerwin BA. Polysorbates 20 and 80 used in the formulation of protein biotherapeutics: Structure and degradation pathways. J Pharm Sci. 2008;97:2924–2935. doi: 10.1002/jps.21190. [DOI] [PubMed] [Google Scholar]
- Khossravi M, Kao YH, Mrsny RJ, Sweeney TD. Analysis methods of polysorbate 20: A new method to assess the stability of polysorbate 20 and established methods that may overlook degraded polysorbate 20. Pharm Res. 2002;19:634–639. doi: 10.1023/a:1015306112979. [DOI] [PubMed] [Google Scholar]
- Kishore RSK, Pappenberger A, Dauphin IB, Ross A, Buergi B, Staempfli A, Mahler HC. Degradation of polysorbates 20 and 80: Studies on thermal autoxidation and hydrolysis. J Pharm Sci. 2011;100:721–731. doi: 10.1002/jps.22290. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Nakajima T, Inoue I. Molecular modeling of the dimeric structure of human lipoprotein lipase and functional studies of the carboxyl-terminal domain. Eur J Biochem. 2002;269:4701–4710. doi: 10.1046/j.1432-1033.2002.03179.x. [DOI] [PubMed] [Google Scholar]
- Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. Genome–wide analysis reveals characteristics of off–target sites bound by the Cas9 endonuclease. Nat Biotechnol. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- Larsson M, Vorrsjo E, Talmud P, Lookene A, Olivecrona G. Apolipoproteins C-I and C-III inhibit lipoprotein lipase activity by displacement of the enzyme from lipid droplets. J Biol Chem. 2013;288:33997–34008. doi: 10.1074/jbc.M113.495366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, McAuley A, Schilke KF, McGuire J. Molecular origins of surfactant-mediated stabilization of protein drugs. Adv Drug Deliv Rev. 2011;63:1160–1171. doi: 10.1016/j.addr.2011.06.015. [DOI] [PubMed] [Google Scholar]
- Lee JS, Kallehauge TB, Pedersen LE, Kildegaard HF. Site-specific integration in CHO cells mediated by CRISPR/Cas9 and homology-directed DNA repair pathway. Sci Rep. 2015;5:8572. doi: 10.1038/srep08572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy NE, Valente KN, Choe LH, Lee KH, Lenhoff AM. Identification and characterization of host cell protein product-associated impurities in monoclonal antibody bioprocessing. Biotechnol Bioeng. 2014;111:904–12. doi: 10.1002/bit.25158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy NE, Valente KN, Lee KH, Lenhoff AM. Host cell protein impurities in chromatographic polishing steps for monoclonal antibody purification. Biotechnol Bioeng. 2015 doi: 10.1002/bit.25882. [DOI] [PubMed] [Google Scholar]
- Lutz EP, Merkel M, Kako Y, Melford K, Radner H, Breslow JL, Bensadoun A, Goldberg IJ. Heparin-binding defective lipoprotein lipase is unstable and causes abnormalities in lipid delivery to tissues. J Clin Invest. 2001;107:1183–1192. doi: 10.1172/JCI11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler HC, Senner F, Maeder K, Mueller R. Surface activity of a monoclonal antibody. J Pharm Sci. 2009;98:4525–4533. doi: 10.1002/jps.21776. [DOI] [PubMed] [Google Scholar]
- Marichal-Gallardo PA, Alvarez MM. State-of-the-art in downstream processing of monoclonal antibodies: process trends in design and validation. Biotechnol Prog. 2012;28:899–916. doi: 10.1002/btpr.1567. [DOI] [PubMed] [Google Scholar]
- McConathy WJ, Gesquiere JC, Bass SH, Tartar A, Fruchart JC, Wang CS. Inhibition of LPL activity by synthetic peptides of apolipoprotein C-III. J Lipid Res. 1992;33:995–1003. [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods. 2015;12:982–988. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson-Ehle P, Garfinkel AS, Schotz MC. Lipolytic enzymes and plasma lipoprotein metabolism. Annu Rev Biochem. 1980;49:667–693. doi: 10.1146/annurev.bi.49.070180.003315. [DOI] [PubMed] [Google Scholar]
- Pereira EB, Zanin GM, Castro HF. Immobilization and catalytic properties of lipase on chitosan for hydrolysis and esterification reactions. Brazilian J Chem Eng. 2003;20:343–355. [Google Scholar]
- Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9:555–568. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- Prasad KN. Butyric acid: A small fatty acid with diverse biological functions. Life Sci. 1980;27:1351–1358. doi: 10.1016/0024-3205(80)90397-5. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert F, Bierau H, Rossi M, Agugiaro D, Soranzo T, Broly H, Mitchell-Logean C. Degradation of an Fc-fusion recombinant protein by host cell proteases: identification of a CHO cathepsin D protease. Biotechnol Bioeng. 2009;104:1132–1141. doi: 10.1002/bit.22494. [DOI] [PubMed] [Google Scholar]
- Ronda C, Pedersen LE, Hansen HG, Kallehauge TB, Betenbaugh MJ, Nielsen AT, Kildegaard HF. Accelerating genome editing in CHO cells using CRISPR Cas9 and CRISPy, a web-based target finding tool. Biotechnol Bioeng. 2014;111:1604–1616. doi: 10.1002/bit.25233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid G, Zilg H, Eberhard U, Johannsen R. Effect of free fatty acids and phospholipids on growth of and product formation by recombinant baby hamster kidney (rBHK) and Chinese hamster ovary (rCHO) cells in culture. J Biotechnol. 1991;17:155–167. doi: 10.1016/0168-1656(91)90006-h. [DOI] [PubMed] [Google Scholar]
- Semb H, Olivecrona T. The relation between glycosylation and activity of guinea pig lipoprotein lipase. J Biol Chem. 1989;264:4195–4200. [PubMed] [Google Scholar]
- Shirai K, Fitzharris TJ, Shinomiya M, Muntz HG, Harmony JA, Jackson RL, Quinn DM. Lipoprotein lipase-catalyzed hydrolysis of phosphatidylcholine of guinea pig very low density lipoproteins and discoidal complexes of phospholipid and apolipoprotein: effect of apolipoprotein C-II on the catalytic mechanism. J Lipid Res. 1983;24:721–30. [PubMed] [Google Scholar]
- Singh SK, Cousens LP, Alvarez D, Mahajan PB. Determinants of immunogenic response to protein therapeutics. Biologicals. 2012;40:364–368. doi: 10.1016/j.biologicals.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Sun T, Li C, Han L, Jiang H, Xie Y, Zhang B, Qian X, Lu H, Zhu J. Functional knockout of FUT8 in Chinese hamster ovary cells using CRISPR/Cas9 to produce a defucosylated antibody. Eng Life Sci. 2015;15:660–666. [Google Scholar]
- Valente KN, Lenhoff AM, Lee KH. Expression of difficult-to-remove host cell protein impurities during extended Chinese hamster ovary cell culture and their impact on continuous bioprocessing. Biotechnol Bioeng. 2015;112:1232–1242. doi: 10.1002/bit.25515. [DOI] [PubMed] [Google Scholar]
- Valente KN, Schaefer AK, Kempton HR, Lenhoff AM, Lee KH. Recovery of Chinese hamster ovary host cell proteins for proteomic analysis. Biotechnol J. 2014;9:87–99. doi: 10.1002/biot.201300190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hunter AK, Mozier NM. Host cell proteins in biologics development: identification, quantitation and risk assessment. Biotechnol Bioeng. 2009;103:446–458. doi: 10.1002/bit.22304. [DOI] [PubMed] [Google Scholar]
- Wicha MS, Liotta LA, Kidwell WR. Effects of free fatty acids on the growth of normal and neoplastic rat mammory epithelial cells. Cancer Res. 1979;19:426–435. [PubMed] [Google Scholar]
- Xu X, Nagarajan H, Lewis NE, Pan S, Cai Z, Liu X, Chen W, Xie M, Wang W, Hammond S, Andersen MR, Neff N, Passarelli B, Koh W, Fan HC, Wang J, Gui Y, Lee KH, Betenbaugh MJ, Quake SR, Famili I, Palsson BO, Wang J. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat Biotechnol. 2011;29:735–741. doi: 10.1038/nbt.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Lookene A, Wu G, Olivecrona G. Calcium triggers folding of lipoprotein lipase into active dimers. J Biol Chem. 2005;280:42580–42591. doi: 10.1074/jbc.M507252200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.