

Abstract
Hepatocellular carcinoma (HCC) is the second-leading cause of cancer-related deaths world-wide with poor survival rate. As many as 40% of HCCs are clonal, with alteration of key tumor suppressor pathways in stem cells is the primary cause of HCC initiation. However, mechanisms that generate metastatic stem cells in pre-neoplastic liver tissue are not well understood. We hypothesized that chronic inflammation is a major driver of the transformation of genetically defective liver stem cells (LSCs) into highly metastatic liver cancer cells in premalignant liver tissue. We developed models of chronic inflammation in wild-type (WT) and β2-spectrin (β2SP)+/- (SPTBN1) mice. CD133+ LSCs derived from pre-neoplastic livers of β2SP+/- mice treated with pIL6 (IL6β2SP+/- LSCs) were highly tumorigenic and metastatic, whereas those derived from WT mice treated with pIL6 (IL6WT LSCs) had significantly less proliferation and no tumorigenic properties. IL6β2SP+/- LSCs not only exhibited nuclear localization of Twist and Slug, markers of epithelial-mesenchymal transition, but also constitutive activation of NFκB (RELA). Knockdown of NFκB decreased the EMT phenotypes and metastatic capacity of these cells. NFκB in IL6β2SP+/- LSCs was activated by TGFβ-activated kinase 1 (TAK1) (MAP3K7), which is associated with poor survival in HCC and IL6 expression. The amount of constitutively activated NFκB increased dramatically from normal to cirrhotic to HCC tissues from human patients. In conclusion, our findings suggest that IL6-mediated inflammation programs constitutive activation of the TAK1-NFκB signaling cascade in CD133+ LSCs and this program interacts with deficient TGFβ signaling, thereby accelerating the transformation of normal LSCs to metastatic cancer stem cells. Indeed, this study is the first to delineate the development of EMT-positive mCSCs in HCC-free liver tissue upon chronic inflammation.
Keywords: EMT, CSCs, metastasis, HCC, NFκB, TAK1, β2SP
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the second leading cause of cancer related deaths worldwide (1). HCC accounts for 70-85% of the total number of primary liver related malignancies (2). Owing to its poor prognosis—HCC patients' 5-year survival rate is less than 5%—identifying new therapeutic options for the disease is warranted (3). The clinical course of HCC is related to a broad spectrum of etiological factors, including hepatitis viruses, fatty diets, chronic alcohol consumption, exposure of hepatotoxins and microorganisms (4). In addition to these etiological factors, repetitive liver injury can initiate chronic inflammation that can lead to fibrosis, cirrhosis, and eventually HCC.
Injured liver tissue has an abundance of the pleiotropic cytokines, transforming growth factor β (TGFβ). The TGFβ signaling pathway, which has a dynamic effect on HCC development, has a pro-fibrogenic role mediated by the activation of hepatic stellate cells. As chronic liver disease progresses to fibrosis, cirrhosis, and finally HCC, TGFβ signaling takes on a tumor suppressive role. This is evidenced by the downregulation of the TGFβ receptor in approximately 25% of large, high-grade HCCs that quickly recur (5, 6). In vitro administration of recombinant human TGFβ2 inhibits the proliferation of HCC cells (7).
For the inhibitory function of TGFβ signaling in HCC progression, the dynamic scaffolding protein β2-spectrin (β2SP) provides key insights into the mechanistic regulation of tumor suppression. β2SP is required to stabilize the SMAD3/SMAD4 complex which transmits TGFβ signaling in the nucleus (8). Loss of β2SP is frequently detected in human HCC (3). Forty to seventy percent of β2SP+/- mice spontaneously develop HCC within 15 months (3). Histological analysis has revealed that tumorigenic liver tissues from β2SP+/- mice have aberrant vascular structures, expansion of endothelial progenitor cells, hyperproliferative hepatocytes, and an upregulation of several oncogenes that promote liver tumorigenesis (3). One recent study demonstrated that β2SP+/- mice have twice as many epithelial cell adhesion molecule+ (EpCAM+) progenitor cells as do WT mice (9). This finding suggests that β2SP inhibits progenitor cells' transformation from a normal to a malignant phenotype in inflamed liver tissue. Also, in vitro and in vivo functional studies have revealed that β2SP negatively regulates cyclin-dependent kinase 4, cyclin D1, and retinoblastoma protein (3, 10), and corroborating β2SP's critical role in TGFβ-induced cell cycle arrest during the G1 phase.
Chronic activation of pro-inflammatory pathways is critical to the progression of hepatitis to liver cancer. Among numerous pro-inflammatory factors, IL6-mediated signaling has been extensively studied in the context of chronic liver diseases and HCC development (11-13). One study found elevated IL6 levels in 40% of liver cancer patients, which suggests that IL6 is associated with HCC progression (14). Indeed, a high IL6 titer is strongly correlated with adverse prognosis in HCC patients (15). Also, the TGFβ pathway induces IL6 secretion to develop chemotherapeutic resistance in HCC (16). As a principal mediator of pro-oncogenesis, IL6 is associated with malignant transformation, expansion of cancer stem cells (CSCs), metastasis, and therapeutic resistance in numerous epithelial cancers (17-19). Together, these observations strongly suggest that IL6 is associated with the development of CSCs in liver cancer. However, it is not investigated on how IL6 driven inflammation interacts with alteration of tumor suppressor pathway to promote CSC initiation.
In injured liver tissue, hepatic stem or progenitor cells must expand to replace damaged hepatocytes which restore normal liver functions. However, chronic inflammation leads to the secretion of cytokines, chemokines, free radicals, and other DNA-damaging molecules, thereby changing the hepatic microenvironment (20). Persistent inflammation during this long-term regenerative process leads to an expansion of hepatic stem and progenitor cells that accumulate genetic and epigenetic alterations. Characterizing the bidirectional interaction between stem cells and the hepatic inflammatory milieu would provide great insight into the early phases of HCC initiation. Also, determining whether epithelial–mesenchymal transition (EMT)-positive CSCs are generated during the early phases of HCC initiation is important. Furthermore, elucidating aberrant signaling pathways in EMT+ CSCs may help to develop specific and individualized therapies for preventing HCC progression.
In this study, we investigated the role of the interaction between deficient TGFβ signaling and IL6-mediated chronic inflammation in reprogramming normal liver stem cells (LSCs) to develop into metastatic liver cells (mCSCs). Our results provide a clinically relevant insight into the early development of mCSCs and suggest a potential therapeutic target to eradicate these in pre-neoplastic liver tissue.
Materials and Methods
Chemical reagents and antibodies
Collagenase type I, DNase I, and Pronase were purchased from Sigma Aldrich. The anti-CD133 antibody was purchased from Miltenyi Biotech. The anti-αSMA, anti-IL6, anti-pNFκB (S536), anti-NFκB, and anti-TAK1 antibodies were purchased from Abcam. The anti-β2SP antibody was a gift from Dr. Lopa Mishra's laboratory. The anti-pYSTAT3, anti-STAT3, anti-TAK1, anti-pIκK α/β and anti-pTAK1 antibodies were purchased from Cell Signaling Technology. The anti-IκKβ was purchased from R&D systems.
Mice
Ten- to twelve-week-old female wild-type (WT) and β2SP+/- mice were provided by Dr. Lopa Mishra's laboratory (8). They were maintained in a specific-pathogen-free facility in accordance with guidelines approved by the Institutional Animal Care and Use Committee of M.D.Anderson Cancer Center. The PiggyBac Transposon System (pB513B transposon and Super PB transposase vector) was purchased from System Biosciences, Inc. Mouse IL6 cDNA was obtained from GE Dharmacon, Inc., and subcloned into the pB513B vector. IL6 cytokine–encoding DNA or control plasmid DNA (5 μg in 1.5 ml of 0.45% saline solution per mouse) was injected hydrodynamically through the tail vein twice per month for 3 months. Eight- to ten-week-old female immunodeficient NOD SCID gamma chain knockout (NSG) mice were purchased from the Jackson Laboratory.
Western blotting
Cell lysis and Western blotting with antibodies against pNFκB, NFκB, pTAK1, TAK1, and GAPDH were performed as described previously (21). For all samples, the total protein concentrations were determined with a bicinchoninic protein assay kit (Pierce). Western blots were detected with an enhanced chemiluminescence kit (Cell Signaling Technologies).
Isolation and culture of primary tumor cell lines
Mice were humanely killed after 3 months of treatment or 15 months old non-treated β2SP+/- mouse and single cell suspension from liver was made as described previously (22). A magnet-activated cell-sorting column with an anti–α-CD133 antibody was used to identify CD133+ LSCs, which were then cultured in poly-D-lysine/laminin–coated plates containing liver-specific supplements in Dulbecco's modified Eagle's medium/F12 media with 10% heat-inactivated serum. The liver specific supplements were rHGF (hepatocyte growth factor) (50ng/ml), rEGF (epidermal growth factor) (20ng/ml), insulin-transferrin-selenium (1×), rFGF (fibroblast growth factor) (20 ng/ml), dexamethasone (1× 10-7 mol/l) and nicotinamide (10 mmol/l) (22).
Immunofluorescent staining of tissue sections
Tissue microarrays (TMAs) of patient liver tissues were purchased from Biomax, Inc., in accordance with a protocol approved by MD Anderson's Institutional Review Board. Antigen retrieval was performed with heat-induced 1× citrate buffer (Sigma Aldrich) for 30 minutes. Tissue staining was performed as described previously (23). Stained TMAs were analyzed with the Vectra automated quantitative pathology imaging system (Perkin Elmer), which is capable of multispectral imaging. Cell segmentation analysis and protein scoring (based on fluorescence or 3, 3′-diaminobenzidine staining) were performed using the inForm software program (Perkin Elmer).
Screening of cell signaling pathways
IL6β2SP+/- LSCs (∼1 × 104 cells/well) were treated with different inhibitors targeting HCC-related cell signaling pathways for 24 or 48 h. All inhibitors were used at biologically effective concentrations. To measure the viability of IL6β2SP+/- LSCs after treating with different inhibitors, 100 μl of CyQUANT NF Cell Proliferation Assay reagent was added to each well after aspiration of medium. After incubation for 1 h at 37°C, fluorescence was measured (excitation 485 nm, emission 538 nm) on a SpectraMax Geminin EM microplate reader (Molecular Devices).
Generation of NFκB-knockdown primary tumor cell line
MISSION shNFκB plasmid was purchased from Sigma Aldrich (NM_009045). 5 μg plasmid was nucleofected in IL6β2SP+/- LSCs using AMAXA nucleofector (program Q25). Cells were maintained in puromycin (10 μg/ml) for 3 weeks to grow the NFκB knockdown cell line. NFκB knockdown was confirmed by Western blotting with an anti–α-NFκB antibody.
Results
HCC tissues have low nuclear β2SP accumulation and high IL6 levels
The adaptor protein β2SP plays an essential role in translocating the SMAD3/SMAD4 complex in the nucleus to drive TGFβ-mediated tumor suppression (24). Thus, the disruption of TGFβ signaling via a reduced accumulation of nuclear β2SP is critical to HCC progression. Given the prominent role of nuclear β2SP in the tumor suppressor activity of TGFβ signaling, the nuclear accumulation of β2SP was compared and quantitated by immunohistochemical staining (IHC) in humans with normal, cirrhotic, and HCC liver tissues. In normal liver tissue, β2SP was strongly expressed in both the nucleus (mean IHC score = 0.188) and the cytoplasm (Fig. 1 a). In contrast, the nuclear accumulation of β2SP was diminished in cirrhotic liver tissue (mean IHC score = 0.15) and significantly decreased in HCC tissue (mean IHC score = 0.097; p < 0.0001) (Fig. 1 c).
Fig. 1. Nuclear β2SP accumulation is inversely related to IL6 level in HCC tissues.
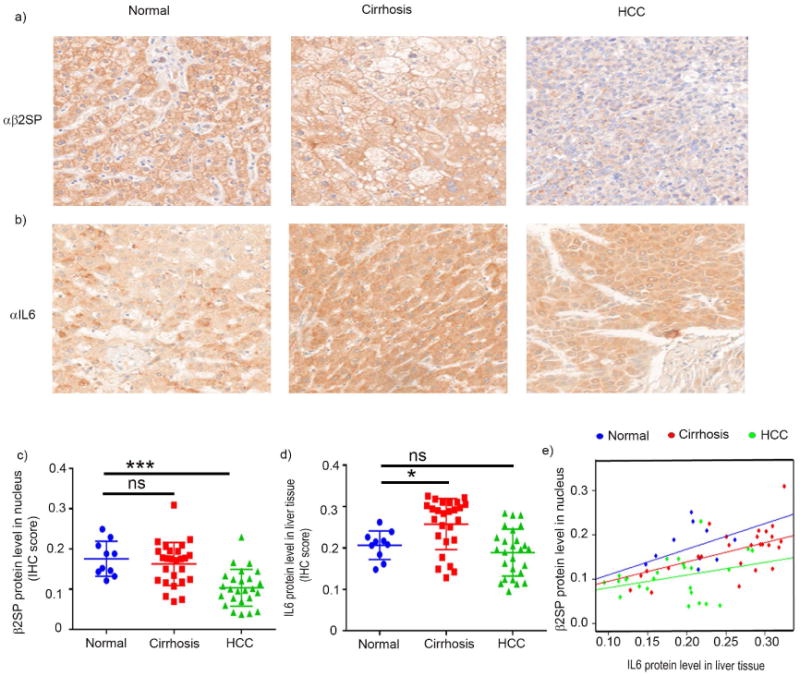
(a and b) Representative photomicrographs of β2SP (a) and IL6 (b) immunohistochemical staining in normal, cirrhotic, and HCC tissues. (c and d) Quantification of staining inetsity. Nuclear β2SP and IL6 protein levels were scored using inFORM software. (e) Robust linear regression using Huber's M estimator (Huber 1981) was applied to compare the correlation between IL6 with nuclear β2SP by cancer stage. Data in panels c–d are the means ± SEMs. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, one-way ANOVA.
Clinical and pre-clinical studies have identified a definite association between chronic inflammatory responses and HCC pathogenesis (25), and accumulating evidence indicates that a high IL6 serum level is associated with the progression of pre-neoplastic liver to HCC (25, 26). To investigate this association, we assessed IL6 protein levels in human normal, cirrhotic, and HCC liver tissues. IHC staining revealed that the IL6 expression level in normal liver tissue (mean IHC score = 0.201) was significantly lower than that in cirrhotic liver tissue (mean IHC score = 0.251; p = 0.05) (Fig. 1 b and d). The difference between IL6 expression levels of cirrhotic and HCC tissues (mean IHC score = 0.190) was not significant.
To investigate a possible connection between IL6 protein level and nuclear localization of β2SP in HCC progression, we performed a regression analysis between IL6 protein level to nuclear β2SP in human normal, cirrhotic, and HCC liver tissues. We found that the slopes of the regression for each cancer stage are significantly different (p=0.0118). For an increase in IL6, we expect a larger increase of β2SP in normal group than cirrhosis group (Δβ=0.1468, p=0.046), or a larger increase of β2SP in cirrhosis group than HCC group (Δβ=0.1448, p=0.0181). Interestingly, our study revealed that higher protein level of IL6 and lower nuclear accumulation of β2SP in cirrhotic patients shifted the distribution towards right of the points of normal subjects. Paradoxically the distribution of majority of points in HCC patients were on the left hand side of the normal subjects. It can be explained by less protein level of IL6 in hepatic tissue of HCC patients and significantly lower accumulation of nuclear β2SP compared to cirrhotic stage of patients.
IL6-mediated chronic inflammation enable to program quiescent CD133+ LSCs to become highly proliferative in β2SP+/- mice
To confirm that reciprocal concurrent IL6 expression and β2SP dysfunction is essential in promoting hepatic tumorigenesis, we established models of IL6-mediated chronic inflammation in both WT and β2SP+/- mice by periodic injection of the IL6 gene via hydrodynamic injection once every 2 weeks for 3 months (Fig. 2 a). Compared with their counterparts treated with control vector, both WT and β2SP+/- mice treated with IL6 had significantly higher percentages of infiltrating immune cells surrounding the central vein areas, indicating liver inflammation (Fig. 2 b). Significantly greater liver inflammation in WT and β2SP+/- mice treated with IL6 than that in the controls was confirmed by histological quantitation (Fig. 2 c). In both WT and β2SP+/- mice, fibrosis intensity (as measured by αSMA expression) was closely associated with IL6 expression following IL6 treatment (Fig. 2 b and d). In hepatic tissue lysates, exogenous IL6 expression was linked to inflammation and fibrosis (Fig. 2 e).
Fig. 2. Chronic IL6 treatment promotes highly proliferative CD133+ LSCs in β2SP+/- mice.

(a) Schematic representation of how chronic pIL6 treatment induces cancer stem cells in β2SP+/- mice. (b–e) Groups of WT and β2SP+/- mice were subdivided into two groups (n = 6 mice per group). Each group was given either control vector (pCtr) or pIL6 by hydrodynamic delivery twice per month for 3 months. (b) Inflammation (top) was evaluated by assessing the infiltration of immune cells, and liver fibrosis (bottom) was evaluated by assessing the expression of the αSMA protein. (c and d) Compared with WT and β2SP+/- mice treated with pCtr, both WT and β2SP+/- mice treated with pIL6 had significantly higher percentages of infiltrating immune cells (P < 0.0001; c) and significantly higher percentages of αSMA protein expression (P < 0.0001; d). (e) The concentration of IL6 in hepatic tissue lysate was measured colorimetrically using an ELISA kit. Compared with those from WT and β2SP+/- mice treated with pCtr, hepatic tissue lysates from both WT and β2SP+/- mice treated with pIL6 had higher concentrations of IL6 (P < 0.0001). (f) CD133+ LSCs from WT and β2SP+/- mice treated with pIL6 appeared in lysine/laminin-coated plates by day 14; only IL6β2SP+/- LSCs were highly proliferative and formed crypt-shaped structures. (g) IL6WT LSCs and IL6β2SP+/- LSCs were cultured in suitable supplements containing medium. Cell proliferation was measured using CyQUANT NF Cell Proliferation Assay reagent at day 3 and 6. (h) Representative immunofluorescent confocal microscopy images of Ki67 expression in IL6WT LSCs (left) and IL6β2SP+/- LSCs (right). Data in panels c–e and g are the means ± SEMs from two independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, one-way ANOVA.
After confirmation of chronic inflammation and liver fibrosis following IL6 treatment in both WT and β2SP+/- mice, we isolated CD133+ stem cells from the mice's livers. Under optimal culture conditions, the CD133+ LSCs from pIL6-treated β2SP+/- mice (IL6β2SP+/- LSCs) were highly proliferative and formed crypt-shaped structures within 2 weeks (Fig. 2 f). Assessing the cells' surface expression of CD133 (Supplementary Fig. 1 a); subjecting the cells to sphere-forming assays and staining the spheres for the stem cell–associated markers Oct4A and Nanog (Supplementary Fig. 1 b and c); and staining the cells for pluripotency markers such as SSEA-1 and TRA1-60 (Supplementary Fig. 1 d) confirmed that IL6β2SP+/- LSCs cells maintained stem-like phenotypes.
Compared with IL6β2SP+/- LSCs, CD133+ stem cells derived from pIL6-treated WT mice (IL6WT LSCs) showed significantly slower proliferation on days 3 and 6 (Fig. 2 g). Also, immunostaining with the proliferation marker Ki67 showed that IL6β2SP+/- LSCs were much more proliferative than IL6WT LSCs were (Fig. 2 h). These data suggest that the partial disruption of β2SP is critical to the acceleration of the development of highly proliferative LSCs subjected to chronic inflammation. Notably, IL6-mediated chronic inflammation for 1 or 2 months did not generate highly proliferative CD133+ stem cells from either WT or β2SP+/- mice.
IL6β2SP+/- LSCs have EMT-positive metastatic CSC phenotypes
To assess the tumor-forming ability of IL6β2SP+/- LSCs, we performed both in vitro and in vivo assays using CD133+ liver tumorigenic stem cells derived from a spontaneous HCC in a β2SP+/- mouse (β2SP+/- CSCs) as a control. Anchorage-independent growth assays showed that IL6β2SP+/- LSCs formed more colonies per field than β2SP+/- CSCs did (Fig. 3 a). In Boyden chamber migration assays, the migratory capacity of IL6β2SP+/- LSCs was 50% greater (p < 0.0001) than that of β2SP+/- CSCs (Fig. 3 b). Similarly, Matrigel-based invasion assays showed IL6β2SP+/- LSCs to be more invasive than β2SP+/- CSCs. Because EMT is associated with increased migration and invasion capacities of tumor cells, we investigated for expression of EMT-related proteins Twist and Slug. IL6β2SP+/- LSCs had very high nuclear accumulation of Twist and Slug, whereas these proteins were undetected or present at only basal levels in β2SP+/- CSCs (Fig. 3 c). These in vitro data suggest that IL6β2SP+/- LSCs are highly aggressive, EMT-positive cells.
Fig. 3. IL6β2SP+/- LSCs have EMT-positive tumorigenic phenotypes.
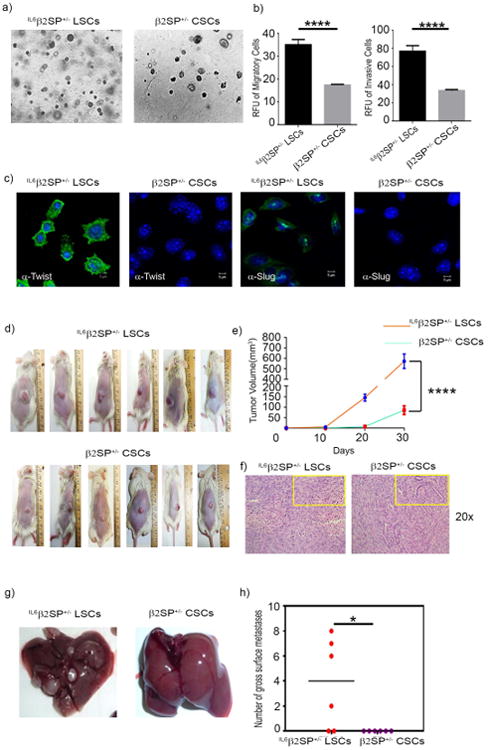
(a) Ten thousand cells/well were overlaid on soft agar. Anchorage-independent colonies were allowed to grow for 2 weeks. (b) For invasion assays, the cells were seeded onto the Matrigel-coated transwell inserts (1 × 105 cells/well) and incubated overnight. Migration assays were performed using transwell inserts. Cells (1 × 104 cells/well) were added to the upper chamber and allowed to migrate toward the lower chamber overnight. Cell invasion and migration were assessed fluorometrically using calcein AM dye. (c) Early-passage IL6β2P+/- and β2P+/- CSCs were stained for the EMT markers Twist and Slug. (d) IL6β2SP+/- LSCs or β2SP+/- CSCs (1 × 105) were subcutaneously injected into NSG mice. (e) Two-way ANOVA with the Tukey post hoc test was used to analyze tumor volume growth rates. (f) Hematoxylin and eosin–stained sections of primary tumor tissues (original magnification ×200). (g) Representative images of spontaneous liver metastases arising from IL6β2SP+/- LSCs (left) and β2SP+/- CSCs (right). (h) Numbers of gross metastatic liver nodules in IL6β2SP+/- LSCs and β2SP+/- CSCs. Data in panels b and h are the means ± SEMs from two independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To corroborate our in vitro results, we assessed the progression and metastasis of tumors formed by IL6β2SP+/- LSCs or β2SP+/- LSCs in immunodeficient mice. IL6β2SP+/- LSCs or β2SP+/- CSCs (∼ 1 ×105) were injected subcutaneously into NOD SCID gamma chain knockout (NSG) mice (n = 6 per cell line). Tumors formed by IL6β2SP+/- LSCs grew faster than tumors formed by β2SP+/- CSCs did (p < 0.0001; Fig. 3 d and e). To determine whether the tumors had the clinicopathological characteristics of HCC, we stained tissue sections with hematoxylin and eosin; IL6β2SP+/- LSCs and β2SP+/- CSCs retained all major HCC histological characteristics, such as granular eosinophilic cytoplasm, rounded nuclei, and prominent nucleoli (Fig. 3 f). Also, some mice injected with IL6β2SP+/- LSCs, but no mice injected with β2SP+/- CSCs, had spontaneous liver metastasis (2-8 nodules per liver) (Fig. 3 g and h). Also, our orthotopic inoculation study showed similar trend where IL6β2SP+/- LSCs inoculated NSG mice showed significantly (p=0.0027) poor survivability compared to β2SP+/- CSCs inoculated NSG mice (Supplementary Fig. 2 a). Interestingly, IL6β2SP+/- LSCs inoculated NSG mice had metastasis in intestinal lymph nodes (Supplementary Fig. 2 b). Together, these data exhibited the metastatic nature and EMT phenotypes of IL6β2SP+/- LSCs.
Constitutive NFκB activation drives the EMT-positive phenotypes and metastatic properties of IL6β2SP+/- LSCs
We then sought to identify the mechanisms underlying the invasive and metastatic properties of IL6β2SP+/- LSCs. Hypothesizing that the disruption of key metastatic/driver pathways inhibits EMT and cellular invasiveness, we treated IL6β2SP+/- LSCs with a panel of pharmacological pathway inhibitors at biologically relevant concentrations for 24 and 48 h. Compared with no treatment, treatment with the NFκB inhibitor QNZ for 24 h significantly reduced the viability of IL6β2SP+/- LSCs (Fig. 4 a). However the control cell line β2SP+/- LSCs did not show any change in viability upon treatment with QNZ for 48 h (data not shown), which suggesting that the constitutive activation of NFκB enables the invasive and metastatic properties of IL6β2SP+/- LSCs. Western blotting confirmed that QNZ treatment dramatically reduced levels of constitutively phosphorylated NFκB (pNFκB) (Fig. 4 b). Immunofluorescent staining confirmed the reduced nuclear accumulation of pNFκB following QNZ treatment (Fig. 4 c). In addition, IL6β2SP+/- LSCs also demonstrated a reduction in nuclear localization and a decrease in the, the transcriptional activity of pNFκB (Fig. 4 d).
Fig. 4. Constitutively activated NFκB drives the tumorigenesis of IL6β2SP+/- LSCs.
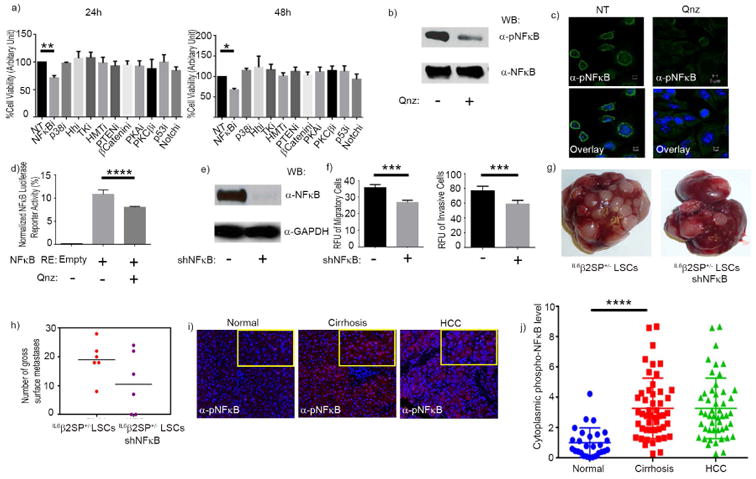
(a) Screening with pharmacological inhibitors revealed that NFκB inhibition decreased cell viability at both 24 and 48 h. (b) Western blotting for pNFκB and total NFκB in untreated cells and cells treated with QNZ (100 nM). (c) Representative confocal microscopy images of immunofluorescent staining for pNFκB in untreated cells and cells treated with QNZ (magnification × 63). Immunofluorescent images were captured using PASCAL software on a Zeiss LSM 510 Meta confocal microscope at 63 × magnifications. (d) IL6β2SP+/- LSCs (1 × 106) were nucleofected. Transfected cells were treated with or without 100 nM QNZ for 48 h. The cells were then lysed, and luciferase activities were measured and normalized. (e) Western blotting confirmed the stable knockdown of NFκB in shNFκB IL6β2SP+/- LSCs. (f) Invasion and migration assays revealed that NFκB knockdown significantly decreased EMT phenotypes. (g) Representative images of liver metastases formed by IL6β2SP+/- LSCs (left) and shNFκB IL6β2SP+/- LSCs (right). (h) Numbers of gross metastatic liver nodules in IL6β2SP+/- LSCs and shNFκB IL6β2SP+/- LSCs. (i) Representative images of immunohistochemical staining of pNFκB (red) in paraffin-embedded specimens of normal, cirrhotic, and HCC tissues. Nuclei were stained with DAPI (blue). (j) The intensity of histological staining was scored by cell segmentation analysis of inForm software. Data in panels a, d, f, h, and j are the means ± SEMs from two independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, one-way ANOVA.
To validate the role of NFκB in driving metastasis, we created IL6β2SP+/- LSCs with stable knockdown of NFκB (shNFκB IL6β2SP+/- LSCs). Knockdown was confirmed by Western blotting (Fig. 4 e). Parental IL6β2SP+/- LSCs and shNFκB IL6β2SP+/- LSCs had identical proliferation rates (data not shown). However, invasion and migration assays revealed that the metastatic capacity of shNFκB IL6β2SP+/- LSCs was significantly less than that of parental IL6β2SP+/- LSCs (P = 0.0007) (Fig. 4 f). Similarly, parental IL6β2SP+/- LSCs injected intraosseously injected into NSG mice yielded more liver metastasis than shNFκB IL6β2SP+/- LSCs did (Fig. 4 g and h). The IL6-mediated constitutive activation of STAT3, which has been demonstrated in certain aggressive tumors (27, 28), is one obvious mechanism underlying the invasive and metastatic nature of IL6β2SP+/- LSCs. However, immunofluorescence staining and Western blotting both showed that STAT3 was not constitutively activated or overexpressed in IL6β2SP+/- LSCs (Supplementary Fig. 3).
To understand the clinical relevance of our findings, we stained human normal, cirrhotic, and HCC tissues with antibodies against NFκB and pNFκB. The findings agree with those in the cellular model, in that NFκB phosphorylation levels in HCC tissues and cirrhotic tissues were significantly higher than those in normal tissues (p < 0.0001) (Fig. 4 i and j).
Constitutively activated TAK1 directly phosphorylates NFκB in IL6β2SP+/- LSCs
To identify the putative serine/threonine kinase(s) responsible for the phosphorylation of the p65 (S536) subunit of NFκB, we treated IL6β2SP+/- LSCs with inhibitors of the kinases TAK1, IKK, RSK1, PKC, and CK2, which are known activators of NFκB (29, 30). Compared with that of untreated control cells, the viability of IL6β2SP+/- LSCs treated with the TAK1 inhibitor was dramatically reduced (p < 0.0001) (Fig. 5 a). IKK inhibition had no noticeable effect on the viability of IL6β2SP+/- LSCs or the phosphorylation status of NFκB in IL6β2SP+/- LSCs (Supplementary Fig. 4). Western blot analyses revealed that TAK1 inhibition decreased the phosphorylation of NFκB dramatically in IL6β2SP+/- LSCs (Fig. 5 b). Also, TAK inhibition did not alter the total NFκB protein level. This study validated the results of our preliminary screening of the putative kinases with pharmacological inhibitors.
Fig. 5. TAK1 mediates the constitutive activation of NFκB in IL6β2SP+/- LSCs.
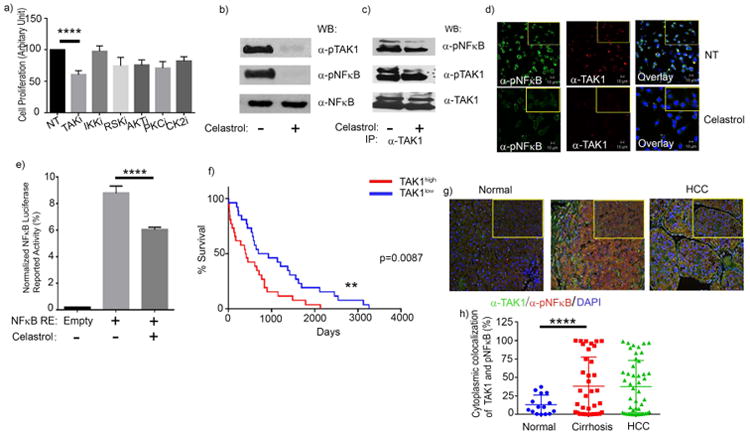
(a) The proliferation of IL6β2SP+/- LSCs treated with a TAK1 inhibitor was significantly lower than that of untreated IL6β2SP+/- LSCs. (b) Western blotting for pTAK1, pNFκB, and NFκB in IL6β2SP+/- LSCs treated with the TAK1 inhibitor celastrol (500 nM) overnight. (c) Co-immunoprecipitation assays revealed that pTAK1 directly interacts with NFκB and mediates NFκB's phosphorylation. Western blotting revealed that overnight treatment with the TAK1 inhibitor celastrol (500 nM) prevents the direct interaction of pTAK1 and pNFκB in IL6β2SP+/- LSCs. (d) Representative confocal microscopy images of immunofluorescent staining for α-pNFκB (green) and α-TAK1 (red) in IL6β2SP+/- LSCs treated with or without celastrol (500 nM) for 24 h. Nuclei were stained with Draq5 (blue). Immunofluorescent images were captured using PASCAL software on a Zeiss LSM 510 Meta confocal microscope at 63× magnification. (e) IL6β2SP+/- LSCs (1 × 106) were nucleofected. Transfected cells were treated with or without 500 nM celastrol for 48 h. The cells were then lysed, and luciferase activities were measured and normalized. (f) The overall survival rate of HCC patients with low TAK1 gene expression levels (TAK1low) was significantly higher than that of HCC patients with high TAK1 gene expression levels (TAK1high; P = 0.0087). (g) Representative images of immunohistochemical staining of TAK1 (green) and pNFκB (red) in paraffin-embedded specimens of normal, cirrhotic, and HCC tissues. Nuclei were stained with DAPI (blue). (h) Cytoplasmic colocalization of TAK1 and pNFκB protein levels were scored using inFORM software. Data in panels a, e, and h are the means ± SEMs from two independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, one-way ANOVA.
To evaluate the direct association between TAK1 and NFκB, we performed co-immunoprecipitation experiments, which revealed interaction of pNFκB and TAK1 in cell lysates (Fig. 5 c). Co-immunoprecipitation of pNFκB upon TAK1 pull-down from the cell lysate revealed that TAK1 inhibition noticeably decreased the interaction of TAK1 with NFκB (Fig. 5 c). TAK1 and pNFκB were colocalized in the cytoplasm of untreated IL6β2SP+/- LSCs, but this colocalization was dramatically decreased in IL6β2SP+/- LSCs treated with TAK1 inhibition (Fig. 5 d). Indeed, the TAK1-mediated phosphorylation of NFκB was essential to NFκB-driven reporter activities in IL6β2SP+/- LSCs (Fig. 5 e). These findings strongly showed that TAK1-mediated direct phosphorylation of NFκB facilitates NFκB's transcriptional activity in the nucleus. Also, we are interested to know the direct role of IL6 to activate TAK1-NFκB pathway in a TGFβ disrupted liver tumor cells (Supplementary Fig. 5). Our recombinant IL6 (rIL6) treatment on β2SP+/- CSCs showed activation of NFκB in 1 h however its activation is alleviated at 4h and 8 h time points. Interestingly rIL6 treatment showed inducible protein expression of NFκB in both 4h and 8h time points. Western blot analysis of phosphorylated TAK1 showed a sustained level of TAK1 activation in β2SP+/- CSCs but its total protein expression did not reflect any significant difference at different points of upon rIL6 treatment.
To substantiate the translational relevance of TAK1 expression with HCC progression, we analyzed TCGA dataset. Mantel-Cox log-rank analysis revealed that the overall survival rate of patients in the TAK1high group was significantly lower than that of patients in the TAK1low group (p = 0.0087) (Fig. 5 f). The median survival times for patients in the TAK1low and TAK1high groups were 813 days and 405 days, respectively. To understand the clinical significance of our findings, we stained human normal, cirrhotic, and HCC tissues with anti-TAK1 and anti-pNFκB antibodies. Our findings agree with those in the cellular model, in that the levels of pNFκB and TAK1 colocalization in advanced HCC tissues and cirrhotic tissues were significantly higher than those in normal tissues (P < 0.0001) (Fig. 5 g and h). Together, these findings establish a strong linkage between TAK1-mediated NFκB phosphorylation and liver tumor progression.
In summary, this study provides novel insight into the mechanism by which IL6-driven chronic liver inflammation reprograms normal stem cells to become mCSCs. Stable expression of IL6 in β2SP-deficient murine liver induces chronic inflammation via resident immune cells. This inflammation constitutively activates the pro-tumorigenic TAK1-NFκB signaling pathway to maintain the metastatic phenotypes of liver CSCs (Fig. 6).
Fig. 6. IL6-mediated chronic inflammation drives the reprogramming of normal LSCs to mCSCs in a TGFβ disrupted background.
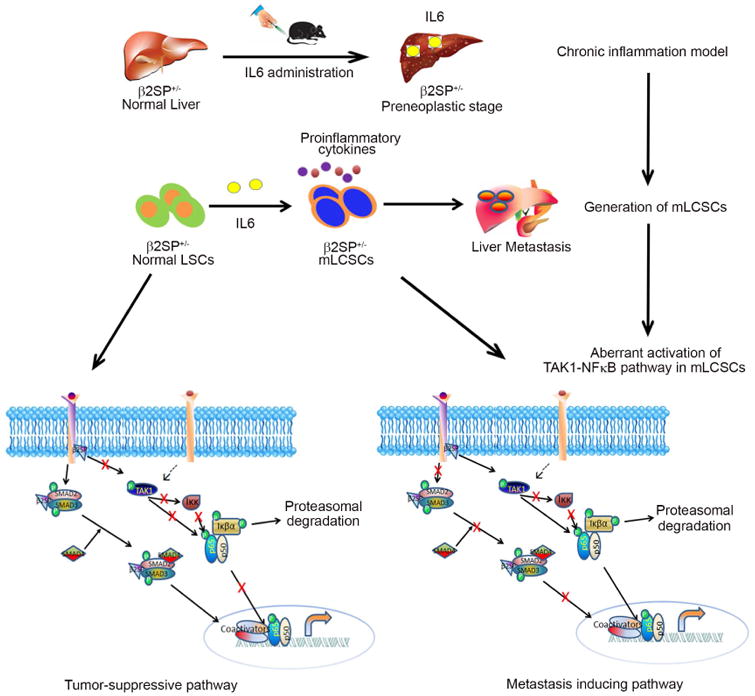
An IL6-encoding Sleeping Beauty transposon was hydrodynamically injected into the livers of β2SP+/- mice twice per month for 3 months. The stable expression of IL6 in liver generates chronic inflammation and subsequent fibrosis in β2SP+/- mice. After 3 months, CD133+ LSCs from these mice had EMT-positive mCSC phenotypes. Pathway screening with small molecule inhibitors and subsequent validation revealed that the constitutive activation of the TAK1-NFκB pathway plays a critical role in maintaining these cells' EMT-positive metastatic phenotypes.
Discussion
The influence of liver inflammation on oncogenesis has been intensively investigated to increase our comprehension of the underlying molecular mechanisms that may link chronic inflammatory responses with tumor initiation (31, 32). The prolonged secretion of cytokines and chemokines during inflammation promotes cancer growth and results in poor host survival. Although the nuclear translocation of β2SP and IL6 levels are inversely related in HCC, metastatic initiation at the pre-neoplastic tissue had not been reported. To our knowledge, this study is the first to demonstrate that EMT+ mCSCs can be generated by inflamed livers. This induction depends on the overexpression of IL6 and the partial disruption of TGFβ signaling, thus our study extends the current knowledge of IL6's role in promoting liver tumorigenesis (11, 33, 34). Multiple studies have shown that IL6 or its downstream signaling is capable of expanding CSCs and inducing EMT phenotypes in cancers of the breast, colon, ovary, lung, brain, and head and neck (35-37). A recent study showed that STAT3, following its IL6-mediated activation, binds to the promoter element of CD133 to induce HCC progression (38). Coculturing IL6-secreting tumor-associated macrophages with CD44+ liver CSCs induces more stemness and tumorigenesis (11). Determining whether IL6 can interact with other oncogenes or defective tumor suppressors to induce similar types of mCSCs is also of interest. In summary, our findings provide important insights into the reciprocal cross-talk between TGFβ signaling and IL6-driven inflammation in pre-neoplastic liver tissues. However, we did not find any visible tumor incidence in these IL6 treated β2SP+/- female mice even after 6 months of post treatment. The possible explanation could be the protective effect of estrogen to restrict IL6 driven HCC development (39).
The variability in the clinical outcomes of HCC patients is associated with numerous populations of CSCs defined by the markers CD13, CD24, CD44, CD90, CD133, EpCAM, DLK1, and CSV (11, 22, 40). The discovery of diverse stem cell markers has increased research into the roles of hepatic stem/progenitor cells in HCC. Liver CSCs are critical to the development of malignant phenotypes, multi-drug resistance, and disease relapse in HCC patients. One study revealed that liver cancer patients with high levels of CD133 expression have shorter overall survival and higher relapse rates than those with low levels of CD133 expression (41). Whether CD133+ cells contribute to malignancy remains unclear. Several studies have shown that CD133+ CSCs in HCC are naturally resistant to cytotoxic chemotherapy and other anti-cancer drugs (42). While most of these studies have focused on identifying and characterizing CSC surface markers, studies that focus on the tumorigenic or metastatic properties of these cells may be warranted. In addition, the influence of the liver immune microenvironment on modulating these normal stem cells' transition to mCSCs must be investigated. Such investigations would have clinical relevance including a significant impact on therapeutic strategies.
In liver cancer, sustained inflammation facilitates the progression of fibrosis to cirrhosis and eventually HCC. By activating hepatic stellate cells, the TGFβ signaling pathway promotes the progression of chronic inflammation to fibrosis (43). Paradoxically, by inhibiting cell cycle regulators and promoting apoptosis, TGFβ is instrumental in tumor suppression during the early stages of tumorigenesis (44, 45). Microarray and proteomic screening of human and mouse HCC tissues have revealed that dysregulated TGFβ signaling is associated with the increased expression of pro-oncogenic pathways such as STAT3, Wnt, and CDK4 (46). Studies have shown that tumors develop in β2SP-haplodeficient mice, and that β2SP expression is significantly reduced in most HCC patients. (8, 9). These findings indicate that TGFβ signaling inhibits the development of HCC. Furthermore, screening of human cancer cell lines has revealed that β2SP expression was diminished in Huh-7 cells and reduced by a factor of 9 in SNU-398 cells (46). One recently published study revealed that β2SP regulates the nuclear localization of β-catenin by increasing the expression of the Wnt inhibitor kallistatin (9). Given the importance of β2SP as a regulator of HCC progression, researchers have also investigated its role in the development of liver CSCs. Studies have shown that β2SP inhibition is required for the induction of stemness associated genes and the genesis of liver CSCs (47). Decreased β2SP expression in HCC cells facilitates sphere formation, tumor development, and aggressive phenotypes (9). Furthermore, β2SP+/- mice demonstrate a 2- to 4-fold expansion of Oct3/4-positive progenitor cells with activated WNT signaling (48, 49). The present study shows for the first time that the TGFβ pathway restrains the development of metastatic clones of HCC CSCs in pre-neoplastic tissues. Thus, β2SP suppresses the transformation of normal stem cells into expandable liver CSCs.
Our investigation of aberrant molecular pathways in liver CSCs uncovered a novel interaction between TAK1 and NFκB that promotes the EMT-driven metastatic process. A previous study showed that the hepatocyte-specific deletion of TAK1 in mice activates the TGFβ signaling pathway to induce spontaneous inflammation, fibrosis, and eventually hepatic tumorigenesis (50). This discovery explains why TAK1 mediated mCSCs occur in livers when TGFβ signaling is disrupted as it plays a tumor suppressor role. Overall, this reciprocal regulation between β2SP and IL6 in the programming of liver CSCs provides insight into the mechanism by which normal stem cells transform into EMT-positive CSCs. Defining the mechanisms by which the loss of β2SP regulates the transition of hepatic stem cells to cells with EMT phenotypes in an inflammation-driven hepatic immune environment may lead to the development of individualized therapies against liver cancer.
Supplementary Material
Acknowledgments
Work in the authors' laboratory was supported by grants from the National Institutes of Health to Dr. Shulin Li (NIH R01DK10276701A1). We are thankful to Dr. Deborah Berry for β2SP and IL6 staining in TMA slides. This research was performed in the Flow Cytometry & Cellular Imaging Facility, which is supported in part by the National Institutes of Health through M. D. Anderson's Cancer Center Support Grant CA016672. The University of Texas MD Anderson Cancer Center is supported in part by the NCI CCSG Core Grant CA16672.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 3.Baek HJ, Lim SC, Kitisin K, Jogunoori W, Tang Y, Marshall MB, Mishra B, et al. Hepatocellular cancer arises from loss of transforming growth factor beta signaling adaptor protein embryonic liver fodrin through abnormal angiogenesis. Hepatology. 2008;48:1128–1137. doi: 10.1002/hep.22460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammerich L, Heymann F, Tacke F. Role of IL-17 and Th17 cells in liver diseases. Clin Dev Immunol. 2011;2011:345803. doi: 10.1155/2011/345803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamazaki K, Masugi Y, Sakamoto M. Molecular pathogenesis of hepatocellular carcinoma: altering transforming growth factor-beta signaling in hepatocarcinogenesis. Dig Dis. 2011;29:284–288. doi: 10.1159/000327560. [DOI] [PubMed] [Google Scholar]
- 6.Mamiya T, Yamazaki K, Masugi Y, Mori T, Effendi K, Du W, Hibi T, et al. Reduced transforming growth factor-beta receptor II expression in hepatocellular carcinoma correlates with intrahepatic metastasis. Lab Invest. 2010;90:1339–1345. doi: 10.1038/labinvest.2010.105. [DOI] [PubMed] [Google Scholar]
- 7.Sun CK, Chua MS, He J, So SK. Suppression of glypican 3 inhibits growth of hepatocellular carcinoma cells through up-regulation of TGF-beta2. Neoplasia. 2011;13:735–747. doi: 10.1593/neo.11664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299:574–577. doi: 10.1126/science.1075994. [DOI] [PubMed] [Google Scholar]
- 9.Zhi X, Lin L, Yang S, Bhuvaneshwar K, Wang H, Gusev Y, Lee MH, et al. betaII-Spectrin (SPTBN1) suppresses progression of hepatocellular carcinoma and Wnt signaling by regulation of Wnt inhibitor kallistatin. Hepatology. 2015;61:598–612. doi: 10.1002/hep.27558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitisin K, Ganesan N, Tang Y, Jogunoori W, Volpe EA, Kim SS, Katuri V, et al. Disruption of transforming growth factor-beta signaling through beta-spectrin ELF leads to hepatocellular cancer through cyclin D1 activation. Oncogene. 2007;26:7103–7110. doi: 10.1038/sj.onc.1210513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, Simeone DM, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147:1393–1404. doi: 10.1053/j.gastro.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang TS, Wu YC, Chi CC, Su WC, Chang PJ, Lee KF, Tung TH, et al. Activation of IL6/IGFIR confers poor prognosis of HBV-related hepatocellular carcinoma through induction of OCT4/NANOG expression. Clin Cancer Res. 2015;21:201–210. doi: 10.1158/1078-0432.CCR-13-3274. [DOI] [PubMed] [Google Scholar]
- 13.Hatting M, Spannbauer M, Peng J, Al Masaoudi M, Sellge G, Nevzorova YA, Gassler N, et al. Lack of gp130 expression in hepatocytes attenuates tumor progression in the DEN model. Cell Death Dis. 2015;6:e1667. doi: 10.1038/cddis.2014.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, Shalapour S, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell. 2013;155:384–396. doi: 10.1016/j.cell.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porta C, De Amici M, Quaglini S, Paglino C, Tagliani F, Boncimino A, Moratti R, et al. Circulating interleukin-6 as a tumor marker for hepatocellular carcinoma. Ann Oncol. 2008;19:353–358. doi: 10.1093/annonc/mdm448. [DOI] [PubMed] [Google Scholar]
- 16.Tang Y, Kitisin K, Jogunoori W, Li C, Deng CX, Mueller SC, Ressom HW, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci U S A. 2008;105:2445–2450. doi: 10.1073/pnas.0705395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, Quraishi AA, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012;47:570–584. doi: 10.1016/j.molcel.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olsen J, Kirkeby LT, Olsen J, Eiholm S, Jess P, Gogenur I, Troelsen JT. High interleukin-6 mRNA expression is a predictor of relapse in colon cancer. Anticancer Res. 2015;35:2235–2240. [PubMed] [Google Scholar]
- 19.Milagre CS, Gopinathan G, Everitt G, Thompson RG, Kulbe H, Zhong H, Hollingsworth RE, et al. Adaptive Upregulation of EGFR Limits Attenuation of Tumor Growth by Neutralizing IL6 Antibodies, with Implications for Combined Therapy in Ovarian Cancer. Cancer Res. 2015;75:1255–1264. doi: 10.1158/0008-5472.CAN-14-1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123:1911–1918. doi: 10.1172/JCI66024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitra A, Ross JA, Rodriguez G, Nagy ZS, Wilson HL, Kirken RA. Signal transducer and activator of transcription 5b (Stat5b) serine 193 is a novel cytokine-induced phospho-regulatory site that is constitutively activated in primary hematopoietic malignancies. J Biol Chem. 2012;287:16596–16608. doi: 10.1074/jbc.M111.319756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitra A, Satelli A, Xia X, Cutrera J, Mishra L, Li S. Cell-surface Vimentin: A mislocalized protein for isolating csVimentin(+) CD133(-) novel stem-like hepatocellular carcinoma cells expressing EMT markers. Int J Cancer. 2015;137:491–496. doi: 10.1002/ijc.29382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitra A, Satelli A, Yan J, Xueqing X, Gagea M, Hunter CA, Mishra L, et al. IL-30 (IL27p28) attenuates liver fibrosis through inducing NKG2D-rae1 interaction between NKT and activated hepatic stellate cells in mice. Hepatology. 2014;60:2027–2039. doi: 10.1002/hep.27392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mishra L, Katuri V, Evans S. The role of PRAJA and ELF in TGF-beta signaling and gastric cancer. Cancer Biol Ther. 2005;4:694–699. doi: 10.4161/cbt.4.7.2015. [DOI] [PubMed] [Google Scholar]
- 25.Budhu A, Wang XW. The role of cytokines in hepatocellular carcinoma. J Leukoc Biol. 2006;80:1197–1213. doi: 10.1189/jlb.0506297. [DOI] [PubMed] [Google Scholar]
- 26.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alas S, Bonavida B. Inhibition of constitutive STAT3 activity sensitizes resistant non-Hodgkin's lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clin Cancer Res. 2003;9:316–326. [PubMed] [Google Scholar]
- 28.Zhao H, Guo Y, Li S, Han R, Ying J, Zhu H, Wang Y, et al. A novel anti-cancer agent Icaritin suppresses hepatocellular carcinoma initiation and malignant growth through the IL-6/Jak2/Stat3 pathway. Oncotarget. 2015;6:31927–31943. doi: 10.18632/oncotarget.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 30.Freudlsperger C, Bian Y, Contag Wise S, Burnett J, Coupar J, Yang X, Chen Z, et al. TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene. 2013;32:1549–1559. doi: 10.1038/onc.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider C, Teufel A, Yevsa T, Staib F, Hohmeyer A, Walenda G, Zimmermann HW, et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut. 2012;61:1733–1743. doi: 10.1136/gutjnl-2011-301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26:549–564. doi: 10.1016/j.ccell.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 33.van der Zee M, Sacchetti A, Cansoy M, Joosten R, Teeuwssen M, Heijmans-Antonissen C, Ewing-Graham PC, et al. IL6/JAK1/STAT3 Signaling Blockade in Endometrial Cancer Affects the ALDHhi/CD126+ Stem-like Component and Reduces Tumor Burden. Cancer Res. 2015;75:3608–3622. doi: 10.1158/0008-5472.CAN-14-2498. [DOI] [PubMed] [Google Scholar]
- 34.Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol Cancer Res. 2015;13:651–658. doi: 10.1158/1541-7786.MCR-14-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C, Lin J. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res. 2011;71:7226–7237. doi: 10.1158/0008-5472.CAN-10-4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu CC, Lin JH, Hsu TW, Su K, Li AF, Hsu HS, Hung SC. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int J Cancer. 2015;136:547–559. doi: 10.1002/ijc.29033. [DOI] [PubMed] [Google Scholar]
- 37.Wang H, Lathia JD, Wu Q, Wang J, Li Z, Heddleston JM, Eyler CE, et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. 2009;27:2393–2404. doi: 10.1002/stem.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Won C, Kim BH, Yi EH, Choi KJ, Kim EK, Jeong JM, Lee JH, et al. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology. 2015;62:1160–1173. doi: 10.1002/hep.27968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi L, Feng Y, Lin H, Ma R, Cai X. Role of estrogen in hepatocellular carcinoma: is inflammation the key? J Transl Med. 2014;12:93. doi: 10.1186/1479-5876-12-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu X, Liu RF, Zhang X, Huang LY, Chen F, Fei QL, Han ZG. DLK1 as a potential target against cancer stem/progenitor cells of hepatocellular carcinoma. Mol Cancer Ther. 2012;11:629–638. doi: 10.1158/1535-7163.MCT-11-0531. [DOI] [PubMed] [Google Scholar]
- 41.Song W, Li H, Tao K, Li R, Song Z, Zhao Q, Zhang F, et al. Expression and clinical significance of the stem cell marker CD133 in hepatocellular carcinoma. Int J Clin Pract. 2008;62:1212–1218. doi: 10.1111/j.1742-1241.2008.01777.x. [DOI] [PubMed] [Google Scholar]
- 42.Ma S, Tang KH, Chan YP, Lee TK, Kwan PS, Castilho A, Ng I, et al. miR-130b Promotes CD133(+) liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell. 2010;7:694–707. doi: 10.1016/j.stem.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 43.Bourd-Boittin K, Bonnier D, Leyme A, Mari B, Tuffery P, Samson M, Ezan F, et al. Protease profiling of liver fibrosis reveals the ADAM metallopeptidase with thrombospondin type 1 motif, 1 as a central activator of transforming growth factor beta. Hepatology. 2011;54:2173–2184. doi: 10.1002/hep.24598. [DOI] [PubMed] [Google Scholar]
- 44.Baek HJ, Pishvaian MJ, Tang Y, Kim TH, Yang S, Zouhairi ME, Mendelson J, et al. Transforming growth factor-beta adaptor, beta2-spectrin, modulates cyclin dependent kinase 4 to reduce development of hepatocellular cancer. Hepatology. 2011;53:1676–1684. doi: 10.1002/hep.24128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L, Roh YS, Song J, Zhang B, Liu C, Loomba R, Seki E. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology. 2014;59:483–495. doi: 10.1002/hep.26698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin L, Amin R, Gallicano GI, Glasgow E, Jogunoori W, Jessup JM, Zasloff M, et al. The STAT3 inhibitor NSC 74859 is effective in hepatocellular cancers with disrupted TGF-beta signaling. Oncogene. 2009;28:961–972. doi: 10.1038/onc.2008.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen CL, Tsukamoto H, Liu JC, Kashiwabara C, Feldman D, Sher L, Dooley S, et al. Reciprocal regulation by TLR4 and TGF-beta in tumor-initiating stem-like cells. J Clin Invest. 2013;123:2832–2849. doi: 10.1172/JCI65859. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Thenappan A, Li Y, Kitisin K, Rashid A, Shetty K, Johnson L, Mishra L. Role of transforming growth factor beta signaling and expansion of progenitor cells in regenerating liver. Hepatology. 2010;51:1373–1382. doi: 10.1002/hep.23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thenappan A, Shukla V, Abdul Khalek FJ, Li Y, Shetty K, Liu P, Li L, et al. Loss of transforming growth factor beta adaptor protein beta-2 spectrin leads to delayed liver regeneration in mice. Hepatology. 2011;53:1641–1650. doi: 10.1002/hep.24111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang L, Inokuchi S, Roh YS, Song J, Loomba R, Park EJ, Seki E. Transforming growth factor-beta signaling in hepatocytes promotes hepatic fibrosis and carcinogenesis in mice with hepatocyte-specific deletion of TAK1. Gastroenterology. 2013;144:1042–1054 e1044. doi: 10.1053/j.gastro.2013.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.