

Abstract
Background
Clinical radiosensitivity is a significant impediment to tumour control and cure, in that it restricts the total doses which can safely be delivered to the whole radiotherapy population, within the tissue tolerance of potentially radiosensitive (RS) individuals. Understanding its causes could lead to personalization of radiotherapy.
Methods
We screened tissues from a unique bank of RS cancer patients for expression defects in major DNA double-strand break repair proteins, using Western blot analysis and subsequently reverse-transcriptase polymerase chain reaction and pulsed-field gel electrophoresis.
Results
We hypothesized that abnormalities in expression of these proteins may explain the radiosensitivity of some of our cancer patients. The cells from one patient showed a reproducibly consistent expression reduction in two complex-forming DNA double-strand break repair protein components (DNA Ligase IV and XRCC4). We also showed a corresponding reduction in both gene products at the mRNA level. Additionally, the mRNA inducibility by ionizing radiation was increased for one of the proteins in the patient’s cells. We confirmed the likely functional significance of the non-homologous end-joining (NHEJ) expression abnormalities with a DNA double strand break (DNA DSB) repair assay.
Conclusions
We have identified a novel biological phenotype linked to clinical radiosensitivity. This is important in that very few molecular defects are known in human radiotherapy subjects. Such knowledge may contribute to the understanding of radiation response mechanisms in cancer patients and to personalization of radiotherapy.
Keywords: Non-homologous end-joining (NHEJ), radiosensitivity cancer, DNA double strand break (DNA DSB)
Introduction
In clinical oncology, radiosensitivity is a significant impediment to tumour control and cure. Fortunately, it is relatively rare, but it has two main consequences. First, it can be considerably morbid for those radiosensitive (RS) patients who suffer it. Second and perhaps even more importantly, it potentially affects many more patients, in that it restricts the total doses which can safely be delivered to the whole radiotherapy population, within the tissue tolerance of the RS individuals. The latter cannot, with the exception of some rare autosomal conditions that have other recognisable clinical manifestations (such as ataxia telangiectasia) be detected prior to therapy.
Multiple genes are known whose dysfunction results in radiosensitivity. These include genes from lower species, many of which have recognisable and often functional human homologues, or genes causing the aforementioned hereditary human conditions with radiosensitivity as one of numerous other clinical features, such as immunodeficiency, neurodegeneration and cancer-proneness (e.g., ataxia telangiectasia) (1). In contrast, very few ‘radiosensitivity genes’ are known in cancer radiotherapy patients. This is a significant impediment to personalization of radiotherapy, used in over 50% of cancer patients (2). Once more ‘radiosensitivity genes’ are known, pre-therapy predictive testing could be envisaged, along the lines of gene expression tests used in cancer management (3).
There is substantial evidence that clinical radiosensitivity in otherwise phenotypically normal cancer patients has a (largely) genetic basis (4,5). We and others have taken different approaches in an attempt to discern which genes may be responsible. One is a candidate molecule approach, where dysfunction of genes and their products or regulators linked to radiosensitivity in other contexts/organisms, are tested for dysfunction in RS cancer patients (6-10). Another are genome-wide association studies (11), which to date have yielded some radiosensitivity susceptibility loci/genes, e.g., TANC1 (12). The vast majority of genetic determinants of clinical radiosensitivity remain unexplained.
DNA double strand breaks (DNA DSBs) are the main ionizing-induced biological lesions (see below). In the present study we used a novel bank of tissues from RS cancer patients and their controls to screen for protein abnormalities (e.g., truncations or reduced amounts) in components of the major DNA DSB pathway in mammalian cells, non-homologous end-joining (NHEJ). Our hypothesis was that any such abnormalities may be responsible for radiosensitivity in some of our cancer patients. If successful, such investigations could lead in the future to a better understanding of the molecular basis of radiation responses and contribute to personalization of cancer radiotherapy.
Methods
Patients
This study received ethics approval from the Ethics Committee of the Peter MacCallum Cancer Centre, Melbourne, Australia (Study Approval Number 96/39). Individual patients also gave consent for the use of their tissues/cells in research.
Control and RS patient characteristics have been previously described (10,13). In summary, clinically RS patients had Radiation Therapy Oncology Group (RTOG) (2) Grade 3 or 4 toxicities, whereas those radiotherapy patients without significant toxicity (RTOG ≤1) were controls. We accessioned pathoclinical details and biological samples from cancer patients who had developed clinical hypersensitivity reactions to radiotherapy (RS cases) (10,13). RS was in the clinical form of either severe acute or severe late reactions, or so-called consequential late effects, which result from severe and persisting acute effects (14).
One RS patient (designated RS33) had received a standard radiotherapy regime for prostate cancer (Figure 1) and developed severe late rectal inflammation (proctitis), clinically manifesting as per rectal bleeding.
Figure 1.
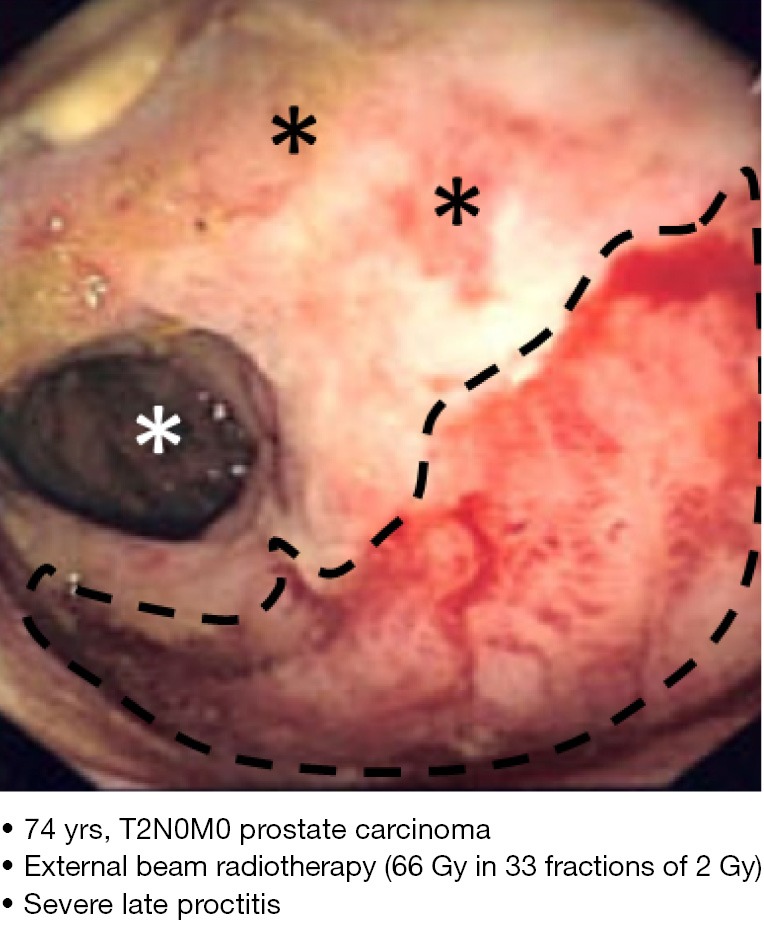
Clinical and treatment characteristics of case RS33. RS33 suffered a severe late radiosensitivity reaction in the tissues adjacent to the radiotherapy target organ (rectum). The dashed line outlines the most severe area of rectal radiosensitivity (proctitis), manifesting vascular engorgement, oedema and haemorrhage. Dark asterisks: other areas of superficial proctitis. White asterisk: upper rectum.
Cell lines and cell culture
Lymphoblastoid cell lines (LCLs) were generated from this patient and other patients’ blood B-cells, as described (13). Human LCLs from non-RS patients, namely CL44, CL39, CL21 and CL7 were from patients that did not develop severe reactions to radiotherapy. C1 was a control cell line from an individual who was not treated for cancer. The Lig IV−/− cell line was renamed from N114-P2 for simplicity; this was a human DNA Ligase IV-mutant pre-B cell line that was previously isolated from its parental cell line, Nalm-6 (15).
All cells except the C1 cell line were grown in RPMI 1640 media, supplemented with 10% FBS and gentamycin, and incubated at 37 °C in a 5% CO2 humidified incubator and fed two to three times per week. C1 cells were treated identically except the media was supplemented with 20% FBS. All cell lines were assayed in log phase growth. All irradiation was performed using a 137Cs source at a dose rate of 0.6 Gy min-1. Cells were irradiated on ice, then immediately incubated at 37 °C for 4 hours, as below.
RNA isolation, reverse transcription and quantitative real time-PCR (RT-PCR)
Total RNA was isolated from cell lines using the RNeasy mini kit (Qiagen, Hilden, Germany) as per the manufacturer’s protocols. Purified RNA was stored at −20 °C. RNA quality and quantity were analyzed using a spectrophotometer, NanoDrop ND-1000 (Nanodrop Technologies, Delaware, USA). One µg of RNA was used for first strand cDNA synthesis. Reverse transcription was carried out with SuperScript III reverse transcriptase (Invitrogen, California, USA) as per the manufacturer’s protocol. Two hundred fifty ng random hexamer primers were used to generate first strand cDNA for use in gene expression studies. First strand cDNA products were diluted 20-fold and stored at −20 °C prior to further analysis.
Gene expression studies
Five control cell lines (CL44, CL39, CL21, CL7, C1), RS33, and a known RS cell line, Lig4−/−, were studied by RT-PCR. XRCC4 and DNA Ligase IV gene expression were investigated. Total RNA was isolated from cell lines prior to and after irradiation at 2 and 10 Gy on ice. Immediately after irradiation, samples were incubated at 37 °C for 4 hours. Subsequently, RNA extractions and first strand cDNA synthesis were carried out as described previously. Quantitative RT-PCR reactions were performed on an ABI Prism 7000 Sequence Detection System (Applied Biosystems, California, USA) using ABsolute QPCR SYBR Green Mix (ABgene, Epsom, UK) as per the manufacturer’s protocol. One µL of diluted cDNA was used. The cycling conditions consisted of (I) 50 °C for 2 mins, (II) 95 °C for 15 minutes and (III) 40 cycles at 95 °C for 15 seconds followed by 60 °C for 1 minute. All cell line expression data were normalized to phosphoglycerate kinase 1 (PGK1), a housekeeping gene. This gene was consistently expressed between unirradiated and irradiated samples. Subsequently, all data were normalized to CL44 at 0 Gy. CL44 was chosen because this cell line showed the least expression variation between duplicate samples. In some cases, pooled controls were analyzed. Primers used for RT-PCR were as follows: PGK, forward (5'-3'): ACACGGAGGATAAAGTCAGCCA; reverse (3'-5'): CACCCCAGGAAGGACTTTACCT; XRCC4, forward: CTGATGGTCATTCAGCATGGA; reverse: TCCATTGCCATGTCATCAGC; DNA Ligase IV, forward: CCGCAGGAAACCATCAAGATC; reverse: TTCTCGTTTAACTGGCCTCGG.
Pulsed field gel electrophoresis (PFGE) DNA damage and repair assay
The kinetics of DNA repair for CL44, CL39, CL21, C1, RS33 and Lig4−/− cell lines were assayed after irradiation using PFGE as described (16), but with different conditions. This assay was carried out over a time course of 240 minutes. Initially cell lines were washed twice with phosphate buffered saline (1.4 M NaCl, 0.02 M KCl, 0.05 M Na2HPO4 and 0.02 M NaH2PO4), then approximately 8.0×105 cells mL−1 from each cell line were plated into 6-well plates. Plates were then irradiated at 40 Gy on ice. Zero dose controls were mock irradiated on ice. After irradiation, plates were incubated at 37 °C to allow repair to occur. At time points 0, 30, 60, 120 and 240 minutes after irradiation, aliquots of 4.0×105 cells were mixed with 0.8% low-melting point (LMP) agarose diluted in PBS, and pipetted into plastic moulds to a final concentration of 1.0×105 cells in 40 µL plugs. Plugs were solidified at 4 °C for 20 minutes. Subsequently, solidified plugs were lysed in ice-cold lysis buffer (2% sarkosyl, 1 mg mL−1 proteinase K (Promega, Wisconsin, USA) in 400 mM Na2EDTA PH 8.0) for 1 h at 4 °C and then incubated at 50 °C for 20 hours. The following day, plugs with lysed cells were washed 3 times in 0.1 M EDTA for 30 minutes each. Finally, plugs were equilibrated in running buffer (45 mM Tris base, 45 mM boric acid, 1 mM Na2EDTA PH 8.0) for 30 minutes prior to insertion of plugs into the wells of the gel. Gels were prepared using 0.8% chromosomal grade PFGE agarose (Bio-Rad, California, USA) and run in 0.5× TBE running buffer. Plugged gels initially were sealed with 0.8% LMP agarose and placed into the PFGE unit, Gene Navigator (Pharmacia Biotech, Uppsala, Sweden) with electrical fields oriented at 120º. Additionally, known quantities of lambda standards were loaded into lanes for normalization during quantitation. Electrophoreses were performed at 6.5 V cm−1 at 14 °C with pulse rates of 45 s for 18 hours. Under these electrophoretic conditions, fragmented DNA up to 2 megabases (mb) are resolved. Following electrophoresis, DNA in gels was stained with SYBR Gold (Invitrogen, California, USA). Fluorescence emitted by DNA chelated with SYBR Gold was imaged using a laser gel reader, Molecular Imager FX (Bio-Rad, California, USA). Fluorescence recorded across different time points or lanes were normalized to the initial damage, t=0, induced in individual cell lines.
Statistical analysis
All data and statistical calculations such as mean, standard error of mean (SEM), two-tailed paired and unpaired t-tests and Fisher’s exact test were performed using Microsoft Excel (Microsoft Corporation, Washington, USA).
Results
DNA DSBs are key radiation-induced cellular lesions (10,16-18). The NHEJ pathway of DNA DSB repair is the fastest and most prominent DNA DSB repair mechanism in mammalian cells [(19) and reviewed in (20)], operative in all cell cycle phases (18). Previously, we took a number of candidate molecule approaches to attempt to elicit the molecular basis of human radiosensitivity (see Introduction). In the present case, we screened for aberrations in the expression of key NHEJ proteins, namely DNA Ligase IV, XRCC4, Ku70 and Ku80, in cells derived from RS cancer patients, with the hypothesis that, because the main target of radiation damage is DNA, their radiosensitivity may result from defects in the DNA damage response.
One of our patients, designated RS33, manifested severe ‘late’ radiosensitivity (Figure 1). He had persistent daily, bright red per rectal bleeding commencing approximately one year after he completed prostate cancer radiotherapy. The per rectal bleeding did not result in anemia and he was subsequently diagnosed with radiation proctitis. He had laser coagulation of rectal mucosal vessels performed, which settled the bleeding. However, two years later he required further laser coagulation, which relieved this symptom.
This patient also had NHEJ protein expression abnormalities (Figure 2). The major finding was that of substantially reduced levels of both XRCC4 and DNA Ligase IV proteins in his lymphoblastoid cells, compared with other RS patients’ cells and control cells. Importantly, these two proteins form a tight functional complex in DNA DSB resolution by NHEJ (see Discussion). The reduced NHEJ protein levels were found reproducibly over many replicate Western blots and were not seen in cells from any other RSs or controls. We are currently performing whole genome sequencing to examine for genomic germline alterations in NHEJ components and their regulators/protein partners, which could explain the observed reduced NHEJ protein levels.
Figure 2.
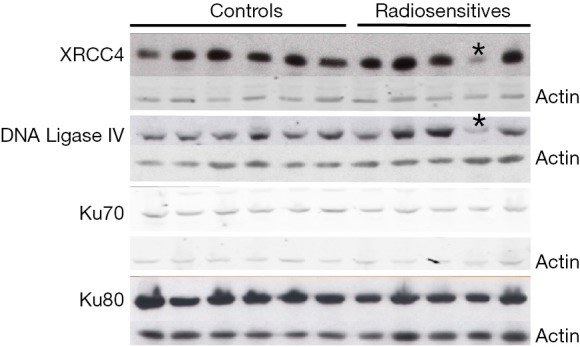
Representative western blot analysis of multiple NHEJ proteins in cells from control and radiosensitive cancer patients. Each vertical lane represents multiple Western blot analyses from an individual cancer patient. In the demonstrated experiment, cells from 6 control, and 5 radiosensitive cases, are shown (horizontal bars). The analysed NHEJ proteins are shown on the left of the figure (XRCC4, DNA Ligase IV, Ku70 and Ku80), whilst the actin controls for each protein blot are shown on the right. Asterisks show low levels of XRCC4 and DNA Ligase IV in cells from the same patient (RS33). The experiment was performed with over five replicates, and low XRCC4/DNA Ligase IV levels were invariantly seen for RS33 but no other cases. NHEJ, non-homologous end-joining.
Reduced levels of XRCC4 and DNA Ligase IV proteins in RS33 prompted us to ask whether this may be due to reduced steady-state mRNA levels of the respective molecules. RT-PCR analysis of XRCC4 mRNA expression in different control cell lines and in RS33 is shown in Figure 3. It confirms that, compared to pooled controls and the DNA Lig IV−/− cell line, not only is XRCC4 protein low in RS33 cells (Figure 2), XRCC4 mRNA also is (white bars). Interestingly, XRCC4 mRNA levels were relatively normal in Lig IV−/− mutant cells.
Figure 3.
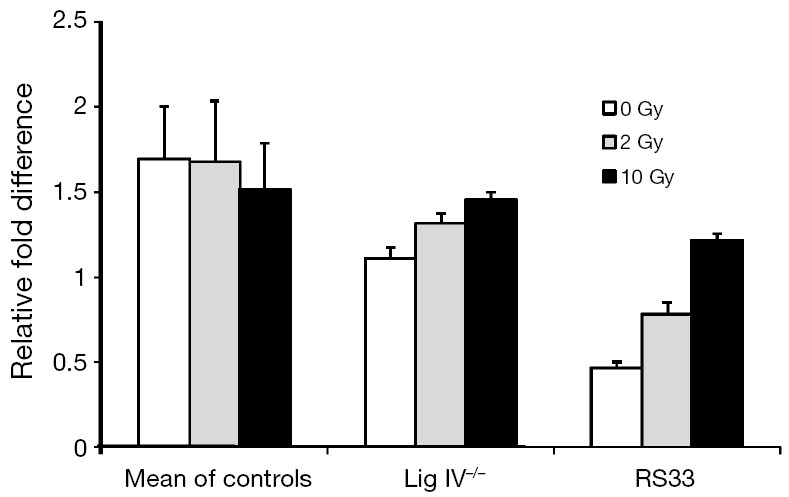
RT-PCR gene expression analysis of XRCC4 mRNA expression in cells from control and radiosensitive cancer patients. XRCC4 levels were determined at baseline and after 2 and 10 Gy ionizing radiation to the different cell lines. Cells from five controls were pooled (‘Mean of Controls’) and XRCC4 expression compared with cells from a radiosensitive patient with a known molecular defect in DNA Ligase IV (‘Lig IV−/−’) and the radiosensitive case we present here (RS33). RT-PCR, real time-PCR.
The DNA damaging agent to which the RS33 patient was sensitive was radiotherapy (ionizing radiation). To determine whether RS33’s low XRCC4/Lig IV could be modulated by radiation, we exposed cells of the three phenotypes to both 2 Gy (a clinically relevant radiation dose) and 10 Gy of ionizing radiation. There was little XRCC4 mRNA modulation by either radiation dose in control and Lig IV−/− cells. However, there was a statistically significant, dose-dependent increase in XRCC4 mRNA after irradiation of RS33 cells (Figure 3).
Because XRCC4 and DNA Ligase IV exist in a tight complex and Ligase IV protein levels were, like XRCC4, also reduced in RS33 cells, we examined steady-state DNA Ligase IV mRNA levels in RS33 cells and controls (Figure 4). As for the DNA Ligase IV protein levels, there was a reduction, albeit slight, in the corresponding mRNA in RS33 cells. None of the three cell groups showed significant ionizing radiation mRNA induction, although there may have been slight induction in RS33 cells. Interestingly, in the DNA Ligase IV mutant cells, DNA Ligase IV mRNA levels were markedly higher than in controls and RS33 cells, possibly representing part of a feedback loop because of the correspondingly low DNA Ligase IV protein levels in these cells.
Figure 4.
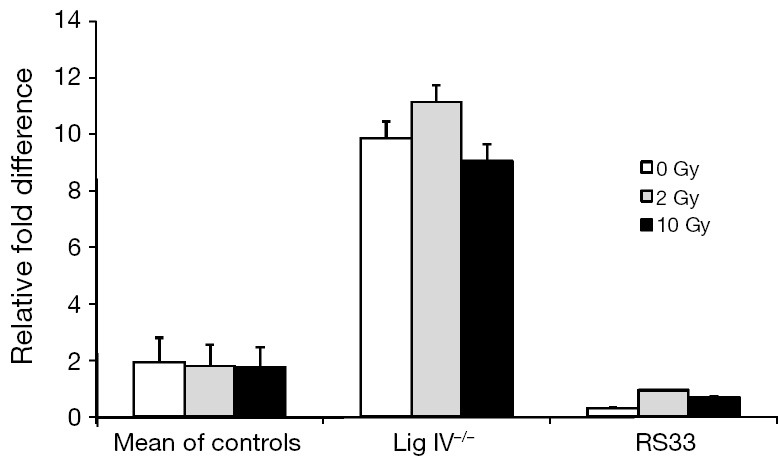
RT-PCR gene expression analysis of DNA Ligase IV mRNA expression in cells from control and radiosensitive cancer patients. DNA Ligase IV levels were determined at baseline and after 2 and 10 Gy ionizing radiation to the different cell lines. Cells from five controls were pooled (‘Mean of Controls’) and DNA Ligase IV expression compared with cells from a radiosensitive patient with a known molecular defect in DNA Ligase IV (‘Lig IV−/−’), which results in reduced constitutive DNA Ligase IV protein levels, and the radiosensitive case we present here (RS33). RT-PCR, real time-PCR.
We next investigated whether there were functional consequences of the reduced NHEJ proteins in RS33 cells. Because of their role in ionizing radiation-induced DNA damage repair, we employed a DNA DSB repair assay utilizing PFGE, to examine DNA DSB repair kinetics in RS33 and control cells (Figure 5A). This assay shows the amount of DNA damage (broken DNA) remaining (reflecting the amount and rate of repair) with time after ionizing radiation exposure. As expected, the DNA Lig IV−/− cells had high levels of remaining DNA DSBs after repair incubation. RS33 cells had an intermediate repair capacity between the DNA Lig IV−/− cells and the five pooled control cell lines, with the median time to rejoin the induced DNA DSBs in RS33 cells being more than double that of the pooled controls (<60 vs. 135 min) (Figure 5A). The RS33 data in Figure 5A were independently confirmed on a separate occasion by a different researcher (data not shown). Figure 5B is a visual representation of the graphical data in Figure 5A. At t=0, the lower molecular weight DNA has migrated out of the gel wells, where high molecular weight DNA remains (arrowheads). It is evident that the repair of DNA fragmentation (stained DNA outside the wells) is impaired in RS33 cells to an intermediate extent between the pooled controls and the DNA Lig IV−/− mutant.
Figure 5.

Pulsed field gel electrophoresis analysis of cells from control and radiosensitive cancer patients. (A) After cells were exposed to 40 Gy ionizing radiation, increasing time periods were allowed for aliquots of cells to repair DNA damage before their DNA was electrophoresed. Cells from five controls were pooled (‘Normal Matched Controls’) for the analysis, and compared with a DNA Ligase IV null control and RS33 cells; (B) visualization, using SYBR Gold, of DNA damage induction and repair. Arrowheads mark high molecular weight DNA retained in gel wells. Top panel: Lig IV−/−; Middle panel: RS33; Lower panel: pooled controls.
Discussion
In this study, we performed a screen of key NHEJ proteins, which mediate the most rapid and prolific DNA DSB repair process in mammalian cells, in cells from a unique bank of RS cancer patients. We hypothesized that abnormalities in expression of these proteins may explain the radiosensitivity of some of our cancer patients. The cells from one patient showed a reproducibly consistent reduction in two complex-forming NHEJ protein components. We also showed a reduction in both gene products at the mRNA level. Additionally, the mRNA inducibility by ionizing radiation was increased for one of the proteins in the patient’s cells. We confirmed the likely functional significance of the NHEJ expression abnormalities with a DNA DSB repair assay.
What might be the molecular defect in RS33 cells? It is very likely a true defect, since (I) XRCC4 and DNA Ligase IV form a tight functional complex during DNA DSB repair and both were reduced in RS33 cells; (II) XRCC4 mRNA was also reduced and showed a dysregulated ionizing radiation response in RS33 cells and (III) we confirmed its likely functional significance with a DNA DSB repair assay.
There may be a mutation in the coding or regulatory regions of the corresponding genes, or other NHEJ components or their positive or negative regulators or associated proteins. For example, prior to NHEJ occurring, there is recognition of DNA DSBs by the MRN (MRE11/Rad50/NBN) complex and recruitment of the Ku heterodimer to broken DNA ends; subsequently, Ku recruits the DNA protein kinase catalytic subunit (DNA-PKcs) with its partner, Artemis. DNA-PKcs is then autophosphorylated and collectively the above events induce association of protein partners XRCC4 and DNA Ligase IV with XLF and PAXX and ligation of the DNA DSB [all reviewed in (20)]. Conceivably, dysfunction of the structure (e.g., truncation) or function of associated proteins may perturb downstream functions such as XRCC4 and DNA Ligase IV expression, as seen here, although a germline mutation in either XRCC4 or DNA Ligase IV is probably the most parsimonious explanation for the reduction of their corresponding mRNA and protein levels in RS33 cells. (Additionally, Ku70 and Ku80 levels were normal in RS33 cells). This is presently being addressed by germline DNA sequencing.
Although reduced, XRCC4 and DNA Ligase IV are not absent in RS33 cells. This is not surprising, since germline ablation in mice of DNA Ligase IV is embryonic lethal (21), and all humans to date with DNA Ligase IV mutations have hypomorphic alleles [reviewed in (22)]. XRCC4 when disrupted in mice is also embryonic lethal (23) and recently, humans with hypomorphic XRCC4 alleles have been identified (24).
The question arises as to why patient RS33 did not manifest other features of XRCC4/DNA Ligase IV impairment, such as immunodeficiency. Presumably, the residual protein levels in this patient were biologically adequate. We speculate that the prostate cancer itself could have been associated with the DNA DSB repair deficient condition, as DNA repair defects are often associated with cancer-proneness in mammals (25).
Why should both XRCC4 and DNA Ligase IV proteins be affected? Less likely, this is due to abnormalities in both proteins or their regulators. As both proteins act in concert it is more probable that reduced levels of one may affect the other. This has indeed been shown in a DNA Ligase IV mutant (26).
XRCC4 and DNA Ligase IV are also mediators of DNA DSB repair during V(D)J recombination, which forms antigen receptor genes, and antibody diversity, by rearrangement of the component gene segments. Class switch recombination further enhances genetic diversity and is also dependent on functional NHEJ (22). We note that there was no overt immunodeficiency in the RS33 patient, although formal testing of immunity was not performed.
It is a potential limitation of this study that the precise molecular defect has not yet been determined in RS33 cells. It will be interesting in the future to evaluate coding and regulatory sequences of known NHEJ factors for DNA sequence variations and to check V(D)J recombination capacity, as well as XRCC4 function (e.g., enhancement of the DNA joining activity of LIG4 (27) and DNA Ligase IV function (e.g., adenylation and recruitment of the XRCC4-DNA Ligase IV complex to chromatin) (27,28) in RS33 cells.
Conclusions
We have identified a novel biological phenotype linked to clinical radiosensitivity. Further work may unveil the molecular defect in this case. This is important in that very few such molecular defects (mutations) are known in human radiotherapy subjects. Such knowledge may contribute to the understanding of radiation response mechanisms in cancer patients and to personalization of radiotherapy.
Acknowledgements
Funding: Australian National Health and Medical Research Grants, Numbers 288713 and 400337.
Ethical Statement: Ethics approval was obtained from the Peter MacCallum Cancer Centre Ethics Committee, Melbourne, Australia, Study Number 96/39. Patients also gave individual consent for study inclusion and use of their cells/tissues.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Rothblum-Oviatt C, Wright J, Lefton-Greif MA, et al. Ataxia telangiectasia: a review. Orphanet J Rare Dis 2016;11:159. 10.1186/s13023-016-0543-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Available online: www.rtog.org
- 3.Marrone M, Stewart A, Dotson WD. Clinical utility of gene-expression profiling in women with early breast cancer: an overview of systematic reviews. Genet Med 2015;17:519-32. 10.1038/gim.2014.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yard BD, Adams DJ, Chie EK, et al. A genetic basis for the variation in the vulnerability of cancer to DNA damage. Nat Commun 2016;7:11428. 10.1038/ncomms11428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnett GC, Coles CE, Elliott RM, et al. Independent validation of genes and polymorphisms reported to be associated with radiation toxicity: a prospective analysis study. Lancet Oncol 2012;13:65-77. 10.1016/S1470-2045(11)70302-3 [DOI] [PubMed] [Google Scholar]
- 6.Fang Z, Kozlov S, McKay MJ, et al. Low levels of ATM in breast cancer patients with clinical radiosensitivity. Genome Integr 2010;1:9. 10.1186/2041-9414-1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Severin DM, Leong T, Cassidy B, et al. Novel DNA sequence variants in the hHR21 DNA repair gene in radiosensitive cancer patients. Int J Radiat Oncol Biol Phys 2001;50:1323-31. 10.1016/S0360-3016(01)01608-X [DOI] [PubMed] [Google Scholar]
- 8.Leong T, Whitty J, Keilar M, et al. Mutation analysis of BRCA1 and BRCA2 cancer predisposition genes in radiation hypersensitive cancer patients. Int J Radiat Oncol Biol Phys 2000;48:959-65. 10.1016/S0360-3016(00)00728-8 [DOI] [PubMed] [Google Scholar]
- 9.Fogarty GB, Muddle R, Sprung CN, et al. Unexpectedly severe acute radiotherapy side effects are associated with single nucleotide polymorphisms of the melanocortin-1 receptor. Int J Radiat Oncol Biol Phys 2010;77:1486-92. 10.1016/j.ijrobp.2009.07.1690 [DOI] [PubMed] [Google Scholar]
- 10.Vasireddy RS, Sprung CN, Cempaka NL, et al. H2AX phosphorylation screen of cells from radiosensitive cancer patients reveals a novel DNA double-strand break repair cellular phenotype. Br J Cancer 2010;102:1511-8. 10.1038/sj.bjc.6605666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kerns SL, Ostrer H, Rosenstein BS. Radiogenomics: using genetics to identify cancer patients at risk for development of adverse effects following radiotherapy. Cancer Discov 2014;4:155-65. 10.1158/2159-8290.CD-13-0197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fachal L, Gómez-Caamaño A, Barnett GC, et al. A three-stage genome-wide association study identifies a susceptibility locus for late radiotherapy toxicity at 2q24.1. Nat Genet 2014;46:891-4. 10.1038/ng.3020 [DOI] [PubMed] [Google Scholar]
- 13.Sprung CN, Chao M, Leong T, et al. Chromosomal radiosensitivity in two cell lineages derived from clinically radiosensitive cancer patients. Clin Cancer Res 2005;11:6352-8. 10.1158/1078-0432.CCR-04-1931 [DOI] [PubMed] [Google Scholar]
- 14.Denham JW, Peters LJ, Johansen J, et al. Do acute mucosal reactions lead to consequential late reactions in patients with head and neck cancer? Radiother Oncol 1999;52:157-64. 10.1016/S0167-8140(99)00107-3 [DOI] [PubMed] [Google Scholar]
- 15.Grawunder U, Zimmer D, Fugmann S, et al. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol Cell 1998;2:477-84. 10.1016/S1097-2765(00)80147-1 [DOI] [PubMed] [Google Scholar]
- 16.McKay MJ, Kefford RF. The spectrum of in vitro radiosensitivity in four human melanoma cell lines is not accounted for by differential induction or rejoining of DNA double strand breaks. Int J Radiat Oncol Biol Phys 1995;31:345-52. 10.1016/0360-3016(94)E0147-C [DOI] [PubMed] [Google Scholar]
- 17.Marnef A, Legube G. Organizing DNA repair in the nucleus: DSBs hit the road. Curr Opin Cell Biol 2017;46:1-8. 10.1016/j.ceb.2016.12.003 [DOI] [PubMed] [Google Scholar]
- 18.Sprung CN, Matthews LJ, McKay MJ. DNA repair deficiencies: connecting carcinogenesis and sensitivity to ionising radiation. Today's Life Science 2002;14:40-4. [Google Scholar]
- 19.McKay MJ, Withana N, Davey DS, et al. Lymphoid and fibroblastic cell lineages from radiosensitive cancer patients: molecular analysis of DNA double strand break repair by major non-homologous end-joining sub-pathways. Asia Pac J Clin Oncol 2011;7:17-26. 10.1111/j.1743-7563.2010.01364.x [DOI] [PubMed] [Google Scholar]
- 20.Lavin MF, Kozlov S, Gatei M, et al. ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor. Biomolecules 2015;5:2877-902. 10.3390/biom5042877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnes DE, Stamp G, Rosewell I, et al. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol 1998;8:1395-8. 10.1016/S0960-9822(98)00021-9 [DOI] [PubMed] [Google Scholar]
- 22.Woodbine L, Gennery AR, Jeggo PA. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst) 2014;16:84-96. 10.1016/j.dnarep.2014.02.011 [DOI] [PubMed] [Google Scholar]
- 23.Yan CT, Kaushal D, Murphy M, et al. XRCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc Natl Acad Sci U S A 2006;103:7378-83. 10.1073/pnas.0601938103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo C, Nakazawa Y, Woodbine L, et al. XRCC4 deficiency in human subjects causes a marked neurological phenotype but no overt immunodeficiency. J Allergy Clin Immunol 2015;136:1007-17. 10.1016/j.jaci.2015.06.007 [DOI] [PubMed] [Google Scholar]
- 25.Bhattacharjee S, Nandi S. Choices have consequences: the nexus between DNA repair pathways and genomic instability in cancer. Clin Transl Med 2016;5:45. 10.1186/s40169-016-0128-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riballo E, Critchlow SE, Teo SH, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol 1999;9:699-702. 10.1016/S0960-9822(99)80311-X [DOI] [PubMed] [Google Scholar]
- 27.Available online: www.genecards.org
- 28.Liu S, Liu X, Kamdar RP, et al. C-Terminal region of DNA ligase IV drives XRCC4/DNA ligase IV complex to chromatin. Biochem Biophys Res Commun 2013;439:173-8. 10.1016/j.bbrc.2013.08.068 [DOI] [PubMed] [Google Scholar]