

Abstract
Background
Pediatric patients with heart failure are treated with medical therapies that were developed for adult patients. These therapies have been shown to be ineffective in pediatric trials, leading to the recognition that new pediatric-specific therapies must be developed. We have previously shown that administration of the recombinant growth factor neuregulin-1 (rNRG1) stimulates heart muscle cell (cardiomyocyte) regeneration in adult mice. We hypothesized that rNRG1 administration may be more effective in the neonatal period, which could provide a new therapeutic paradigm for treating heart failure in pediatric patients.
Methods
We used a cryoinjury model to induce myocardial dysfunction and scar formation for evaluating the effectiveness of rNRG1-administration in neonatal mice. We evaluated the ability of rNRG1 to stimulate cardiomyocyte proliferation in intact cultured myocardium from pediatric patients.
Results
After cryoinjury in neonatal mice, early administration of rNRG1 from birth for 34 days improved myocardial function and reduced the prevalence of transmural scars. In contrast, late administration of rNRG1 from 4 to 34 days after cryoinjury transiently improved myocardial function. The mechanisms of early administration involved cardiomyocyte protection (38%) and proliferation (62%). rNRG1 induced cardiomyocyte proliferation in myocardium from infants with heart disease less than 6 months of age.
Conclusion
Our results identify a more effective time period within which to execute future clinical trials of rNRG1 for stimulating cardiomyocyte regeneration.
Introduction
Congenital heart disease (CHD) is the leading cause of birth defect-related morbidity and mortality (1, 2). Although corrective surgery enables young patients to survive most forms of CHD, they frequently develop heart failure (3). The available medical therapies are based on disease paradigms developed for adult patients but subsequently were shown to be ineffective in controlled pediatric trials (4–6). There is an increasing awareness that the underlying mechanisms of heart failure are different in pediatric patients and that new therapeutic paradigms have to be developed (4).
The goal of cardiac regeneration is to provide new functionally integrated heart muscle cells (cardiomyocytes). Three principal strategies are being pursued: Stem cell transplantation; Differentiation of fibroblasts into cardiomyocytes; and Stimulating the proliferation of endogenous cardiomyocytes (7, 8). Although human adults show little or no endogenous cardiomyocyte proliferation, this does not be seem true for younger people (9, 10). We have already shown that cardiomyocyte proliferation contributes to physiologic heart growth in young humans (10). Cardiomyocyte proliferation during post-natal heart growth has also been demonstrated in mouse and rat pups, but it declines significantly during the first week of life (11, 12). The rate of endogenous cardiomyocyte proliferation is higher in neonatal mice enabling them to completely regenerate (13–15) in contrast to pre-adolescent mice, which only partially regenerate (16, 17). Although endogenous cardiomyocyte proliferation can be detected in humans without heart disease up to 20 years of age (10), it is likely that the presence of heart disease influences the rate cardiomyocyte proliferation.
Neuregulin-1 (NRG1), a member of the epidermal growth factor (EGF) family, is required for cardiac development (18, 19) and for limiting the injury after myocardial ischemia (20).
Administration of recombinant rNRG1 preparations is beneficial in a variety of small and large animal models of acquired heart disease (21–23). rNRG1 is currently being pursued as an investigational new drug (IND) for the treatment of heart failure (24, 25) and has been shown to be effective in adult patients with left ventricular ejection fraction (EF) < 40% (26, 27). Thus, recombinant rNRG1 is available and suitable for administration in humans.
Different mechanisms of action have been proposed to explain the beneficial effects of rNRG1 administration in heart failure (24, 25). We (22) and others (28–30) have previously demonstrated that rNRG1 stimulates cardiomyocyte proliferation. Thus, administration of rNRG1 stimulates a cellular mechanism that may show a large benefit in pediatric patients.
Here, we combine regeneration experiments in neonatal mice with cell biological experiments in intact human myocardium to characterize the period of cardiomyocyte proliferation, the ability to stimulate it with rNRG1, and the relationship between the timing of rNRG1-administration and myocardial repair.
Results
Development and validation of a cryoinjury method
We used cryoinjury to create a neonatal mouse model of myocardial dysfunction and scarring. We emulated key elements of previously published amputation and cryoinjury methods in zebrafish (31–34) and in neonatal mice (14, 35). We fashioned a metal cryoprobe, cooled it in liquid nitrogen for approximately 20 min, and applied it to the surface of the heart for 2 sec. Using this technique, we performed over 500 surgeries. We observed a 24-hr mortality of 20% and between 24 hr and 7 days post injury (dpi) a mortality of 25%. Mortality was largely associated with maternal cannibalism. The cryoinjury method also showed robustness and reproducibility in an external lab (BJH and JMP).
Having implemented a feasible and survivable injury technique, we examined the extent and degree of tissue injury achieved by cryoinjury. Bright field microscopy showed superficial hemorrhages at 1 dpi (Fig. 1A). Vital staining with triphenyltetrazolium chloride (TTC) demarcated the injured region on the surface of the heart (Fig. 1B). We examined the extent of myocardial death visualized by TUNEL staining on histological sections (Fig. 1C). The TUNEL-positive myocardial volume at 1 dpi was 0.58 ± 0.02 mm3 (n = 6), corresponding to approximately 18% of the myocardium (Fig. 1D). Staining with a sarcomeric marker revealed disorganized sarcomeres and an overall decreased abundance of sarcomeric proteins in the cryoinjured area (Suppl. Fig. S1).
Figure 1. Cryoinjury induces cell death, myocardial dysfunction, and decreased cardiomyocyte cell cycle activity in neonatal mice.
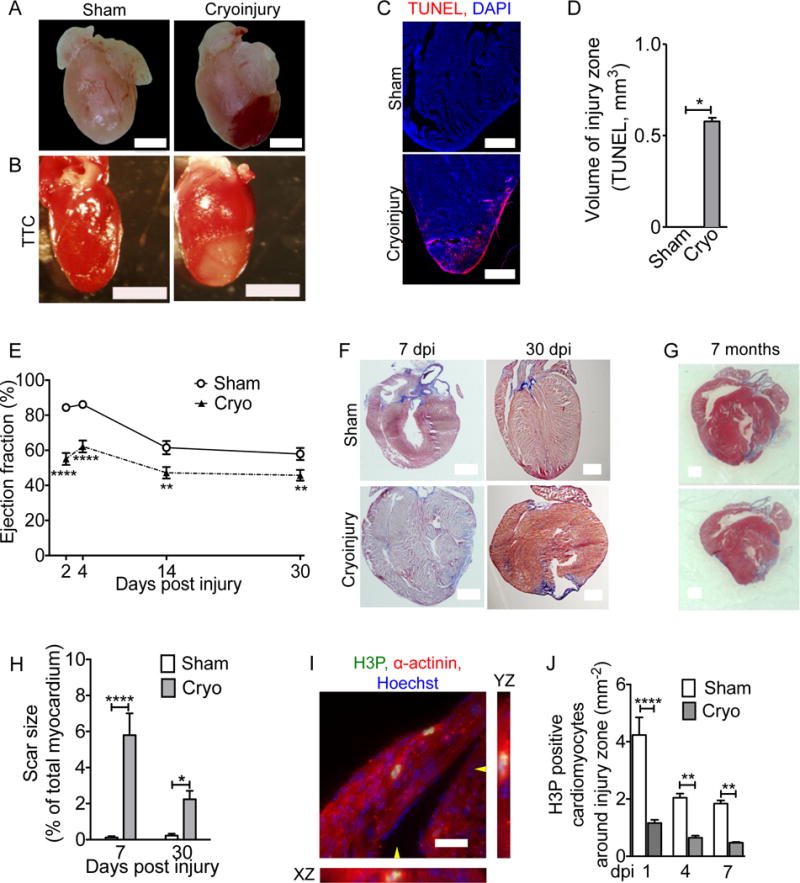
Mice underwent sham surgery or cryoinjury on day of life 1 (P1). (A) Hematoma at the injury site. (B) Vital staining with triphenyltetrazolium chloride (TTC) shows injury zone. (C,D) Myocardial cell death visualized by TUNEL staining. (E) Cryoinjury induces a sustained decrease in the ejection fraction. (F,G) AFOG-stained sections show that scar (blue) is formed within 7 dpi and present 30 days later (F). (G) Cyoinjury-induced scars, visualized on two sections of the same heart (500 μm apart) by Masson Trichrome staining, persist to 7 months after injury. (H) Quantification of scar size. (I) Two cardiomyocytes in M-phase visualized with antibodies against phosphorylated histone H3 (H3P) and α-actinin. The position of orthogonal reconstructions of the cardiomyocyte in the center are indicated with yellow arrowheads. (J) Quantification of M-phase cardiomyocytes in the region around the injury zone shows significant and sustained reduction after cryoinjury. Scale bars 1 mm (A,B,F,G), 20 μm (C,I). Statistical test by t-test (D) and ANOVA Bonferroni’s Multiple Comparison Test (E, H, J) * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Cryoinjury induces myocardial dysfunction and scarring
We performed echocardiography to examine the degree and time-course of myocardial dysfunction. The left ventricular ejection fraction (EF) decreased in the control hearts between 4 and 14 dpi (Fig. 1E), consistent with the previously reported decrease of contractility at this age (36). Cryoinjury reduced the EF significantly at all examined time points. At 30 dpi the EF in the cryoinjury group was 45.7 ± 3% (n = 19) compared to 57.9 ± 3.5% (n = 19, P < 0.05, ANOVA) in sham mice. Thus, cryoinjury induced significant and sustained myocardial dysfunction.
We examined the extent and time-course of myocardial injury. This revealed fibrin deposition at 1 dpi and significant fibrosis at 7 dpi, which matured into transmural scars at 30 dpi (Fig. 1F, Suppl. Fig. S2). We followed a group of mice for 7 months after cryoinjury and they showed transmural scars (n = 6, Fig. 1G, Suppl. Fig. S3). Morphometric analysis revealed that the relative scar volume after neonatal cryoinjury was comparable to values obtained after ligation of the left anterior descending artery in adult mice (Fig. 1H)(37). In summary, cryoinjury in neonatal mouse hearts on day of life 1 (P1) induced persistent myocardial dysfunction and scar formation.
Cryoinjury reduces cardiomyocyte cycling
The data demonstrate a lack of significant regeneration at the structural (scar, Fig. 1F) and functional level (echocardiography, Fig. 1E) and raise the question whether there was evidence for regeneration at the cellular level. Because cardiomyocyte cell cycle activity is increased with myocardial regeneration in zebrafish (38) and neonatal mice (13, 14), we evaluated this mechanism by visualizing cardiomyocytes in karyokinesis using an anti-phospho-histone H3 (H3P) antibody. In sham-operated and cryoinjured hearts the overall number of H3P-positive cardiomyocytes decreased significantly between 1 and 7 dpi (Fig. 1I,J), consistent with prior studies showing that cardiomyocyte cell cycle activity decreases in the first week of life (11, 12). However, the numbers of H3P-positive cardiomyocytes in the injury and border zones were significantly lower in cryoinjured hearts at 1, 4, and 7 dpi compared with the corresponding region in sham hearts (Fig. 1J). In summary, these results indicate that cryoinjury in neonatal mice inhibits endogenous cardiomyocyte cell cycle activity. Taken together, these results indicate that neonatal hearts do not regenerate to the same degree after cryoinjury as reported following myocardial resection or ligation of the left anterior descending coronary artery (14, 15).
Administration of rNRG1 improves myocardial function
Administration of rNRG1 stimulates cardiomyocyte cycling and proliferation in vitro and myocardial repair in vivo (22, 28–30). We used rNRG1 to investigate the effect of stimulating cardiomyocyte cycling. We performed two independent preclinical studies in which only the beginning of rNRG1 therapy was varied (schematic shown in Fig. 2A). In both experiments, cryoinjury was induced one day after birth in all animals, with control animals receiving daily BSA and test animals rNRG1 (100 ng/g) injections. In the first study (referred to as Early Administration from here on), therapy began at birth and continued every day for 34 days. These mice showed a significant improvement of the ejection fraction (EF, measured by echocardiography) beginning at 5 dpi (Fig. 2B). The absolute improvement of the EF was increased by 14 dpi and persisted for 30 days (64 dpi) after cessation of rNRG1 administration. Cardiac MRI (cMRI), performed by a blinded core laboratory at 64 dpi, showed an increase of the EF from 44.3 ± 2.7 (n = 6) in BSA-treated mice to 58.3 ± 1 (n = 5, P < 0.0001, t-test) in rNRG1-treated mice, in agreement with the echocardiography results (Fig. 2C, Suppl. Fig. S4, Suppl. Movies S1–S2). The relative heart weights were significantly lower in rNRG1-treated mice at 64 dpi (Fig. 2D), indicating that rNRG1-administration induced beneficial changes in the myocardium that persisted for 30 days after cessation of therapy.
Figure 2. Early administration of rNRG1 improves myocardial function and structure.
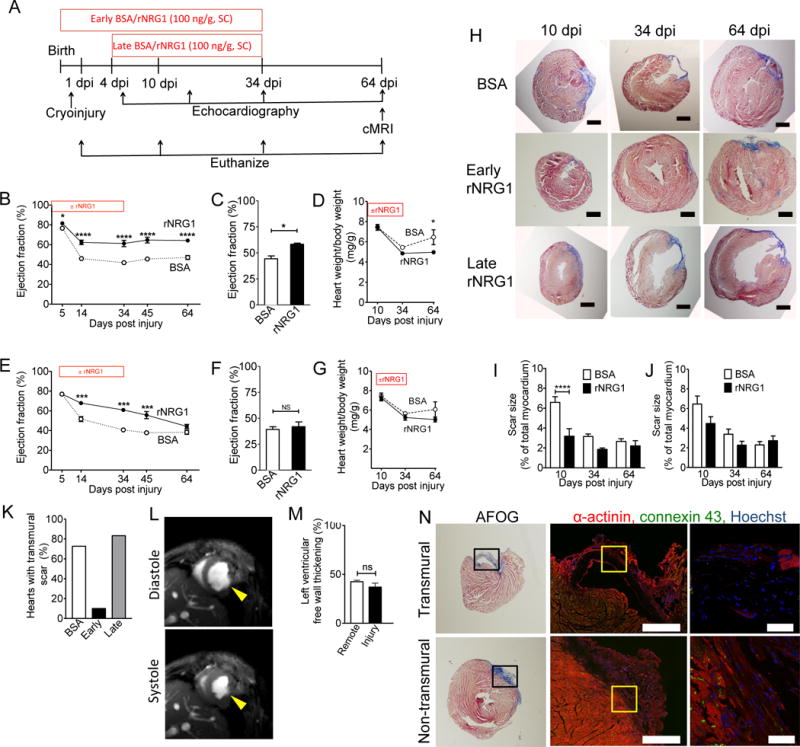
(A) Experimental design of mouse pre-clinical trials. (B–K) Mice underwent cryoinjury on day 1 of life (P1) and were treated with BSA or rNRG1 from day of birth (P0, early administration, B–D), or from P5 (late administration, E–G). (B,C) Prolonged improvement in myocardial function after early administration shown by echocardiography (B) and cMRI at 64 dpi (C). (E,F) Late administration of rNRG1 resulted in transient improvement of myocardial function measured by echocardiography (E) and cMRI at 64 dpi (F). (D,G) Indexed heart weights showed early rNRG1 administration reduced cardiac hypertrophy at 64 dpi. (H) Time-series of AFOG-stained section shows scar (blue) is formed within 10 dpi and still present at 64 dpi. Note transmural scars after cryoinjury in BSA and late rNRG1 treatment groups. (I,J) Quantification of scar size after AFOG-staining shows transient and significant scar reduction after early rNRG1 administration (I). (K) Early administration reduces the percent of transmural scars at 34 and 64 dpi. (L) Non-transmural injury site thickens in systole (64 dpi, early administration) (M) Relative thickening of non-transmural scars is similar to remote LV free wall myocardium. (N) Transmural and non-transmural scars were identified by AFOG sections (left panels). Black rectangles indicate photomicrographs shown in the middle panels. Non-transmural scars have cardiomyocytes connected by gap junctions visualized with Connexin 43 staining (64 dpi, early administration, middle and right panels). Yellow squares indicate zoomed areas (Right panels). Scale bars 1 mm (H), 500 μm (N, center panel), 50 μm (N, far right). dpi: days post injury; BSA: bovine serum albumin; SC: subcutaneous injection: P0: day of birth. Statistical significance was tested with t-test (C,F,M) and ANOVA Bonferroni's Multiple Comparison Test (B,D,E,G,I,J,K) * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
In the second study (referred to as Late Administration study from here on), we began therapy at 5 days after birth (4 dpi) and kept the remainder of the experimental design the same.
Echocardiography showed a significant increase of the EF, first observed at 14 dpi (Fig. 2E). However, at 64 dpi, i.e. 30 days after cessation of rNRG1 injections, echocardiography and cardiac MRI showed that the EF was no longer different between BSA- and rNRG1-treated mice (Fig. 2E, F, Suppl. Fig. S5, Suppl. Movies S3–S4). There were no significant changes in heart weight after late administration (Fig. 2G). In summary, early administration of rNRG1 yields sustained improvements in cardiac function, leading to the conclusion that the timing of rNRG1 therapy is important.
Early administration of rNRG1 prevents transmural scar formation
We examined histological sections for scar formation at 10, 34, and 64 dpi (Fig. 2H, Suppl. Figs. S6–S7). Quantification of the scar size at 10 dpi showed a statistically significant decrease in rNRG1-treated mice after early but not after late administration (Fig. 2I,J). The relative scar size at 64 dpi, i.e. when mice reached adult age, was approximately 2.5% after early and late administration (Fig. 2I,J), which is comparable to our measurements in adult mice after LAD ligation (22). At 34 and 64 dpi after early administration of rNRG1, only one heart showed a transmural scar (n = 10, Fig. 2K). In contrast, 76.5% of mice starting rNRG1 late (n = 12) or receiving BSA (n = 22) showed transmural scars at 34 and 64 dpi (P = 0.0003, Fisher’s exact test on raw data, Fig. 2K). In conclusion, early rNRG1 administration prevents transmural scar formation without affecting the total volume of scar tissue.
Non-transmural scars contract during systole and contain cardiomyocytes with electromechanical connections
We examined cMRI scans to evaluate the functionality of the non-transmural scars after early administration (Fig. 2L). We measured the thickness of non-transmural scars in diastole and systole and calculated relative systolic thickening (Fig. 2M). The systolic thickening of non-transmural injury sites was similar to corresponding remote LV free wall myocardium (Fig. 2M). In contrast, the thickness of transmural scars in the late rNRG1 and BSA groups was so low that it could not be measured, even after enhancement by gadolinium injection (Suppl. Fig. S8).
We examined to what extent cardiomyocytes in non-transmural scars were electromechanically connected with gap junctions. We visualized gap junctions with an antibody against connexin 43 (Cx-43, Fig. 2N). Transmural scars did not contain gap junctions (Fig. 2N, upper panels, Suppl. Movie S5). In contrast, non-transmural scars had intact myocardium and gap junctions (Fig. 2N, lower panels, Suppl. Movie S5). These results show that early administration of rNRG1 induces formation of functionally active myocardium.
Early administration of rNRG1 preserves myocardium
Because early administration of rNRG1 began prior to cryoinjury, we considered a cardioprotective effect. The size of the hematoma demarcating the contact site of the myocardium with the cryoprobe was similar in control and rNRG1-treated mice, indicating that the extent of initial injury was not affected by rNRG1 (Fig. 3A, B). However, at 1 dpi mice treated with BSA had 0.88 ± 0.04 mm3 (n = 6) and rNRG1-treated mice 0.51 ± 0.07 mm3 (n = 5) TUNEL-positive myocardium, representing a significant decrease (P = 0.0009, t-test, Fig. 3C, D). The difference, i.e. 0.37 mm3, is the myocardium protected by rNRG1 and corresponds to 13.6% of the heart at 1 dpi. Assuming that approximately 1×106 cardiomyocytes exist in the heart at birth (17), rNRG1 rescues approximately 136,000 cardiomyocytes at 1 dpi (Suppl. Results S1–S2). This suggests that the first two injections of rNRG1 of the early administration protocol elicit a cardioprotective effect.
Figure 3. Early administration of rNRG1 reduces myocardial death and stimulates cardiomyocyte proliferation.
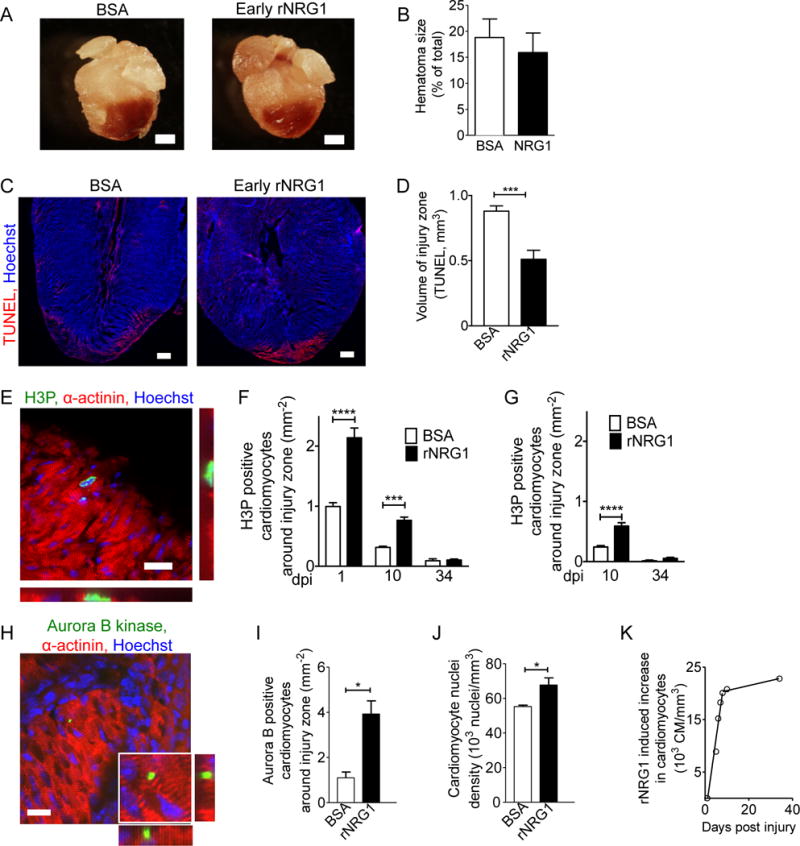
(A,B) Hematomas are present at the zone of injury at 1 dpi (A). Hematoma size quantification shows no change after early administration of rNRG1 (B). (C,D) Photomicrographs (C) and quantification (D) of myocardial cell death visualized by TUNEL staining at 1 dpi after early administration. (E) Cardiomyocytes in M-phase were visualized with an antibody against phosphorylated histone H3 (H3P). (F,G) H3P-positive cardiomyocytes were quantified around injury zone after early (F) and late (G) administration. Treatment with rNRG1 increases cardiomyocyte cell cycle activity, and early administration captures the regenerative phase (F). (H,I) Cardiomyoctyes in cytokinesis were visualized with an antibody against Aurora B kinase (H) and quantified around the injury zone after early administration at 1 dpi (I). (J) Cardiomyocyte nuclear density is increased after early administration of rNRG1 (34 dpi). (K) Early administration of rNRG1 increases the cardiomyocyte density by ~62,000 cardiomyocytes/mm3 within the first 8 days, compared to BSA controls. Scale bars 1 mm (A), 20 μm (C,E,H). Statistical significance was tested with t-test (B,D,I,J) and ANOVA (F,G) * P < 0.05, *** P < 0.001, **** P < 0.0001.
Administration of rNRG1 stimulates cardiomyocyte cycling
Because rNRG1 was shown to increase cardiomyocyte cycling in vitro (28, 29) and in vivo (37), we examined the effect of rNRG1 on cardiomyocyte cell cycling by quantifying the number of H3P-positive cardiomyocytes on histological sections (Fig. 3E–G). Early administration of rNRG1 induced a 2-fold increase of H3P-positive cardiomyocytes compared with BSA-injected animals at 1 and 10 dpi (Fig. 3F). Late administration of rNRG1 showed a similar effect at 10 dpi (Fig. 3G), however, it should be noted this effect started 5 days later. We examined histological sections for evidence of cardiomyocyte mitosis at 1 dpi and found a significant increase in the number of aurora B-kinase positive cardiomyocytes in early rNRG1-treated animals (Fig. 3H, I). In conclusion, rNRG1 increases cardiomyocyte mitosis.
Early administration of rNRG1 stimulates significant cardiomyocyte regeneration in the first 10 days of life
To quantify cardiomyocytes directly, we performed stereology on α-actinin and Hoechst stained tissue. Early administration of rNRG1 induced an increase of the cardiomyocyte nuclei density by 18% compared to BSA-treated mice at 64 dpi (Fig. 3J). We correlated these directly determined stereology results with the number of cardiomyocytes predicted to be regenerated using our cell cycle data (H3P) using a linear regression model (Suppl. Results S3). We then calculated the difference of the cardiomyocyte volume density (in mm−3) between BSA- and rNRG1-treated mice for six age bins between birth and 34 days of age (Suppl. Results S5) and plotted the rNRG1-induced increase for each bin (Fig. 3K, Suppl. Results S6). Between 1 and 10 dpi, there is a steep slope of the rNRG1-induced increase of the number of cardiomyocytes (slope = 3,661 cardiomyocytes/mm3/day, Fig. 3K). In other words, rNRG1 stimulates the generation of approximately 36,610 additional cardiomyocytes per mm3 during the first 10 days of life. This accounts for approximately 78% of the total number of new cardiomyocytes generated during this period. After 10 days of life, the rate of rNRG1-stimulated cardiomyocyte proliferation declines significantly to approximately 96 cardiomyocytes/mm3/day, resulting in 2,300 new cardiomyocytes/mm3 generated between day 10 to 34. These calculations indicate that rNRG1-stimulated cardiomyocyte proliferation in mice is most active in the first 10 days of life (Fig. 3K). In summary, both the timing of the rNRG1 administration and the animal age (P0-P5) are critical determinates of the ability to regenerate myocardium.
ErbB4-expression in cardiomyocytes is required for rNRG1 stimulated cardiomyocyte cycling
Although we have previously shown that rNRG1 stimulates cycling of adult cardiomyocytes via ErbB4 as receptor (22), it is possible that in neonatal hearts, rNRG1 may act on stem or progenitor cells. To explore this possibility, we used ErbB4 floxed mice to inactivate the NRG1-receptor gene in cardiomyocytes. We administered tamoxifen in α-MHC-Mer-Cre-Mer; ErbB4F/wt (control group) and in α-MHC-Mer-Cre-Mer; ErbB4F/F (test group) from day of birth (P0) to P3, leading to significant reduction of ErbB4 mRNA levels (Fig. 4A). We assayed cardiomyocyte H3P at P12 of life (Fig. 4B). rNRG1 increased H3P positive cardiomyocytes in control mice (n=3) but not in test mice (n=3), indicating that ErbB4 is required for rNRG1 stimulated cardiomyocyte cycling (Fig. 4C). Collectively, these data indicate that rNRG1 acts directly on cardiomyocytes via ErbB4 to induce cycling.
Figure 4. rNRG1 acts via ErbB4 on cardiomyocytes in neonatal mouse hearts in vivo.

(A–C) Experiments were performed in α-MHC-Mer-Cre-Mer+/+; ErbB4F/wt (control) and in α-MHC-Mer-Cre-Mer+/+; ErbB4F/F (test). rNRG1 or BSA was administered from day of birth (P0) until P12. ErbB4 inactivation was induced with tamoxifen administration on days P1–3, which caused a significant down-regulation of ErbB4 mRNA levels (A). Representative example of a cardiomyocyte in M-phase with orthogonal reconstructions (H3P, B). rNRG1 increased cardiomyocyte cell cycle activity in ErbB4F/wt, but not in ErbB4F/F mice (C). Statistical test by t-test (A) and ANOVA Bonferroni's Multiple Comparison Test (C) * P < 0.05, scale bar 50 μm.
Cryoinjury and rNRG1 administration induce gene regulation patterns that parallel the structural changes
To assess possible changes in gene expression profiles induced by cryoinjury and rNRG1 administration, we examined gene expression profiles. We induced cryoinjury on day 1 of life (P1) and performed RNAseq after early administration of BSA (n = 5) or rNRG1 (n = 5). Compared with sham (n = 5), cryoinjury (n = 5) changed the regulation of 2,867 genes (P < 0.05). We examined a heat map of 622 genes that were significantly regulated between the BSA and rNRG1-treated groups (P < 0.05, Fig 5A, Suppl. Tab. S1). Genes that were significantly up-regulated (P < 0.05) by rNRG1 treatment included cyclic AMP-dependent transcription factor 3 (ATF3), Geminin (Gmnn), centromere protein A (CENPA), growth differentiation factor 15 (GDF15), connective tissue growth factor (CTGF), and apelin (APLN). Gap junction protein γ2 (GJC2) was up-regulated in rNRG1-treated hearts, consistent with our finding that early administration of rNRG1 induced formation of functional myocardium. Genes that were significantly down-regulated (P < 0.05) included collagen 23α1 (Col23a1), suppressor of cytokine signaling-1 (SOCS1), bone-morphogenic protein 10 (BMP-10), interleukin 6 (IL-6), and Gdpd3. Functional annotation clustering revealed that the genes changing with rNRG1 treatment encoded for cytokine activity, cell growth and division regulators, and transcription factors (Fig. 5B). In summary, the broad changes in gene regulation parallel the functional and structural changes.
Figure 5. Cryoinjury and rNRG1 administration induce gene regulation patterns that parallel structural and functional changes.
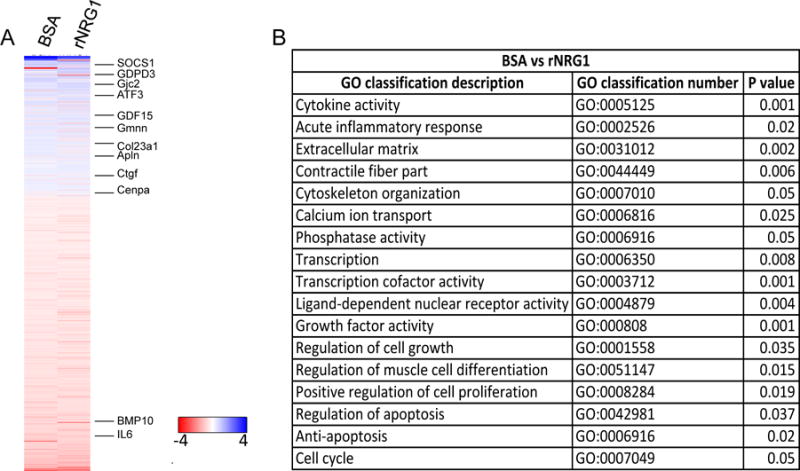
Mice underwent cryoinjury on day 1 of life (P1) and were treated with BSA or rNRG1 according to the early administration protocol. Expression profiling was performed at 10 dpi with 5 mice per group and normalized to sham (n = 5). (A) Heat map shows 622 genes that were significantly regulated between BSA and rNRG1 treated mice (P<0.05). Selected genes discussed in the text are indicated. The color chart indicates fold change of expression using a log2 scale. (B) Functional annotation clustering of differentially expressed genes shows significant regulation of multiple pathways by rNRG1. GO, gene ontology.
Cardiomyocyte cell cycling in pediatric patients with heart disease is decreased
The results from our mouse studies suggest that rNRG1 therapy is most effective during a therapeutic period. To determine whether a similar trend is present in humans, we examined myocardium from human patients (for clinical characteristics, refer to Suppl. Tab. S2–S3). Although our prior work suggested that humans without heart disease show cardiomyocyte proliferation up to 20 years of age, this is likely affected by the presence of heart disease. We isolated cardiomyocytes from patient samples and stained them with antibodies against pancadherin to determine cell quality (Fig. 6A). We then used flow cytometry to quantify the population of cardiomyocytes that were H3P-positive (Fig. 6B). Cardiomyocytes from patients with heart disease exhibited lower cell cycle activity compared to non-diseased myocardium (Fig. 6C). We detected H3P-positive cardiomyocytes in patients less than 6 months old, but not in patients older than that. In conclusion, pediatric patients with heart disease show lower activity and earlier cessation of cardiomyocyte cell cycling than controls.
Figure 6. Pediatric patients with heart disease show decreased cardiomyocyte cell cycle activity.
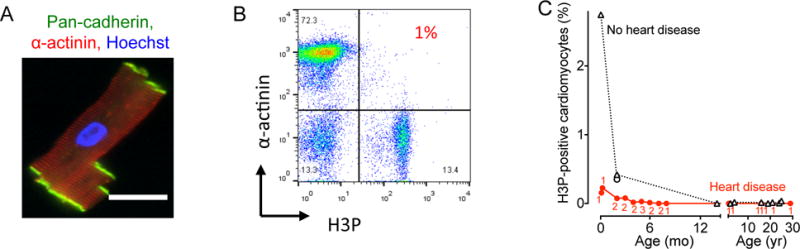
Cardiomyocytes from patients were isolated, stained, and analyzed by flow cytometry. (A) Isolated human cardiomyoctes were intact as evidenced by staining with antibodies against pan-cadherin and α-actinin. (B) Representative double marker plot of a 3 month old patient showing flow cytometry analysis of cardiomyocyte cell cycle activity using cardiomyocyte (α-actinin) and cell cycle markers (H3P). (C) Summary graph shows that patients with heart disease exhibit decreased cycling compared to age-matched controls without heart disease. Numbers of patients per data point are indicated in red, no heart disease were 1 patient per data point. Circles represent RV and triangles LV samples. Scale bar: 50 μm.
Infants with heart disease have the capacity for stimulated cardiomyocyte cycling
We tested whether rNRG1 therapy could stimulate cycling of cardiomyocytes from patients with heart disease. To accomplish this, we modified an organotypic cell culture system for primary human myocardium (39). Sheet-like parallel arrangement of cardiomyocytes, sarcomeric striations (Fig. 7A), and gap junctions (Fig. 7B) were present after 72 hours of organotypic culture. In conclusion, organotypic culture represents a 3-dimensional multicellular model of the human myocardium and a suitable platform for studies of cardiomyocyte cell cycle stimulation. This enabled us to test molecular manipulations mimicking therapeutic interventions and to determine the effect on cardiomyocyte proliferation in situ, without disrupting the native myocardial tissue architecture. Myocardial samples were collected at the time of surgery and maintained with either 100 ng/mL rNRG1, the same concentration that stimulated cycling of cultured cardiomyocytes (22, 28, 40), or fetal calf serum (1% or 20%) for 72 hours. Tissue samples were subsequently sectioned, stained with antibodies against α-actinin and H3P (Fig. 7C), and the number of mitotic cardiomyocytes quantified manually using fluorescence microscopy (Fig. 7D). Compared with controls (1% FCS, n = 8, all < 6 months old), rNRG1 induced an increase in the M-phase cardiomyocyte population by 13– to 31–fold in myocardium from patients 2 to 5 months of age (n = 8 hearts studied, Fig. 7D). In samples from 14 patients ≥ 6 months of age (age range 6 months – 66 years (Fig. 7D), we did not detect M-phase cardiomyocytes. Taken together, these results indicate that infants with heart disease < 6 months of age have cardiomyocytes that can be induced with rNRG1 to re-enter the cell cycle.
Figure 7. rNRG1 stimulates cardiomyocyte cycling in myocardium from infants with heart disease younger than 6 months of age.
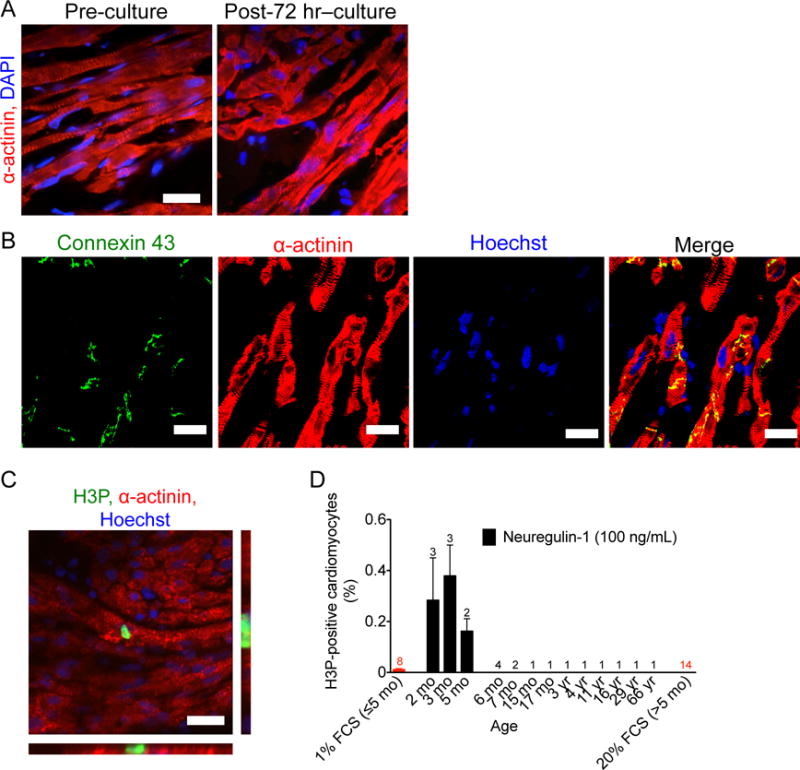
For organotypic culture, chunks of myocardium were maintained in the presence of 1% FCS or rNRG1 for 3 days, fixed, and analyzed by immunofluorescence microscopy. (A,B) Organotypic culture for 3 days does not change microscopic architecture (A). Gap junctions, electromechanical connections, were identified by connexin 43 staining were present after 72 hours of organotypic culture (B). (C,D) rNRG1 stimulates cardiomyocytes to enter M-phase in a 2-month old patient with ToF (C). Quantitative analysis showed that rNRG1 increased M-phase cardiomyocytes in an age-dependent manner (D). Numbers of patients per data point are indicated (D). Scale bars 20 μm (A,C), 50 μm (B).
Infants with heart disease show the capacity for stimulated cardiomyocyte proliferation
We used a non-genetic labeling technique with the fluorescent dye carboxyfluorescein succinimidyl ester (CFSE, Suppl. Fig. S9) to identify proliferating human cardiomyocytes. Using our organotypic culture system, we labeled myocardium with CFSE, whereby fluorescein– protein adducts are retained by cells and are diluted by half with every cell division. We used the CFSE assay to identify cardiomyocytes that have divided and detected them by flow cytometry. We used a forward and side scatter gating strategy to enrich for large cells (cardiomyocytes, Fig. 8A, left panel). Doublets and cell aggregates were excluded (Fig. 8A, middle panel) (16). Dead cells were excluded with 7-AAD dye (Fig. 8A, right panel). CFSE populations were quantified in the Fl-1 channel and revealed two distinct populations (CFSElo and CFSE hi, Fig. 8B). Figure 8B shows a representative example of a 3 months old infant exhibiting a CFSElo population of 4.1%. The CFSElo population represents cardiomyocytes that have undergone several cell divisions; whereas the CFSEhi population represents quiescent cardiomyocytes. A CFSEhi population was found in every heart (Suppl. Fig. S10). We isolated the CFSElo population for lineage reanalysis and stained it with isotype antibody to examine nonspecific binding (Fig. 8C, left panel). Staining with an antibody against troponin T showed that 94.7% of this population were cardiomyocytes (Fig. 8C, right panel). These cardiomyocytes had similar forward and side-scatter characteristics (Fig. 8D) compared to the original forward and side scatter gate settings (Fig. 8A, left panel). These experiments demonstrate that the CFSElo cells (Fig. 8B) are indeed cardiomyocytes.
Figure 8. rNRG1 stimulates cardiomyocyte proliferation in myocardium from patients with heart disease younger than 6 months of age.
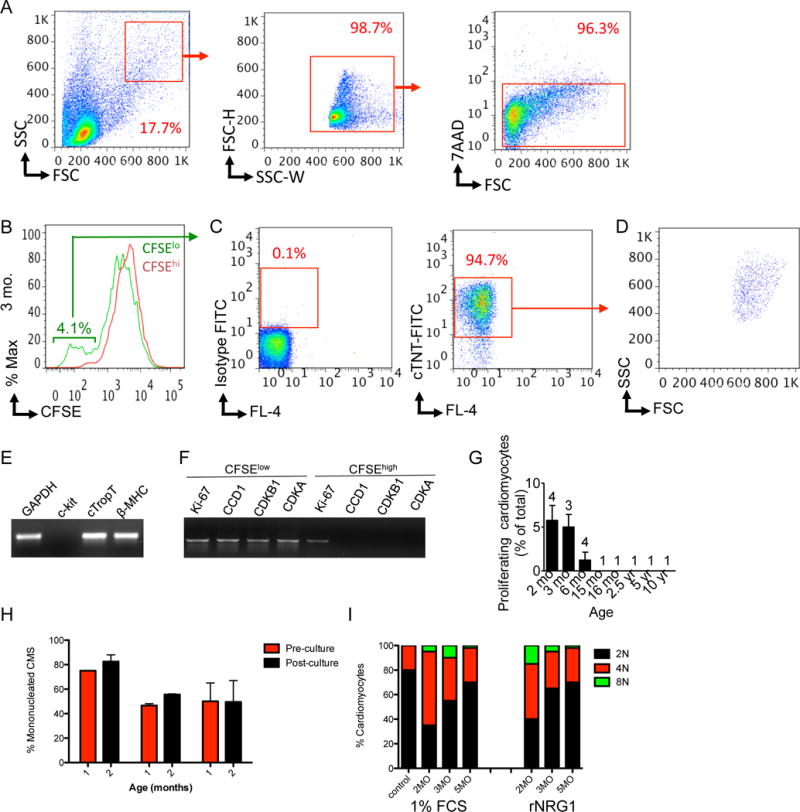
Organotypic cultures of human myocardium were metabolically labeled with CFSE and then maintained in the presence of 1% FBS or rNRG1 for 3 days. Cardiomyocytes were analyzed and isolated by FACS. (A) FACS strategy for enrichment by size (left panel), doublet discrimination (middle panel), and viability (right panel). (B) Flow cytometry analysis of a 3 months-old reveals a CFSElo population of 4.1%. (C–D) After fixation, this population was stained with isotype control (C, left panel) and antibodies against troponin T (C, right panel). Analysis by flow cytometry shows that 94.7% of this population were cardiomyocytes (C, right panel) with a forward and side scatter characteristics (D) similar to (A). (E,F) RT-PCR showed that CFSElo cardiomyocytes expressed markers of mature differentiated cardiomyocytes (E) and cell-cycle associated genes (F). (G) Graph of proportion of CFSElo populations shows that stimulation of cardiomyocyte proliferation in patients with heart disease is age-dependent. Numbers of patients per data point are indicated. (H,I) Laser scanning cytometry shows that administration of rNRG1 in organotypic culture did not change the overall percentage of mononucleated cardiomyocytes (H) or the ploidy pattern of mononucleated cardiomyocytes (I).
To further characterize the CFSElo population, we bulk sorted the CFSElo cells, isolated RNA and performed RT-PCR analysis to examine expression of marker genes. CFSElo cardiomyocytes expressed cardiac troponin T (cTNT) and β-myosin heavy chain (β-MHC), but not c-kit, suggesting that this population contained mature differentiated CMs and no stem cells (41) (Fig. 8E). Furthermore, cell cycle genes were expressed in the CFSElo but not in the CFSEhi population (Fig. 8F).
After this extensive validation of the CFSE assay, we examined the CFSElo population by flow cytometry in patients with heart disease (n = 16). Quantification showed that the CFSElo population was 5.8 ± 1.7% (n = 4) at 2 months, 5 ± 1.4% (n = 3) at 3 months, and 1.2 ± 0.9% (n = 4) at 6 months (Fig. 8G and Suppl. Fig. S10). CFSElo populations were not detectable over 6 months of age, indicating that patient age is an important determinate in the proliferative response to rNRG1 (Fig. 8G).
We examined the possibility that rNRG1 stimulation increased the proportion of cardiomyocytes with higher DNA contents, which may arise by endomitosis, endocycling, or binucleation. Addition of rNRG1 did not change the proportion of mononucleated cardiomyocytes (Fig. 8H) or ploidy in the mononucleated portion (Fig. 8I). This supports the conclusion that rNRG1 stimulates cardiomyocyte division in intact human myocardium.
Discussion
This paper advances a strategy for inducing myocardial repair in pediatric patients. The key conclusions of the work are: [1] a resident population of proliferation-competent cardiomyocytes exists in both neonatal mice and in young (< 6 month old) humans; [2] rNRG1 therapy is cardioprotective and enhances endogenous physiological growth mechanisms by stimulating proliferation of cardiomyocytes in neonatal mice; and [3] a therapeutic window is identified in human myocardium whereby rNRG1-stimulation of cardiomyocyte proliferation is most effective below the age of 6 months.
Myocardial regeneration experiments in neonatal mice were not feasible until the recent introduction of appropriate techniques for inducing injury (14, 15, 42). Ligation of the left anterior descending coronary artery (LAD (13, 15)) and amputation injury (14) in neonatal mice were reported to lead to scarless repair, although the latter results have been challenged (43). In our hands, cryoinjury is a technically feasible method that was independently reproduced in another lab (BH, JP). Our finding of scar formation and dysfunction is in line with the scar formation and delayed repair process after cryoinjury in zebrafish (31, 33, 34) and a prior report in neonatal mice (35). Our results indicate that cryoinjury in neonatal mice is a useful model because it recapitulates scar formation, dysfunction, and decrease of cardiomyocyte cell cycle activity frequently seen in young patients with heart disease.
A surprising finding of this study is that cryoinjured mice and human infants with heart disease have decreased cardiomyocyte cell cycle activity. The decrease of cardiomyocyte cell cycle activity after cryoinjury contrasts with the reported increase of cardiomyocyte cycling after LAD ligation (13) and amputation (14), possibly due to technical differences in the scoring of cardiomyocyte cell cycle events (44). Our results suggest that a population of proliferation-competent cardiomyocytes exists in young mammals, which is inhibited or depleted by heart disease. Administration of rNRG1 rescues this inhibition to near normal levels (~2-fold increase) and stimulates the generation of new heart muscle in neonatal mice following cryoinjury and in organotypic cultures of myocardium from infants with heart disease (13– to 31–fold in myocardium from patients 2 to 5 months of age). Future basic research should focus on the molecular characterization of proliferation-competent cardiomyocytes to be able to track these.
Our model of the mechanisms activated by early administration of rNRG1 in mice involves myocardial protection and regeneration. What are their relative contributions to the overall reparative process? Our comparison of the extent of myocardial death between BSA- and rNRG1-treated mice in the early administration group at 1 dpi showed a difference of ~14%, corresponding to approximately 136,000 cardiomyocytes. Based on our H3P analyses, 0.04% of these 136,000 protected cardiomyocytes will actively cycle, resulting in ~26,000 new cardiomyocytes over a 34-day period (Suppl. Results S1, S2, and S4). On the other hand, direct quantification of cardiomyocyte nuclear density at 34 dpi revealed 3.1 × 104 and 3.8 × 104 cardiomyocytes/mm3 for BSA- and rNRG1-treated animals, respectively, corresponding to a difference of 7,000 cardiomyocytes/mm3. Subtracting the number of cardiomyocytes contributed by protection, this corresponds to an additional ~224,000 new cardiomyocytes in rNRG1-treated animals in the early administration regimen. From this we calculated that the cardiomyocytes generated by stimulated proliferation of uninjured cardiomyocytes account for 198,000/224,000 = 89% and by proliferation of protected cardiomyocytes for 26,000/224,000 = 11%. We calculated the relative contributions of rNRG1-induced cardiomyocyte protection as 136,000/(334,000 + 26,000) = 38%, with the remaining 62% resulting from rNRG1-stimulated cardiomyocyte proliferation (Suppl. Results S7–S8). In addition to the increase in the number of cardiomyocytes, rNRG1 may activate other beneficial mechanisms, as suggested by the transient improvement of myocardial function after late administration. The broad range of genes regulated by rNRG1 administration (Fig. 5) is consistent with the function of the neuregulin gene in sustaining the cardiac gene regulatory network during development (19). The rNRG1-regulated genes may hold clues for a deeper understanding of the mechanism that can be activated with rNRG1.
We identified a therapeutic window in neonatal mice, and found that a similar window exists in human infants. In mice, this period ends within the first 5 days of life, and is similar to the transient period of scarless repair recently demonstrated by us (15) and others (13) after LAD ligation. In humans, this therapeutic window ends around 6 months of age (Fig. 7D), which is significantly shorter than would have been predicted from our results in humans without heart disease (10). Since our mechanistic model involves cardiomyocyte protection, rNRG1 could be administered in infants with congenital heart disease prior to surgical repair, which is feasible because most of these surgeries are scheduled electively.
It is intriguing to note that human cardiomyocyte cell cycle re-entry was not stimulated by rNRG1 above 6 months (Fig. 7D, 8G). We have previously demonstrated that rNRG1 stimulates cardiomyocyte division in young adult mice (22). Setting aside potential species-differences, how could this apparent discrepancy be reconciled? Our results in Figure 6C show premature cessation of cardiomyocyte cycling in the presence of heart disease, indicating that heart disease in infants, as was the case in all of our patients, drives proliferation-competent cardiomyocytes out of the cell cycle and into a permanently quiescent phenotype. In contrast, the mice that we studied in Bersell et al. 2009 (22) were young adult mice that were free from heart disease until 8 weeks of life. In other words, these mice were not exposed to signals driving cardiomyocytes prematurely into a permanently quiescent phenotype. As a result, cardiomyocytes with proliferative potential remained present into adulthood and could be stimulated by rNRG1.
Collectively, the present study shows that rNRG1-induced cardiomyocyte proliferation is a conserved mechanism in neonatal mice and myocardium from human infants and maybe a useful therapeutic strategy for treating pediatric patients with heart disease. rNRG1 therapy may have risks associated with it’s potential effects in other organs. We have demonstrated that long-term administration of rNRG1 in neonatal mice does not induce somatic, organ, or neoplastic growth [Note to editor and reviewers: “Please refer to companion paper by Ganapathy et al.”]. Although one cannot predict these outcomes in children, clinical studies investigating the safety and efficacy of rNRG1 as a cardiac therapeutic in adult heart failure patients provided no evidence of uncontrolled growth effects (26, 27). Furthermore, the systemic administration of other recombinant growth factors (i.e. insulin, granulocyte-macrophage colony-stimulating factor, erythropoetin, thrombopoetin, and insulin-like growth factor 1) that modify the activity of well-defined signaling pathways is a successful therapeutic paradigm and has a good track record for safety in pediatric patients (45). In summary, our results identify a therapeutic period for future clinical trials of rNRG1 in pediatric patients.
Materials and Methods
Cryoinjury
Mouse experiments were approved by Boston Children’s Hospital and Institute of Molecular Biotechnology. ICR mice were used. Pups born after 5 pm were considered to be P0 the following day and subjected to cryoablation the next day (P1). Pups received a subcutaneous injection with 0.1% bupivacain, were placed inside a sleeve and put in an ice-water bath until they were non-responsive to the paw reflex. Ventrolateral thoracotomy was performed between the 4–5th ribs, the pericardium removed, and the heart exposed. A 1.5 mm-diameter vanadium probe was equilibrated in liquid nitrogen for 20 min and applied to the LV epicardium for 2 sec. Sham injury consisted of opening the chest and removing the pericardium. The chest was closed with 6-0 Prolene sutures and the skin sealed with Webglue (Webster Veterinary) or 8-0 Prolene sutures. Pups were recovered under a heating lamp and placed on a warming blanket until they became responsive, rubbed with bedding, and returned to their mothers.
Visualization of injury by triphenyltetrazolium chloride (TTC)
Freshly-excised mouse hearts were washed in ice-cold 50 mM KCl in PBS, immediately placed in 1% TTC (wt/vol, in phosphate buffer pH 7.4) at 37 °C for 20 min, and then fixed in 10% phosphate-buffered formaldehyde (PBF) overnight. Photomicrographs were taken using a Nikon SMZ1000 stereomicroscope at 0.8×, 2×, or 3× magnification and an Olympus DP70 CCD camera.
Characterization of scar structure and function
We defined a transmural scar as the presence of transmural blue collagen on any of 6–12 AFOG-stained sections per heart. We measured the radial thickness of non-transmural scars and remote LV myocardium in diastole and systole on cMRI images using Image J and calculated relative systolic thickening as percent systolic change = (length in systole – length in diastole)/length in systole.
Cardiomyocyte-specific deletion of ErbB4
Experiments were performed in α-MHC-Mer-Cre-Mer+/+; ErbB4F/wt (control) and in α-MHC-Mer-Cre-Mer+/+; ErbB4F/F (test). ErbB4 inactivation was induced with tamoxifen on days P1-3 (30ug/g body weight sc, dissolved in peanut oil). rNRG1 or BSA was administered from day of birth (P0) for 12 days (P12).
Quantification of myocardial function in vivo by echocardiography and cMRI
We performed transthoracic echocardiography under anesthesia with a Vevo 2100 device (VisualSonics). cMRI was performed under isoflurane anesthesia with a 9.4 T small-animal MRI scanner (Bruker Biospin MRI). Details of imaging are provided in Supplemental Materials and Methods.
RNA-Seq sample preparation and data analysis
Total RNA was extracted using the RNeasy Micro kit (Qiagen). cDNA libraries were prepared using the TruSeq Stranded mRNA kit (Illumina). Sequencing was performed on NextSeq 500 instruments (single-strand, single-end indexed, 75 bp per read) at a depth of 35 × 106 per sample. Single reads were mapped to the mouse genome (m10) using STAR in a strand-specific manner. Cufflinks was used to determine FPKM levels for each gene from the STAR alignment and was used as input for Cuffdiff. All treatment groups were normalized to sham. Differential gene expression was calculated using Cuffdiff. Read counts were normalized between all samples. Genes with significant change of expression were defined by an adjusted P-value < 0.05 (Maverix Biomics). Functional clustering was performed with David online software. Supplemental Materials and Methods provide additional information.
Human myocardial samples
Discarded and de-identified human myocardial samples were collected from patients undergoing heart surgery (IRB protocol # Z06-10-0489). Clinical information was retrieved through an honest broker (IRB-P00000126). Normal human heart samples, including 1 fetal heart, were provided by the Sydney Heart Bank at the University of Sydney (Project No: 2012/2814). When known, patients with a 22q11 microdeletion were excluded. For detailed information, please refer to Supplemental Tables S2–S5.
Human cardiomyocyte dissociation
Myocardial tissue was washed and resuspended in cold isolation buffer (130 mM NaCl; 5 mM KCl; 1.2 mM KH2PO4; 6 mM HEPES; 5 mM NaHCO3; 1 mM MgCl2; 5 mM glucose) pH 7.4. Isolation buffer was supplemented with 0.36 mM CaCl2 for enzyme activity. Cardiac tissue was incubated for 15–20 mins in isolation buffer supplemented with collagenase IV (Sigma). After each incubation step, the supernatants were transferred to a tube and centrifuged at 600 rpm for 4 min. Cell pellets were resuspended in ice-cold isolation buffer. Several rounds of digestion were performed until tissue was fully digested.
Laser scanning cytometry
Myocardial cell preparations were prepared as described above and LSC analysis for nucleation and ploidy performed as previously described (10).
CFSE assay
Carboxyfluorescein succinimidyl ester (CFSE) is a fluorescein-derived intracellular fluorescent label, which is divided between daughter cells upon cell division. Human myocardial samples were washed twice in warm PBS, incubated in PBS/2% FBS containing 0.5 μM CFSE (Molecular Probes C1157), at 37°C for 30 minutes, and washed with 10-times volume of ice-cold PBS/2% FBS. Samples were washed once with warm DMEM medium and incubated in supplemented DMEM for 72 hours. Cardiomyocytes were then isolated as described in human cardiomyocyte dissociation section and analyzed on an ARIAII (BD Bioscience) in the Fl-1 channel. Viable populations were distinguished by 7-aminoactinomycin D (7-AAD). For lineage re-analysis of the CFSElo population, cells were gated according to CFSE-loaded but rNRG-1 un-stimulated controls and 10,000 cells bulk sorted into PBS/5% FCS from two patient samples. Cells were fixed in 4% PFA for 15 mins, washed, and stained with either primary antibodies to cardiac troponin-T (Neomarkers) or isotype control (IgG1), conjugated to FITC fluorophore. Cells were analyzed on a FACSAriaII.
Flow cytometry and FACS
Dissociated cardiomyocytes were prepared as previously described (10), washed, and pelleted for antibody staining. For cell-cycle analysis sarcomeric α-actinin (Sigma) antibody was conjugated to a FITC secondary antibody and phosphorylated histone H3 (Upstate) antibody was conjugated to a Pacific Blue secondary antibody (both Invitrogen, Molecular Probes monoclonal antibody conjugation kits). Cells were fixed in 4% paraformaldehyde and washed twice with PBS/5% FCS. Cells were stained in PBS/5% FCS and antibody cocktail for 1 hour at 4°C, washed twice and resuspended in PBS/5% FCS. Samples were acquired on a FACSAria Cell Sorter (BD Biosciences) and data analyzed with Flowjo software.
Quantitative RT-PCR
Cardiomyocytes were isolated and sorted with a FACSAriaII (100 μm) nozzle into Trizol buffer and frozen at −80°C. RNA extraction was performed with an RNeasy Micro kit (Qiagen) according to the manufacturer’s instructions, including on-column DNase I digestion. cDNA was synthesized from 200 ng total RNA. Eluted RNA samples were reverse transcribed using SuperScript II and random hexamers (Invitrogen). PCR was performed using iQ5 Real-time PCR thermal cycler or Bio-Rad CFX384 Touch thermal cycler and iQ SYBER Green Supermix (Bio-Rad) or iTaq Universal SYBR Green Supermix. The normalized values of each biological replicate were averaged before the calculation of fold change in expression levels. The primer sequences are provided in the Supplemental Table S10–S11.
Statistical analyses
Numerical results are presented as means ± SEM. Continuous outcomes were compared with analysis of variance (ANOVA) followed by Bonferroni post-hoc testing. Statistical significance was achieved with a two-sided P value ≤ 0.05. Statistical analyses were performed with GraphPad Prism, version 6.
Supplementary Material
One Sentence Summary.
Administration of the recombinant growth factor neuregulin during a therapeutic period stimulates heart muscle repair.
Acknowledgments
We thank H. Sadek and M. Ahmad (UTSW, Dallas) for training in mouse surgery, M. Farley and D. Burstein (BIDMC, Boston) for help with cardiac MRI, and members of the Kuhn lab for helpful discussions. We are grateful to the cardiac surgeons at Boston Children’s Hospital for providing de-identified human myocardium (Drs. Frank Pigula, Francis Fynn-Thompson, Sitaram Emani, John Mayer, Christopher Baird, and Pedro Del Nido). Clinical information was retrieved by Ellen McCusty (Boston Children’s Hospital). We are grateful for the assistance provided by the staff at the laser scanning cytometer (LSC) core at the department of transfusion medicine, including Leslie Silberstein and Shin-Young Park, and for FACS assistance by Noreen Francis and Dick Bennett (Boston Children’s Hospital). We are grateful for the assistance by William Horne and Annabel Ferguson at the Rangos Genomics Facility at Children’s Hospital of Pittsburgh.
Funding: This research was supported by the Department of Cardiology, the Translational Research Program, and the Office of Faculty Development (all Boston Children’s Hospital), and the NIH (R01HL106302, K08HL085143, T32HL007572 and a Shared Instrumentation Grant RR028792 to BIDMC Boston). BG, NA, and BK were supported by the Richard King Mellon Institute for Pediatric Research (Children’s Hospital of Pittsburgh). JMP and BJH were funded by a Fondation Leducq Transatlantic Network of Excellence Award.
Footnotes
Author contributions: BDP, BG, SW, and BK designed experiments. BDP, BG, SW, SC, NA, DB, and BH performed and analyzed experiments. BDP, BG, SW, and BK wrote and all authors edited the manuscript.
Competing interests: The authors declare no conflicting financial interests.
References
- 1.Tennant PW, Pearce MS, Bythell M, Rankin J. 20-year survival of children born with congenital anomalies: a population-based study. Lancet. 2010;375:649. doi: 10.1016/S0140-6736(09)61922-X. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 3.Bolger AP, Coats AJ, Gatzoulis MA. Congenital heart disease: the original heart failure syndrome. Eur Heart J. 2003;24:970. doi: 10.1016/s0195-668x(03)00005-8. [DOI] [PubMed] [Google Scholar]
- 4.Burns KM, Byrne BJ, Gelb BD, Kuhn B, Leinwand LA, Mital S, Pearson GD, Rodefeld M, Rossano JW, Stauffer BL, Taylor MD, Towbin JA, Redington AN. New mechanistic and therapeutic targets for pediatric heart failure: report from a National Heart, Lung, and Blood Institute Working Group. Circulation. 2014;130:79. doi: 10.1161/CIRCULATIONAHA.113.007980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaddy RE, Boucek MM, Hsu DT, Boucek RJ, Canter CE, Mahony L, Ross RD, Pahl E, Blume ED, Dodd DA, Rosenthal DN, Burr J, LaSalle B, Holubkov R, Lukas MA, Tani LY. Carvedilol for children and adolescents with heart failure: a randomized controlled trial. JAMA: the journal of the American Medical Association. 2007;298:1171. doi: 10.1001/jama.298.10.1171. [DOI] [PubMed] [Google Scholar]
- 6.Hsu DT, Zak V, Mahony L, Sleeper LA, Atz AM, Levine JC, Barker PC, Ravishankar C, McCrindle BW, Williams RV, Altmann K, Ghanayem NS, Margossian R, Chung WK, Border WL, Pearson GD, Stylianou MP, Mital S. Enalapril in infants with single ventricle: results of a multicenter randomized trial. Circulation. 2010;122:333. doi: 10.1161/CIRCULATIONAHA.109.927988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garbern JC, Lee RT. Cardiac stem cell therapy and the promise of heart regeneration. Cell stem cell. 2013;12:689. doi: 10.1016/j.stem.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin Z, Pu WT. Strategies for cardiac regeneration and repair. Sci Transl Med. 2014;6:239rv1. doi: 10.1126/scitranslmed.3006681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, Silberstein LE, Remedios CG Dos, Graham D, Colan S, Kuhn B. Cardiomyocyte proliferation contributes to heart growth in young humans. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1446. doi: 10.1073/pnas.1214608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:H2183. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- 12.Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996;28:1737. doi: 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- 13.Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:187. doi: 10.1073/pnas.1208863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haubner BJ, Adamowicz-Brice M, Khadayate S, Tiefenthaler V, Metzler B, Aitman T, Penninger JM. Complete cardiac regeneration in a mouse model of myocardial infarction. Aging (Albany NY) 2012;4:966. doi: 10.18632/aging.100526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walsh S, Ponten A, Fleischmann BK, Jovinge S. Cardiomyocyte cell cycle control and growth estimation in vivo–an analysis based on cardiomyocyte nuclei. Cardiovasc Res. 2010;86:365. doi: 10.1093/cvr/cvq005. [DOI] [PubMed] [Google Scholar]
- 17.Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J, Kesteven SH, Holman SR, Matsuda T, Lovelock JD, Howard WW, Iismaa SE, Chan AY, Crawford BH, Wagner MB, Martin DI, Lefer DJ, Graham RM, Husain A. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell. 2014;157:795. doi: 10.1016/j.cell.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- 19.Lai D, Forrai A, Liu X, Wolstein O, Michalicek J, Ahmed I, Garratt AN, Birchmeier C, Zhou M, Hartley L, Robb L, Feneley MP, Fatkin D, Harvey RP. Neuregulin 1 Sustains the Gene Regulatory Network in Both Trabecular and Nontrabecular Myocardium. Circ Res. 2010;107:715. doi: 10.1161/CIRCRESAHA.110.218693. [DOI] [PubMed] [Google Scholar]
- 20.Hedhli N, Huang Q, Kalinowski A, Palmeri M, Hu X, Russell RR, Russell KS. Endothelium-derived neuregulin protects the heart against ischemic injury. Circulation. 2011;123:2254. doi: 10.1161/CIRCULATIONAHA.110.991125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Gu X, Li Z, Li X, Li H, Chang J, Chen P, Jin J, Xi B, Chen D, Lai D, Graham RM, Zhou M. Neuregulin-1/erbB-activation improves cardiac function and survival in models of ischemic, dilated, and viral cardiomyopathy. J Am Coll Cardiol. 2006;48:1438. doi: 10.1016/j.jacc.2006.05.057. [DOI] [PubMed] [Google Scholar]
- 22.Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 23.Jay SM, Murthy AC, Hawkins JF, Wortzel JR, Steinhauser ML, Alvarez LM, Gannon J, Macrae CA, Griffith LG, Lee RT. An engineered bivalent neuregulin protects against doxorubicin-induced cardiotoxicity with reduced proneoplastic potential. Circulation. 2013;128:152. doi: 10.1161/CIRCULATIONAHA.113.002203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wadugu B, Kuhn B. The role of neuregulin/ErbB2/ErbB4 signaling in the heart with special focus on effects on cardiomyocyte proliferation. Am J Physiol Heart Circ Physiol. 2012;302:H2139. doi: 10.1152/ajpheart.00063.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parodi EM, Kuhn B. Signalling between microvascular endothelium and cardiomyocytes through neuregulin. Cardiovascular research. 2014;102:194. doi: 10.1093/cvr/cvu021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao R, Zhang J, Cheng L, Wu X, Dong W, Yang X, Li T, Liu X, Xu Y, Li X, Zhou M. A phase II, randomized, double-blind, multicenter, based on standart therapy, placebo-controlled study of th efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J Am Coll Cardiol. 2010;55:1907. doi: 10.1016/j.jacc.2009.12.044. [DOI] [PubMed] [Google Scholar]
- 27.Jabbour A, Hayward CS, Keogh AM, Kotlyar E, McCrohon JA, England JF, Amor R, Liu X, Li XY, Zhou MD, Graham RM, Macdonald PS. Parenteral administration of recombinant human neuregulin-1 to patients with stable chronic heart failure produces favourable acute and chronic haemodynamic responses. Eur J Heart Fail. 2010 doi: 10.1093/eurjhf/hfq152. [DOI] [PubMed] [Google Scholar]
- 28.Zhao YY, Sawyer DR, Baliga RR, Opel DJ, Han X, Marchionni MA, Kelly RA. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J Biol Chem. 1998;273:10261. doi: 10.1074/jbc.273.17.10261. [DOI] [PubMed] [Google Scholar]
- 29.Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen JE, Purcell BP, MacArthur JW, Jr, Mu A, Shudo Y, Patel JB, Brusalis CM, Trubelja A, Fairman AS, Edwards BB, Davis MS, Hung G, Hiesinger W, Atluri P, Margulies KB, Burdick JA, Woo YJ. A Bioengineered Hydrogel System Enables Targeted and Sustained Intramyocardial Delivery of Neuregulin, Activating the Cardiomyocyte Cell Cycle and Enhancing Ventricular Function in a Murine Model of Ischemic Cardiomyopathy. Circ Heart Fail. 2014 doi: 10.1161/CIRCHEARTFAILURE.113.001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Rosa JM, Martin V, Peralta M, Torres M, Mercader N. Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Development. 2011;138:1663. doi: 10.1242/dev.060897. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez-Rosa JM, Mercader N. Cryoinjury as a myocardial infarction model for the study of cardiac regeneration in the zebrafish. NatProtoc. 2012;7:782. doi: 10.1038/nprot.2012.025. [DOI] [PubMed] [Google Scholar]
- 33.Chablais F, Veit J, Rainer G, Jazwinska A. The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Dev Biol. 2011;11:21. doi: 10.1186/1471-213X-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schnabel K, Wu CC, Kurth T, Weidinger G. Regeneration of cryoinjury induced necrotic heart lesions in zebrafish is associated with epicardial activation and cardiomyocyte proliferation. PLoS One. 2011;6:e18503. doi: 10.1371/journal.pone.0018503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jesty SA, Steffey MA, Lee FK, Breitbach M, Hesse M, Reining S, Lee JC, Doran RM, Nikitin AY, Fleischmann BK, Kotlikoff MI. c-kit+ precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:13380. doi: 10.1073/pnas.1208114109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bose AK, Mathewson JW, Anderson BE, Andrews AM, Martin Gerdes A, Benjamin Perryman M, Grossfeld PD. Initial experience with high frequency ultrasound for the newborn C57BL mouse. Echocardiography. 2007;24:412. doi: 10.1111/j.1540-8175.2007.00402.x. [DOI] [PubMed] [Google Scholar]
- 37.Bersell K, Choudhury S, Mollova M, Polizzotti BD, Ganapathy B, Walsh S, Wadugu B, Arab S, Kuhn B. Moderate and high amounts of tamoxifen in alpha-MHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Dis Model Mech. 2013 doi: 10.1242/dmm.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 39.Brandenburger M, Wenzel J, Bogdan R, Richardt D, Nguemo F, Reppel M, Hescheler J, Terlau H, Dendorfer A. Organotypic slice culture from human adult ventricular myocardium. Cardiovascular research. 2012;93:50. doi: 10.1093/cvr/cvr259. [DOI] [PubMed] [Google Scholar]
- 40.Engel FB, Hauck L, Boehm M, Nabel EG, Dietz R, von Harsdorf R. p21(CIP1) Controls proliferating cell nuclear antigen level in adult cardiomyocytes. Mol Cell Biol. 2003;23:555. doi: 10.1128/MCB.23.2.555-565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zaruba MM, Soonpaa M, Reuter S, Field LJ. Cardiomyogenic potential of C-kit(+)-expressing cells derived from neonatal and adult mouse hearts. Circulation. 2010;121:1992. doi: 10.1161/CIRCULATIONAHA.109.909093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahmoud AI, Porrello ER, Kimura W, Olson EN, Sadek HA. Surgical models for cardiac regeneration in neonatal mice. Nat Protoc. 2014;9:305. doi: 10.1038/nprot.2014.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andersen DC, Ganesalingam S, Jensen CH, Sheikh SP. Do Neonatal Mouse Hearts Regenerate following Heart Apex Resection? Stem Cell Reports. 2014;2:406. doi: 10.1016/j.stemcr.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ang KL, Shenje LT, Reuter S, Soonpaa MH, Rubart M, Field LJ, Galinanes M. Limitations of conventional approaches to identify myocyte nuclei in histologic sections of the heart. Am J Physiol Cell Physiol. 2010;298:C1603. doi: 10.1152/ajpcell.00435.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khwaja OS, Ho E, Barnes KV, O’Leary HM, Pereira LM, Finkelstein Y, Nelson CA, 3rd, Vogel-Farley V, DeGregorio G, Holm IA, Khatwa U, Kapur K, Alexander ME, Finnegan DM, Cantwell NG, Walco AC, Rappaport L, Gregas M, Fichorova RN, Shannon MW, Sur M, Kaufmann WE. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:4596. doi: 10.1073/pnas.1311141111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frahm J, Haase A, Matthaei D. Rapid NMR imaging of dynamic processes using the FLASH technique. Magn Reson Med. 1986;3:321. doi: 10.1002/mrm.1910030217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.